
Nano-zirconia as an excellent nano support for immobilization of sulfonic acid: a new, efficient and highly recyclable heterogeneous solid acid nanocatalyst for multicomponent reactions†
Ali Amoozadeh*,
Salman Rahmani,
Mehrnoosh Bitaraf,
Fatemeh Bolghan Abadi and
Elham Tabrizian
Faculty of Chemistry, Semnan University, Semnan 35131-19111, Iran. E-mail: aamozadeh@semnan.ac.ir; Fax: +98 (23) 33354110; Tel: +98 (23) 33366177
First published on 17th November 2015
Abstract
Nano-zirconia-supported sulfonic acid [nano-ZrO2-SO3H (n-ZrSA)] is synthesized by immobilizing sulfonic acid groups on the surface of nano zirconium dioxide to produce a novel heterogeneous reusable solid acid nanocatalyst. This new nanocatalyst is characterized by FT-IR spectroscopy, thermogravimetric analysis (TGA), X-ray diffraction (XRD), field emission scanning electron microscopy (FE-SEM), transmission electron microscopy (TEM), the Hammett acidity function and pH analysis. The introduced nano-zirconia-supported sulfonic acid is used as an efficient and recyclable catalyst for different heterocyclic multicomponent reactions such as the synthesis of hexahydroquinoline, 1,8-dioxo-decahydroacridine, polyhydroquinoline and 1,8-dioxo-octahydroxanthene derivatives. Optimization of the reaction conditions was studied using a central composite design (CCD) which is one of the most widely used response surface methodologies. The newly prepared heterogeneous solid acid nanocatalyst is easily separated and reusable for five cycles without any apparent loss of its catalytic activity, which confirmed the stability of the covalent bonding of the sulfonic acid groups. n-ZrSA has advantages such as its low cost, low toxicity, ease of preparation, good stability, high reusability and operational simplicity.
1. Introduction
Nowadays, heterogeneous solid acid catalysts are widely used for chemical reactions and processes. Acid catalyzed organic synthesis is an important way to produce numerous organic products. Sulfuric, nitric, hydrochloric, phosphoric and fluorhydric acids are common homogeneous acid catalysts that are widely used in chemical syntheses and industrial processes.1,2 However, these catalysts are not environmentally benign because of their problems such as difficult separation from the products and corrosive properties. These disadvantages of homogeneous acid catalysts cause different problems such as using specialized reaction equipment, increasing operational difficulties, high consumption of energy and the formation of large amounts of waste products. In recent years, due to these drawbacks, the development of novel, low cost, nontoxic, well separable and recyclable heterogeneous catalysts has been the most important challenge for scientists.3–8 Heterogenization of homogeneous catalysts by immobilization of sulfonic acid on various solid supports leads to the production of novel heterogeneous solid acid catalysts. This topic has been the focus of extensive research during the past decade.9–14 Transition metal nanoparticles are used as efficient catalysts for various synthetic organic transformations due to their high surface area-to-volume ratio and coordination sites which are mainly responsible for their catalytic activity.15 These properties lead to their wide application as excellent supports to produce new, efficient, nontoxic, environmentally friendly and reusable heterogeneous solid acid nanocatalysts.Zirconium is one of the most important transition metals in the earth’s crust (130 mg kg−1), which is not found in nature as a native metal. The silicate mineral of zirconium, known as zircon (ZrSiO4), is its principal source. Furthermore, zirconium dioxide (ZrO2) is commercially available. It is mainly used as a refractory material and opacifier, although it is used in small amounts as an alloying agent due to its strong resistance to corrosion. Considering the multitude of potential applications, zirconium dioxide is commonly used in numerous research fields as laboratory crucibles, metallurgical furnaces, refractory materials,16 oxygen sensors, fuel cell membranes, in ceramic production,17 as a protective coating on particles of titanium dioxide pigments,17 for strontium adsorption,18 acylation of 1,n-diols19 and for other applications.
Given the excellent properties of ZrO2 and the significance of heterogeneous solid acid catalysis, in a continuation of our last studies about heterogeneous solid acid nanocatalysts,20–22 we decided to investigate nano-ZrO2 as a new nano support. As sulfonation with chlorosulfonic acid is a convenient, fast and efficient method for heterogenization of homogeneous catalysts according to Zolfigol's report,23 the catalytic properties of nano-ZrO2 have been improved by its reaction with chlorosulfonic acid to produce nano-ZrO2-supported sulfonic acid (nano-ZrO2-SO3H) (Scheme 1).
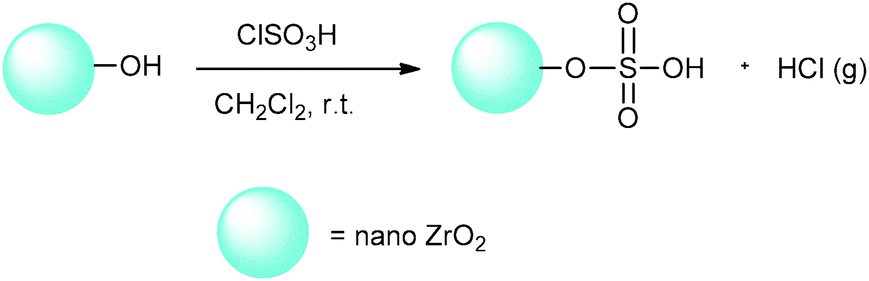 | ||
| Scheme 1 Preparation of nano-ZrO2-SO3H (n-ZrSA). | ||
2. Experimental
2.1. Materials and instruments
Chemicals were purchased from Merck chemical companies. Thin-Layer Chromatography (TLC) on commercial plates of silica gel 60 F254 was used to monitor the progress of the reactions. The products were characterized using FT-IR spectrometry, 1HNMR, 13CNMR and a CHN analyzer. 1H and 13C NMR spectra were recorded on a Bruker Avance Spectrometer 400 & 500 MHz using CDCl3-d and DMSO-d6 as solvents. The chemical shifts are expressed in parts per million (ppm) and tetramethylsilane (TMS) was used as an internal reference. Elemental analyses were performed using a Perkin Elmer CHN analyzer, 2400 series II. Melting points were recorded on a THERMO SCIENTIFIC 9100 apparatus. The wide angle X-ray diffraction spectrum was obtained by a Siemens D5000 (Siemens AG, Munich, Germany) X-ray diffractometer using Cu-Kα radiation of wavelength 1.54 Å. Field emission scanning electron microscopy (FE-SEM) was carried out using a Philips XL30 field emission scanning electron microscope (Royal Philips Electronics, Amsterdam, The Netherlands) operating at 20 kV. The sample was mounted on a double sided adhesive carbon disk and sputter-coated with a thin layer of gold to prevent sample charging problems. The Fourier transform infrared spectroscopy (FT-IR) spectrum was recorded on a Shimadzo FT-IR 8400 instrument. The n-ZrSA sample was mixed with a KBr powder and compressed into a pellet, wherein, the n-ZrSA powder was evenly dispersed. Thermogravimetric analysis (TGA) was conducted on a DuPont 2000 thermal analysis apparatus under an air atmosphere at a heating rate of 5 °C min−1.2.2. Preparation of nano-ZrO2
The zirconium dioxide nanoparticles were prepared using the chemical precipitation method. 10 g of ZrOCl2·8H2O was dissolved in 100 mL bidistilled water using a hot plate magnetic stirrer. The desired volume of 2 M NaOH was added to the above mentioned precursor solution until the pH value became 10. After 15 minutes, the precipitate was filtered off, washed and dried at 120 °C overnight. The dried ZrO2·nH2O was calcined at different temperatures from 500 to 1200 °C at a rate of 10 °C min−1 and kept at the respective temperature for 1 h.242.3. Preparation of nano-ZrO2-SO3H (n-ZrSA)
Chlorosulfonic acid (0.5 mL, 7.5 mmol) (Caution: this is highly corrosive and a water absorbent. Be careful when using this liquid. Protective gloves, protective clothing and eye and face protection equipment are also needed.) was added dropwise over a period of 30 min at room temperature to nano-ZrO2 (3.08 g, 25 mmol) in dry CH2Cl2 (20 mL). A suction flask equipped with a constant-pressure dropping funnel and a gas inlet tube for conducting the HCl gas over an adsorbing solution (i.e., water) was used. Stirring was continued until HCl evolution was finished. Then, the mixture was shaken for 30 min. A light cream powder of nano-zirconia-supported sulfonic acid was obtained. Afterward, CH2Cl2 was removed under a reduced pressure and the solid powder was washed with ethanol (10 mL) and dried at 100 °C.2.4. Investigation of n-ZrSA as a solid acid nanocatalyst for multicomponent one-pot synthesis of heterocyclic compounds
3. Results and discussion
3.1. Characterization of nano-ZrO2-SO3H
Nano-zirconia-supported sulfonic acid was synthesized by immobilizing sulfonic acid groups on the surface of nano zirconium dioxide and was characterized by FT-IR spectroscopy, thermogravimetric analysis (TGA), X-ray diffraction (XRD), field emission scanning electron microscopy (FE-SEM) and transmission electron microscopy (TEM), Hammett acidity function and pH analysis.FT-IR spectroscopy is employed to monitor the immobilization process by comparing the support and modified surface (Fig. 1). In graph (a) of Fig. 1, the absorbance bands around 520, 580 and 727 cm−1 were due to Zr–O–Zr vibration, and the absorbance bands around 3400–3500 cm−1 were due to the adsorbed water (Fig. 1, graph (a) and (b)) which is consistent with the reported IR spectra for nano-ZrO2.25 In graph (b) of Fig. 1, the absorption range of 1165–1248 and 1007–1065 cm−1 was due to O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SO asymmetric and symmetric stretching modes, respectively. The S–O stretching mode lies in the 560–608 cm−1 range. The obtained absorption and stretching bands completely differ from stretching vibrations of the OSO and S–O groups in sulfated zirconia.26 According to the obtained results, the presence of the sulfonic acid group has been proven which is consistent with the reported IR spectra of –SO3H.27
SO asymmetric and symmetric stretching modes, respectively. The S–O stretching mode lies in the 560–608 cm−1 range. The obtained absorption and stretching bands completely differ from stretching vibrations of the OSO and S–O groups in sulfated zirconia.26 According to the obtained results, the presence of the sulfonic acid group has been proven which is consistent with the reported IR spectra of –SO3H.27
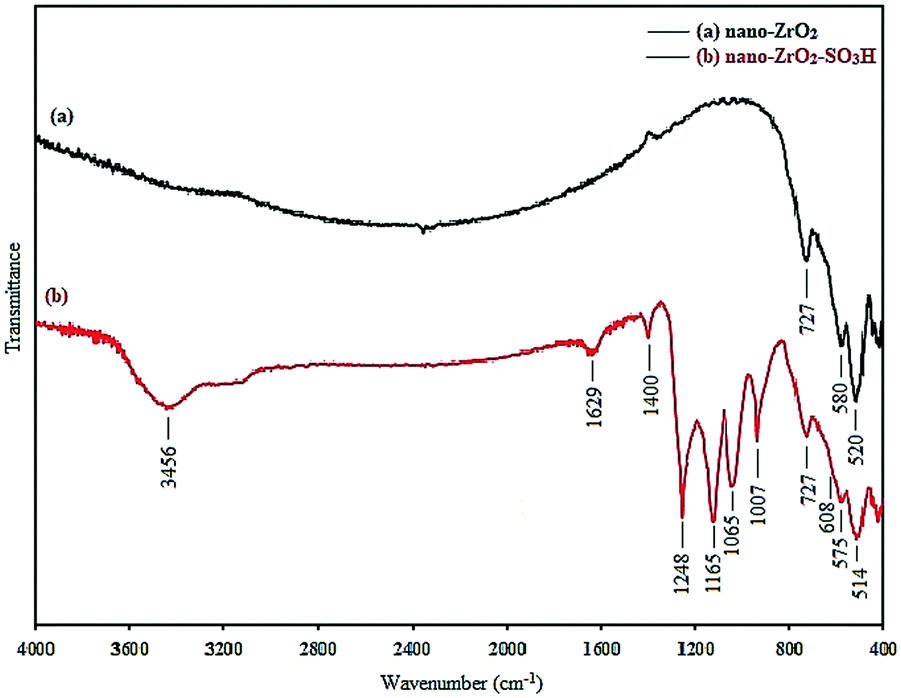 | ||
| Fig. 1 The FT-IR spectra of (a) the nano-ZrO2 powder and (b) nano-ZrO2-SO3H. | ||
Thermogravimetric analysis (TGA) was used to determine the percentage of the chemisorbed sulfonic acid group on the surface of nano-ZrO2. The TGA curve of nano-ZrO2 (Fig. 2a) displays a weight loss (5 wt%) below 100 °C which corresponds to the loss of the physically adsorbed water. Likewise, there is a slight weight loss (4 wt%) between 100 °C and 800 °C, which may contribute to the dehydroxylation of nano-ZrO2.
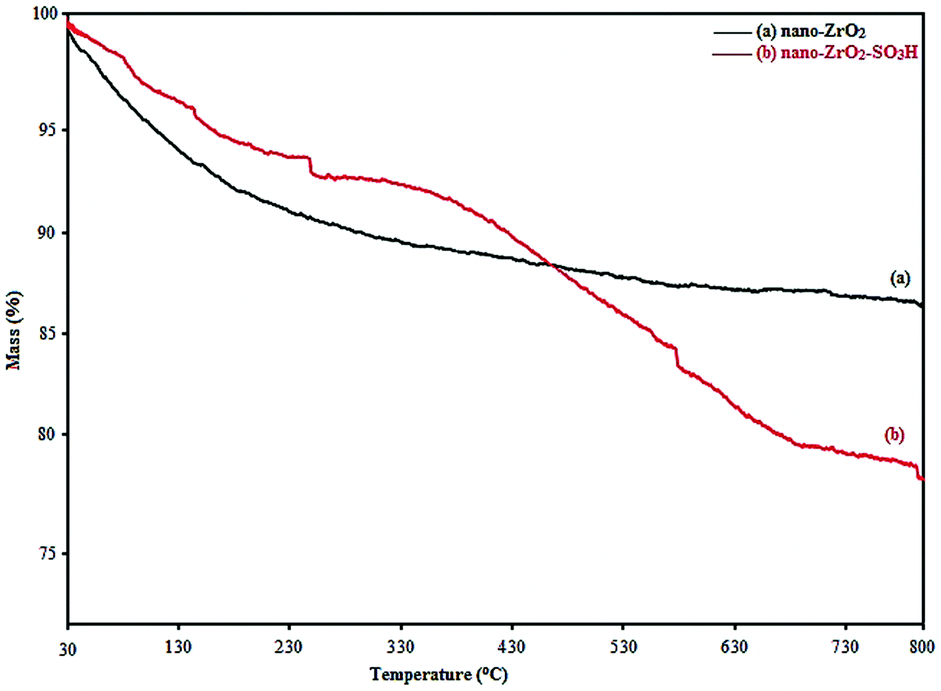 | ||
| Fig. 2 TGA curve of (a) nano-ZrO2 and (b) nano-ZrO2-SO3H. | ||
In the TGA curve of nano-ZrO2-SO3H (Fig. 2b), a three-stage decomposition is seen corresponding to different mass loss ranges. In the first region, a mass loss of approximately 4% weight occurred below 100 °C that was attributable to the loss of the trapped water from the catalyst. A mass loss of approximately 4% weight occurred between 100 and 330 °C that was related to the slow mass loss of the SO3H groups. Finally, a mass loss of approximately 14% weight occurred between 330–700 °C that was related to the sudden mass loss of the SO3H groups.27,28 From TGA, it can be concluded that nano-ZrO2-SO3H could be safely used in organic reactions due to its high thermal stability (up 150 °C).
The crystal phases of nano-ZrO2 and n-ZrSA were examined using the powder X-ray diffraction technique. Peak signals with Miller indices (100), (011), (110), (111), (111),(002), (200), (021), (211), (121), (112), (202), (022), (220), (122), (202), (013), (130), (310), (131), (131), (113), (213), (311), (023) and (222), as shown in Fig. 3a, confirm the formation of a nano zirconium dioxide monoclinic crystal phase which coincides with the JCPD 07-0343 standard. The average diameter of the nano-ZrO2 powder was also determined from the X-ray pattern using the Scherrer formula given as t = 0.9λ/B1/2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ, where t is the average crystal size, λ the X-ray wavelength used (1.54 Å), B1/2 the angular line width at half maximum intensity and θ is Bragg's angle. The average crystal size of the nano-ZrO2 powder for 2θ = 27.875° is calculated to be around 21.28 nm and average crystal size of the nano-ZrO2-SO3H powder for 2θ = 27.745° is calculated to be around 21.27 nm. According to the PXRD data for the peak at 27.875° (Fig. 3a), the maximum crystal growth of nano-ZrO2 is 554 nm. Fig. 3b illustrates the XRD patterns of modified nano-ZrO2. As shown in Fig. 3b, the peak intensities of nano-ZrO2-SO3H (n-ZrSA) are almost the same as those of nano-ZrO2 (Fig. 3a) indicating a retention of the monoclinic crystal phase structure during functionalization of nano-ZrO2.
cosθ, where t is the average crystal size, λ the X-ray wavelength used (1.54 Å), B1/2 the angular line width at half maximum intensity and θ is Bragg's angle. The average crystal size of the nano-ZrO2 powder for 2θ = 27.875° is calculated to be around 21.28 nm and average crystal size of the nano-ZrO2-SO3H powder for 2θ = 27.745° is calculated to be around 21.27 nm. According to the PXRD data for the peak at 27.875° (Fig. 3a), the maximum crystal growth of nano-ZrO2 is 554 nm. Fig. 3b illustrates the XRD patterns of modified nano-ZrO2. As shown in Fig. 3b, the peak intensities of nano-ZrO2-SO3H (n-ZrSA) are almost the same as those of nano-ZrO2 (Fig. 3a) indicating a retention of the monoclinic crystal phase structure during functionalization of nano-ZrO2.
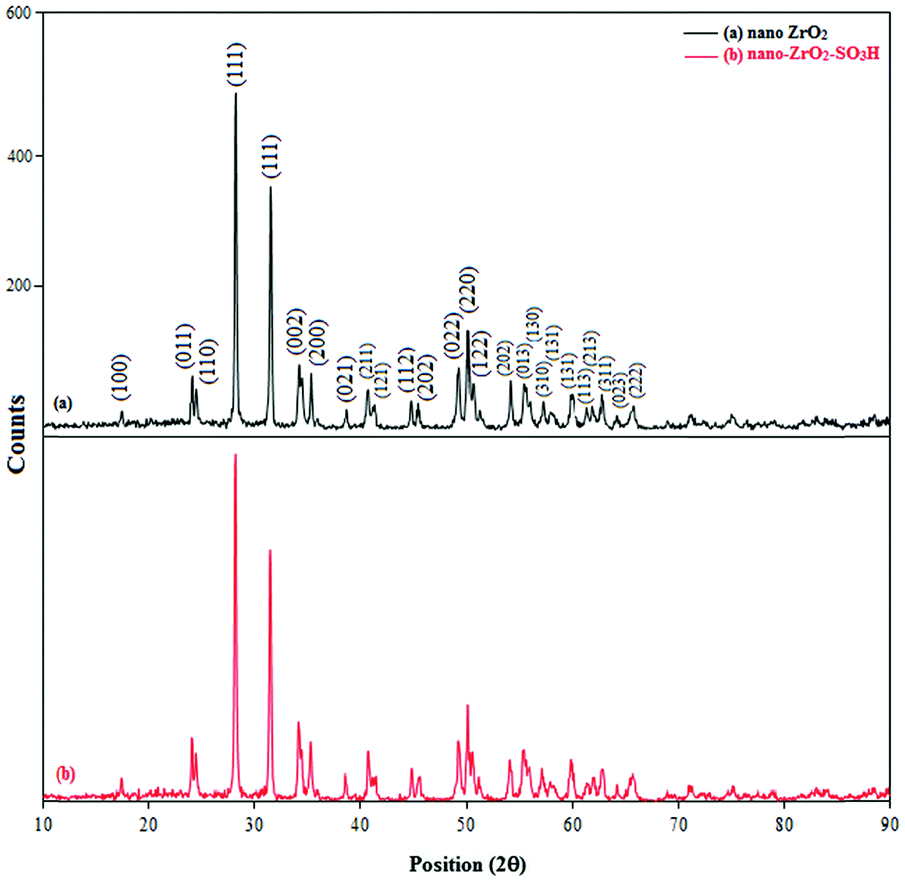 | ||
| Fig. 3 The X-ray diffraction pattern of (a) the nano-ZrO2 powder and (b) nano-ZrO2-SO3H. | ||
The morphology of nano-ZrO2 is shown in Fig. 4. The field emission scanning electron microscopy (FE-SEM) images of the nano-ZrO2 powder ascertain that the spherical nano-ZrO2 powder has a minimum particle size of about 30–40 nm (Fig. 4a and b). Meanwhile the FE-SEM images of n-ZrSA show nearly spherical nanoparticles with a minimum diameter of about 35–40 nm (Fig. 4c and d). It is clear that the modification process has been performed successfully.
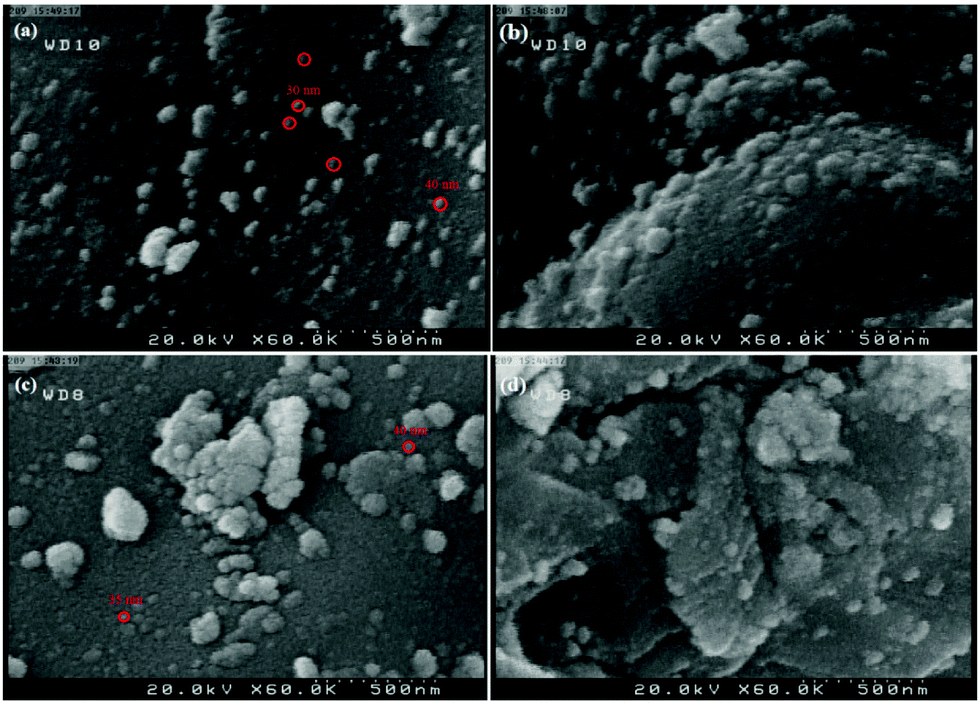 | ||
| Fig. 4 The FE-SEM of (a and b) nano-ZrO2 and (c and d) nano-ZrO2-SO3H. | ||
Fig. 5 displays the TEM images of nano-ZrO2-SO3H which confirm spherical-like and nanometer-sized particles of the catalyst.
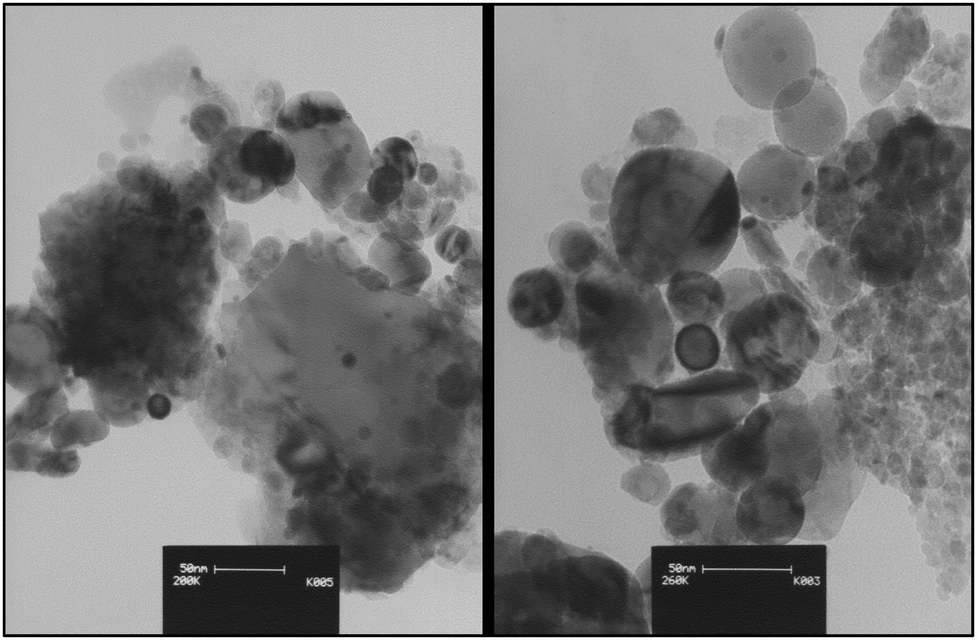 | ||
| Fig. 5 The TEM images of nano-ZrO2-SO3H. | ||
The acid strength of a catalyst can be effectively expressed using the Hammett indicator method29 which can be calculated using the following equation:
| H0 = pK(I)aq + log([I]s/[IH+]s), |
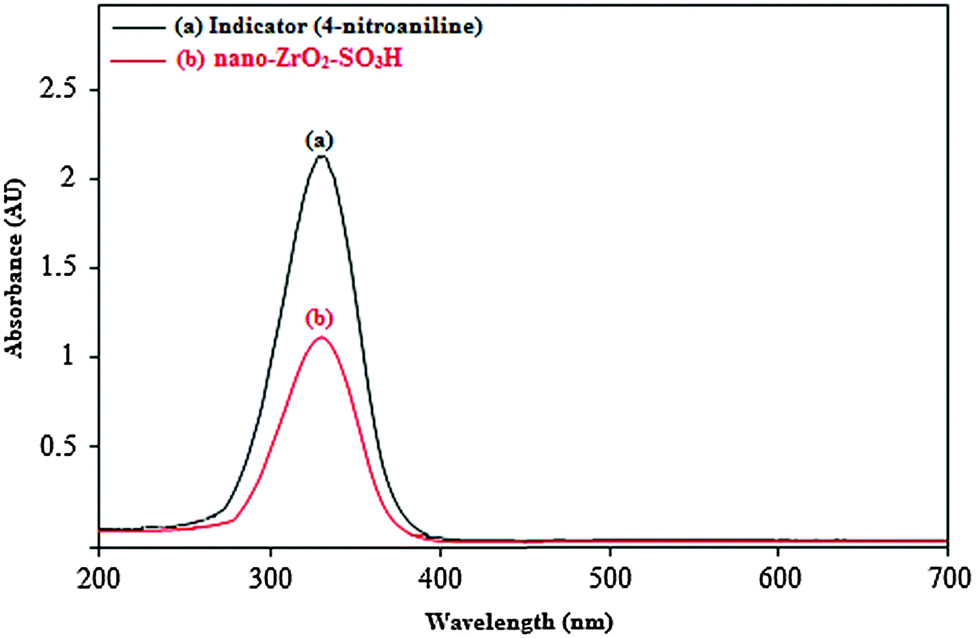 | ||
| Fig. 6 Absorption spectra of (a) 4-nitroaniline (indicator) and (b) nano-ZrO2-SO3H (catalyst) in CCl4. | ||
The obtained results are listed in Table 1, which shows the acidity strength of nano-ZrO2-SO3H. The results of the Hammett acidity function (H0) also confirm the synthesis of a new catalyst with a good density of acid sites (–SO3H groups) on the surface of nano-ZrO2 (Table 1).
| Entry | Catalyst | A max | [I]s (%) | [IH+]s (%) | H 0 |
|---|---|---|---|---|---|
| Condition for the UV-visible spectrum measurement: solvent, CCl4; indicator, 4-nitroaniline (pK(I)aq = 0.99), 1.44 × 10−4 mol L−1; catalyst, n-ZrSA (20 mg), 25 °C. | |||||
| 1 | — | 2.135 | 100 | 0 | — |
| 2 | n-ZrSA | 1.112 | 52.18 | 47.82 | 1.30 |
The acid capacities of n-ZrSA were determined by acid–base potentiometric titration of the aqueous suspension of a weighed amount of thoroughly washed catalyst. For this purpose, the prepared n-ZrSA was stored in a vacuum desiccator over an anhydrous silica gel, then, was dried at 120 °C for 6 hours. Next, the surface acidic protons of nano-ZrO2-SO3H were ion-exchanged with a saturated solution of NaCl (10 mL) by sonication. This process was repeated twice more, yielding 30 mL of the proton-exchanged brine solution and the obtained solution was titrated using a NaOH (0.01 M) solution in the presence of a pH meter. The loading of the acid sites on the synthesized catalyst was realized to about 3.9 mmol H+ per g of n-ZrSA.
The obtained results from the characterization methods showed that the sulfonic acid groups (–SO3H) were supported on the surface of nano-ZrO2. It is notable that nano-ZrO2-SO3H acts as a Lewis and a Brønsted acid simultaneously, in which nano-ZrO2 functions as a Lewis acid and sulfonic acids function as the Brønsted acid. Brønsted acids can usually provide a hydrogen bond which can initiate the catalytic procedure. According to the obtained results, we expected better catalytic activity for the nano-ZrO2-supported sulfonic acid compared with nano-ZrO2. In order to prove this hypothesis, we have decided to investigate its catalytic activity in some organic reactions.
3.2. Investigation of nano-ZrO2-SO3H in one-pot multicomponent reactions
Considering our research toward the development of solid acid catalysts and evaluating their catalytic activity for the synthesis of organic compounds,30–32 nano-titania-supported sulfonic acid20 and nano-WO3-supported sulfonic acid21 as novel and efficient nanocatalysts were recently reported. On the basis of the information obtained from the above mentioned studies, in this context, the catalytic activity of nano-ZrO2-supported sulfonic acid as a highly efficient, low cost, heterogeneous, reusable and inexpensive solid acid nanocatalyst was examined in multicomponent reactions for the synthesis of heterocyclic compounds such as hexahydroquinoline, 1,8-dioxo-decahydroacridine, polyhydroquinoline and 1,8-dioxo-octahydroxanthene derivatives. The structures of some of the final products were well characterized using spectral (IR, 1H NMR, 13C NMR) data.However, many of these methods suffer from disadvantages such as long reaction times, unsatisfactory yields, harsh reaction conditions, expensive reagents, hazardous and toxic solvents or tedious work-ups. Therefore, the introduction of efficient and economical catalysts that solve these drawbacks is desirable. For this purpose, we decided to examine the applicability of n-ZrSA in the promotion of the synthesis of hexahydroquinolines. To study the role of the catalyst, the reaction was carried out in catalyst free conditions. The obtained result was not desirable (25%). So, the prepared nano-ZrO2-SO3H was tested for the synthesis of hexahydroquinolines by a multicomponent one-pot reaction between dimedone (1) (1 mmol), benzaldehyde (2) (1 mmol), ammonium acetate (3) (1 mmol) and malononitrile (4) (1 mmol) under solvent free conditions as a model reaction (Scheme 2).
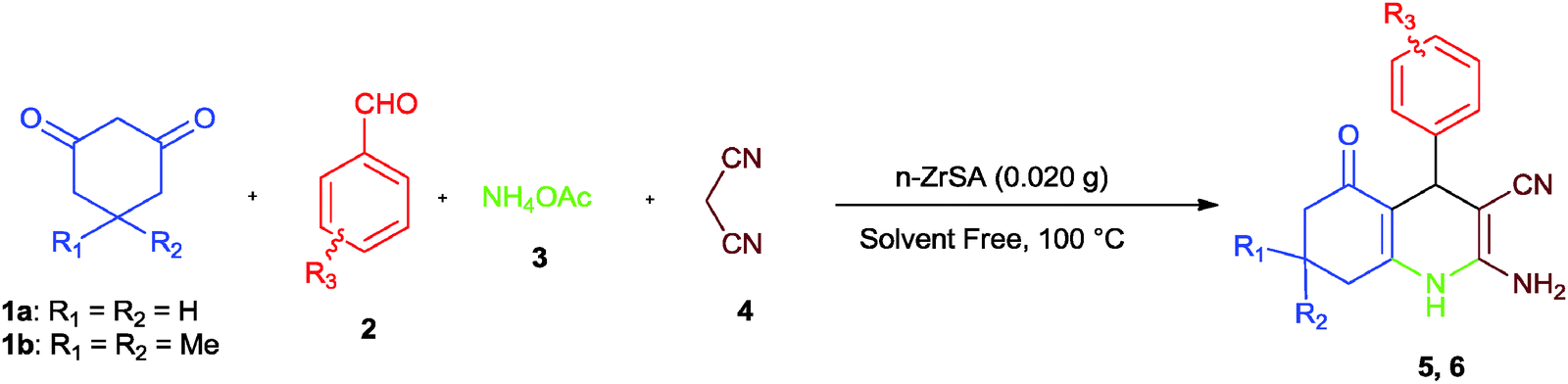 | ||
| Scheme 2 Synthesis of hexahydroquinoline derivatives by nano-ZrO2-SO3H. | ||
In order to find the optimal conditions for the synthesis of hexahydroquinoline derivatives, the central composite design (CCD), one of the most applicable types of the response surface model (RSM), was applied. It has been accepted as a more effective optimization method to attain improved responses via a fitted quadratic model which can be expressed by the following equation:
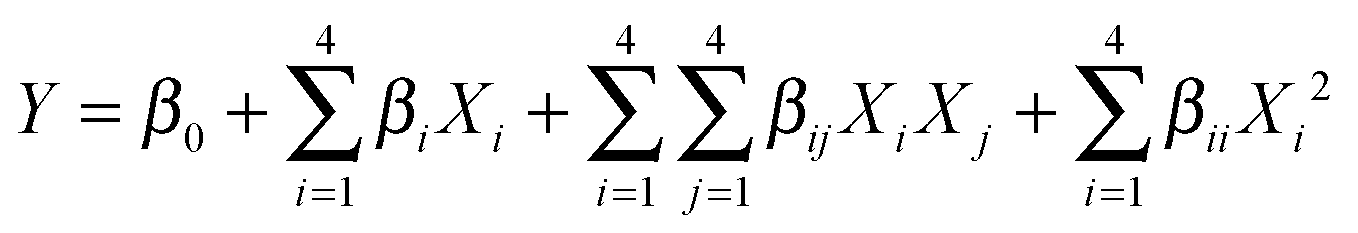
First of all, preliminary experiments were carried out to investigate the appropriate parameters and to determine the experimental domain. Considering these experiments, the effects of the catalyst amount (X1), temperature (X2), and reaction time (X3) were assessed with the reaction yield as the response. A five-level CCD, with three independent variables and their corresponding values, is shown in Table S1 (in the ESI†). Based on the CCD method, the total number of experiments was found to be 20 which consisted of eight full factorial points, six axial points and six central points. The conditions of the 20 trials along with their respective yields are shown in Table S2 (in the ESI†).
An analysis of variance (ANOVA) performed on the model can give valuable information on the significance of the fitted model and its terms. From the ANOVA, as shown in Table 2, p-values of the model and the lack of fit are lower and higher than 0.05, respectively, which means that the fitted model is significant at a confidence level of 95% and it does not require a reduction to lower orders. Also, the values for R-squared and adj. R-squared are above 0.9 and close to each other which indicate that the fitted model has high accuracy and reliability for the prediction of the reaction yield. In other words, there is good agreement between the experimental and predicted responses which confirms the suitability of the following fitted model in terms of being significantly linear and quadratic as well as the interaction of the terms on the basis of their p-values:
| Y = −544.56 + 21.00X1 + 5.59X2 + 14.23X3 + 0.04X1X2 − 0.55X12 − 0.03X22 − 0.37X32 |
| Source | Sum of squares | df | Mean square | F value | p-value prob > F |
|---|---|---|---|---|---|
| Model | 4188.54 | 9 | 465.39 | 129.79 | <0.0001 |
| X 1-catalyst amount | 713.91 | 1 | 713.91 | 199.09 | <0.0001 |
| X 2-temperature | 43.42 | 1 | 43.42 | 12.11 | 0.0059 |
| X 3-time | 1228.25 | 1 | 1228.25 | 342.53 | <0.0001 |
| X 1 X 2 | 18.24 | 1 | 18.24 | 5.09 | 0.0477 |
| X 1 X 3 | 8.86 | 1 | 8.86 | 2.47 | 0.1470 |
| X 2 X 3 | 0.097 | 1 | 0.097 | 0.027 | 0.8728 |
| X 1 2 | 1134.36 | 1 | 1134.36 | 316.34 | <0.0001 |
| X 2 2 | 135.38 | 1 | 135.38 | 37.75 | 0.0001 |
| X 3 2 | 1224.22 | 1 | 1224.22 | 341.40 | <0.0001 |
| Residual | 35.86 | 10 | 3.59 | ||
| Lack of fit | 24.67 | 5 | 4.93 | 2.20 | 0.2030 |
| Pure error | 11.19 | 5 | 2.24 | ||
| Cor. total | 4224.40 | 19 | |||
| R 2 = 0.99 | Adj.-R2 = 0.98 | Pred.-R2 = 0.95 | |||
It can be seen from the response equation that X1 has a more linear effect on the product yield, so the amount of catalyst can be considered as the main factor in the progression of the reaction.
In order to investigate the main interaction effects between two parameters on the yield of the reaction, three-dimensional profiles of the yield versus a pair of parameters were applied (Fig. 7). According to their p-values, the interaction of X1X2 is significant, which means that the simultaneous increase of the catalyst amount and reaction temperature causes the product yield to increase up until 0.020 g of the catalyst was used and a reaction temperature of 100 °C was reached. Meanwhile, higher loading of the catalyst and also the temperature, not only did not improve the yield to a higher extent, but also decreased the product yields most probably due to the formation of undesirable by-products.
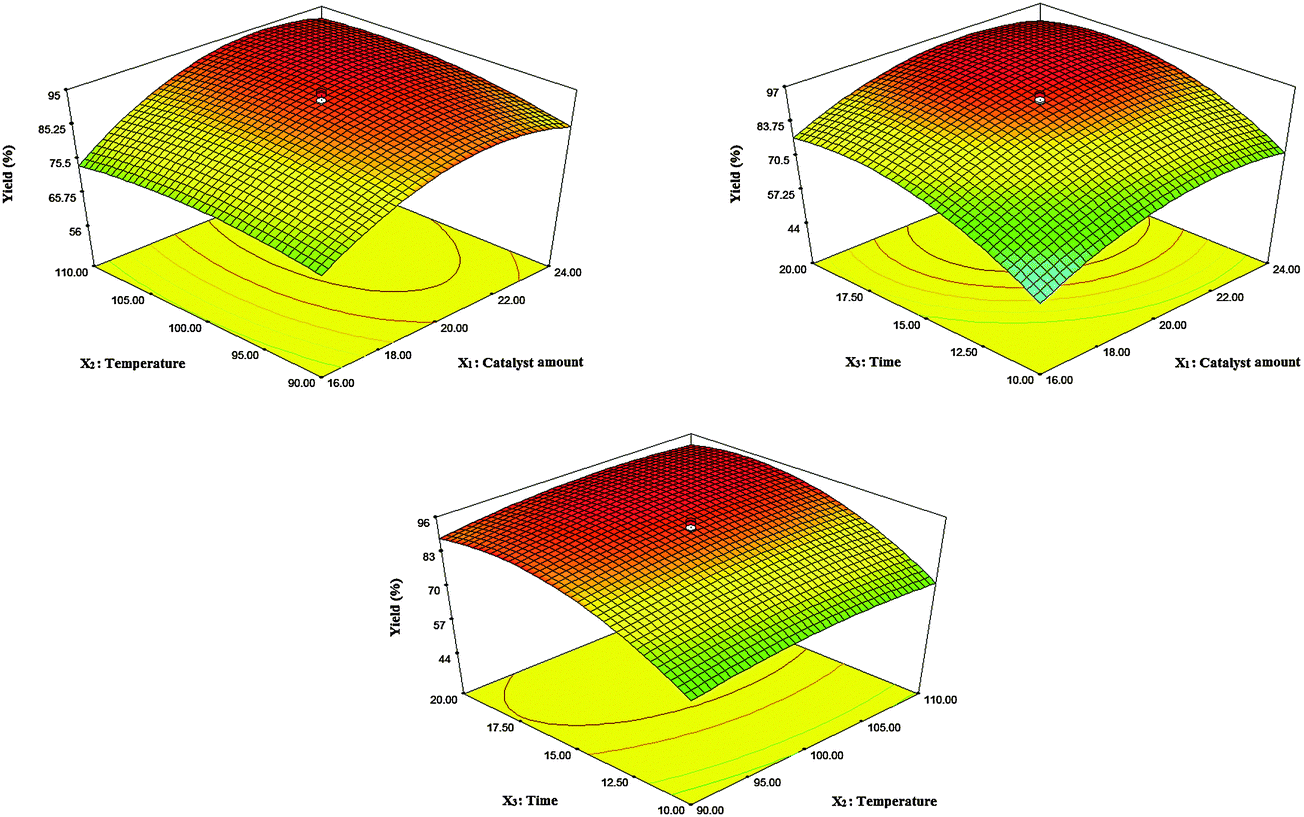 | ||
| Fig. 7 Three dimensional response surfaces for the effect of factors on the reaction yield. | ||
The main goal of this design was to optimize and maximize the yield of the reaction corresponding to the conditions of the experiment in which the response equation was maximized. In this work, determination of the optimal conditions was done with the aid of the desirability function using Design-Expert 7.0.0. First, the desired goals were selected for each factor and for the response to obtain the maximum product yield with a high desirability function (close to one). Then, conditions possessing high desirability were tested three times. A negligible difference between the average yields and the prediction values of the software confirms the high accuracy and precision of the optimal conditions. The results showed that 0.020 g of the catalyst, a 100 °C reaction temperature, and a 16 min reaction time were the optimal conditions for the synthesis of 2-amino-7,7-dimethyl-5-oxo-4-phenyl-1,4,5,6,7,8-hexahydroquinoline-3-carbonitrile.
To compare the efficiency of the solvent-free versus solvent conditions, the model reaction was performed under optimal conditions. The results showed that a higher yield and shorter reaction time were obtained when the reaction was carried out under solvent-free conditions.
After optimizing the conditions, the generality of the method was successfully studied by using various aromatic aldehydes (including aldehydes with electron-releasing substituents, electron-withdrawing substituents and halogens on the aromatic ring) and cyclic 1,3-diketone compounds. The obtained results are summarized in Table 3.
| Entry | Ar | R1 = R2 | Product | Time (min) | Yieldb (%) | Melting point (°C) | Literature m.p. (°C) |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: 1 (1 mmol), 2 (1 mmol), 3 (1 mmol), 4 (1 mmol), n-ZrSA (0.020 g) under solvent-free conditions at 100 °C. b Isolated yield. | |||||||
| 1 | –C6H5 | H | 5a | 15 | 90 | 256–259 | 25736 |
| 2 | o-Cl-C6H4 | H | 5b | 13 | 91 | 286–288 | 28636 |
| 3 | p-Cl-C6H4 | H | 5c | 15 | 93 | 288–289 | 28936 |
| 4 | p-F-C6H4 | H | 5d | 14 | 93 | 285–286 | 28636 |
| 5 | p-H3C-C6H4 | H | 5e | 23 | 89 | 258–260 | 25836 |
| 6 | p-H3CO-C6H4 | H | 5f | 25 | 87 | 275–278 | 27836 |
| 7 | m-O2N-C6H4 | H | 5g | 15 | 91 | 242–243 | 24336 |
| 8 | p-O2N-C6H4 | H | 5h | 13 | 93 | 259–262 | 26036 |
| 9 | –C6H5 | CH3 | 6a | 16 | 94 | 234–237 | 275–27737 |
| 10 | p-H3C-C6H4 | CH3 | 6b | 22 | 91 | >300 | 294–29537 |
| 11 | p-H3CO-C6H4 | CH3 | 6c | 22 | 90 | 287–290 | 289–29337 |
| 12 | m-O2N-C6H4 | CH3 | 6d | 14 | 93 | 280–284 | 282–28338 |
| 13 | o-Cl-C6H4 | CH3 | 6e | 12 | 94 | 273–275 | 273–27637 |
| 14 | p-Cl-C6H4 | CH3 | 6f | 14 | 95 | 288–291 | 290–29138 |
| 15 | p-F-C6H4 | CH3 | 6g | 12 | 95 | 298–301 | 299–30038 |
| 16 | p-Br-C6H4 | CH3 | 6h | 14 | 94 | 295–297 | 295–29638 |
| 17 | p-(CH3)2N-C6H4 | CH3 | 6i | 25 | 90 | >300 | >30038 |
| 18 | 2,6-Cl2-C6H3 | CH3 | 6j | 18 | 92 | >300 | >30038 |
The present method not only affords the products in excellent yields but also avoids the problems associated with catalyst cost, handling, safety and pollution.
As indicated in Table 3, the reaction works easily for a vast variety of aromatic aldehydes with both electron-donating and electron-withdrawing groups and different cyclic di-ketones to give corresponding hexahydroquinoline derivatives in good to excellent yields. In almost all cases, the reactions proceeded smoothly within 12–25 minutes.
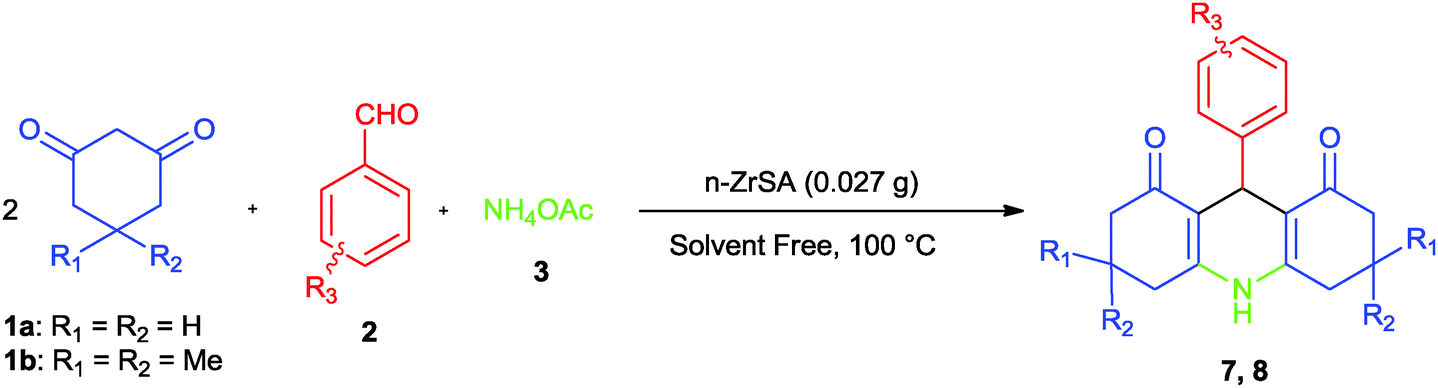 | ||
| Scheme 3 Synthesis of 1,8-dioxo-decahydroacridine derivatives by nano-ZrO2-SO3H. | ||
Due to the effect of n-ZrSA, the synthesis of 1,8-dioxo-decahydroacridine was studied in catalyst free conditions where the obtained result illustrates that the yield was low (8%). The activity of n-ZrSA is compared with nano-ZrO2. The obtained results showed that n-ZrSA (91% yield) was a more suitable option rather than nano-ZrO2 (6% yield).
To examine the limitation and the scope of the reaction, the synthesis of different 1,8-dioxo-decahydroacridine derivatives has been investigated. The obtained results are summarized in Table 4.
| Entry | Ar | R1 = R2 | Product | Time (min) | Yieldb (%) | Melting point (°C) | Literature m.p. (°C) |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: 1 (2 mmol), 2 (1 mmol), 3 (1 mmol), n-ZrSA (0.027 g) under solvent free conditions at 100 °C. b Isolated yield. | |||||||
| 1 | –C6H5 | H | 7a | 50 | 89 | 280–282 | 279–28042 |
| 2 | p-Cl-C6H4 | H | 7b | 45 | 92 | 295–297 | 297–29842 |
| 3 | p-Br-C6H4 | H | 7c | 45 | 91 | 310–312 | 310–31242 |
| 4 | p-H3C-C6H4 | H | 7d | 55 | 87 | 254–256 | 255–25742 |
| 5 | p-HO-C6H4 | H | 7e | 65 | 84 | 304–305 | 303–30542 |
| 6 | p-H3CO-C6H4 | H | 7f | 60 | 86 | 302–304 | 303–30542 |
| 7 | m-O2N-C6H4 | H | 7g | 45 | 93 | 280–282 | 282–28442 |
| 8 | –C6H5 | CH3 | 8a | 45 | 91 | 290–291 | 290–29142 |
| 9 | p-H3CO-C6H4 | CH3 | 8b | 55 | 88 | 274–276 | 276–27842 |
| 10 | p-HO-C6H4 | CH3 | 8c | 60 | 86 | 307–308 | 303–30542 |
| 11 | p-H3C-C6H4 | CH3 | 8d | 52 | 89 | 269–270 | 269–27143 |
| 12 | p-F-C6H4 | CH3 | 8e | 40 | 93 | 292–294 | 292–29443 |
| 13 | p-Cl-C6H4 | CH3 | 8f | 40 | 93 | 299–302 | 298–30042 |
| 14 | o-Cl-C6H4 | CH3 | 8g | 38 | 92 | 221–224 | 220–22243 |
| 15 | p-Br-C6H4 | CH3 | 8h | 40 | 92 | 308–311 | 313–31543 |
| 16 | p-O2N-C6H4 | CH3 | 8i | 35 | 94 | 312–314 | 290–29243 |
| 17 | m-O2N-C6H4 | CH3 | 8j | 37 | 93 | 282–284 | 293–29543 |
| 18 | 2-Naphthaldehyde | CH3 | 8k | 50 | 88 | 358–359 | 265–26743 |
| 19 | p-(CH3)2N-C6H4 | CH3 | 8l | 50 | 89 | 264–267 | 220–22243 |
As indicated in Table 4, the new conditions are very suitable for a vast variety of aromatic aldehydes with both electron-donating and electron-withdrawing groups to give corresponding 1,8-dioxo-decahydroacridine derivatives in good to excellent yields. In nearly all cases, the reactions proceeded smoothly within 35–65 minutes. However, it is notable that the substituted aromatic aldehydes with electron-withdrawing groups increase the rate of the reaction (Table 4, entries 2, 3, 7 and 12–17) probably by activating the carbonyl group as an electrophilic center. Conversely, in the case of the electron-donating groups, the reaction proceeded more slowly (Table 4, entries 4–6 and 9–11).
Synthesis of polyhydroquinolines was performed by a three component condensation between dimedone (1) (1 mmol), benzaldehyde (2) (1 mmol), ammonium acetate (3) (1 mmol) and ethylacetoacetate (9) (1 mmol) with 0.019 g of n-ZrSA at 100 °C under solvent free conditions as a model reaction (Scheme 4).
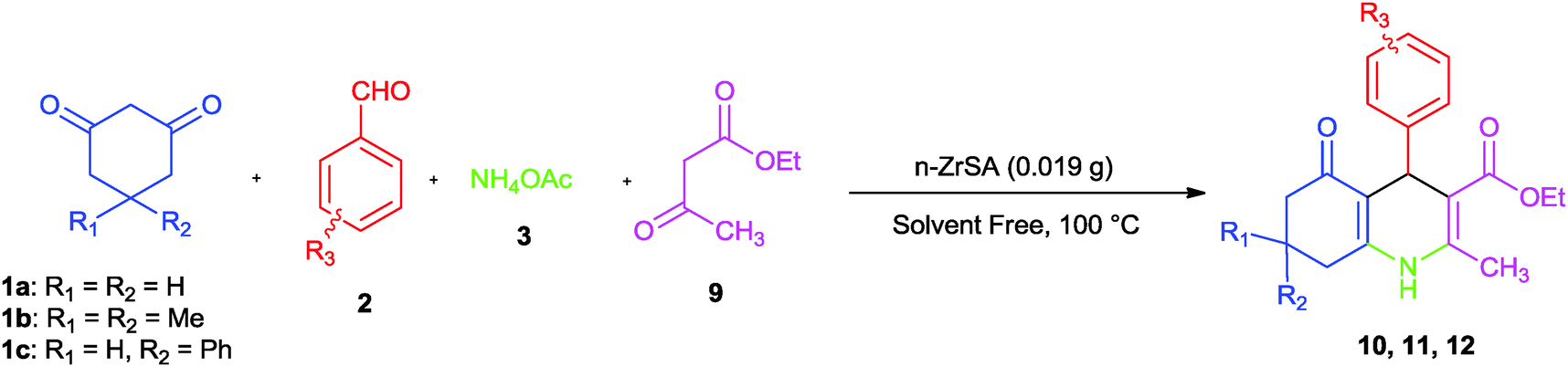 | ||
| Scheme 4 Synthesis of polyhydroquinoline derivatives by nano-ZrO2-SO3H. | ||
According to the low yield of the product reaction under catalyst free conditions (22%), the catalytic activity of n-ZrSA was compared with nano-ZrO2. The obtained results showed that n-ZrSA (94% yield) was a more suitable option rather than nano-ZrO2 (18% yield). To examine the versatility of the optimal reaction conditions and the tolerance of functional groups for the Hantzsch reaction, the synthesis of different polyhydroquinolines has been examined. The obtained results are summarized in Table 5.
| Entry | Ar | R1, R2 | Product | Time (min) | Yieldb (%) | Melting point (°C) | Literature m.p. (°C) |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: 1 (1 mmol), 2 (1 mmol), 3 (1 mmol), 9 (1 mmol), n-ZrSA (0.019 g) as the catalyst under solvent-free conditions at 100 °C. b Isolated yields. | |||||||
| 1 | –C6H5 | H | 10a | 37 | 91 | 239–241 | 240–24246 |
| 2 | p-H3C-C6H4 | H | 10b | 42 | 93 | 242–244 | 241–24247 |
| 3 | p-H3CO-C6H4 | H | 10c | 45 | 94 | 192–194 | 193–19547 |
| 4 | p-F-C6H4 | H | 10d | 33 | 91 | 244–245 | 243–24447 |
| 5 | p-Cl-C6H4 | H | 10e | 32 | 86 | 236–237 | 234–23547 |
| 6 | p-HO-C6H4 | H | 10f | 50 | 84 | 220–221 | 220–22247 |
| 7 | o-O2N-C6H4 | H | 10g | 30 | 90 | 191–193 | 190–19147 |
| 8 | m-O2N-C6H4 | H | 10h | 32 | 89 | 199–201 | 198–20047 |
| 9 | p-O2N-C6H4 | H | 10i | 30 | 89 | 203–204 | 204–20547 |
| 10 | –C6H5 | CH3 | 11a | 35 | 94 | 210–211 | 202–20446 |
| 11 | o-O2N-C6H4 | CH3 | 11b | 30 | 96 | 203–205 | 207–20848 |
| 12 | m-O2N-C6H4 | CH3 | 11c | 32 | 94 | 170–171 | 182–18448 |
| 13 | p-O2N-C6H4 | CH3 | 11d | 28 | 96 | 207–209 | 202–20348 |
| 14 | o-Cl-C6H4 | CH3 | 11e | 30 | 94 | 207–209 | 208–20946 |
| 15 | p-Cl-C6H4 | CH3 | 11f | 30 | 95 | 243–245 | 245–24746 |
| 16 | p-F-C6H4 | CH3 | 11g | 30 | 95 | 180–181 | 184–18648 |
| 17 | p-Br-C6H4 | CH3 | 11h | 30 | 95 | 250–251 | 250–25148 |
| 18 | p-HO-C6H4 | CH3 | 11i | 45 | 89 | 232–233 | 232–23446 |
| 19 | p-H3CO-C6H4 | CH3 | 11j | 45 | 90 | 255–256 | 258–25946 |
| 20 | p-H3C-C6H4 | CH3 | 11k | 40 | 91 | 283–284 | 260–26248 |
| 21 | p-(CH3)2N-C6H4 | CH3 | 11l | 45 | 89 | 263–265 | 263–26446 |
| 22 | 2,6-DiCl-C6H3 | CH3 | 11m | 30 | 90 | 243–245 | 244–24646 |
| 23 | 2-HO–5-Br-C6H3 | CH3 | 11n | 50 | 88 | 246–248 | 24746 |
| 25 | –C6H4 | H, Ph | 12a | 35 | 93 | 213–215 | 213–21546 |
| 26 | p-H3CO-C6H4 | H, Ph | 12b | 40 | 88 | 236–238 | 236–23846 |
| 27 | p-Cl-C6H4 | H, Ph | 12c | 32 | 92 | 190–192 | 190–19246 |
As indicated in Table 5, the n-ZrSA catalyzed Hantzsch reaction was very suitable for the synthesis of a vast variety of polyhydroquinoline derivatives by condensation between different aromatic aldehydes with both electron-donating and electron-withdrawing groups and cyclic di-ketones to give a corresponding product in good to excellent yields.
The synthesis of 1,8-dioxo-octahydroxanthene derivatives was studied by condensation between dimedone (1) (2 mmol) and benzaldehyde (2) (1 mmol) under solvent free conditions as a model reaction (Scheme 5). In order to explore the efficiency of n-ZrSA, the model reaction was carried out under the catalyst free conditions and compared with n-ZrSA and nano-ZrO2. The obtained results showed a higher yield for n-ZrSA (94% yield) compared to catalyst free (21% yield) and nano-ZrO2 (25% yield).
 | ||
| Scheme 5 Synthesis of 1,8-dioxo-octahydroxanthene derivatives by nano-ZrO2-SO3H. | ||
The scope of octahydroxanthene synthesis in the presence of nano-ZrO2-SO3H was successfully studied using various aromatic aldehydes (including aldehydes with electron-releasing substituents, electron-withdrawing substituents and halogens on the aromatic ring) and cyclic 1,3-diketone compounds. The results are summarized in Table 6.
| Entry | Ar | R1 | R2 | Product | Time (h) | Yieldb (%) | Melting point (°C) | Literature m.p. (°C) |
|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: 1 (2 mmol), 2 (1 mmol), n-ZrSA (0.039 g) under solvent-free conditions at 100 °C. b Isolated yield. | ||||||||
| 1 | –C6H5 | H | H | 13a | 2.1 | 92 | 213–215 | 213–21521 |
| 2 | p-H3C-C6H4 | H | H | 13b | 2.1 | 88 | 244–246 | 244–24621 |
| 3 | p-H3CO-C6H4 | H | H | 13c | 2.2 | 87 | 190–191 | 190–19121 |
| 4 | p-O2N-C6H4 | H | H | 13d | 1.8 | 93 | 234–236 | 234–23621 |
| 5 | p-Br-C6H4 | H | H | 13e | 1.7 | 90 | 222–225 | 222–22521 |
| 6 | –C6H5 | CH3 | CH3 | 14a | 2 | 94 | 203–204 | 205–20654 |
| 7 | o-H3C-C6H4 | CH3 | CH3 | 14b | 2.1 | 88 | 230–232 | 230–23221 |
| 8 | p-H3C-C6H4 | CH3 | CH3 | 14c | 2.2 | 89 | 212–214 | 220–22252 |
| 9 | p-H3CO-C6H4 | CH3 | CH3 | 14d | 2.2 | 90 | 242–244 | 250–25152 |
| 10 | p-HO-C6H4 | CH3 | CH3 | 14e | 2.3 | 86 | 246–247 | 246–24855 |
| 11 | p-F-C6H4 | CH3 | CH3 | 14f | 1.6 | 94 | 206–207 | 208–20955 |
| 12 | p-Cl-C6H4 | CH3 | CH3 | 14g | 1.6 | 93 | 237–239 | 236–23752 |
| 13 | p-Br-C6H4 | CH3 | CH3 | 14h | 1.7 | 90 | 264–267 | 263-26556 |
| 14 | o-O2N-C6H4 | CH3 | CH3 | 14i | 1.8 | 94 | 244–245 | 246–24855 |
| 15 | m-O2N-C6H4 | CH3 | CH3 | 14j | 1.8 | 92 | 168–169 | 167–16854 |
| 16 | p-O2N-C6H4 | CH3 | CH3 | 14k | 1.7 | 95 | 222–224 | 229–23052 |
| 17 | C6H5–CHCH– |
CH3 | CH3 | 14l | 2.3 | 84 | 256–258 | 256–25821 |
| 18 | 2-Naphthaldehyde | CH3 | CH3 | 14m | 2.1 | 90 | 234–236 | 234–23521 |
| 19 | 2-OH–5-Br-C6H3 | CH3 | CH3 | 14n | 2.3 | 84 | 268–271 | 268–27121 |
| 20 | –C6H5 | H | Ph | 15a | 2.1 | 90 | 196–198 | 196–19821 |
| 21 | p-Cl-C6H4 | H | Ph | 15b | 2.2 | 89 | 234–236 | 234–23621 |
| 22 | p-H3C-C6H4 | H | Ph | 15c | 2.2 | 89 | 204–208 | 204–20821 |
| 23 | p-H3CO-C6H4 | H | Ph | 15d | 1.9 | 91 | 212–215 | 212–21521 |
As shown in Table 6, fortunately the new catalyst also works very well for a vast variety of 1,8-dioxo-octahydroxanthene derivatives.
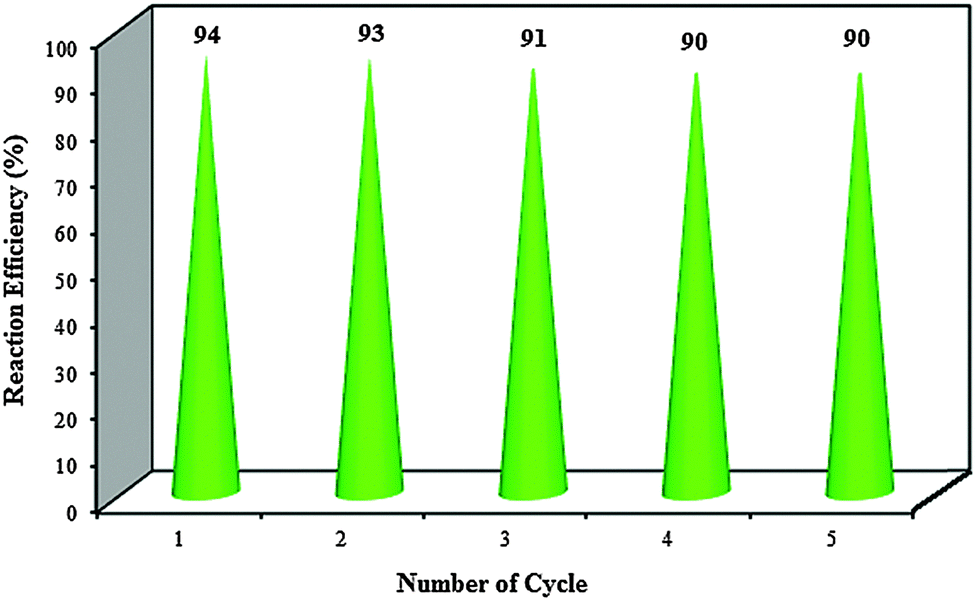 | ||
| Fig. 8 Reusability of nano-ZrO2-SO3H. | ||
4. Conclusion
In summary, for the first time, the preparation and characterization of n-ZrO2-SO3H as an active and efficient heterogeneous acidic nanocatalyst were described. The catalytic activity of the catalyst was probed through the synthesis of hexahydroquinoline, 1,8-dioxo-octahydroacridine, polyhydroquinoline and 1,8-dioxo-octahydroxanthene derivatives by a one-pot multicomponent reaction strategy under solvent-free conditions. All the reactions work easily for a variety of aldehydes with both electron-donating and electron-withdrawing groups to give corresponding products in excellent yields. The catalyst was reused for five consecutive cycles with consistent activity. Easy preparation and separation, high reusability and excellent catalytic performance are the main advantages of the synthesized catalyst.Acknowledgements
We gratefully acknowledge the Department of Chemistry of Semnan University for supporting this work.References
- M. D. González, Y. Cesteros, J. Llorca and P. Salagre, J. Catal., 2012, 290, 202–209 CrossRef.
- J. A. Melero, R. van Grieken and G. Morales, Chem. Rev., 2006, 106, 3790–3812 CrossRef CAS PubMed.
- N. Ahmed and Z. N. Siddiqui, J. Mol. Catal. A: Chem., 2014, 387, 45–56 CrossRef CAS.
- R. D. Andrei, M. I. Popa, F. Fajula and V. Hulea, J. Catal., 2015, 323, 76–84 CrossRef CAS.
- M. Cao, D. Wu, W. Su and R. Cao, J. Catal., 2015, 321, 62–69 CrossRef CAS.
- S. Dabral, S. Nishimura and K. Ebitani, ChemSusChem, 2014, 7, 260–267 CrossRef CAS PubMed.
- R. Pagadala, S. Maddila, V. D. B. C. Dasireddy and S. B. Jonnalagadda, Catal. Commun., 2014, 45, 148–152 CrossRef CAS.
- S. Shabbir, Y. Lee and H. Rhee, J. Catal., 2015, 322, 104–108 CrossRef CAS.
- C.-H. Kuo, A. S. Poyraz, L. Jin, Y. Meng, L. Pahalagedara, S.-Y. Chen, D. A. Kriz, C. Guild, A. Gudz and S. L. Suib, Green Chem., 2014, 16, 785–791 RSC.
- A. Mobaraki, B. Movassagh and B. Karimi, Appl. Catal., A, 2014, 472, 123–133 CrossRef CAS.
- R. Parella and S. A. Babu, Catal. Commun., 2012, 29, 118–121 CrossRef CAS.
- Z. N. Siddiqui and N. Ahmed, Appl. Organomet. Chem., 2013, 27, 553–561 CAS.
- H. Wan, Z. Wu, W. Chen, G. Guan, Y. Cai, C. Chen, Z. Li and X. Liu, J. Mol. Catal. A: Chem., 2015, 398, 127–132 CrossRef CAS.
- M. A. Zolfigol, A. Khazaei, M. Safaiee, M. Mokhlesi, R. Rostamian, M. Bagheri, M. Shiri and H. G. Kruger, J. Mol. Catal. A: Chem., 2013, 370, 80–86 CrossRef CAS.
- J. Deng, L.-P. Mo, F.-Y. Zhao, L.-L. Hou, L. Yang and Z.-H. Zhang, Green Chem., 2011, 13, 2576–2584 RSC.
- D. R. Lide, CRC Handbook of Chemistry and Physics 4, CRC Press, New York, 2007–2008, p. 42 Search PubMed.
- R. Nielsen, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2005 Search PubMed.
- H. Tel, Y. Alta, M. Eral, S. Sert, B. Cetinkaya and S. Inan, Chem. Eng. J., 2010, 161, 151–160 CrossRef CAS.
- V. Singh, V. Sapehiyia and G. L. Kad, J. Mol. Catal. A: Chem., 2004, 210, 119–124 CrossRef CAS.
- S. Rahmani, A. Amoozadeh and E. Kolvari, Catal. Commun., 2014, 56, 184–188 CrossRef CAS.
- A. Amoozadeh and S. Rahmani, J. Mol. Catal. A: Chem., 2015, 306, 96–107 CrossRef.
- A. Amoozadeh, S. Golian and S. Rahmani, RSC Adv., 2015, 5, 45974–45982 RSC.
- M. A. Zolfigol, Tetrahedron Lett., 2001, 57, 9509–9511 CrossRef CAS.
- K. Geethalakshmi, T. Prabhakaran and J. Hemalatha, Inter. Schol. Scien. Res. Innov., 2012, 6(4), 150–153 Search PubMed.
- M. Ranjbar, M. Yousei, M. Lahooti and A. Malekzadeh, Int. J. Nanosci. Nanotechnol., 2012, 8, 191–196 Search PubMed.
- G. D. Yadav, N. P. Ajgaonkar and A. Varma, J. Catal., 2012, 292, 99–110 CrossRef CAS.
- H. R. Shaterian, M. Ghashang and M. Feyzi, Appl. Catal., A, 2008, 345, 128–133 CrossRef CAS.
- M. A. Zolfigol, V. Khakyzadeh, A. R. Moosavi-Zare, G. Chehardoli, F. Derakhshan-Panah, A. Zare and O. Khaledian, Sci. Iran., 2012, 19(6), 1584–1590 CrossRef CAS.
- H. Xing, T. Wang, Z. Zhou and Y. Dai, J. Mol. Catal. A: Chem., 2007, 264, 53–59 CrossRef CAS.
- A. Amoozadeh, E. Tabrizian and S. Rahmani, C. R. Chim., 2015, 18, 848–857 CrossRef CAS.
- E. Tabrizian, A. Amoozadeh, S. Rahmani, E. Imanifar, S. Azhari and M. Malmir, Chin. Chem. Lett., 2015, 26, 1278–1282 CrossRef CAS.
- E. Kolvari, A. Amoozadeh, N. Koukabi, S. Otokesh and M. Isari, Tetrahedron Lett., 2014, 55, 3648–3651 CrossRef CAS.
- D. Doube, M. Bloun, C. Brideau, C. Chan, S. Desmarais, D. Eithier, J. P. Falgueyeret, R. W. Friesen, M. Girad, Y. Girad, J. Guay, P. Tagari and R. N. Yong, Bioorg. Med. Chem. Lett., 1998, 8, 1225–1230 CrossRef.
- M. P. Maguire, K. R. Sheets, K. Mcvety, A. P. Spada and A. Ziberstein, J. Med. Chem., 1994, 37, 2129–2137 CrossRef CAS PubMed.
- G. Roma, M. D. Braccio, G. Grossi and M. Chia, Eur. J. Med. Chem., 2000, 35, 1021–1035 CrossRef CAS PubMed.
- A. R. Gholap, K. S. Toti, F. Shirazi, R. Kumari, M. K. Bhat, M. V. Deshpande and K. V. Srinivasan, Bioorg. Med. Chem. Lett., 2007, 15, 6705–6715 CrossRef CAS PubMed.
- S. Kumar, P. Sharma, K. K. Kapoor and M. S. Hunda, Tetrahedron, 2008, 64, 536–542 CrossRef CAS.
- S. Tu, J. Zhang, X. Zhu, Y. Zhang, Q. Wang, J. Xu, B. Jiang, R. Jia, J. Zhang and F. Shi, J. Heterocycl. Chem., 2006, 43, 985–988 CrossRef CAS.
- J.-C. Xu, W.-M. Li, H. Zheng, Y.-F. Lai and P.-F. Zhang, Tetrahedron, 2011, 67, 9582–9587 CrossRef CAS.
- O. Berkan, B. Sarac, R. Simsek, S. Yildirim, Y. Sariogli and C. Safak, Eur. J. Med. Chem., 2002, 37(6), 519–523 CrossRef CAS PubMed.
- P. W. Groundwater and M. A. Munawar, Adv. Heterocycl. Chem., 1997, 70, 90–162 CrossRef.
- S. Khaksar, S. M. Vahdat, M. Akbari and S. Baghery, Arabian J. Chem., 2014 DOI:10.1016/j.arabjc.2014.10.026.
- M. M. Masoud Nasr-Esfahani and T. Abdizadeh, C. R. Chim., 2015, 18, 547–553 CrossRef.
- A. Srivastava and C. Nizamuddin, Indian J. Heterocycl. Chem., 2004, 13(3), 261–264 CAS.
- M. J. Wainwright, J. Antimicrob. Chemother., 2011, 47(1), 1–13 CrossRef.
- S. Otokesh, N. Koukabi, E. Kolvari, A. Amoozadeh, M. Malmir and S. Azhari, S. Afr. J. Chem., 2015, 68, 15–20 CrossRef CAS.
- S. Ko, M. N. V. Sastry, C. Lin and C.-F. Yao, Tetrahedron Lett., 2005, 46, 5771–5774 CrossRef CAS.
- B. Janardhan, B. Ranjitha and P. A. Crooks, J. Saudi Chem. Soc., 2014, 5, 722–726 CrossRef.
- A. N. Dadhania, V. K. Raval and D. K. Raval, J. Saudi Chem. Soc., 2014 DOI:10.1016/j.jscs.2013.12.003.
- R. Giri, J. R. Goodell and C. Xing, Bioorg. Med. Chem., 2010, 18, 1456–1463 CrossRef CAS PubMed.
- N. Mulakayala, P. V. N. S. Murthy and D. Rambabu, Bioorg. Med. Chem. Lett., 2012, 22, 2186–2191 CrossRef CAS PubMed.
- S. Rostamizadeh, A. M. Amani, G. H. Mahdavinia, G. Amiri and H. Sepehrian, Ultrason. Sonochem., 2010, 17, 306–309 CrossRef CAS PubMed.
- P. P. Salvi, A. M. Mandharea, A. S. Sartape, D. K. Pawar, S. H. Han and S. S. Kolekar, C. R. Chim., 2011, 14, 883–886 CrossRef CAS.
- K. Venkatesan, S. S. Pujari, R. J. Lahoti and K. V. Srinivasan, Ultrason. Sonochem., 2008, 15, 548–551 CrossRef CAS PubMed.
- T. S. Jin, J. S. Zhang, A. Q. Wang and T. S. Li, Ultrason. Sonochem., 2006, 13, 220–223 CrossRef CAS PubMed.
- S. Kantevari, R. Bantu and L. Nagarapu, J. Mol. Catal. A: Chem., 2007, 269, 53–60 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nj02430g |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |