
Influences of the protonic state of an imidazole-phenanthroline ligand on the luminescence properties of copper(I) complexes: experimental and theoretical research†
Xinfang
Liu
a,
Rongfang
Li
a,
Lufang
Ma
a,
Xun
Feng
*a and
Yuqiang
Ding
*b
aCollege of Chemistry and Chemical Engineering, Luoyang Normal University, Luoyang, Henan, P. R. China. E-mail: fengx@lynu.edu.cn
bThe Key Laboratory of Food Colloids and Biotechnology, Ministry of Education, School of Chemical and Material Engineering, Jiangnan University, Wuxi 214122, China. E-mail: yding@jiangnan.edu.cn
First published on 11th November 2015
Abstract
Two ionic (1a and 1b) and two neutral (2a and 2b) Cu(I) complexes containing an un-deprotonated or a deprotonated nitrogen ligand {2-(4-methyl phenyl) imidazole[4,5-f]-1,10-phenanthroline, MHPIP} and different phosphine ligands (bis[2-(diphenylphosphino) phenyl]ether and PPh3) have been synthesized and characterized by elemental analysis, 1H NMR spectroscopy and X-ray crystallography (1b, 2a and 2b). The complexes adopt a distorted tetrahedral geometry constructed by MHPIP (or MPIP−) and phosphine ligands. The emission spectra show that the ionic complexes exhibit almost ignorable luminescence. However, the deprotonation of the nitrogen ligand makes the neutral complexes exhibit orange or yellow emission both in solution and solid-powder states. Considering the different luminous characters of the neutral complexes, density functional theory (DFT) calculations have been performed at the B3LYP/6-31G** level to provide information about the impact of phosphine ligands on the frontier orbital.
Introduction
In recent decades, much effort has been devoted to the design and synthesis of copper(I) complexes due to the low cost and stable supply of the raw materials and their potential applications as luminescent materials.1 In addition, the emission energies of the copper(I) complexes can cover the entire visible region with the change of the ligand,2 so the ligand strategy plays an important role in designing luminescent species. The 1,10-phenanthroline (1,10-phen) ligand has been extensively utilized to prepare metal complexes with attractive luminescence features.3 However, [Cu(1,10-phen)2]+ is a weakly emissive compound because a distortion from tetrahedral toward square planar geometry may occur and accelerate non-radiative decay of the excited-state.4 Replacing one 1,10-phen with a hindered phosphine ligand is an effective method to reduce excited state relaxation.5 Another fruitful strategy involves the chemical functionalization of 1,10-phen at various ring positions.6 For example, copper(I) (2,9-dialkyl-1,10-phenanthroline) units have been widely used to construct photo- and electroactive devices.7However, such ligands are almost 2,9-substituted 1,10-phen and the chemical functionalization at other ring positions is still under development. Furthermore, the ligands are mostly used to prepare ionic complexes, more general copper(I) neutral complexes are rarely reported. Given this, we turn to 2-(4-methylbenzene) imidazo[4,5-f]-1,10-phen (MHPIP), a 5,6-substituted-1,10-phen-imidazole ligand, which combines the versatile structural and chemical properties of imidazole and 1,10-phen. So far, such ligands have been mainly used as neutral ligands.8 Occasionally, they are deprotonated to produce neutral complexes.9 However, the impacts of the protonic state on the luminescence properties of complexes are rarely comparatively studied except the synthesis of Ruthenium(II) complexes by Aukauloo et al.8 We are curious whether the deprotonation of the 1,10-phen-imidazole ligand will influence the luminescence properties of copper(I) complexes, and how the ancillary ligand influences the emission characters.
Herein, we prepared two ionic and two neutral copper(I) complexes with MHPIP (or deprotonated MPIP) and DPEphos (or PPh3) as a N- and a P-containing ligand, respectively, namely, [Cu(MHPIP)(DPEphos)](BF4) (1a), [Cu(MHPIP)(PPh3)2](BF4) (1b), Cu(MPIP)(DPEphos) (2a) and Cu(MPIP)(PPh3)2 (2b). Besides the synthesis and structural characterization, the absorption and emission spectra of the complexes have also been studied. Considering the different luminescent characters, density functional theory (DFT) calculations of the neutral complexes have been performed at the B3LYP/6-31G** level to provide information about the frontier orbital.
Results and discussion
Syntheses
The synthesis of the mononuclear copper(I) complexes is outlined in Scheme 1. Reaction of the precursor Cu(CH3CN)4·BF4 with HMPIP and phosphine ligands gives ionic complexes [Cu(MHPIP)(DPEphos)]·BF4 (1a) and [Cu(MHPIP)(PPh3)2]·BF4 (1b). By treating 1a and 1b with strong base (KOH), neutral complexes Cu(MPIP)(DPEphos) (2a) and Cu(MPIP) (PPh3)2 (2b) can be prepared and crystallized from anhydrous ether. The neutral complexes can also be prepared by the one-step reaction of the precursor, ligands and KOH (see Scheme 1). However, the one-step method prepares the neutral complexes in a lower yield than the two-step method because the precursor prefers to react with KOH which has been proven by the observation of a black Cu2O powder. During the deprotonation of MHPIP, KOH has been proven to be a better candidate than NaOH because K+ matches well with BF4− in radius. | ||
| Scheme 1 The synthesis route of the ionic and neutral complexes. | ||
Crystal structures
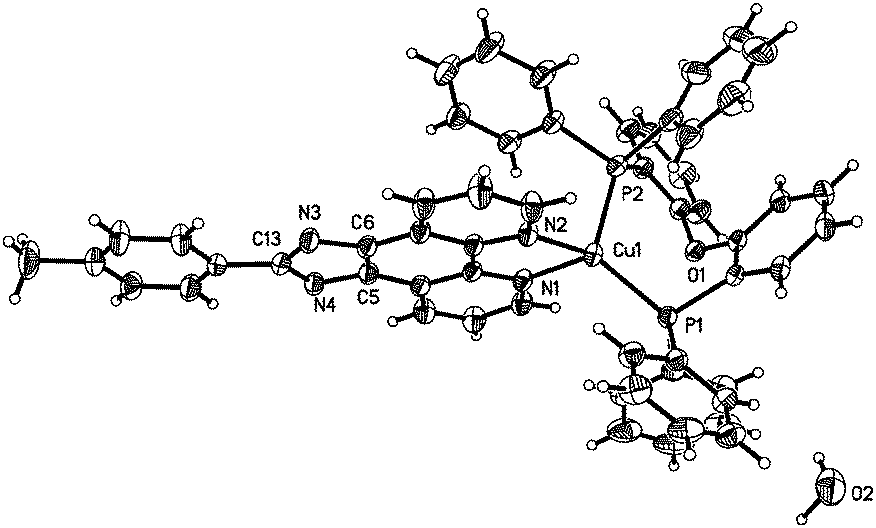
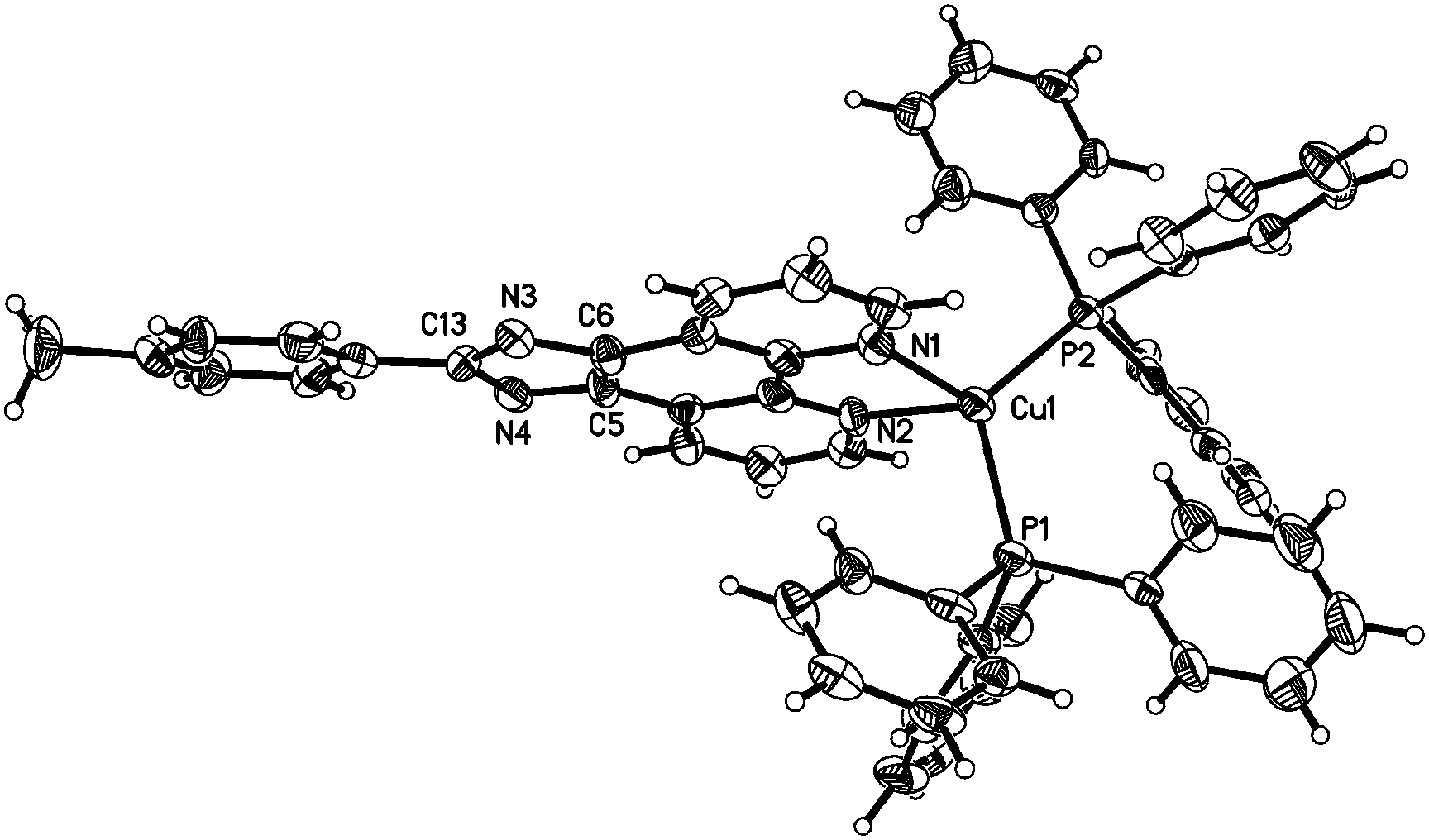
To unambiguously elucidate the structures of the copper(I) complexes, X-ray diffraction analyses of complexes 1b, 2a and 2b are carried out and the crystal structures are shown in Fig. 1–3. The selected bond distances and bond angles are shown in Table 1. The Cu(I) center in all three complexes has a distorted tetrahedral geometry formed by two phosphorus atoms from two PPh3 ligands (1b and 2b) (or one DPEphos ligand, 2a) and two nitrogen atoms from MHPIP (1b) (or MPIP anion, 2a and 2b). The Cu–N and Cu–P bond lengths are in the range of 2.060(2)–2.099(3) and 2.236(6)–2.297(7) Å, respectively, which are consistent with previously studied copper(I) complexes with the same or similar ligands.10 | ||
| Fig. 1 (a) ORTEP diagram of 1b with thermal ellipsoids shown at the 30% probability level. (b) Hydrogen bonds and π–π interactions in 1b. | ||
 | ||
| Fig. 2 ORTEP diagram of complex 2a with thermal ellipsoids shown at the 30% probability level. | ||
 | ||
| Fig. 3 ORTEP diagram of complex 2b with thermal ellipsoids shown at the 30% probability level. | ||
| 1b | 2a | 2b | |
|---|---|---|---|
| Bond length (Å) | |||
| Cu(1)–N(1) | 2.074(3) | 2.060(2) | 2.092(7) |
| Cu(1)–N(2) | 2.099(3) | 2.078(2) | 2.097(8) |
| Cu(1)–P(1) | 2.265(1) | 2.236(6) | 2.247(2) |
| Cu(1)–P(2) | 2.286(1) | 2.297(7) | 2.284(2) |
| N(3)–C(13) | 1.325(5) | 1.346(3) | 1.370(11) |
| N(3)–C(5) | 1.384(5) | 1.369(3) | 1.382(10) |
| N(4)–C(13) | 1.363(5) | 1.359(3) | 1.362(11) |
| N(4)–C(6) | 1.365(5) | 1.366(3) | 1.366(10) |
| C(5)–C(6) | 1.374(6) | 1.392(4) | 1.362(11) |
| Bond angles (°) | |||
| N(1)–Cu(1)–N(2) | 80.57(1) | 80.85(8) | 80.1(3) |
| N(1)–Cu(1)–P(1) | 112.2(1) | 118.7(6) | 118.6(2) |
| N(1)–Cu(1)–P(2) | 109.3(9) | 106.6(6) | 109.6(2) |
| P(2)–Cu(1)–P(1) | 125.3(4) | 115.8(2) | 120.8(9) |
| P(2)–Cu(1)–N(2) | 108.3(1) | 101.7(6) | 105.3(2) |
| P(1)–Cu(1)–N(2) | 112.2(1) | 127.2(6) | 114.6(2) |
For ionic complex 1b, a [Cu(PPh3)2(MHPIP)]+ ion cluster is formed when PPh3 and MHPIP coordinate with the Cu(I) center. Two Cu(I) ion clusters are linked by two water and CH3OH molecules through intermolecular hydrogen bonds (N4–H4⋯O1, 1.95(4) Å, 165(4)°; O1–H1D⋯O2, 1.96 Å, 163.1°; O2–H2⋯N3#1, 2.08 Å, 165.7°) to form a dimer (Fig. 1b). In addition, strong intermolecular π–π interactions between the imidazole and benzene rings of the adjacent ion clusters (perpendicular distance: 3.378(9) Å; centroid distance: 3.916(1) Å) also play an important role in stabilizing the dimer unit.11 The counter anion, BF4−, is linked to adjacent free water by weak intermolecular hydrogen bonds (O1–H1C⋯F4A, 2.01 Å, 173.3°; O1–H1C⋯F3B, 2.07 Å, 153.1°). And the BF4− anions stack with the ion clusters through ionic bonds.
For the neutral complexes (2a and 2b) involving the same MPIP and different phosphine ligands, the bond lengths in Table 1 indicate that DPEphos meshes with MPIP better than PPh3 because the average Cu–N distance of 2a (2.069(2) Å) is distinctly shorter than that of 2b (2.095(8) Å). In addition, the P–Cu–P angle in 2a (115.8(2)°) presents a smaller distortion than that of 2b (120.8(9)°) due to the presence of ether linkage of the DPEphos ligand. These results are all consistent with the findings of the previous studies.12
The purities of complexes 1b, 2a and 2b are determined by comparison of the simulated and experimental X-ray powder diffraction patterns, and the results are reported in Fig. S1 (ESI†). The peak positions of the experimental patterns nearly matched with the simulated ones generated from the crystal structures, which indicates that the phases of three complexes are pure. The differences in intensity may be due to the preferred orientation of the microcrystalline powder samples.
Absorption and emission spectra
The normalized absorption of the complexes in anhydrous ether is shown in Fig. 4a. These complexes display two intense absorption bands from 230 to 320 nm, which are ascribed to ligand-to-ligand charge transfer (LLCT) transitions or intraligand charge transition (ILCT). The neutral complexes display an absorption peak (shown as a shoulder for 2b) at about 335 nm, while the peak of the ionic complexes is unobserved or obscure, indicating the influence of deprotonation on the absorption spectra. A weak low-energy band with maxima ranging from 380 to 480 nm is found. The absorption bands are obscure (especially for 2b) owing to their small transition probability, i.e. the rather low absorptivity, which are analogous to those reported for copper(I) complexes.13 Different from the absorption bands before mentioned, the absorption peak between 380 and 480 nm changes with the P-, N-containing ligand. As suggested by TD-DFT studies (vide infra), the lowest-lying absorption bands of 2a and 2b are mainly assigned to LLCT and ILCT transition, respectively. The different transition may influence the emissive behaviour of the neutral complexes. | ||
| Fig. 4 (a) Absorbance and emission spectra of the complexes in anhydrous ether. (b) Emission spectra of the complexes in powder states. | ||
Emission spectra are measured at room temperature in different solvents. The results show that the complexes are not emissive in CH2Cl2 and CH3OH. However, neutral complex 2b exhibits an emission λmax at 576 nm (yellow region) and 2a shows a broad peak centered at 609 nm (orange region) (Fig. 4a). The emission peak shifts toward higher energy with the ligand changing from PPh3 to DPEphos. The trend of emission peaks is consistent with that of the low-energy absorptions shown in Fig. 4a, illustrating that both spectra may originate from the same electronic excited state. According to the literature, the tendency of the emission energies (2b > 2a) is contrary to the electron-donating ability of the phosphine ligands (PPh3 < DPEphos) and the complexes have the same order as the P–Cu–P bond angles (120.8(9)° for 2b and 115.8(2)° for 2a, see Table 1).1,14 The ionic complexes are slightly soluble in anhydrous ether, so it is difficult to determine their luminescence properties in solution.
Finally, the emission spectra in solid-powder states are explored to exclude the influences of the solvent (Fig. 4b). The results show that the neutral complexes exhibit luminescence at 612 nm for 2a and at 580 nm for 2b, which are actually close to their solution emissions. The emission peak of 2a shows a remarkable red-shift (∼32 nm) relative to that of 2b. The red-shift may be caused by the introduction of the electron-donating ether oxygen in DPEphos, raising the LUMO level, less influencing the HOMO energy (see Fig. 5), thus decreasing the HOMO–LUMO gap.15 The ionic complexes exhibit green luminescence (570 and 529 nm for 1a and 1b, respectively), while the emission intensity is very weak.
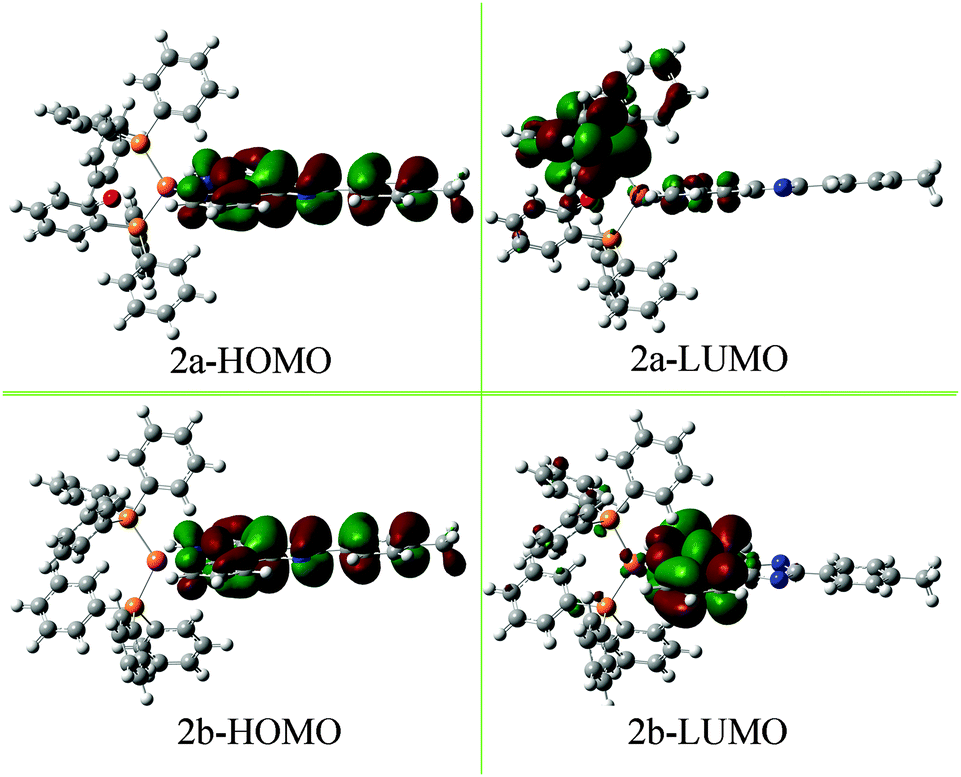 | ||
| Fig. 5 HOMO and LUMO diagrams of complexes 2a and 2b. | ||
DFT calculations
To gain insight into the relationship between the electronic structure and luminescence properties of the neutral complexes, considering their excellent luminescence properties and their different emissive characters, density functional theory (DFT) calculations with the B3LYP/6-31G** method were performed.To analyse the electronic structure in more detail, the Mulliken and the NBO charges have been determined15 (Table S1 and Fig. S2, ESI†). These charges do not represent absolute charges but the trends among the complexes can impress the electronic effects. When compared with 2b, the charges of the Phen-N atoms (N1 and N2) in 2a are in average 0.007 (Mulliken charge) or 0.018 (NBO charge) more negative. In addition, the P atoms in 2a are more positive (0.007 for Mulliken charge and 0.023 for NBO charge on average) than those of 2b. The results preliminarily suggest that LLCT transition (MPIP → DPEphos) more likely occurs in 2a which will be further illustrated by the following analysis of the frontier orbital.
The frontier orbital is shown in Fig. 5, more figures of TD-DFT studies are provided in the Supporting Information (see Fig. S3 and S4, ESI†). The HOMO of 2a and 2b is mainly over the MPIP ligand (97.43% and 97.30%, respectively), and the HOMO−1, HOMO−2 and HOMO−3 are almost resident on MPIP. It is worth noting that the LUMO of 2a and 2b shows large difference. For example, the LUMO of 2a is mainly on DPEphos (71.62%) while partly on MPIP (22.40%) and Cu (5.98%); the LUMO of 2b is mainly located on MPIP (83.18%), and less parts on PPh3 (9.95%) and Cu (6.87%). Hence, the calculated lowest-energy absorption of 2a at 458 nm from HOMO−1 → LUMO transition is of a significant LLCT transition (MPIP → DPEphos) character mixed with a certain degree of the intraligand (ILCT) transition and a small amount of LMCT transition (MPIP → Cu).16 Similarly, the lowest-energy absorption of 2b is mainly assigned to ILCT transition and partly assigned to LLCT transition (MPIP → PPh3) and LMCT transition (MPIP → Cu). In exactly the same way, the next two absorption bands of 2a and 2b have been assigned and listed in Table 2. Some subtle differences are observed about the molecular orbital configurations and assignment (Table 2) as well as the frontier molecular orbitals (Fig. S3 and S4, ESI†) of 2a and 2b, which can provide evidence for the different absorption and emission spectra before mentioned.
| λ (nm)/E (eV) | f | Main configurations | Assignment | |
|---|---|---|---|---|
| 2a | 498/2.84 | 0.0346 | HOMO−1 → LUMO (75.5%) | LLCT/ILCT/LMCT |
| 357/2.97 | 0.2305 |
HOMO−3 → LUMO (8.8%)
HOMO−1 → LUMO+6 (24.9%) HOMO → LUMO+8 (15.9%) |
LLCT/ILCT/LMCT LLCT LLCT |
|
| 302/3.66 | 0.4888 | HOMO−2 → LUMO+5 (81.6%) | LLCT/ILCT | |
| 2b | 495/3.12 | 0.0278 | HOMO−1 → LUMO (81.7%) | ILCT/LLCT/LMCT |
| 350/3.54 | 0.3270 |
HOMO−2 → LUMO (38.6%)
HOMO → LUMO+5 (15.5%) |
ILCT/LLCT/LMCT LLCT/ILCT | |
| 295/4.20 | 0.7752 | HOMO−1 → LUMO+1 (70.1%) | LLCT | |
Conclusions
We have synthesized two ionic and two neutral mononuclear Cu(I) complexes bearing different protonic states of the 1,10-phen-imidazole ligand. This paper describes the differences in the emission properties of the neutral and ionic complexes at ambient temperature. The emissive properties of the neutral complexes can change with the ancillary phosphine ligands as supported by detailed density functional theory calculations. Theoretical calculations can provide new insight into the synthesis of different colours of copper(I) phosphorescent materials via well modulating the structures of ligands.Experimental section
Materials
CuCl, DPEphos, PPh3, 4-methyl benzaldehyde, 1,10-phen monohydrate, fluoroboric acid (HBF4) and other chemicals were obtained from commercial sources and used without further purification. All solvents were dried by common methods and freshly distilled prior to use. 2-(4-methyl phenyl) imidazo[4,5-f]-1,10-phen was prepared according to the reported literature procedures.17 [Cu(CH3CN)4]BF4 was synthesized via the described procedure.18Experimental details
The synthetic procedures involving the reaction of Cu(I) species were carried out under a nitrogen atmosphere using a standard schlenk flask, considering the oxidative stability of the copper(I) complexes in the reactions.Synthesis procedure
Characterization methods
1H NMR (400 MHz) spectra were collected on a Bruker ACF-400 spectrometer and the NMR chemical shifts were recorded with deuterated CH2Cl2 or DMSO as a solvent and tetramethylsilane as an internal standard. Elemental analyses (C, H, O and N) were determined using a Bio-Rad elemental analysis system. The powder X-ray diffraction (PXRD) patterns were measured using a Bruker D8 Advance powder diffractometer at 40 kV and 40 mA with Cu Kα radiation (λ = 1.5418 Å), with a scan speed of 0.2 s per step and a step size of 0.02 (2θ). Absorption spectra were measured using a TU-1901 UV/vis spectrophotometer. PL spectra were obtained using an RF-5301PC spectrofluorimeter connected to a photomultiplier tube with a xenon lamp as the excitation source.X-Ray crystallography
X-ray data for complexes 1b, 2a and 2b were collected using a Bruker SMART APEX II CCD area detector diffractometer using graphite monochromated Mo Kα (λ = 0.71073 Å). The crystal structure was solved by direct methods and refined on F2 by full-matrix least-squares methods using the SHELXTL-97 program.19 The hydrogen atom positions were calculated theoretically and included in the final cycles of refinement in a riding model along with attached carbon atoms. Complexes 1b and 2a belong to the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , and 2b belongs to the orthorhombic space group Pna21. The BF4 anion in 1b was found to display some degree of disorder and was refined successfully. A summary of the key crystallographic information is listed in Table 3. More detailed crystallographic data have been given in their cif files.
, and 2b belongs to the orthorhombic space group Pna21. The BF4 anion in 1b was found to display some degree of disorder and was refined successfully. A summary of the key crystallographic information is listed in Table 3. More detailed crystallographic data have been given in their cif files.
| Complex | 1b | 2a | 2b |
|---|---|---|---|
| Formula | C57H50BCuF4N4O2P2 | C56H43CuN4O2P2 | C56H43CuN4P2 |
| Formula weight | 1035.30 | 929.42 | 897.42 |
| Crystal form | Yellow, block | Orange, block | Red, block |
| Crystal system | Triclinic | Triclinic | Orthorhombic |
| Space group |
P |
P |
Pna21 |
| a/Å | 10.3872(6) | 13.0549(2) | 19.491(4) |
| b/Å | 14.4087(10) | 13.6031(2) | 25.892(5) |
| c/Å | 18.4400(10) | 14.2237(2) | 8.9679(18) |
| α (°) | 95.95(2) | 73.0790(10) | 90.00 |
| β (°) | 102.000(14) | 75.2490(10) | 90.00 |
| γ (°) | 109.680(13) | 87.8950(10) | 90.00 |
| V/Å3 | 2496.2(3) | 2335.04(6) | 4525.7(16) |
| Z/F(000) | 2/1072 | 2/964 | 4/1864 |
| D c/mg m−3 | 1.377 | 1.322 | 1.317 |
| μ/mm−1 | 0.564 | 0.584 | 0.597 |
| Crystal size/mm | 0.20 × 0.40 × 0.58 | 0.21 × 0.21 × 0.19 | 0.20 × 0.27 × 0.40 |
| θ range | 3.02–25.01 | 1.61–25.10 | 3.15–25.00 |
| Index range | −12 ≦ h ≦ 10 | −15 ≦ h ≦15 | −23 ≦ h ≦ 13 |
| −17 ≦ k ≦ 17 | −16 ≦ k ≦ 15 | −30 ≦ k ≦ 28 | |
| −20 ≦ l ≦ 21 | −16 ≦ l ≦ 16 | −10 ≦ l ≦ 10 | |
| Reflections collected/unique [R(int)] | 8481/6597 | 8270/7165 | 6495/4136 |
| R, wR2 (I > 2σ(I)) | 0.0654, 0.1504 | 0.0459, 0.0974 | 0.0660, 0.0890 |
| Goodness of fit, F2 | 0.979 | 1.056 | 0.904 |
| Largest diff. peak, hole/e Å3 | 0.461, −0.343 | 0.802, −0.302 | 0.553, −0.351 |
Theoretical calculations
The geometry optimization of the neutral copper(I) complexes 2a and 2b was performed using the Gaussian 03 suite where the crystal structure data were used as the starting geometry. The calculations were carried out at the B3LYP/6-31G** level of the density functional theory (DFT). A 6-31G basis set was employed for C, H, O and N atoms, and a LANL2DZ basic set was implemented for Cu atoms.Acknowledgements
The authors are grateful for the financial support from the National Natural Science Foundation of China (No. 21302082 and 21271098), the Scientific Research Fund of Henan Provincial Education Department (No. 14B150057) and the Program for Innovative Research Team (in Science and Technology) in the University of Henan Province (14IRTSTHN008).References
- M. J. Leitl, F. R. Küchle, H. A. Mayer, L. Wesemann and H. Yersin, J. Phys. Chem. A, 2013, 117(46), 11823 CrossRef CAS PubMed; T. Hofbeck, U. Monkowius and H. Yersin, J. Am. Chem. Soc., 2015, 137(1), 399 CrossRef PubMed; Y. Sevryugina, O. Hietsoi and M. A. Petrukhina, Chem. Commun., 2007, 3853 RSC; O. Hietsoi, C. Dubceac, A. S. Filatov and M. A. Petrukhina, Chem. Commun., 2011, 47(24), 6939 RSC; O. Hietsoi, A. S. Filatov and M. A. Petrukhina, Dalton Trans., 2011, 40(34), 8598 RSC; R. Starosta, M. Florek, J. Król, M. Puchalska and A. Kochel, New J. Chem., 2010, 34(7), 1441 RSC.
- C. Kutal, Coord. Chem. Rev., 1990, 99, 213 CrossRef CAS; P. C. Ford, E. Cariati and J. Bourassa, Chem. Rev., 1999, 99(12), 3625 CrossRef PubMed; D. V. Scaltrito, D. W. Thompson, J. A. O'Callaghan and G. J. Meyer, Chem. Rev., 2000, 208(1), 243 Search PubMed; H. Araki, K. Tsuge, Y. Sasaki, S. Ishizaka and N. Kitamura, Inorg. Chem., 2005, 44(26), 9667 CrossRef PubMed; V. W. W. Yam and K. M. C. Wong, Chem. Commun., 2011, 47(42), 11579 RSC; M. J. Leitl, F. R. Küchle, H. A. Mayer, L. Wesemann and H. Yersin, J. Phys. Chem. A, 2013, 117(46), 11823 CrossRef PubMed; T. Gneuß, M. J. Leitl, L. H. Finger, N. Rau, H. Yersin and J. Sundermeyer, Dalton Trans., 2015, 44(18), 8506 RSC.
- A. Bencini and V. Lippolis, Coord. Chem. Rev., 2010, 254(17), 2096 CrossRef CAS; X. Ma, X. Li, Y. E. Cha and L. P. Jin, Cryst. Growth Des., 2012, 12(11), 5227 Search PubMed.
- N. Armaroli, Chem. Soc. Rev., 2001, 30(2), 113 RSC; M. Hashimoto, S. Igawa, M. Yashima, I. Kawata, M. Hoshino and M. Osawa, J. Am. Chem. Soc., 2011, 133(27), 10348 CrossRef CAS PubMed.
- D. G. Cuttell, S. M. Kuang, P. E. Fanwick, D. R. McMillin and R. A. Walton, J. Am. Chem. Soc., 2002, 124, 6 CrossRef CAS PubMed; R. B. Hou, T. H. Huang, X. J. Wang, X. F. Jiang, Q. L. Ni, L. C. Gui, Y. J. Fan and Y. L. Tan, Dalton Trans., 2011, 40, 7551 RSC.
- G. Accorsi, A. Listorti, K. Yoosaf and N. Armaroli, Chem. Soc. Rev., 2009, 38(6), 1690 RSC.
- C. E. A. Palmer and D. R. McMillin, Inorg. Chem., 1987, 26, 3837 CrossRef CAS; M. T. Miller, P. K. Gantzel and T. B. A. Karpishin, J. Am. Chem. Soc., 1999, 121, 4292 CrossRef; D. G. Cuttell, S. M. Kuang and P. E. Fanwick, J. Am. Chem. Soc., 2001, 124(1), 6 CrossRef; A. Kaeser, M. Mohankumar, J. Mohanraj, F. Monti, M. Holler, J. Cid, O. Moudam, I. Nierengarten, L. Karmazin-Brelot, C. Duhayon, B. Delavaux-Nicot, N. Armaroli and J. Nierengarten, Inorg. Chem., 2013, 52, 12140 CrossRef PubMed; R. Starosta, M. Puchalska, J. Cybińska, M. Barys and A. V. Mudring, Dalton Trans., 2011, 40(11), 2459 RSC.
- Y. Xiong, X. F. He, X. H. Zou, J. Z. Wu, X. M. Chen, L. N. Ji, R. H. Li, J. Y. Zhou and K. B. Yu, J. Chem. Soc., Dalton Trans., 1999, 19 RSC; Q. Zhao, S. J. Liu, M. Shi, F. Y. Li, H. Jing, T. Yi and C. H. Huang, Organometallics, 2007, 26, 5922 CrossRef CAS; A. Quaranta, F. Lachaud, C. Herrero, R. Guillot, M. F. Charlot, W. Leibl and A. Aukauloo, Chem. – Eur. J., 2007, 13, 8201 CrossRef PubMed; X. L. Wang, Y. Q. Chen, Q. Gao, H. Y. Lin, G. C. Liu, J. X. Zhang and A. X. Tian, Cryst. Growth Des., 2010, 10(5), 2174 Search PubMed; Y. Wei, K. Wu, J. He, W. Zheng and X. Xiao, CrystEngComm, 2011, 13, 52 RSC; L. Wang, L. Ni and J. Yao, Polyhedron, 2013, 59, 115 CrossRef.
- H. Chao, Y. X. Yuan and L. N. Ji, Transition Met. Chem., 2004, 29, 774 CrossRef CAS; Y. J. Huang and L. Ni, Chin. J. Inorg. Chem., 2011, 27(8), 1649 Search PubMed; R. O. Bonello, I. R. Morgan, B. R. Yeo, L. E. L. Jones, B. M. Kariuki, I. A. Fallis and S. J. A. Pope, J. Organomet. Chem., 2014, 749, 150 CrossRef.
- C. Janiak, Dalton Trans., 2000, 3885 RSC; J. Min, Q. Zhang, W. Sun, Y. Cheng and L. Wang, Dalton Trans., 2011, 40, 686 RSC; C. W. Hsu, C. C. Lin, M. W. Chung, Y. Chi, G. H. Lee, P. T. Chou, C. H. Chang and P. Y. Chen, J. Am. Chem. Soc., 2011, 133, 12085 CrossRef CAS PubMed.
- A. Bhattacharyya, P. K. Bhaumik, A. Bauzá, P. P. Jana, A. Frontera, M. G. B. Drew and S. Chattopadhyay, RSC Adv., 2014, 4, 58643 RSC; H. L. S. Wong, D. R. Allan, N. R. Champness, J. McMaster, M. Schröder and A. J. Blake, Angew. Chem., Int. Ed., 2013, 52(19), 5093 CrossRef CAS PubMed; R. Banik, S. Roy, A. Bauza, A. Frontera and S. Das, RSC Adv., 2015, 5, 10826 RSC.
- S. M. Kuang, D. G. Cuttell, D. R. McMillin, P. E. Fanwick and R. A. Walton, Inorg. Chem., 2002, 41(12), 3313 CrossRef CAS PubMed.
- H. Nitadori, L. Ordronneau, J. Boixel, D. Jacquemin, A. Boucekkine, A. Singh, M. Akita, I. Ledoux, V. Guerchais and H. L. Bozec, Chem. Commun., 2012, 48, 10395 RSC; X. L. Li, Y. B. Ai, B. Yang, J. Chen, M. Tan, X. L. Xin and Y. H. Shi, Polyhedron, 2012, 35(1), 47 CrossRef CAS; R. Marion, F. Sguerra, F. D. Meo, E. Sauvageot, J. F. Lohier, R. Daniellou, J. L. Renaud, M. Linares, M. Hamel and S. Gaillard, Inorg. Chem., 2014, 53(17), 9181 CrossRef PubMed.
- T. McCormick, W. L. Jia and S. Wang, Inorg. Chem., 2006, 45, 147 CrossRef CAS PubMed; S. Sakaki, H. Mizutani, Y. I. Kase, K. J. Inokuchi, T. Arai and T. Hamada, J. Chem. Soc., Dalton Trans., 1996, 1909 RSC.
- E. D. Hedegård, J. Bendix and S. P. Sauer, THEOCHEM, 2009, 913(1), 1 CrossRef.
- G. F. Manbeck, W. W. Brennessel and R. Eisenberg, Inorg. Chem., 2011, 50, 3431 CrossRef CAS PubMed; M. G. Crestani, G. F. Manbeck, W. W. Brennessel, T. M. McCormick and R. Eisenberg, Inorg. Chem., 2011, 50, 7172 CrossRef PubMed; S. Perruchas, C. Tard, X. F. Le Goff, A. Fargues, A. Garcia, S. Kahlal, J. Y. Saillard, T. Gacoin and J. P. Boilot, Inorg. Chem., 2011, 50, 10682 CrossRef PubMed.
- E. A. Steck and A. R. Day, J. Am. Chem. Soc., 1943, 65(3), 452 CrossRef CAS; H. Xu, K. C. Zheng, H. Deng, L. J. Lin, Q. L. Zhang and L. N. Ji, New J. Chem., 2003, 27(8), 1255 RSC; J. Liu, W. J. Mei, L. J. Lin, Q. L. Zhang, H. Chao, F. C. Yun and L. N. Ji, Inorg. Chim. Acta, 2004, 357(1), 285 CrossRef.
- G. J. Kubas, B. Monzyk and A. L. Crumbliss, Inorg. Synth., 1979, 19, 90 CrossRef CAS.
- G. M. Sheldrick, SHELXTL, version 5.1, Bruker Analytical X-ray Systems, Inc., Madision, WI, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Calculated Mulliken and NBO charges, the frontier orbital of 2a and 2b, 1H NMR spectra of 1a, 1b, 2a and 2b. CCDC 1057666–1057668. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj02100f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |