
Photoinduced electron transfer from the oxo–MoIVselenolato complex to oxygen†
Joyee
Mitra‡
*a and
Sabyasachi
Sarkar
*b
aDepartment of Chemistry, Indian Institute of Technology Kanpur, Kanpur 208 016, Uttar Pradesh, India
bDepartment of Chemistry, Indian Institute of Engineering Science & Technology Shibpur (IIEST), Howrah 711 103, West Bengal, India. E-mail: abya@iitk.ac.in
First published on 12th November 2015
Abstract
[NBu4][MoO(bpy)(mnt)(SePh)]·CH2Cl2 (1) and [NBu4][MoO(phen)(mnt)(SePh)]·CH2Cl2 (2) complexes; (bpy = 2,2′-bipyridine, phen = 1,10-phenanthroline, mnt = maleonitriledithiolate ion) stabilized by aromatic diimine have been synthesized and characterized. These complexes under aerobic conditions on irradiation with sunlight (or with light from a tungsten lamp) in solution generate superoxide radicals concomitant to the formation of paramagnetic Mo(V) species due to a single electron transfer from the Mo(IV) complex to oxygen. The Mo(V) produced under photo-irradiation was characterized by EPR spectroscopy and the generated superoxide radical has been shown to transform nitrobluetetrazoleum (NBT) chloride to blue diformazan. Both 1 (and 2) are phosphorescent having an emission lifetime of 6–8 μs. Ground state and time dependent density functional theoretical calculations confirm that the electronic transitions from predominantly metal based molecular orbitals are responsible for the low-lying charge transfer excited states and also for the observed interaction with oxygen.
Introduction
Metallo-dithiolenes have gained importance due to their potential utility as Q-switch laser dyes, as light conversion materials, in non-linear optics and as catalysts.1 Transition metal complexes with aromatic diimine ligands like 2,2′-bipyridine, 1,10-phenanthroline and their derivatives are known to behave as photoactive energy or electron transfer agents depending on their ancillary ligands.2 The ligand combination of diimine as the pull ligand and dithiolene as the push ligand has been studied in square planar Ni, Pd and Pt complexes3 but such combination with other metals having octahedral geometry is rare.Many physiological processes involve stepwise electron transfer to activate dioxygen, producing free radical intermediates.4 The limiting step in such oxidation processes is the first electron transfer forming superoxide radicals. Synthetic models emulating such processes usually involve redox-active transition metal complexes for reductive activation of dioxygen to superoxo, peroxo or oxo species associated with an increase in the formal oxidation state of the metal. Monoxo molybdenum complexes are of some interest as they have been reported to exhibit photo-redox reactions.5,6
In this article, we report the chemistry of two octahedral oxo–MoIV(diimine)(dithiolene)(selenolato) complexes bearing the push–pull ligand combination. The properties of the synthesized complexes are dominated by both ligands, one of which is easily oxidized and the other easily reduced.3 We observed light induced electron transfer by these newly synthesized complexes, which have been studied by EPR, electrochemistry and by using density functional theoretical (DFT) and time dependent DFT calculations.7
Experimental
All reactions and manipulations were performed under argon using the modified Schlenk technique. Solvents were dried and distilled according to standard procedures. [Na2(mnt)]8 and [NBu4]2[Mo2O4(mnt)2]9 were prepared following reported procedures.Elemental analyses for carbon, hydrogen and nitrogen were carried out using a Perkin-Elmer 2400 micro analyzer. FTIR spectra were recorded on a Bruker Vertex70 FT-IR spectrometer as pressed KBr pellets. Electronic spectra were recorded on a Perkin-Elmer Lambda35 UV-visible spectrophotometer. Mass spectra (negative ion) were recorded on a Micromass Quattro triple quadrupole mass spectrometer with an analytical electrospray source. The ESI capillary was set at 3.5 kV and the cone voltage was 20 V. Electron paramagnetic resonance spectra were recorded on a Bruker Biospin EMX EPR X-band spectrometer with a frequency of 9.856 GHz. Samples for the chemically oxidized species of 1 (and 2) were prepared using a previously reported method.10 A small amount of I2 (less than the stoichiometric amount for one electron oxidation) was placed with 1 (and 2) in the EPR tube under an inert atmosphere. Few drops of dry, degassed DCM were allowed to move down the inner wall of the tube and as soon as the solid mixture of the complex and iodine was dissolved, the tube was cooled in liq. N2 and placed in the pre-cooled Dewar attached to the EPR machine to record the spectra at 120 K. The sample was thawed in argon to record the room temperature EPR data. For experiments with oxygen, the thawed EPR sample was exposed to air briefly before refreezing. Spin quantitation was carried out using 1,3-bisdiphenylene-2-phenylallyl (BDPA) free radical as the standard. Cyclic voltammetric measurements of 10−3 M DCM solution of the compounds were recorded with a glassy carbon electrode as the working electrode with 0.2 M [Bu4N][PF6] as the supporting electrolyte, Ag/AgCl electrode as the reference electrode, and a platinum auxiliary electrode using BASi Epsilon, EC Bioanalytical Systems, Inc. All electrochemical experiments were carried out under argon at 298 K. Potentials are referenced against ferrocene/ferrocenium (Fc/Fc+) and are reported relative to the Ag/AgCl electrode (E1/2(Fc+/Fc)) 0.459 V vs. Ag/AgCl electrode.
X-Ray crystallography
Diffraction-quality single crystals were glued to glass fibers and mounted on a BRUKER SMART APEX diffractometer equipped with a CCD area detector. Data were collected using graphite-monochromated Mo Kα radiation (λ = 0.71069 Å) at low temperature (100 K). Cell constants were obtained from the least-squares refinement of three dimensional centroids through the use of CCD recording of narrow ω rotation frames, completing almost all-reciprocal space in the stated θ range. All data were collected with SMART 5.628 (BRUKER 2003), and were integrated with the BRUKER SAINT program.11 Empirical absorption correction was applied using the SADABS program. The structure was solved using SIR9712 and refined using SHELXL-97.13 Crystal structures were viewed using ORTEP.14 The respective space group of the compounds was determined on the basis of the lack of systematic absence and intensity statistics. Full-matrix least squares/difference Fourier cycles were performed which located the remaining non-hydrogen atoms. All non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were added at idealized positions with a uniform Uiso value. 2 possess one molecule in its asymmetric unit. In the asymmetric unit of 1, there are two independent monomeric units of which one is structurally discussed with its ORTEP plot. Additional symmetry for 1 has been checked through PLATON, which did not suggest the presence of any additional symmetry in 1. A c/2 pseudo-symmetry was found in 1. Both 1 and 2 contain a DCM molecule in their crystal lattice.Computational details
Density-functional theory calculations were performed using the Gaussian 03 (Revision B.04) package.15 Becke's three-parameter hybrid-exchange functional, the nonlocal correlation provided by the Lee, Yang, and Parr expression, and the Vosko, Wilk and Nuair 1980 local correlation functional (III) (B3LYP)16 were employed. The 6-31G*+ basis set was used for C, N, Se, S, O and H. For another set of calculations, the BP8617 functional was used instead of B3LYP. The LANL2DZ basis set18 and the LANL2 pseudopotential of Hay and Wadt were used for Mo.19 Klamt's conductor reaction field (COSMO)20 solvent model was used for calculations with DCM solvent.Synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O 924; elemental analysis found (calculated) in % for C37H51Cl2MoN5OS2Se; C 50.02 (49.83); H 5.93 (5.76); N 7.56 (7.85) m/z = 612.37, anionic mass corresponding to C20H13MoN4O4S2Se−.§
O 927; elemental analysis found (calculated) in % for C39H51Cl2MoN5OS2Se; C 50.48 (51.15); H 5.73 (5.61); N 7.32 (7.65) m/z = 636.40, anionic mass corresponding to C22H13MoN4O4S2Se−.§
O 924; elemental analysis found (calculated) in % for C37H51Cl2MoN5OS2Se; C 50.02 (49.83); H 5.93 (5.76); N 7.56 (7.85) m/z = 612.37, anionic mass corresponding to C20H13MoN4O4S2Se−.§
O 927; elemental analysis found (calculated) in % for C39H51Cl2MoN5OS2Se; C 50.48 (51.15); H 5.73 (5.61); N 7.32 (7.65) m/z = 636.40, anionic mass corresponding to C22H13MoN4O4S2Se−.§
Results and discussion
Structure description
The {MoV2O4} core in dioxo-bridged complexes can easily be protonated via the formation of intermediate monooxo-bridged species {MoV2O3}9 (Scheme 1) before complete cleavage of the bridged structure. This monooxo-bridged species is reverted back to the starting {MoV2O4} core under favorable conditions via a monomeric intermediate {OMoV(OH)} moiety.9,21 In the present synthesis selenol serves multiple roles: a proton source, a reducing agent and also as a coordinating ligand in the reaction with the {MoV2O4} core to yield monomeric oxo–MoIV(selenolato) species in the presence of aromatic diimines as chelating ligands. These selenolato complexes are synthesized faster compared to the iso-structural thiolato complexes,10 owing to the rapid protonation of the {MoV2O4} core by more acidic selenol (pKa of selenophenol is 4.6 and that of thiophenol is 6.0).22 The monooxo-bridged dimeric Mo(V) intermediate once formed responds to its reduction with excess of selenol yielding the stable monomeric Mo(IV) species coordinated to selenolato and to diimine as the co-ligand (Scheme 1). Such complexes are found to react with molecular oxygen under visible light illumination. The use of a robust chelating ligand like aromatic diimine is required to block the 5th and 6th coordination sites around molybdenum thereby negating the possibility of μ-oxo dimer formation, which is thermodynamically more stable compared to the monomeric pentavalent species.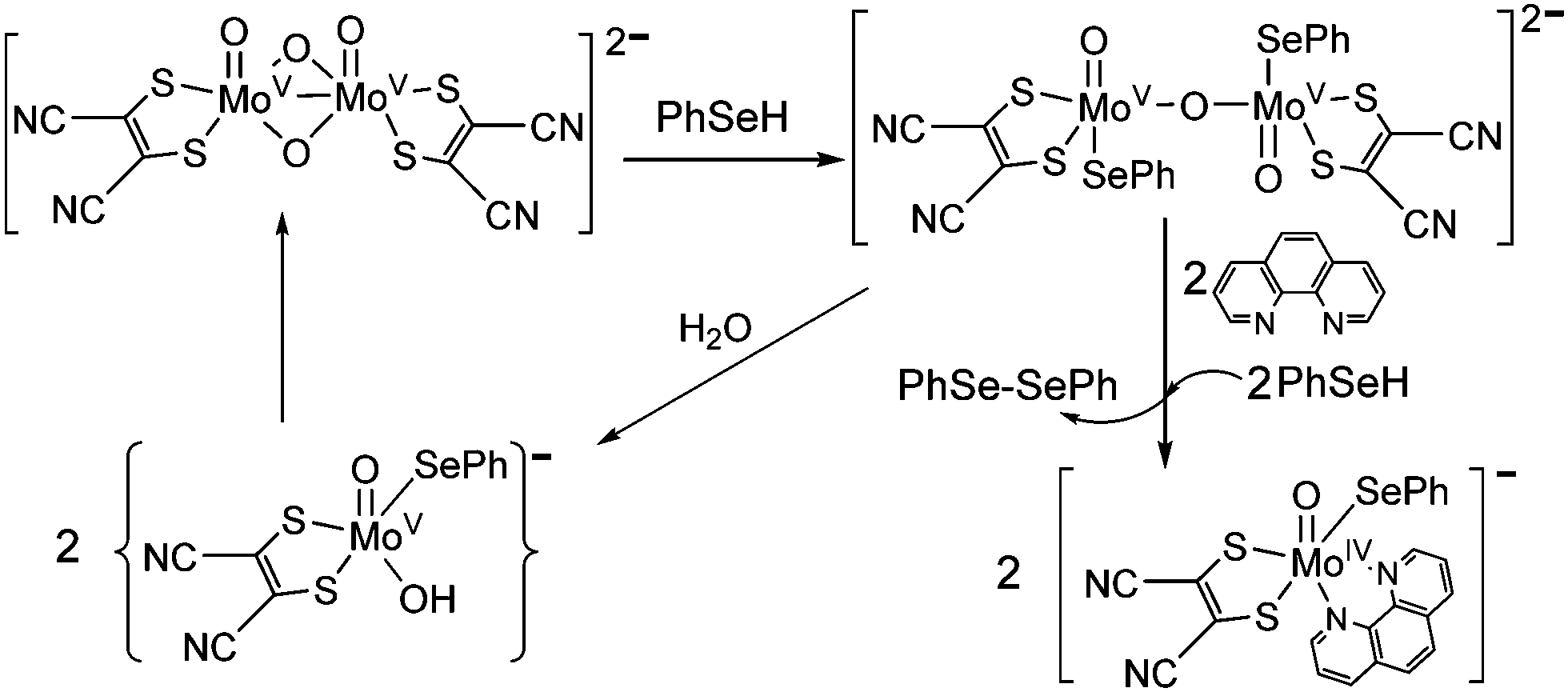 | ||
| Scheme 1 Protonation of the {MoV2O4} core and subsequent reduction and formation of 2 in the presence of PhSeH and 1,10-phenanthroline. | ||
Fig. 1a and b show the crystal structures of the anions of 1·CH2Cl2 and 2·CH2Cl2 respectively. The molybdenum center is coordinated to a terminal oxo, two sulfur atoms from the dithiolene ligand, two nitrogen atoms from the diimine and a selenolato moiety in each case. As expected the Mo atom is raised above the basal plane comprising two sulfur, one selenium and one nitrogen atom by ∼0.34 Å for both 1 and 2 adopting a distorted octahedral structure. The Mo–Se bond distance of 2.558(1) Å is slightly longer compared to other reported hexacoordinated complexes23 indicating a single bond character between Mo and Se atoms. The MoO distance of 1.693(4) Å is similar to other reported MoIV–oxo species.10,23 The two diimine nitrogen atoms are coordinated to the molybdenum center at a distance of 2.231(3) and 2.310(5) Å respectively. The Mo–N bond trans to oxo is longer by ∼0.08 Å due to the strong trans effect of the molybdenyl group. OMo–N4 and OMo–N3 angles are 86.2(2)° (86.1(2)°) and 155.8(2)° (156.4(2)°) respectively for 1 (and 2). The observed OMo–Se angles are 101.62(15)° and 102.66(14)° for 1 and 2 respectively. C–S distances of 1.746(6) and 1.737(7) Å and the average CC bond distance of 1.383(9) Å are in good agreement with reported values.23 Diimine bite 70.00(18)° (70.55(18)° for 2) and dithiolene bite 84.37(6)° and (84.24(5)°) are observed for 1 (and 2) respectively. The pertinent bond distances and angles are provided in Table S2 of the ESI.†
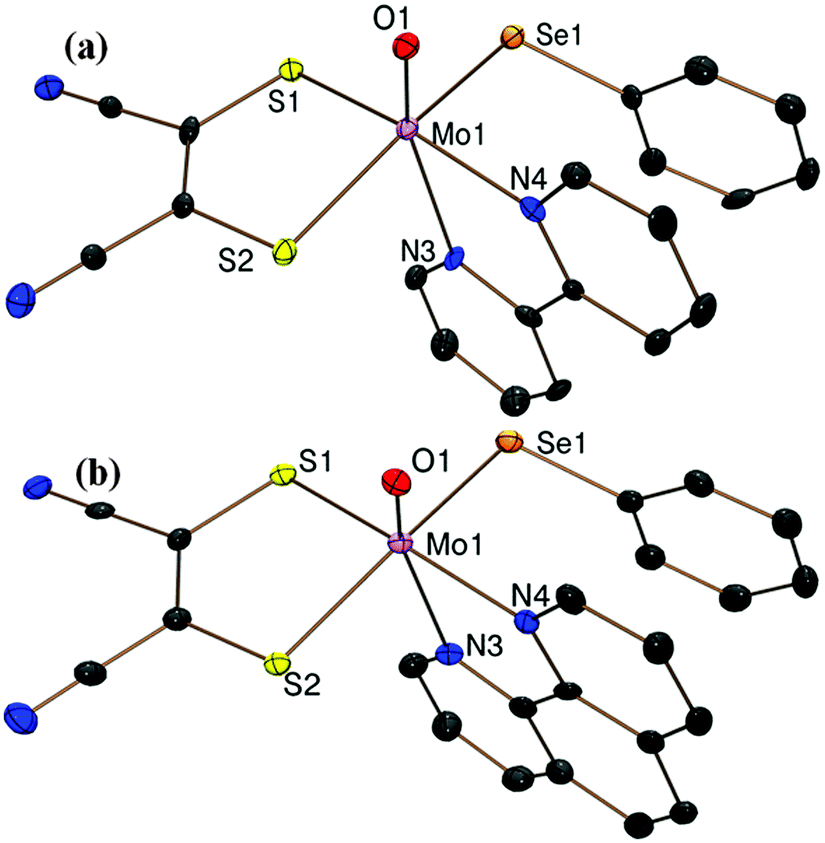 | ||
| Fig. 1 ORTEP plots of the crystal structures of the anions of (a) 1 and (b) 2 at 50% probability of thermal ellipsoids with a partial atom labeling scheme. | ||
Spectroscopic behavior
A low energy solvatochromic band appeared around 600 nm and another more intense band around 390 nm for both 1 and 2 (Fig. 2). A substantial shift to longer wavelength (24–28 nm) is observed in acetone compared to DCM indicating the charge transfer character of the electronic transition (Fig. S1, ESI†). It is known that due to a transient charge separation in the excited state, metal complexes with diimine ligands exhibit a light induced charge transfer transition.24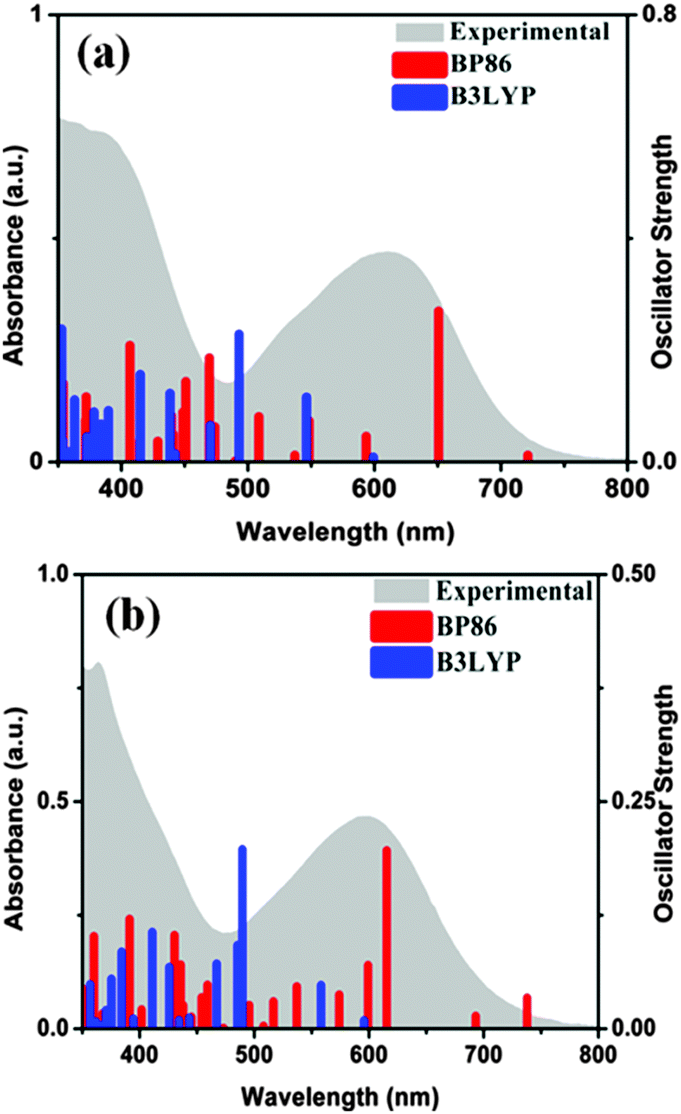 | ||
| Fig. 2 Electronic spectra of 10−4 M solutions of (a) 1 and (b) 2 in DCM (grey shade) and the TDDFT calculated electronic transitions. Red and blue bars are related to the use of BP86 and B3LYP functionals respectively in DCM as the solvent. | ||
TDDFT calculations were carried out to identify the main features of the absorption spectra.3,25 Both B3LYP and BP86 functionals were employed in calculations incorporating DCM as solvent. The electronic spectra are characterized by a high density of states between 350 and 500 nm assigned mainly to intra-ligand transitions involving n → π* and π → π* transitions of both diimine and dithiolene. The excited states between 550 and 700 nm have considerable metal contribution and can be assigned as MLCT transitions (HOMO → LUMO (LUMO+1)) (Fig. S2, S3 and Tables S3, S4, ESI†). Time dependent density functional theoretical calculations (TDDFT) support the involvement of metal d-orbital in the transition to diimine excited state. Such an assignment is common in M(diimine)(dithiolene) complexes.3,25 Our experimental data are more in agreement with the BP86 functional derived results for the MLCT transitions. Calculated MLCT transitions with the B3LYP function are at much lower wavelength compared to experimental data. Similar observations of failure of the B3LYP functional have been reported elsewhere.26 Deviations from the experimental results are often reported for TDDFT, as the calculations are dependent on basis sets, exchange–correlation functional, solvent models, etc.27 The MLCT state resulted in chemical change that may be best explained by considering an intermediate resembling an anion radical (oxidized metal and reduced ligand).28
Photo-physical properties of {MoO} depend on their oxidation state; pentavalent complexes are fluorescent (lifetime ∼110 ns for OMoCl4(CH3CN)−)5 while tetravalent complexes sometimes show phosphorescence.6
On excitation of degassed DCM solutions of 1 (and 2) at 390 nm, a strong emission is observed around ∼470 nm with a less intense peak at ∼650 nm (Fig. 3). The oxo–MoIV complexes are known to be phosphorescent in solution having a lifetime of ∼2.5 μs (±10%) for [MoOCl(CN-tBu)4][BPh4].6 The enhancement of the lifetime (6.21 μs and 8.72 μs for 1 and 2 respectively) in comparison to the literature value may be due to the presence of chromophoric diimine in these complexes. The lifetime values are consistent with emission from the triplet state. The singlet excited states of 1 (and 2) are populated on photoexcitation, followed by a fast inter-system crossing (ISC) resulting in the formation of MLCT excited state with a dominant triplet character. This state is responsible for the observed phosphorescence. Close lying π–π* and MLCT states along with the presence of a heavy metal result in an increased spin–orbit coupling and thus the efficiency of the radiative deactivation pathway increases.29 Quenching of emission is observed in the presence of dioxygen similar to other phosphorescent complexes. These results are supported by TDDFT calculations (Table S5, ESI†).
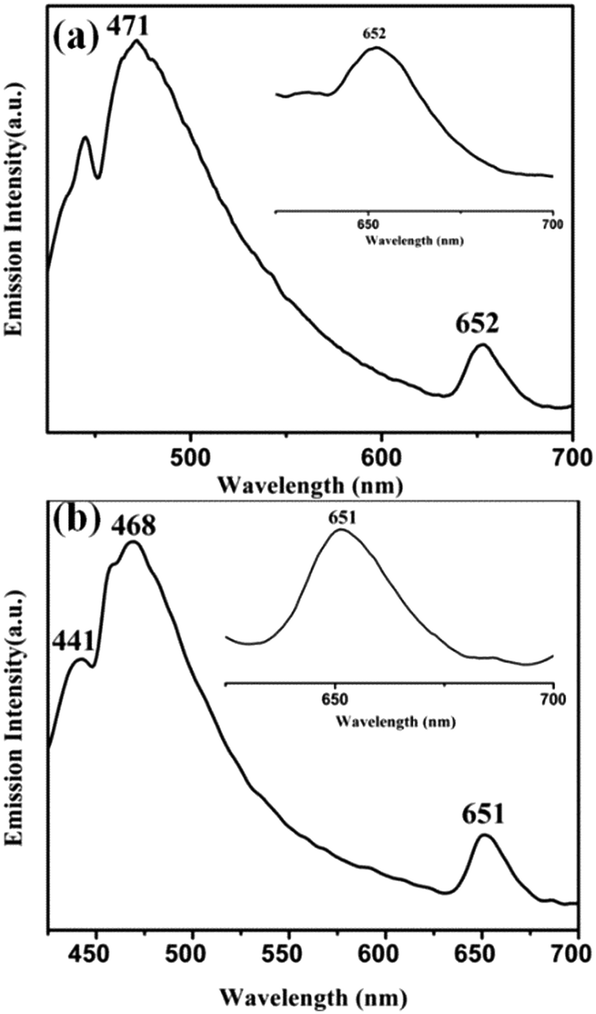 | ||
| Fig. 3 Phosphorescent emission spectra of (a) 1 and (b) 2 in DCM (λex, 390 nm). The inset shows the emission at ∼650 nm. | ||
EPR analysis
EPR silent Mo(IV) complexes 1 (and 2) were chemically oxidized by one electron under an inert atmosphere at room temperature (and at 120 K) to record the X-band EPR spectrum. The characteristic Mo(V) EPR spectrum was observed with six hyperfine lines and g = 1.992 (Fig. 4a). The EPR spectrum of 1 (and 2) in DCM at 120 K is axial with g∥ = 2.028 and g⊥=1.992 (Fig. 4b). The high g value of 1 (and 2) can be attributed to the Mo–Se coordination, resulting in enhanced delocalization of the unpaired electron over selenium p-orbitals. Hyperfine interaction of the Mo based unpaired electron with the Se nucleus is considerably smaller than the line width of the spectrum, and thus no splitting is observed due to 77Se (I = 1/2, 7.63% natural abundance, possessing low magnetic moment 0.53506, and its contribution to electron density in SOMO is very small (Fig. 7)) in the EPR spectrum. The spectrum does not exhibit super-hyperfine coupling to the N (I = 1) center in support of the absence of unpaired electron density on the diimine ligands in HOMO (SOMO). Spin quantitation of the Mo(V) signal using BDPA radical as the integration standard gave 0.75 ± 0.05 spins per molecule for both 1 and 2 (i.e. approximately 1 spin per monomeric molybdenum species).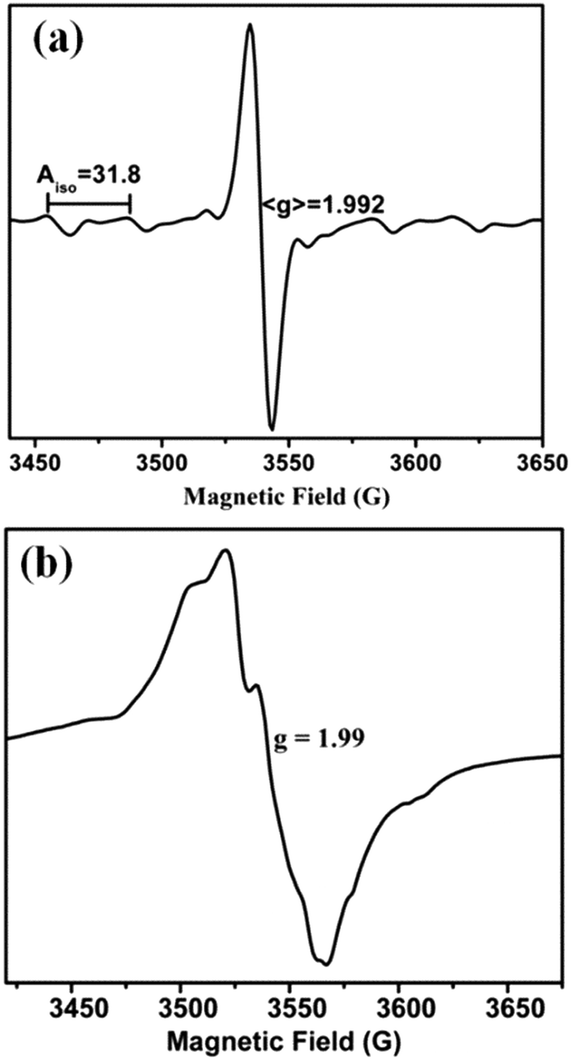 | ||
| Fig. 4 X-band EPR spectra of chemically oxidized 1 in DCM solution (a) at room temperature and (b) at 120 K. | ||
Interestingly, a similar EPR signal was observed on irradiating a solution of 1 (and 2) in air using a 100 W tungsten lamp (Fig. 5a). Around 20 min of irradiation resulted in the appearance of an EPR signal with g = 1.992 from the initial EPR silent solution. However, this signal was found to disappear on prolonged exposure to light. Suitable color filters were employed to identify the wavelength range most suitable for photo-excitation. Such filters accompanied with cooling of the light exposed solution took care of the heat generated by the tungsten lamp. We observed that the complexes responded sluggishly with 0.1 M K2Cr2O7 solution (yellow-orange filter) to generate EPR signals compared to 0.1 M CuSO4 solution (blue) or 0.1 M CoCl2 solution (pink-red filter). The EPR spectrum observed in all three cases consisted of one intense g = 1.992 signal at room temperature. 1 (or 2) has strong absorbance in the visible region. Consequently, their photo-induced electron transfer could be initiated by selective excitation with low energy visible light. Irradiation of deoxygenated solutions of 1 (or 2) does not show any EPR activity supporting the involvement of dissolved oxygen in generating the observed Mo(V) EPR signal. Also, solution of 1 (or 2) on standing in the dark did not show any EPR signal when exposed to air. It should be mentioned that the isostructural thiolato complexes10 do not show any photo-activation under similar conditions.
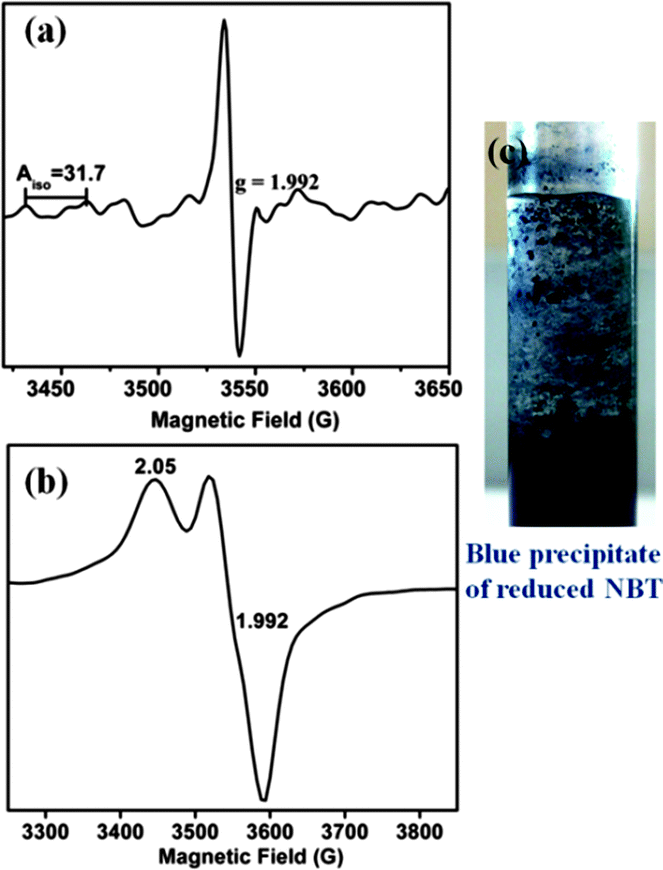 | ||
| Fig. 5 X-band EPR spectrum of 1 in solution under light irradiation in air (a) at room temperature and (b) at 120 K. (c) Blue precipitate of diformazan from reduced NBT confirming superoxide radical formation with a solution of 1 in air and on exposure to sunlight. | ||
On flash freezing the solution of 1 (or 2) after light irradiation, a broad anisotropic EPR signal was observed which consisted of two components g∥ = 2.05 and g⊥=1.992 (Fig. 5b). These could be merged signals of Mo(V) species and the superoxide radical. The presence of superoxide radicals has been identified previously in frozen solutions from the characteristic EPR signal having g∥ 2.08 (broad line) and g⊥ 2.00, detected during oxidation by molecular oxygen.30,31 On warming this solution to room temperature, only a symmetric six-line signal characteristic of Mo(V) was observed. The MLCT excited state generated on excitation of the singlet ground state of 1 (and 2) followed by a fast ISC can be quenched by molecular O2via electron transfer to generate O2˙−. Superoxide anion radicals are known to be temperature sensitive.30 Due to rapid rotation in solution, an extremely broad signal for O2− is expected which is inversely related to its spin relaxation time and thus precludes its detection by EPR spectroscopy at room temperature. Detection is possible only in frozen solutions where the orbital degeneracy is lifted.32 Quantification of the superoxide radical was difficult as it is known to react with Mo(V) converting it to Mo(VI)33 and also with DCM in a multistep process consuming one or more of the radical species per chloride to yield oxygenated products albeit at a sluggish rate.34
The generation of superoxide radicals (O2˙) upon photo-induced single electron transfer from 1 (and 2) in air has been confirmed chemically using the nitrobluetetrazolium (NBT) test.35 Reduction of the pale yellow NBT dye to blue colored diformazan was used to detect the presence of superoxide radicals36 (Fig. 5c). The diformazan was extracted into aqueous phosphate buffer and quantified using a known concentration of [Me4N][O2]37 to record an average of ∼60% conversion. No change in color from yellow NBT to blue diformazan was observed under an inert atmosphere. Thus, 1 and 2 generate superoxide radical ions upon irradiation with visible light concomitant with the oxidation of the complexes to Mo(V) where dioxygen acts as the external oxidant. A scheme for the photo-induced reaction of 1 (and 2) with oxygen is presented in Scheme 2.
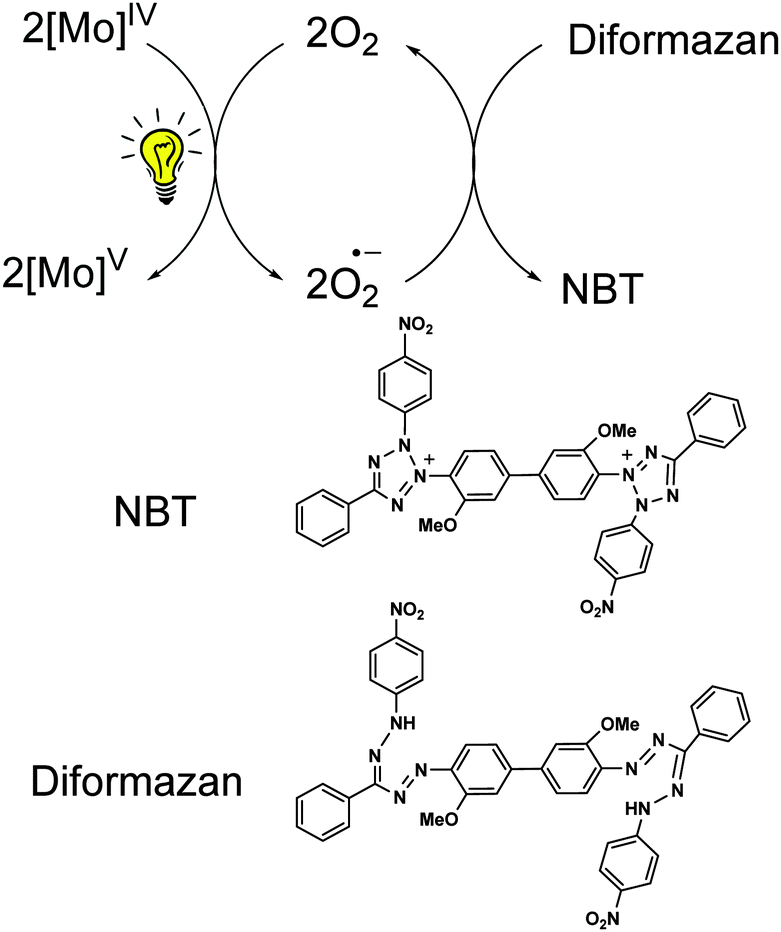 | ||
| Scheme 2 Reaction scheme for the generation of superoxide radicals on photoexcitation of Mo(IV) species in 1 concomitant to its oxidation to Mo(V). The superoxide radical further reacts with NBT forming diformazan. | ||
Electrochemistry
The change of the equatorial ligand from thiolate to selenolate has a marked effect on the redox potential in these complexes.10 One electron oxidation of both 1 and 2 occurs at −0.017 V for 1 and at −0.028 V for 2 respectively (vs. Ag/AgCl) (Fig. 6). The one electron oxidized species loses another electron almost simultaneously following successive two steps of one electron oxidation processes. Both the oxidation waves are due to the loss of an electron from the molybdenum centre for 1 (and 2). This has been supplemented by the observation of Mo dxy based HOMO (and SOMO) for the complexes and their one electron oxidized species (vide computational analysis). Two oxidative processes become clearer on increasing the scan rate to 0.5 V s−1 (Fig. S4 and S5, ESI†). Both these oxidations are quasi-reversible, supporting the very short lifetime of the intermediate oxidized species. A peak at −0.6 V appears on repeated scanning and is not characteristic of 1 and 2. It may appear due to species adsorbed on the electrode surface or may be due to some follow-up electrochemical reactions.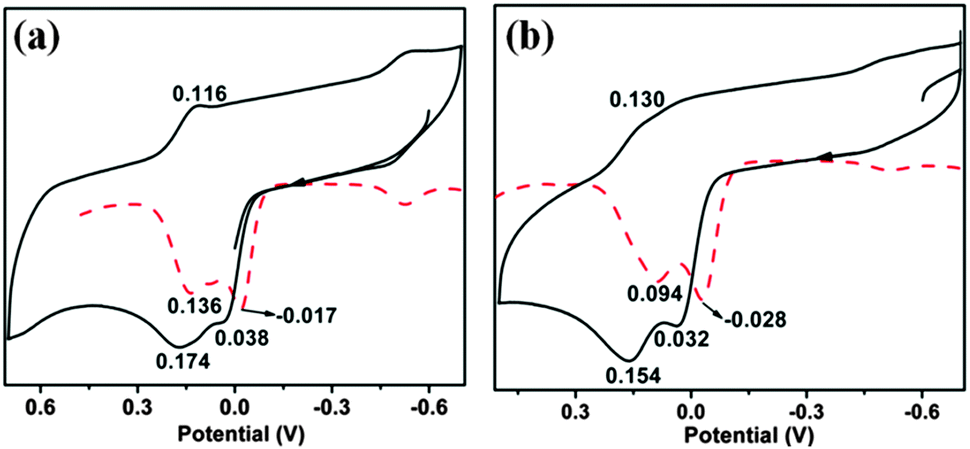 | ||
| Fig. 6 Cyclic voltammograms of (a) 1 and (b) 2 in DCM at a scan rate of 100 mV s−1. Differential pulse polarograms are shown as dashed traces. | ||
Computational analysis
Geometry optimization reproduced the structural features from the crystallographic analysis of 1 and 2. Average deviations of 0.02–0.03 Å in the bond lengths were observed. Such a small difference between crystallographic and optimized geometries is not unusual and may originate from crystal packing effects.A predominantly Mo dxy based HOMO and a HOMO−1 delocalized on S and Se p orbitals are obtained. The LUMO and LUMO+1 are delocalized over the diimine ligand, supporting MLCT transition as the origin of the low energy band in the electronic spectra. On one electron oxidation of the complexes, the HOMO retains its metal character (Fig. 7). A decrease in the HOMO–LUMO gap is observed for both 1 and 2 with BP86 calculations compared to B3LYP due to destabilization of HOMO and stabilization of LUMO. The HOMO–LUMO gap decreases in Mo(V) species as compared to the corresponding Mo(IV) species in both the functionals. This is more prominent in the calculations with BP86. HOMOs of Mo(IV) and Mo(V) species are energetically similar for both the complexes as calculated using the BP86 functional (calculated orbital energies for Mo(IV) and Mo(V) are −90.86 kcal mol−1 and −86.01 kcal mol−1 respectively) as compared with the results from the B3LYP functional (calculated orbital energies for Mo(IV) and Mo(V) are −109.99 kcal mol−1 and −88.87 kcal mol−1 respectively) (Fig. 7 and Table S6, ESI†). Thus, results from the BP86 functional are consistent with the experimentally observed closely spaced electrochemical responses.
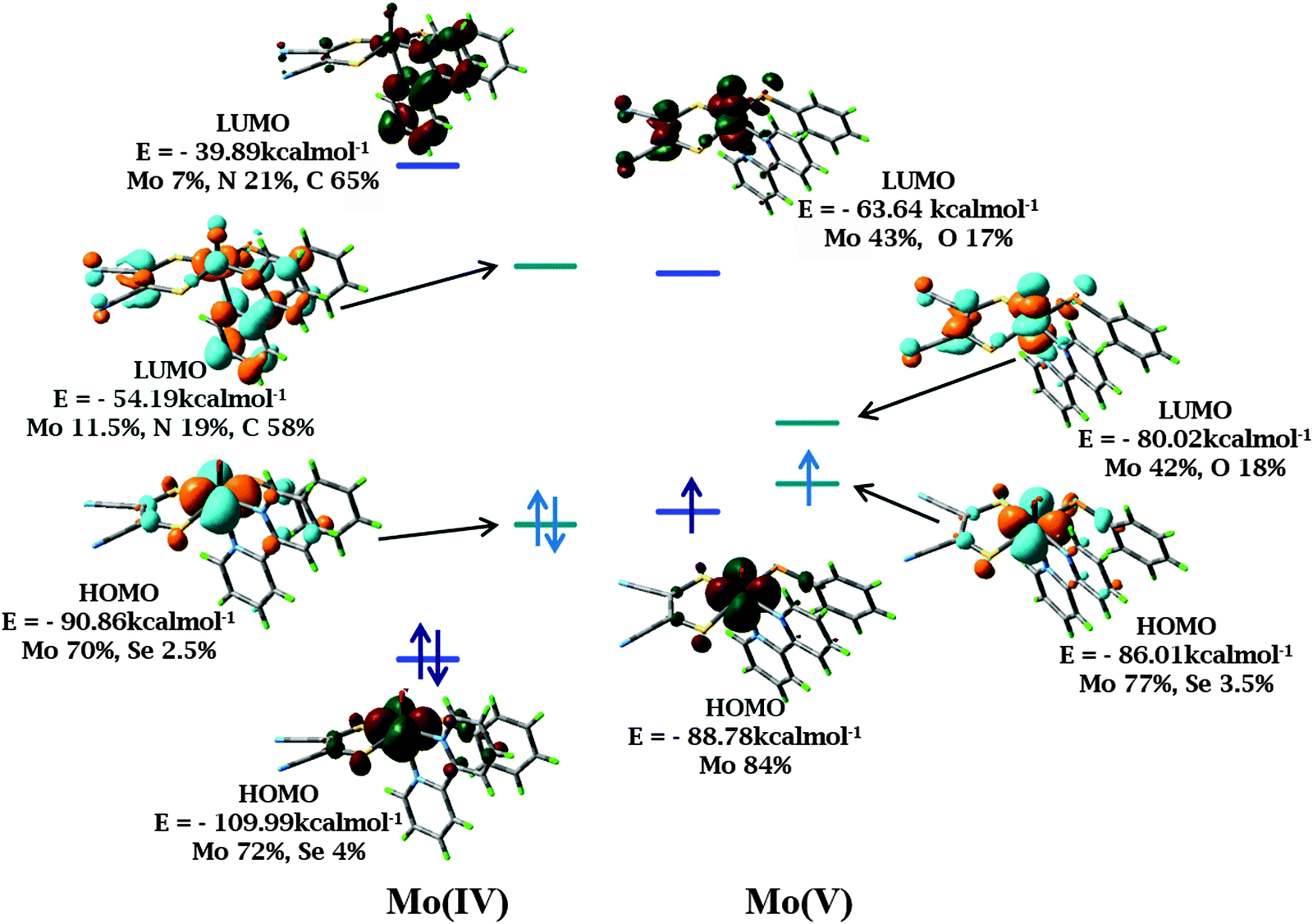 | ||
| Fig. 7 Frontier orbital images of 1 and its one electron oxidized species in DCM using B3LYP (red and green) and BP86 (orange and cyan) functionals respectively. The iso-surface cutoff value is 0.04. The energy levels corresponding to B3LYP and BP86 have been depicted in blue and cyan respectively. | ||
Charge analysis on metal and surrounding ligand atoms revealed a positive charge on Mo which increases slightly upon one electron oxidation, consistent with the removal of an electron from the metal center. According to Mulliken spin population analysis, the unpaired electron is localized on the Mo center, consistent with the oxidation of Mo(IV) (d2 system) to Mo(V) (d1 system). The percentage of metal d orbital in HOMO is of great importance as the nature of the frontier orbitals and electronic transitions are helpful in analyzing the photo-activity and electrochemistry of the complexes. Thus, an appropriate choice of diimine and dithiolene would allow predetermination of the nature of lowest energy transitions and the emitting states.
It is known that a change in the geometry of the species concerned can alter the population of molecular orbitals. To address this aspect, a single point energy calculation has also been performed with the one electron oxidized species without geometry optimization, which essentially yields almost the same population of molecular orbitals thereby eliminating the possibility of severe alteration of population in the molecular orbitals due to a change in the geometry.
Conclusions
Oxo–MoIV(diimine)(dithiolene)(selenolato) complexes participate in facile photo-induced single electron transfer to dioxygen producing superoxide radicals concomitant to the generation of EPR active Mo(V) species. Generation of superoxide radicals has been supported from the classical biochemical NBT test. Significantly increased lifetime was recorded for both the phosphorescent complexes compared to previously reported oxo–MoIV species, attributed to the presence of aromatic diimine ligands. Time dependent density functional theoretical calculations were employed conjointly with ground state calculations to support the experimentally observed photo-activation of the synthesized complexes and to rationalize the observed metal based single electron transfer.Acknowledgements
SS acknowledges DST, India, for funding. JM is thankful for the DST-INSPIRE Faculty award.Notes and references
- V. P. Mueller-Westerhoff and D. I. Yoon, Tetrahedron, 1991, 47, 440 CrossRef CAS; M. Hissler, J. E. McGarrah, W. B. Connick, D. K. Geiger, S. D. Cummings and R. Eisenberg, Coord. Chem. Rev., 2000, 208, 115 CrossRef; N. Robertson and L. Cronin, Coord. Chem. Rev., 2002, 227, 93 CrossRef.
- G. A. Crosby, Acc. Chem. Res., 1975, 8, 231 CrossRef CAS; L. Spiccia, G. B. Deacon and C. M. Kepert, Coord. Chem. Rev., 2004, 248, 1329 CrossRef; J. H. Alstrum-Acevedo, M. K. Brennaman and T. J. Meyer, Inorg. Chem., 2005, 44, 6802 CrossRef PubMed; F. G. Gao and A. J. Bard, J. Am. Chem. Soc., 2000, 122, 7426 CrossRef.
- C. A. Mitsopoulou, Coord. Chem. Rev., 2010, 254, 1448 CrossRef CAS; S. D. Cummings and R. Eisenberg, Inorg. Chem., 1995, 34, 2007 CrossRef.
- D. T. Sawyer, Oxygen Chemistry, Oxford University, New York, 1991 Search PubMed.
- J. R. Winkler and H. B. Gray, Comments Inorg. Chem., 1981, 1, 257 CrossRef CAS; A. K. Mohammed and A. W. Maverick, Inorg. Chem., 1992, 31, 4441 CrossRef.
- R. A. Isotovich, A. S. Beadle, F. R. Fronczek and A. W. Maverick, Inorg. Chem., 1998, 37, 4258 CrossRef.
- C. Jamorski, M. E. Casida and D. R. Salahub, J. Chem. Phys., 1996, 104, 5134 CrossRef CAS.
- E. I. Stiefel, L. E. Bennet, Z. Dori, T. H. Crawford, C. Simo and H. B. Gray, Inorg. Chem., 1970, 9, 281 CrossRef CAS.
- K. Pal, R. Maiti, P. K. Chaudhury and S. Sarkar, Inorg. Chim. Acta, 2007, 360, 2721 CrossRef CAS.
- J. Mitra and S. Sarkar, Inorg. Chem., 2013, 52, 3032 CrossRef CAS PubMed.
- SAINT, version 5.6, Bruker AXS Inc., Madison, WI, 2000 Search PubMed.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115 CrossRef CAS.
- G. M. Sheldrick, SHELX97. Programs for Crystal Structure Analysis (Release 97-2), University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, G. A. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision B.04, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS; J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822 CrossRef.
- G. A. Patersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081 CrossRef.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270 CrossRef CAS; W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284 CrossRef.
- F. Eckert and A. Klamt, AIChE J., 2002, 48, 369 CrossRef CAS.
- J. Mitra and S. Sarkar, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2011, 50, 401 Search PubMed.
- J. Huang, C. Li, S. P. Nolan and J. L. Petersen, Organometallics, 1998, 17, 3516 CrossRef CAS.
- H. Tano, R. Tajima, H. Miyake, S. Itoh and H. Sugimoto, Inorg. Chem., 2008, 47, 7465 CrossRef CAS PubMed; B. S. Lim and R. H. Holm, J. Am. Chem. Soc., 2001, 123, 1920 CrossRef PubMed; A. Majumdar and S. Sarkar, Coord. Chem. Rev., 2011, 255, 1039 CrossRef; R. H. Holm, E. I. Solomon, A. Majumdar and A. Tenderholt, Coord. Chem. Rev., 2011, 255, 993 CrossRef.
- J. K. Nagle, J. S. Bernstein, R. C. Young and T. J. Meyer, Inorg. Chem., 1981, 20, 1760 CrossRef CAS; K. Kalyanasundaram, Coord. Chem. Rev., 1982, 46, 159 CrossRef.
- A. Vlček Jr and S. Zálǐs, Coord. Chem. Rev., 2007, 251, 258 CrossRef CAS; M. Buhl, C. Reimann, D. A. Pantazis, T. Bredow and F. Neese, J. Chem. Theory Comput., 2008, 4, 1449 CrossRef.
- B. Champagne, M. Guillaume and F. Zutterman, Chem. Phys. Lett., 2006, 425, 105 CrossRef CAS; N. A. Besley and F. A. Asmuruf, Phys. Chem. Chem. Phys., 2010, 12, 12024 RSC.
- C. Adamo and D. Jacquemin, Chem. Soc. Rev., 2013, 42, 845 RSC; A. D. Laurent and D. Jacquemin, Int. J. Quantum Chem., 2013, 113, 2019 CrossRef CAS.
- P. P. Zarnegar and D. G. Whitten, J. Am. Chem. Soc., 1971, 93, 3776 CrossRef CAS; P. P. Zarnegar, C. R. Bock and D. G. Whitten, J. Am. Chem. Soc., 1973, 95, 4367 CrossRef.
- F. D. Angelis, S. Fantacci, N. Evans, C. S. M. Klein, C. Zakeeruddin, J.-E. Moser, K. Kalyanasundaram, H. J. Bolink, M. Gratzel and M. Nazeeruddin, Inorg. Chem., 2007, 46, 5989 CrossRef PubMed.
- R. N. Bagchi, A. M. Bond, F. Scholz and R. Stosser, J. Am. Chem. Soc., 1989, 111, 8270 CrossRef CAS.
- P. F. Knowles, J. F. Gibson, F. M. Pick and R. C. Bray, Biochem. J., 1969, 111, 53 CrossRef CAS PubMed.
- D. T. Sawyer, G. Chiercato, Ch. T. Angelis, E. J. Nanni and T. Tsuchiya, Anal. Chem., 1982, 54, 1720 CrossRef CAS.
- Y. Sasaki, Bull. Chem. Soc. Jpn., 1977, 50, 1939 CrossRef CAS.
- J. L. Roberts Jr and D. T. Sawyer, J. Am. Chem. Soc., 1981, 103, 712 CrossRef.
- B. H. J. Bielski, G. G. Shiue and S. Bajuk, J. Phys. Chem., 1980, 84, 830 CrossRef CAS; J. E. Ryer-Powder and H. Forman, J. Free Radicals Biol. Med., 1989, 6, 513 CrossRef.
- K. V. Rajagopalan and P. Handler, J. Biol. Chem., 1964, 239, 2022 Search PubMed; R. W. Miller, Can. J. Biochem., 1970, 48, 935 CrossRef CAS PubMed; C. O. Beauchamp and I. Fridovich, Anal. Biochem., 1971, 44, 276 CrossRef PubMed.
- D. Bhattacharya, S. Maji, K. Pal and S. Sarkar, Inorg. Chem., 2008, 47, 5036 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of X-ray crystallography, spectroscopic data and DFT calculations. CCDC 826727 and 826728. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj02494c |
| ‡ Present address: DST – INSPIRE Faculty, DIMC, CSIR-CSMCRI Bhavnagar, Gijubhai Badheka Marg, Bhavnagar 364 002, Gujarat, India. E-mail: http://joyeemitra@csmcri.org |
§ Crystallographic data for 1: empirical formula C37H51Cl2MoN5OS2Se, formula wt: 891.77, crystal system, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 9.562(5) Å, b = 19.281(5) Å, c = 23.109(5) Å, α = 79.293(5)°, β = 83.660(5)°, γ = 77.619(5)°, V = 4077.88 Å3, Z = 4. Crystallographic data for 2: empirical formula C39H51Cl2MoN5OS2Se, formula wt: 915.79, crystal system, triclinic, space group P, a = 9.288(5) Å, b = 11.771(5) Å, c = 19.492(5) Å, α = 83.300(5)°, β = 84.385(5)°, γ = 83.738(5)°, V = 2096.03, Z = 2. CCDC 826727 (1) and 826728 (2). , a = 9.562(5) Å, b = 19.281(5) Å, c = 23.109(5) Å, α = 79.293(5)°, β = 83.660(5)°, γ = 77.619(5)°, V = 4077.88 Å3, Z = 4. Crystallographic data for 2: empirical formula C39H51Cl2MoN5OS2Se, formula wt: 915.79, crystal system, triclinic, space group P, a = 9.288(5) Å, b = 11.771(5) Å, c = 19.492(5) Å, α = 83.300(5)°, β = 84.385(5)°, γ = 83.738(5)°, V = 2096.03, Z = 2. CCDC 826727 (1) and 826728 (2). |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |