
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel indenyl ligands bearing electron-withdrawing functional groups†
Iva
Honzíčková
a,
Jaromír
Vinklárek
a,
Carlos C.
Romão
b,
Zdeňka
Růžičková
a and
Jan
Honzíček
*c
aDepartment of General and Inorganic Chemistry, Faculty of Chemical Technology, University of Pardubice, Studentská 573, Pardubice, Czech Republic
bInstituto de Tecnologia Química e Biológica António Xavier, Universidade Nova de Lisboa, Av. Da República, 2780-157 Oeiras, Portugal
cInstitute of Chemistry and Technology of Macromolecular Materials Faculty of Chemical Technology, University of Pardubice, Studentská 573, Pardubice, Czech Republic. E-mail: jan.honzicek@upce.cz; Fax: +420 46603 7068
First published on 27th October 2015
Abstract
A series of molybdenum complexes bearing new ligands is reported. The study covers a series of molybdenum compounds with the η5-coordinated indenyl ligand substituted with acyl-, ester- and amide-functions. This portfolio was extended by adding one representative with a η3-coordinated ester-substituted indenyl ligand. The functionalized indenes, necessary for the assembly, were prepared by convenient routes starting from inexpensive and readily available materials, enabling their production on a multigram scale. All structural types presented in this experimental study were supported by X-ray crystallographic data.
Introduction
The attachment of functional groups at cyclopentadienyl (Cp) rings of transition metal complexes can change considerably their electronic properties as well as the range of potential applications.1,2 So far, many different substitution patterns have been developed for the Cp ligand but only a few of them cover electron-withdrawing substituents and are applicable for ligands with extended π-systems such as indenyl (Ind) or fluorenyl (Flu). These congeners of the Cp bearing annulated benzene rings show unique properties with consequent implications in the design of new catalytic systems.3 Hence, the replacement of the Cp ligand with a larger π-system often has a strong effect on reactivity.4 This so-called “indenyl effect” accelerates the reaction rates or switches the reaction pathway due to a lower energetic barrier of the haptotropic rearrangement of the π-ligand.5 Recent studies have further revealed that a subtle modification of the indenyl ligand (e.g. replacement of one hydrogen with a methyl group) may play a crucial role in activation of coordinated ligands.2,6,7The modification of the indenyl ligand with electron-withdrawing substituents is rather rare. In 2000, Deck et al. extended a synthetic route giving perfluoroaryl-functionalized cyclopentadienides8 on the indenes and described their rhenium complexes.9 Some other strong electron-withdrawing substituents such as –F,10 –CN,11 –COOR,12,13 –CONHR14,15 and –COR,11,12,16 have been successfully attached on the Cp ring of various transition metal complexes using different strategies. However, the synthesis of the indenyl congeners has not been reported, although some of the suitable indene precursors are already known for several decades.17
The aim of this study is to extend a family of indenyl ligands with ester-, amide- and acyl-functionalized derivatives. It will cover a series of modified indenes and unprecedented indenyl complexes. It is expected that a strong electron-withdrawing power of the attached functional groups will lead to more electron deficient metal centers that should be beneficial for their future application in catalysis. We decided to start our investigations with a familiar and well defined fragment, (η3-C3H5)Mo(CO)2, before moving into unexplored areas. The allyl molybdenum scaffold is accessible from the halide precursor [(η3-C3H5)Mo(CO)2(NCMe)2X], which is an excellent starting material for the incorporation of a monoanionic π-ligand into the molybdenum coordination sphere.15,16,18–20
Results and discussion
Ester-functionalized indenes
Three approaches for the synthesis of ester-functionalized indenes were examined. The obvious one using the reaction of sodium indenide with one equivalent of chloroformate was found to be ineffective since the 1,1-disubstituted derivative appears as a major product. In case of the methyl ester, 3-(MeOCO)C9H7 (2) and 1,1-(MeOCO)2C9H6 (2a) were isolated in 2% and 15% yields, respectively (Scheme 1).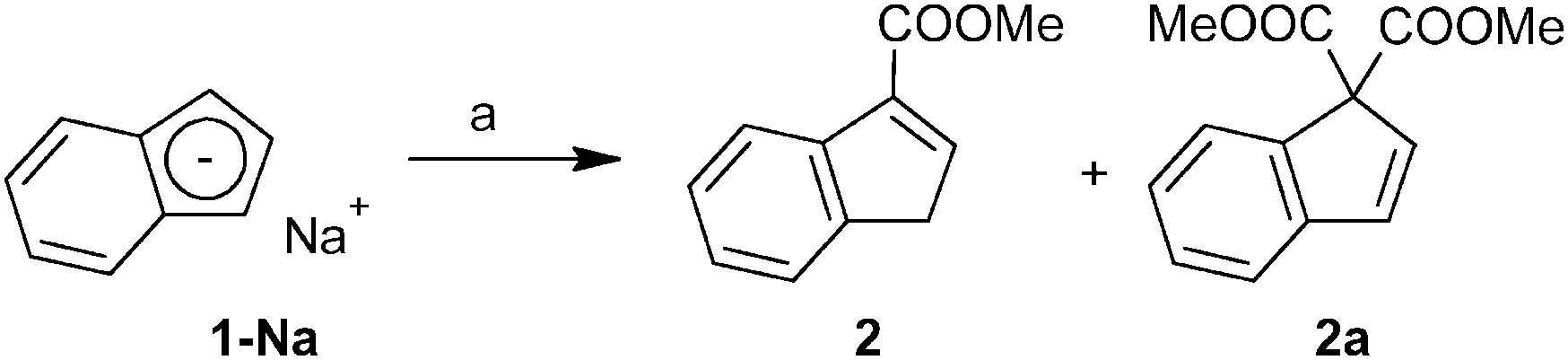 | ||
| Scheme 1 Reaction of sodium indenide (1-Na) with chloroformate. Reagent: ClCOOMe. | ||
Alternatively, a series of ester-functionalized indenes (3–5) was prepared by an esterification of indene-3-carboxylic acid according to a protocol reported for methyl ester 2, see Scheme 2.21 Since availability of the starting 3-(HOCO)C9H7 is a limiting factor of the procedure, an alternative strategy was used for the preparation of 3-(MeOCO)C9H7 (2) on a larger scale. The reaction of sodium indenide (1-Na) with dimethyl carbonate gives the functionalized indene 2 in one step and about 41% isolated yield without the need of a chromatographic purification step (Scheme 2).
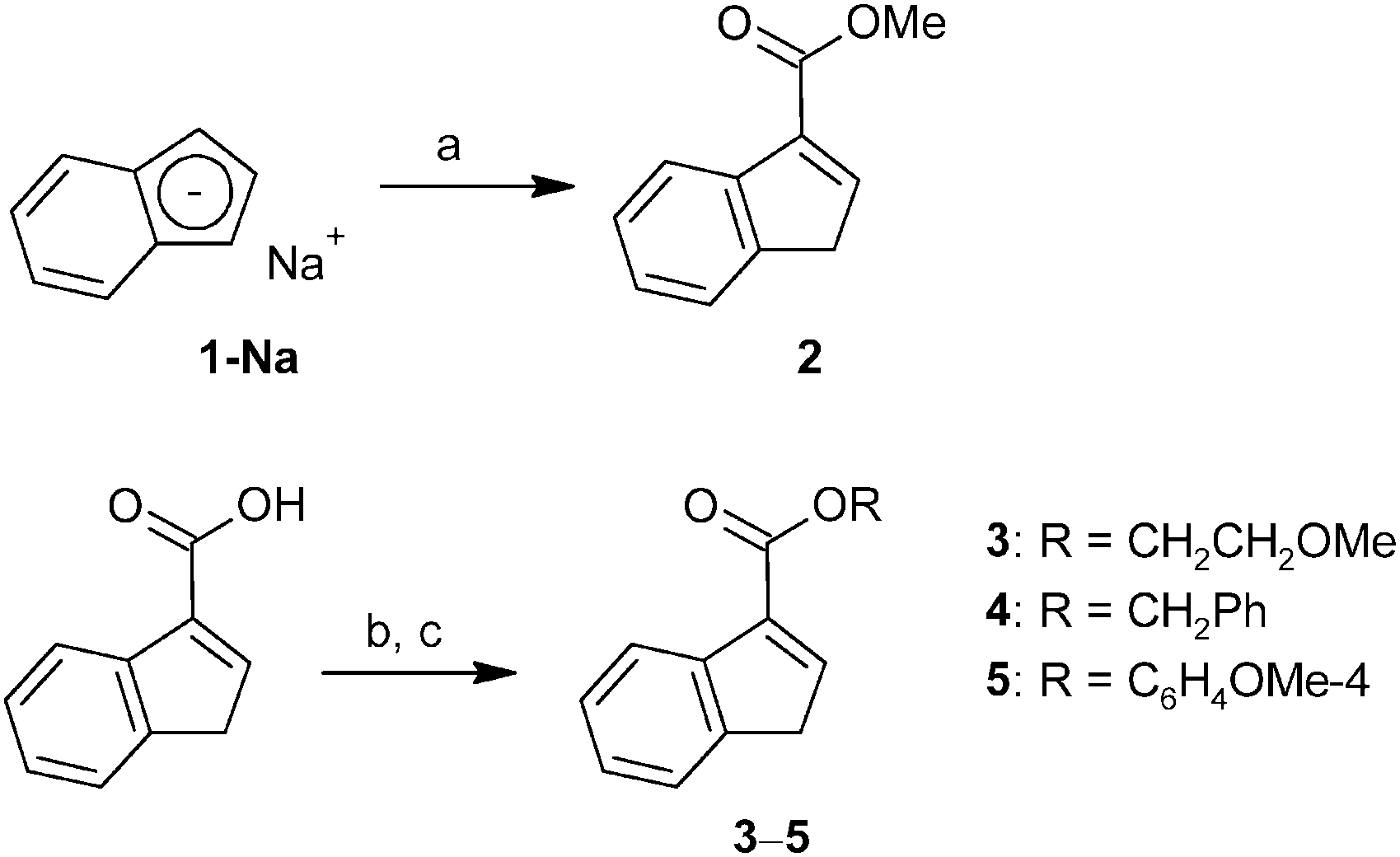 | ||
| Scheme 2 Synthesis of ester-functionalized indenes. Reagents: (a) OC(OMe)2, (b) SOCl2, (c) ROH. | ||
Ester-functionalized indenyl molybdenum compounds
The ability of ester-functionalized indenyl ligands to form η5-coordination compounds was evidenced for the following molybdenum species. The series of ester-substituted indenes (3–5) was deprotonated with n-butyl lithium. The reaction of indenides (3-Li–5-Li) formed in situ with the halide complex [(η3-C3H5)Mo(CO)2(NCMe)2Cl] (6), accompanied by a loss of the stabilizing MeCN ligands, resulted in the formation of η5-indenyl compounds [(η3-C3H5){η5-1-(ROCO)C9H6}Mo(CO)2] (7–10), see Scheme 3.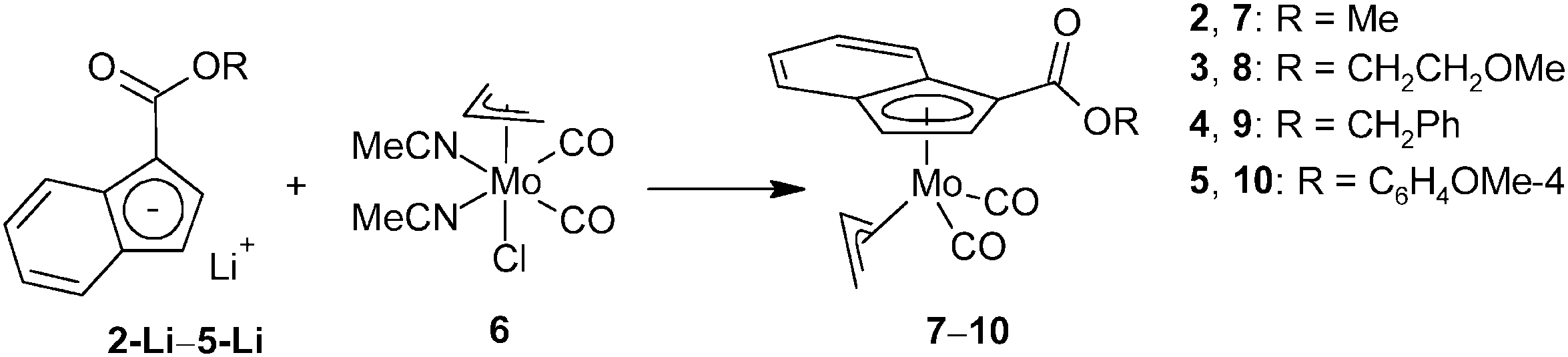 | ||
| Scheme 3 Synthesis of molybdenum complexes bearing ester-functionalized indenyl. | ||
The infrared and Raman spectra of compounds 7–10 show two CO stretching bands in a range typical for terminal carbonyl ligands, see Table 1. The CO stretching bands of the ester groups vary between 1702 and 1722 cm−1, thus revealing a very low delocalization of π-electrons of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group and excluding an alternative κ-O-coordination mode of the functionalized indenyl ligand. The 1H NMR spectra of compounds 7–10 show the presence of two conformers arising from a different orientation of the η3-bonded allyl ligand. The signals of the allyl ligand were assigned to a conformer with the allyl ligand eclipsed with OC–Mo–CO (exo) and that with a staggered conformation (endo) according to data published for the unsubstituted analogue.18 At room temperature, the exo-conformer predominates over the endo-conformer. The molar ratio (exo/endo) was found to be 3
O group and excluding an alternative κ-O-coordination mode of the functionalized indenyl ligand. The 1H NMR spectra of compounds 7–10 show the presence of two conformers arising from a different orientation of the η3-bonded allyl ligand. The signals of the allyl ligand were assigned to a conformer with the allyl ligand eclipsed with OC–Mo–CO (exo) and that with a staggered conformation (endo) according to data published for the unsubstituted analogue.18 At room temperature, the exo-conformer predominates over the endo-conformer. The molar ratio (exo/endo) was found to be 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for compounds 7–10 correlating well with data published for monosubstituted derivatives of [(η3-C3H5)(η5-Ind)Mo(CO)2].7
:1 for compounds 7–10 correlating well with data published for monosubstituted derivatives of [(η3-C3H5)(η5-Ind)Mo(CO)2].7
The solid state structure of compound 7 was determined by the single crystal X-ray analysis. The molecule has a distorted tetrahedral structure with allyl, indenyl and two carbonyl ligands around molybdenum in the formal oxidation state II, see Fig. 1. The geometric parameters describing the coordination sphere of molybdenum are listed in Table 2. The η3-coordinated allyl ligand is positionaly disordered and splits into exo and endo conformations with an occupancy of about 7:3. The substituted indenyl ligand is η5-coordinated as evidenced by a low value of the envelope fold angle [Ω = 4.5(3)°] and also by Δ(M–C) [0.119(3) Å]. The ester group is almost coplanar with the indenyl framework. The dihedral angle between the C5 ring of indenyl and a plane defined by atoms C1, C10, O1 and O2 is 1.28(16)°.
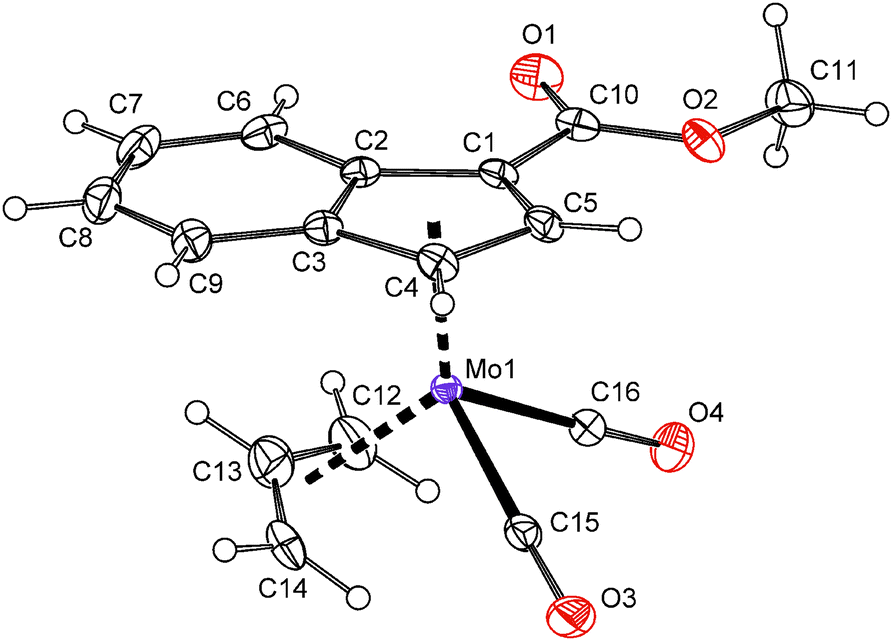 | ||
| Fig. 1 ORTEP drawing of the molybdenum compound [(η3-C3H5){η5-1-(MeOCO)C9H6}Mo(CO)2] present in the crystal structure of 7. Thermal ellipsoids are drawn at the 30% probability level. Only the major conformation of the positionaly disordered allyl ligand is shown for clarity. | ||
| 7 | 12 | 14 | 20 | 22·MeOH | |
|---|---|---|---|---|---|
| a Distances are given in Å; angles and dihedral angles are given in °. b Ω is the envelope fold angle defined for the indenyl ligand as the angle between planes defined by C1, C2 and C3 and that of C1, C3, C8 and C9.22 c Δ(M–C) represents the differences in the metal–carbon bonds. It is defined for the indenyl compounds as the difference between the averages of the metal–carbon distances M–C8 and M–C9 and those of M–C1, M–C2, and M–C3.22 d Data for Ind. e Data for 1-(MeOCO)C9H6. | |||||
| Mo–Cg(C5) | 2.0358(12) | 2.0203(10) | 2.0366(11) | 2.0229(10) | 2.0369(11) |
| Mo–Cg(C3) | 2.044(4) | 2.1423(19) | 2.053(3) | 2.047(3) | 2.049(3) |
| Mo–C(CO) | 1.936(3) | 1.943(2) | 1.933(3) | 1.931(2) | 1.939(3) |
| 1.950(3) | 1.952(2) | 1.934(3) | 1.954(2) | 1.943(3) | |
| Cg(C5)–Mo–Cg(C3) | 126.06(13) | 131.62(6) | 127.88(10) | 126.69(11) | 127.09(10) |
| C(CO)–Mo–C(CO) | 80.26(12) | 79.22(9) | 79.15(13) | 82.79(9) | 78.04(11) |
| Ω | 4.5(3) | 4.4(2)d | 3.4(3) | 4.5(2) | 3.7(3) |
| 18.3(2)e | |||||
| Δ(M–C)c | 0.119(3) | 0.114(2)d | 0.106(3) | 0.104(2) | 0.105(3) |
| 0.765(2)e | |||||
Having obtained a series of molybdenum compounds with η5-bonded ester-substituted indenyl ligands, we sought to extend the portfolio of such compounds by (i) adding a representative with a η3-coordinated ester-substituted indenyl ligand, (ii) introducing an acetyl substituent as a representative of a stronger electron-withdrawing substituent and (iii) developing a versatile route for amide-functionalized derivatives.
On the first count, we chose the mixed-indenyl species of type [(η3-Ind)(η5-Ind)Mo(CO)2]. The methyl ester derivative [{η3-1-(MeOCO)C9H6}(η5-Ind)Mo(CO)2] (12) was prepared by a reaction of 2-Li with one equivalent of [{(η5-Ind)Mo(CO)2(μ-Cl)}2] (11) in a moderate isolated yield (43%), see Scheme 4. Coordination of the indenyl ligand was evidenced by infrared spectroscopy, which shows the stretching bands of the carbonyl ligands at frequencies (νa: 1943 cm−1; νs: 1876, 1828 cm−1) similar to those reported for the unsubstituted parent compound [(η3-Ind)(η5-Ind)Mo(CO)2].23 The appearance of two bands of the νs(CO) is due to a vibration coupling of the carbonyl ligands in the crystal lattice. The CO stretching band of the ester group appears at a lower frequency (1694 cm−1) than that of compound 7 thus implying a more electrophilic character of the molybdenum center. The 1H NMR spectrum shows broadened signals indicating a fluxional structure in solution due to a fast hapticity exchange of the indenyl rings. This observation correlates well with the properties of the unsubstituted analogue [(η3-Ind)(η5-Ind)Mo(CO)2] that gives only one set of signals for both indenyl ligands.24
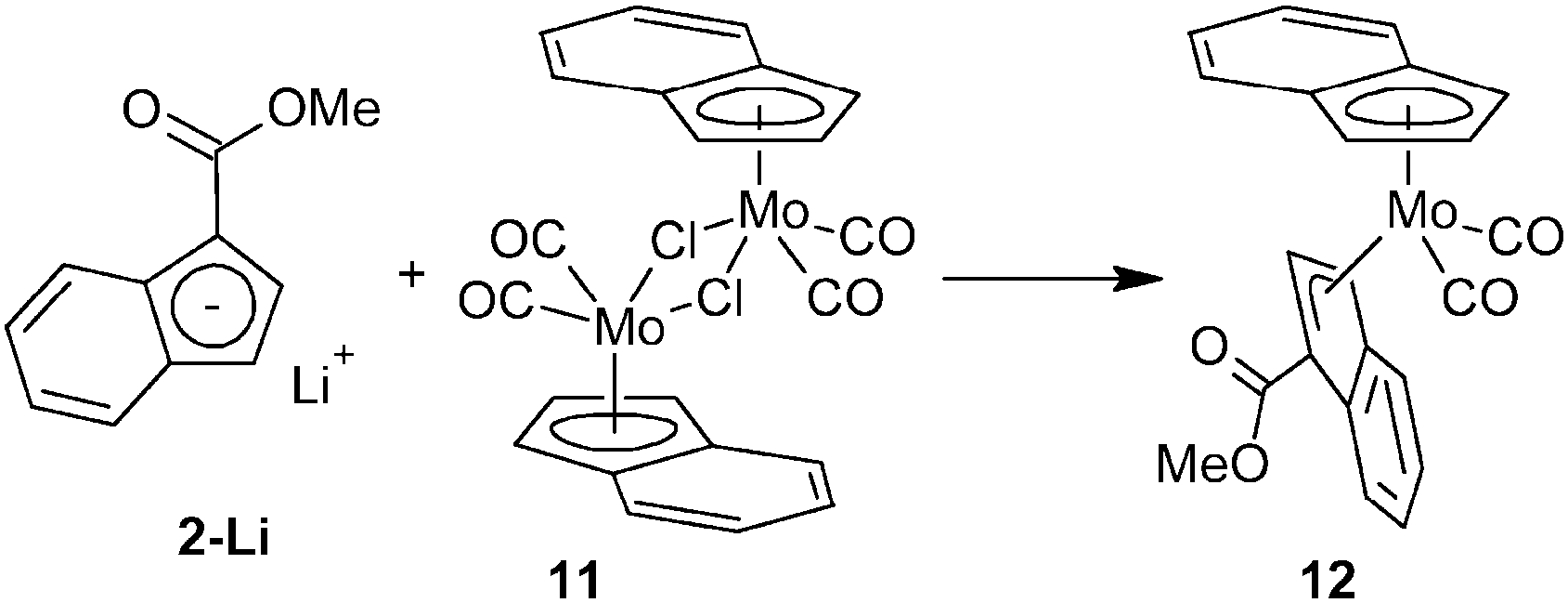 | ||
| Scheme 4 Synthesis of a mixed indenyl molybdenum complex. | ||
The X-ray diffraction analysis reveals that the less donating ester-substituted indenyl ligand adopts the η3-coordination mode in the solid state, whereas the electron richer unsubstituted indenyl stays η5-coordinated, see Fig. 2. The hapticity of indenyl ligands is clearly elucidated from the slip parameters listed in Table 2. Hence, the substituted indenyl has considerably higher values of Ω and Δ(M–C) then the unsubstituted one. Compound 12 adopts a conformation, similar to the unsubstituted counterpart,23 with the η3-indenyl ligand in exo-conformation and the C6-ring of the η5-indenyl facing away from the carbonyl ligands thereby avoiding repulsive interactions. The ester group is almost coplanar with the η3-coordinated π-system of the indenyl framework. The dihedral angle between a plane defined by three carbon atoms of Ind (C1, C4 and C5) and a plane defined by atoms C1, C10, O1 and O2 is 3.8(2)°.
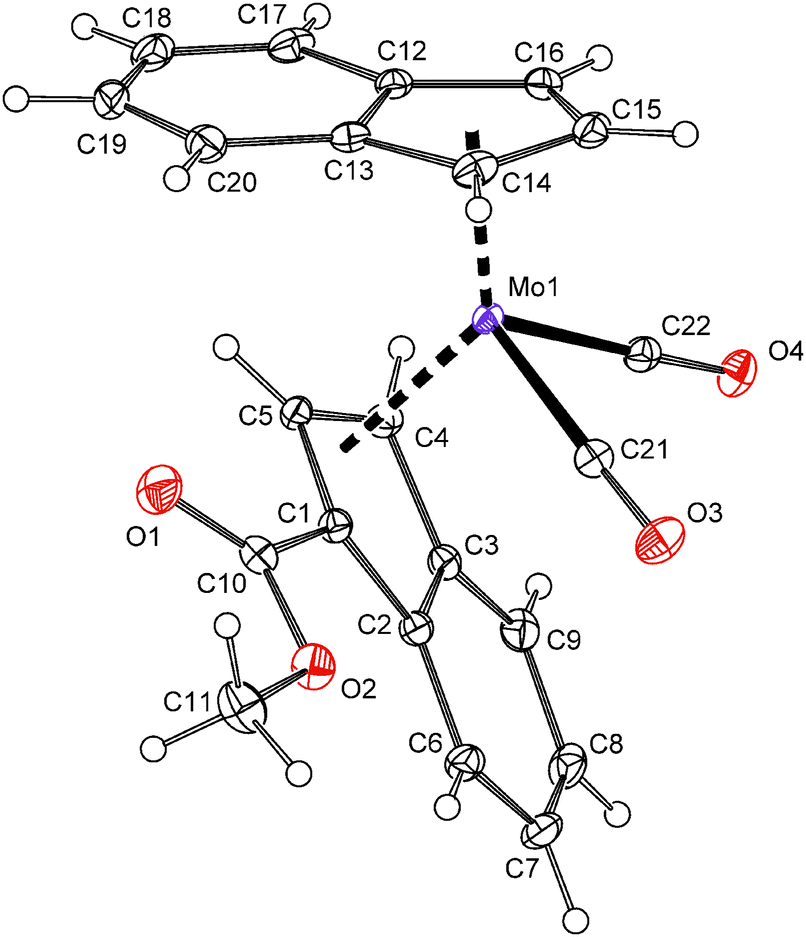 | ||
| Fig. 2 ORTEP drawing of the molybdenum compound [{η3-1-(MeOCO)C9H6}(η5-Ind)Mo(CO)2] present in the crystal structure of 12. Thermal ellipsoids are drawn at the 30% probability level. | ||
The crystal structure of 12 is stabilized by a sandwich π–π stacking involving the unsubstituted indenyl ligands of two neighboring molecules. The distance between the centroid of the five-membered ring [Cg(C12–C16)] and a plane defined by the parallel five-membered ring [Pl(C12′–C16′)] is 3.3075(10) Å. A T-shaped interaction between the face of the six-membered ring of the substituted indenyl ligand (C2–C6) and the six-membered ring vertex of the unsubstituted indenyl (C17′–H17′) connects the molecules into zig-zag chains along the b-axis. The distance between the centroid Cg(C2–C6) and the carbon atom C17′ is 3.532(2) Å.
Acetyl-functionalized indenyl molybdenum compound
A similar strategy as described for indene 2 was utilized for the synthesis of the acetyl-functionalized derivative 3-(MeCO)C9H7 (13). Hence, the reaction of sodium indenide with ethyl acetate produced indene 13 in 38% isolated yield including a vacuum distillation step (Scheme 5). The protocol utilized inexpensive and readily available starting materials providing the product on a multigram scale. The vibrational spectra show a strong band of CO stretching in a range typical for the keto-group (IR: 1669 cm−1; Raman: 1666 cm−1). The 1H and 13C{1H} NMR spectra reveal the appearance of an isomer with the acetyl function in the 3-position of the indene framework. This observation is in line with the X-ray analysis of a single crystal obtained by recrystallization from hexane at low temperature. Hence, the acetyl group is attached to the sp2 carbon atom of the indene framework, see Fig. 3. The bond length between the carbon atom C1 and the oxygen atom O1 is 1.222(2) Å that is within a range typical for the CO double bond.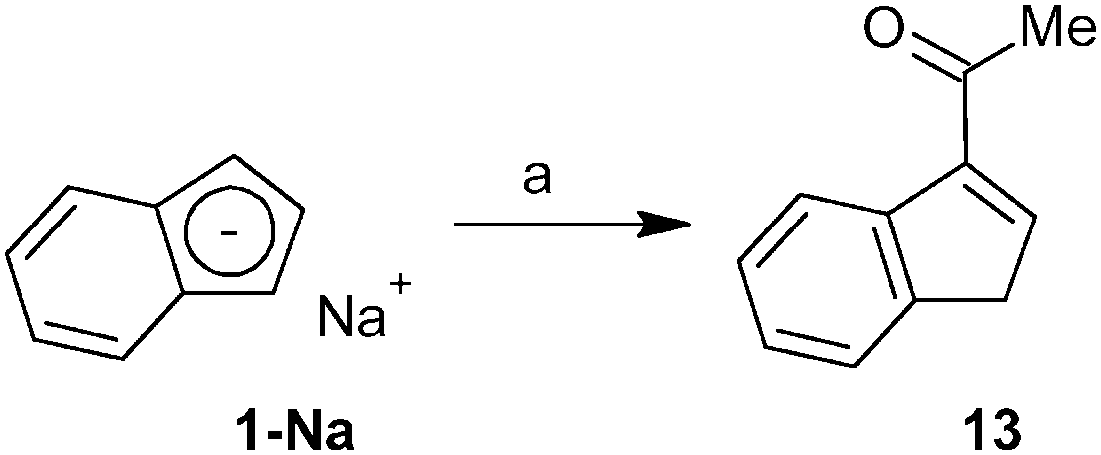 | ||
| Scheme 5 Synthesis of acetyl-functionalized indene 13. Reagent: (a) MeCOOEt. | ||
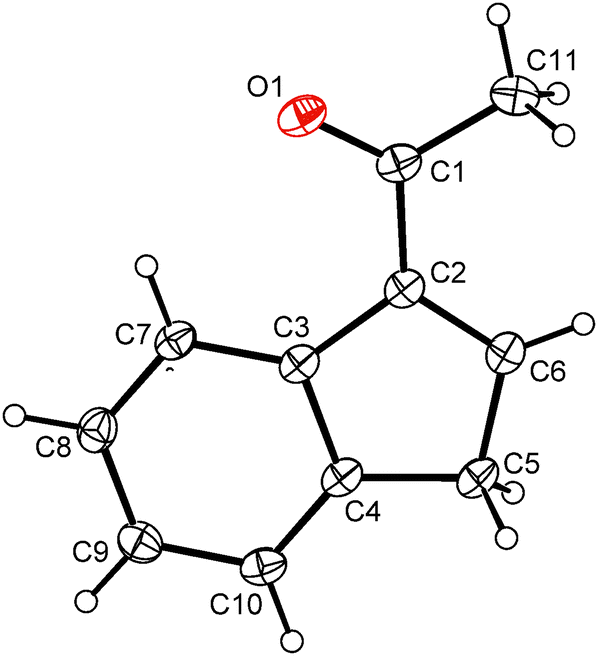 | ||
| Fig. 3 ORTEP drawing of 3-(MeCO)C9H7 present in the crystal structure of 13. Thermal ellipsoids are drawn at the 30% probability level. | ||
Deprotonation of indene 13 with n-butyl lithium followed by addition of the allyl complex [(η3-C3H5)Mo(CO)2(NCMe)2Cl] (6) yields the desired η5-indenyl complex [(η3-C3H5){η5-1-(MeCO)C9H6}Mo(CO)2] (14), see Scheme 6.
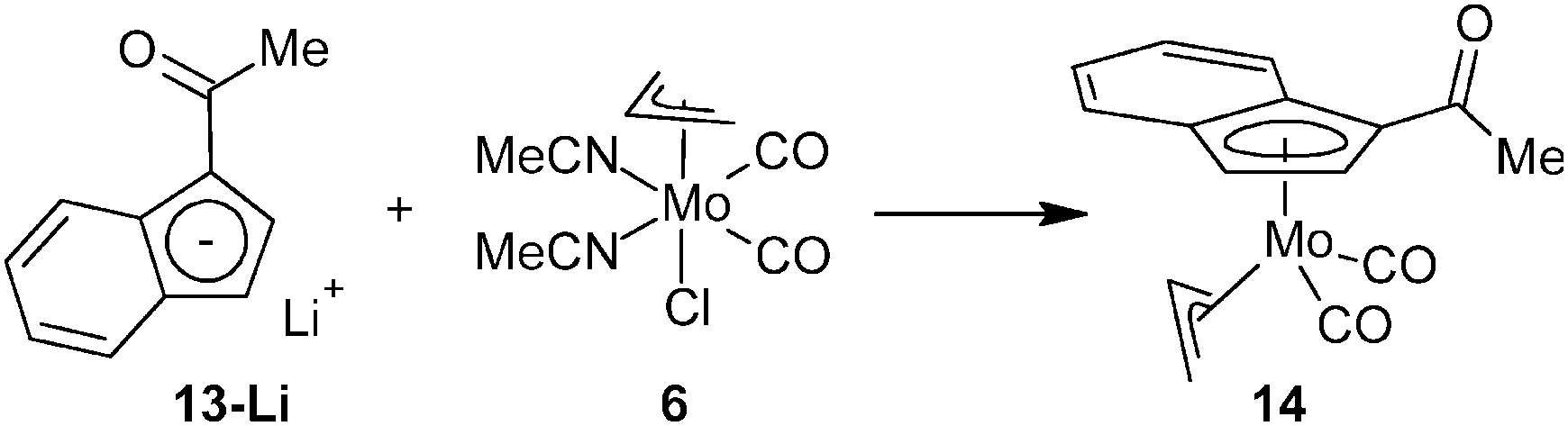 | ||
| Scheme 6 Synthesis of an acetyl-functionalized indenyl molybdenum complex. | ||
Infrared, Raman and 1H NMR spectroscopy confirmed the successful assembly of the acetyl-functionalized indenyl molybdenum framework. Hence, the vibrational spectra of compound 14 show, in addition to stretching bands of the carbonyl ligands, a characteristic band of the CO stretching at similar frequencies as observed for indene precursor 13, see Table 3. The 1H NMR spectrum of compound 14 features a similar pattern as observed for the ester derivatives. The molar ratio of exo- and endo-conformers (3:1) is virtually the same as observed for the ester derivatives. This observation indicates that the abundance of given conformers is directed by the bulkiness of the modified indenyl ligand while electronic properties of the substituents play only a minor role.
| Infrared | Raman | |||
|---|---|---|---|---|
ν(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O) O) |
ν(CO) |
ν(CO) |
ν(CO) |
|
| a The stretching frequencies are given in cm−1. | ||||
| 14 | 1936 | 1660 | 1932 | 1666 |
| 1858 | 1836 | |||
| 20 | 1932 | 1635 | 1931 | 1637 |
| 1848 | 1850 | |||
| 21 | 1932 | 1633 | 1933 | 1635 |
| 1859 | 1866 | |||
| 22 | 1940 | 1667 | 1948 | 1664 |
| 1852 | 1848 | |||
X-ray crystallographic structure determination confirmed that the acetyl-substituted molybdenum compound 14 is isostructural with ester-derivative 7. The acetyl function is coplanar with the indenyl framework. The dihedral angle between the C5 ring of indenyl and a plane defined by atoms C1, C10, O1 and O2 is 2.15(17)° (Fig. 4).
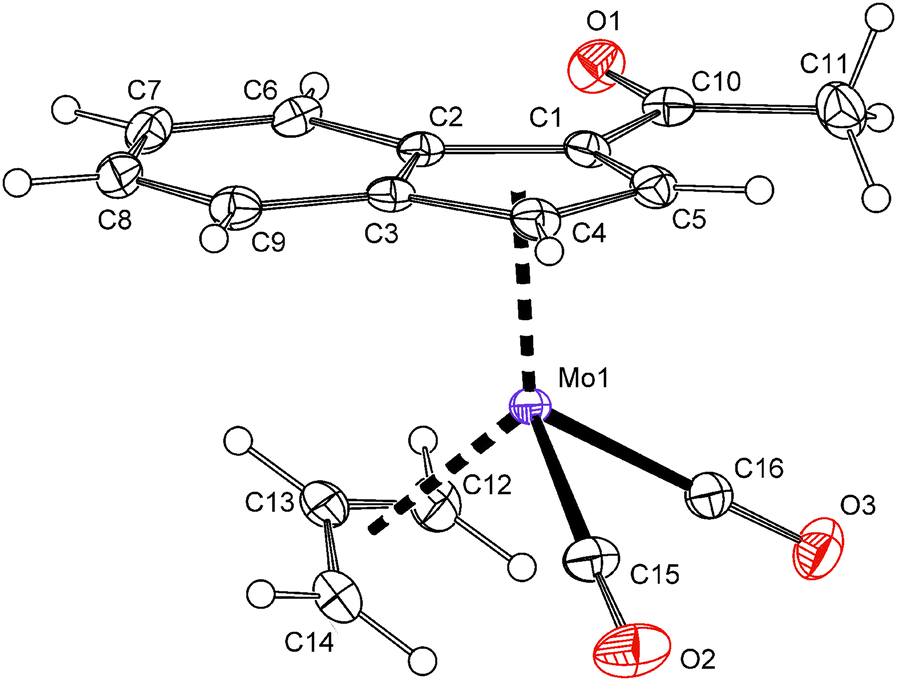 | ||
| Fig. 4 ORTEP drawing of the molybdenum compound [(η3-C3H5){η5-1-(MeCO)C9H6}Mo(CO)2] present in the crystal structure of 14. Thermal ellipsoids are drawn at the 30% probability level. | ||
Amide-functionalized indenyl molybdenum compounds
Finally, we have also investigated the scope of our synthetic protocol for compounds with amide-functions. The starting amide-substituted indenes were synthesized using two strategies. tert-Butyl derivative 15 was prepared by a reaction of lithium indenide (1-Li) with tert-butyl isocyanate, see Scheme 7. Rather inexpensive starting materials and an acceptable isolated yield (65%) predestinate this procedure for the use on a multigram scale.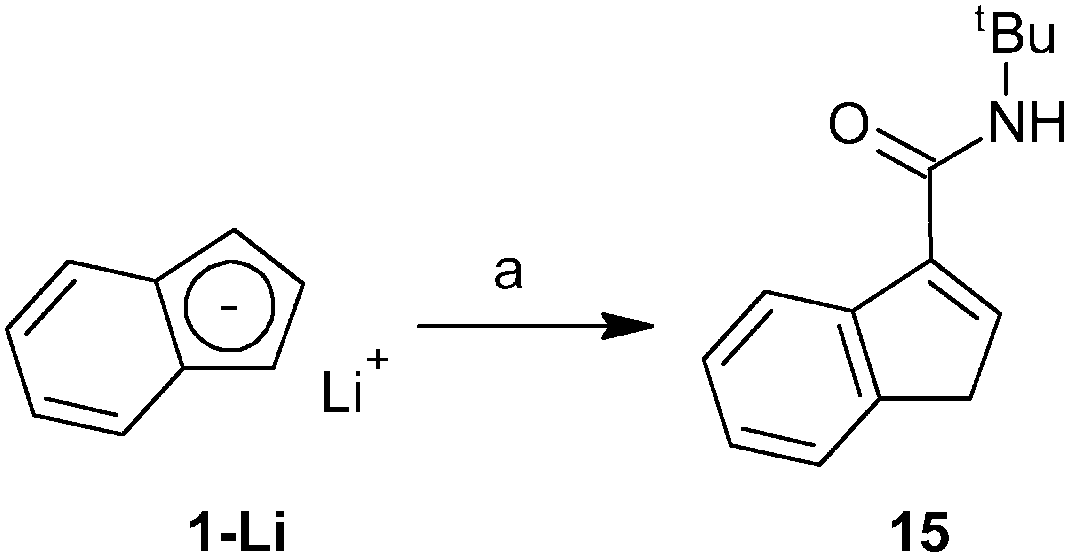 | ||
| Scheme 7 Synthesis of carboxamide-functionalized indene 15. Reagent: (a) tBuNCO. | ||
Unfortunately, the higher reactivity of aryl isocyanates precludes their use for the synthesis of aryl amides such as 16 and 19 (for structures, see Scheme 9). In fact, the reaction of phenyl isocyanate gives the acyl-substituted N,N-diphenyl urea 17 and the 1,3-disubstituted indene 18 as major outcomes in about 22% and 8% yields, respectively (based on indene). The desired 3-(PhNHCO)C9H7 (16) appears only as a minor product (in ∼4% yield) that was separated from the reaction mixture by column chromatography (Scheme 8).
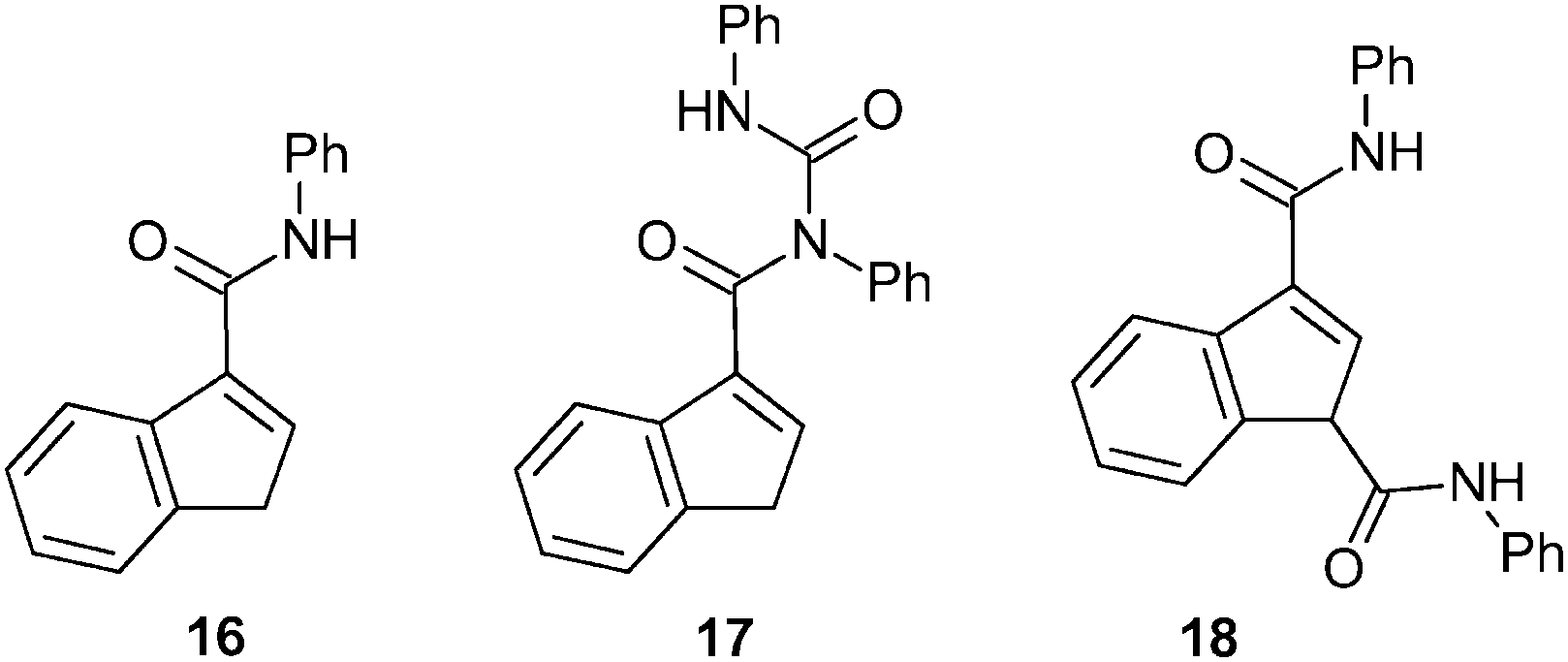 | ||
| Scheme 8 Product of the reaction between 1-Li and PhNCO. | ||
 | ||
| Scheme 9 Synthesis of carboxamide-functionalized indenes 16 and 19. Reagents: (a) SOCl2, (b) RNCO. | ||
This led us to use 3-(ClCO)C9H7 prepared in situ for the synthesis of aryl amides 16 and 19, see Scheme 9. The carboxylic acid, 3-(HOCO)C9H7, was treated with thionyl chloride and the product reacted with anilines to give carboxamides 16 and 19 in satisfactory isolated yields.
Indenyl molybdenum compounds bearing the amide function group are accessible using the aforementioned protocol successfully used for the ester-derivatives. Deprotonation with n-butyl lithium followed by a metathesis reaction gave indenyl molybdenum compounds 20–22 in moderate isolated yields, see Scheme 10. The compounds were characterized by infrared, Raman and 1H NMR spectroscopy. The structures of compounds 20 and 22 were determined by X-ray diffraction analysis. The vibration spectra of compounds 20–22 show stretching bands of the carbonyl ligands at frequencies similar to those observed for ester-derivatives 7–10 (cf. data in Tables 1 and 3). Frequencies of N–H and CO stretching bands of the amide functions are also consistent with the desired η5-coordination mode of the substituted indenyl ligands.
 | ||
| Scheme 10 Synthesis of amide-functionalized indenyl molybdenum complexes. | ||
In case of compounds 20 and 22, a successful assembly of the [(η3-C3H5)(η5-Ind)Mo(CO)2] moiety was further confirmed by the X-ray diffraction analysis, see Fig. 5 and 6. The carboxamide-functionalized indenyl ligands are η5-coordinated to the molybdenum as evidenced by ring slip parameters (Table 2). Surprisingly, only a very weak hydrogen bond (N1–H1⋯O1′) was observed in the crystal lattice of compound 20 probably as a result of the steric hindrance of the bulky t-butyl group. The amide functions of neighboring molecules are connected into zig-zag chains along the c-axis. The distance between the nitrogen atom (N1) and the oxygen atom (O1′) of the neighboring amide function is 3.295(2) Å. In case of 22·MeOH, the amide functions and the hydroxyl group of methanol are connected into chains along the a-axis by considerably stronger hydrogen bonds N1–H1⋯O2 and O2–H2⋯O1′. The distances N1⋯O2 and O2⋯O1′ are 2.936(3) and 2.714(3) Å, respectively.
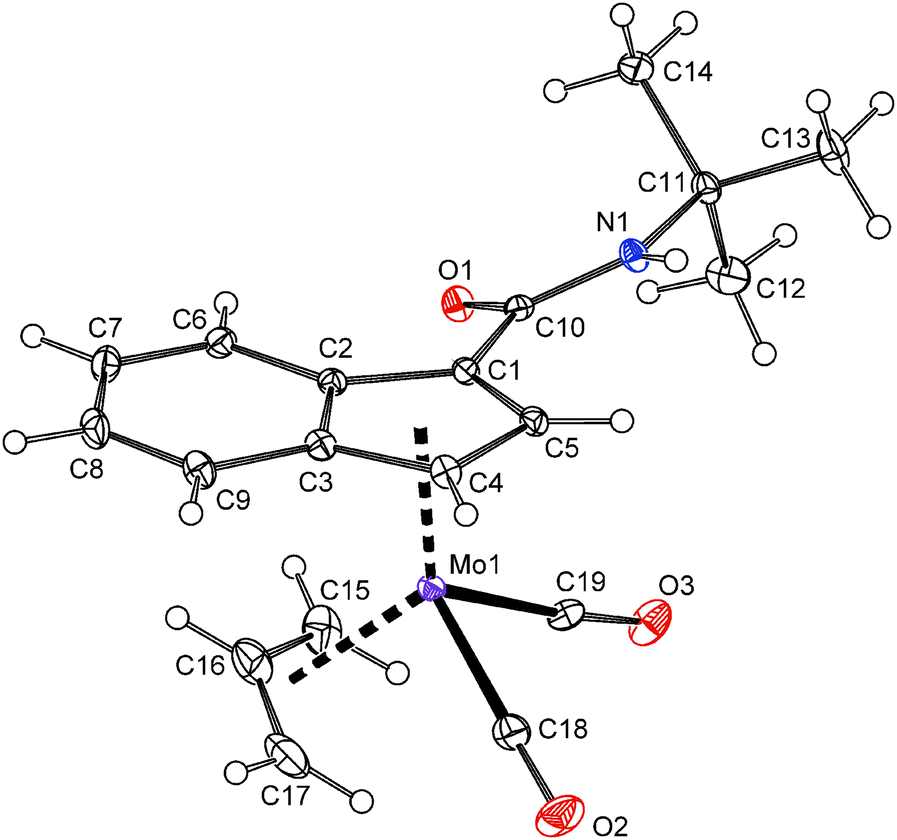 | ||
| Fig. 5 ORTEP drawing of the molybdenum compound [(η3-C3H5){η5-1-(tBuNHCO)C9H6}Mo(CO)2] present in the crystal structure of 20. Thermal ellipsoids are drawn at the 30% probability level. | ||
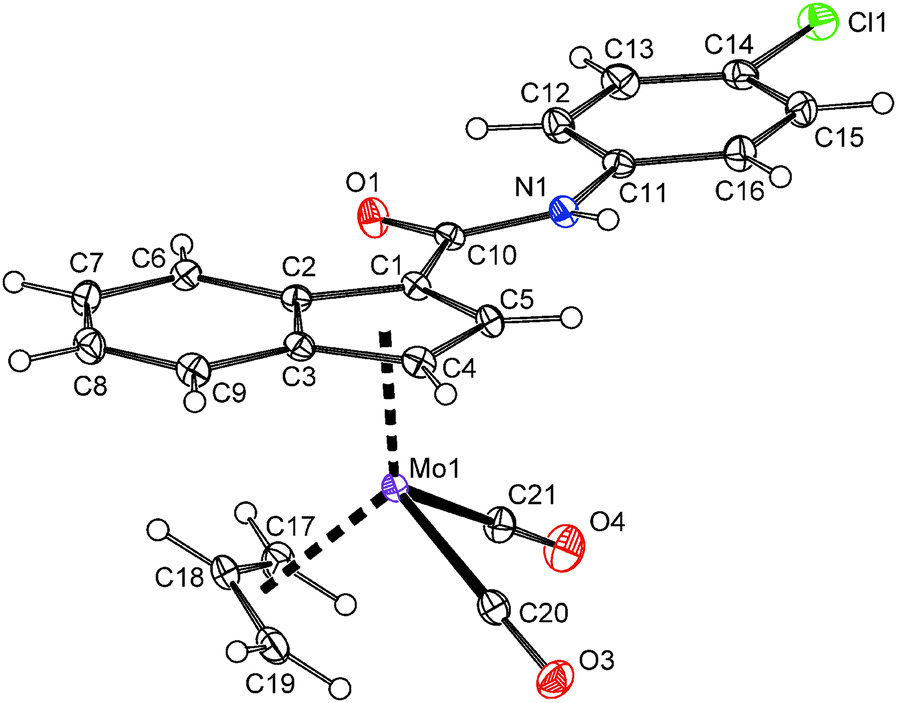 | ||
| Fig. 6 ORTEP drawing of the molybdenum compound [(η3-C3H5){η5-1-(4-ClC6H4NHCO)C9H6}Mo(CO)2] present in the crystal structure of 22·MeOH. Thermal ellipsoids are drawn at the 30% probability level. | ||
Conclusions
The present study established a new family of ring-substituted indenyl complexes with an unprecedented substitution pattern. The derivatives of [(η3-C3H5)(η5-Ind)Mo(CO)2] bearing a strong electron-withdrawing functional group attached to the indenyl framework were prepared by a convenient route starting from functionalized lithium indenides. Since the availability of the ligand precursor is often a limiting factor of a synthetic protocol and consequent applications, attention was further given to low-cost pathways giving the ester-, acyl- and amide-functionalized indenes without the necessity of a tedious chromatographic purification step. On that count, one representative from each group was synthesized on the 6–14 g scale starting from common and inexpensive starting materials.The herewith established substitution of the indenyl ligands with polar functional groups opens a novel pathway for chemical modifications of transition metal complexes and may accelerate their use in organic synthesis or catalysis.
Experimental section
Methods and materials
All operations were performed under nitrogen using conventional Schlenk-line techniques. The solvents were purified and dried by standard methods.25 The starting materials were available commercially or prepared according to literature procedures: 3-(HOCO)C9H7,21 [(η3-C3H5)Mo(CO)2(NCMe)2Cl] (6),18 [{(η5-Ind)Mo(CO)2(μ-Cl)}2] (11).20Measurements
Infrared and Raman spectra were measured on a Nicolet iS50 FTIR spectrometer equipped with a Raman module. The infrared spectra were recorded in the 4000–400 cm−1 region (resolution 1 cm−1) using a Diamond Smart Orbit ATR. Raman spectra were recorded in the 4000–100 cm−1 region (resolution 2 cm−1) in glass capillaries. The excitation source consisted of a Nd:YAG laser emitting at 1064 cm−1. 1H and 13C{1H} NMR spectra were measured on a Bruker Avance 400 and a Bruker Avance 500 spectrometers, respectively, at room temperature. The chemical shifts are given in ppm relative to TMS.Synthesis of 3-(MeOCO)C9H7 (2)
Sodium indenide (1-Na), freshly prepared from indene (1; 11.7 mL, 0.10 mol) and sodium hydride (2.4 g, 0.10 mmol) in THF (150 mL), was treated with dimethyl carbonate (8.4 mL, 0.10 mol) and heated under reflux overnight. After cooling to room temperature, the reaction mixture was poured on an ice/water mixture and extracted with diethyl ether. The combined organic phases were washed with water, dried with magnesium sulfate and volatiles were vacuum evaporated on a rotavapor. The crude product was vacuum distilled (120 °C, 660 Pa). Yield: 7.2 g (41 mmol, 41%). Pale yellow oil. Anal. Calc. for C11H10O2: C: 75.84; H: 5.79. Found: C: 75.72; H: 5.85. 1H NMR (CDCl3; 400 MHz; δ ppm): 7.94 (d, 3J(1H,1H) = 7.7 Hz, 1H, C9H7), 7.28 (d, 3J(1H,1H) = 7.5 Hz, 1H, C9H7), 7.24 (s, 1H, C9H7), 7.21 (t, 3J(1H,1H) = 7.5 Hz, 1H, C9H7), 7.10 (t, 3J(1H,1H) = 7.5 Hz, 1H, C9H7), 3.73 (s, 3H, COOCH3), 3.26 (s, 2H, C9H7). 13C{1H} NMR (CDCl3; 101 MHz; δ ppm): 164.3 (1C, Cq, COOMe), 144.4 (1C, CH, C9H7), 143.3, 140.6, 135.8 (3 × 1C, Cq, C9H7), 126.5, 125.4, 123.6, 122.3 (4 × 1C, CH, C9H7), 51.3 (1C, CH3, COOCH3), 38.2 (1C, CH2, C9H7). IR (ATR, cm−1): 1713s [ν(CO)CO]. Raman (capillary, cm−1): 1715(4) [ν(CO)CO].
Reaction of 1-Na with ClCOOMe
Sodium indenide (1-Na), freshly prepared from indene (1, 11.7 mL, 0.10 mol) and sodium hydride (2.4 g, 0.10 mol) in THF (150 mL), was treated with methyl chloroformate (7.7 mL, 0.1 mol) and stirred at room temperature overnight. The reaction mixture was poured on an ice/water mixture and extracted with diethyl ether. The combined organic phases were dried with magnesium sulfate and volatiles were evaporated on a rotavapor. Fraction distillation of the crude product gave 2 as a minor product (yield: 0.4 g, 2.3 mmol, 2%) and 2a as the major product (yield: 3.5 g, 15 mmol, 15%). 1,1-(MeOCO)2C9H6 (2a): colorless crystals. Bp = 95 °C (10 Pa). Mp = 84 °C. Anal. Calc. for C13H12O4: C: 67.23; H: 5.21. Found: C: 67.38; H: 5.14. 1H NMR (CDCl3; 400 MHz; δ ppm): 7.69 (d, 3J(1H,1H) = 7.4 Hz, 1H, C9H6), 7.36–7.24 (m, 3H, C9H6), 6.91 (d, 3J(1H,1H) = 5.5 Hz, 1H, C9H6), 6.56 (d, 3J(1H,1H) = 5.5 Hz, 1H, C9H6), 3.73 (s, 6H, COOCH3). 13C{1H} NMR (CDCl3; 101 MHz; δ ppm): 168.5 (2C, Cq, COOMe), 143.8, 139.9 (2 × 1C, Cq, C9H6), 135.1, 133.5, 129.4, 126.6, 125.6, 121.9 (6 × 1C, CH, C9H6), 53.5 (2C, CH3, COOCH3). IR (ATR, cm−1): 1743s [νa(CO)CO], 1724s [ν(CO)CO]. Raman (capillary, cm−1): 1740(2) [νa(CO)CO], 1724(2) [νs(CO)CO].
Synthesis of 3-(MeOCH2CH2OCO)C9H7 (3)
3-(HOCO)C9H7 (0.80 g; 5.0 mmol) was dissolved in the excess of thionyl chloride (5 mL) and stirred at room temperature for 1 h and then heated at 60 °C for 10 min. The volatiles were vacuum evaporated. The sticky solid was treated with the excess of MeOCH2CH2OH (3.8 g, 50 mmol) and stirred for 2 h. The reaction mixture was diluted with diethyl ether and the reaction was quenched with addition of aqueous solution sodium carbonate. The organic phase was separated and the water phase was washed with an additional portion of diethyl ether. The combined organic phases were washed with water, dried with magnesium sulfate. The volatiles were vacuum evaporated on a rotavapor and the crude product was purified by column chromatography on silica (diethyl ether/hexane = 1:9). Yield: 0.84 g (3.8 mmol, 77%). Pale yellow oil. Anal. Calc. for C13H14O3: C: 71.54; H: 6.47. Found: C: 71.59; H: 6.45. 1H NMR (CDCl3; 400 MHz; δ ppm): 8.04 (d, 3J(1H,1H) = 7.7 Hz, 1H, C9H7), 7.49 (t, 3J(1H,1H) = 2.0 Hz, 1H, C9H7), 7.46 (d, 3J(1H,1H) = 7.5 Hz, 1H, C9H7), 7.34 (t, 3J(1H,1H) = 7.5 Hz, 1H, C9H7), 7.25 (td, 3J(1H,1H) = 7.5 Hz, 4J(1H,1H) = 1.2 Hz, 1H, C9H7), 4.45 (t, 3J(1H,1H) = 4.7 Hz, 2H, CH2), 3.72 (t, 3J(1H,1H) = 4.7 Hz, 2H, CH2), 3.51 (d, 3J(1H,1H) = 2.0 Hz, 2H, C9H7), 3.42 (s, 3H, OCH3). 13C{1H} NMR (CDCl3; 101 MHz; δ ppm): 164.2 (1C, Cq, COOMe), 145.0 (1C, CH, C9H7), 143.5, 140.9, 136.2 (3 × 1C, Cq, C9H7), 126.9, 125.7, 124.0, 122.7 (4 × 1C, CH, C9H7), 70.8, 63.7 (2 × 1C, CH2, CH2), 59.2 (1C, CH3, OCH3), 38.6 (1C, CH2, C9H7). IR (ATR, cm−1): 1713s [ν(CO)CO]. Raman (capillary, cm−1): 1717(3) [ν(CO)CO].
Synthesis of 3-(PhCH2OCO)C9H7 (4)
The steps of synthesis followed the procedure for compound 3. Reagents: 3-(HOCO)C9H7 (0.80 g, 5.0 mmol), PhCH2OH (5.4 g, 50 mmol). The crude product was purified by column chromatography on silica (diethyl ether/hexane = 1:9). Yield: 1.0 g (4.0 mmol, 80%). Pale yellow oil. Anal. Calc. for C17H14O2: C: 81.58; H: 5.64. Found: C: 81.51; H: 5.80. 1H NMR (CDCl3; 400 MHz; δ ppm): 8.06 (d, 3J(1H,1H) = 7.7 Hz, 1H, C9H7), 7.69 (t, 3J(1H,1H) = 1.8 Hz, 1H, C9H7), 7.49–7.32 (m, 2H of C9H7 and 5H of C6H5), 7.26 (t, 3J(1H,1H) = 7.4 Hz, 1H, C9H7), 5.37 (s, 2H, CH2Ph), 3.52 (d, 3J(1H,1H) = 1.8 Hz, 2H, C9H7). 13C{1H} NMR (CDCl3; 101 MHz; δ ppm): 164.1 (1C, Cq, COOMe), 145.1 (1C, CH, C9H7), 143.5, 140.9, 136.3 (3 × 1C, Cq, C9H7), 128.8 (2C, C6H5), 128.4 (1C, C6H5), 128.3 (2C, C6H5), 126.9, 125.8, 124.0, 122.7 (4 × 1C, CH, C9H7), 66.4 (1C, CH2, CH2), 38.6 (1C, CH2, C9H7). IR (ATR, cm−1): 1713s [ν(CO)CO]. Raman (capillary, cm−1): 1717(2) [ν(CO)CO].
Synthesis of 3-(4-MeOC6H4OCO)C9H7 (5)
The steps of synthesis followed the procedure for compound 3. Reagents: 3-(HOCO)C9H7 (0.80 g, 5.0 mmol), 4-MeOC6H4OH (1.1 g, 9 mmol). The crude product was purified by column chromatography on silica (diethyl ether/hexane = 1:9). Yield: 0.72 g (2.7 mmol, 54%). Colorless crystals. Mp = 110 °C. Anal. Calc. for C17H14O3: C: 76.68; H: 5.30. Found: C: 76.75; H: 5.33. 1H NMR (CDCl3; 400 MHz; δ ppm): 8.08 (d, 3J(1H,1H) = 7.6 Hz, 1H, C9H7), 7.67 (t, 3J(1H,1H) = 2.0 Hz, 1H, C9H7), 7.50 (d, 3J(1H,1H) = 7.4 Hz, 1H, C9H7), 7.36 (t, 3J(1H,1H) = 7.6 Hz, 1H, C9H7), 7.28 (td, 3J(1H,1H) = 7.5 Hz, 4J(1H,1H) = 1.2 Hz, 1H, C9H7), 7.13 (d, 3J(1H,1H) = 9.1 Hz, 2H, C6H4), 7.93 (d, 3J(1H,1H) = 9.1 Hz, 2H, C6H4), 3.81 (s, 3H, OCH3), 3.60 (d, 3J(1H,1H) = 2.0 Hz, 2H, C9H7). 13C{1H} NMR (CDCl3; 101 MHz; δ ppm): 162.9 (1C, Cq, COOMe), 157.6 (1C, Cq, C6H4), 146.3 (1C, CH, C9H7), 144.3 (1C, Cq, C6H4), 143.5, 140.7, 136.0 (3 × 1C, Cq, C9H7), 127.0, 126.0, 124.1, 122.7 (4 × 1C, CH, C9H7), 122.7, 114.8 (2 × 2C, CH, C6H4), 55.9 (1C, CH3, OCH3), 38.9 (1C, CH2, C9H7). IR (ATR, cm−1): 1725s [ν(CO)CO]. Raman (capillary, cm−1): 1727(8) [ν(CO)CO].
Synthesis of [(η3-C3H5){η5-1-(MeOCO)C9H6}Mo(CO)2] (7)
3-(MeOCO)C9H7 (2; 0.87 g, 5 mmol) was dissolved with 30 mL of THF, cooled at 0 °C and treated dropwise with 3.1 mL of nBuLi (1.6 mol L−1). The reaction mixture was stirred for 1 h and then added dropwise to the THF solution of [(η3-C3H5)Mo(CO)2(NCMe)2Cl] (6; 1.55 g, 5 mmol) precooled to −80 °C. The reaction mixture was stirred at room temperature overnight and then vacuum evaporated to dryness. The solid residue was washed with cold hexane (10 mL) and extracted several times with hot hexane. The volatiles were vacuum evaporated and product was vacuum dried. Yield: 1.24 g (3.4 mmol, 68%). Yellow powder. Mp = 89 °C. Anal. Calc. for C16H14MoO4: C: 52.47; H: 3.85. Found: C: 52.29; H: 3.80. 1H NMR (CDCl3, 400 MHz, δ ppm; 3:1 mixture of 7a (exo-C3H5) and 7b (endo-C3H5)): 7.86–7.80 (m, 1H of a and 1H of b, H4,7 of C9H6), 7.30–7.05 (m, 3H of a and 3H of b, H4–7 of C9H6), 6.21 (s-br, 1H of a, H2,3 of C9H6), 6.11 (s-br, 1H of b, H2,3 of C9H6), 6.05 (s-br, 1H of a, H2,3 of C9H6), 5.98 (s-br, 1H of b, H2,3 of C9H6), 3.94 (s, 3H of a and 3H of b, CH3), 3.50 (s-br, 1H of b, C3H5), 3.31 (s-br, 2H of b, C3H5), 2.33 (d, 3J(1H,1H) = 6.0 Hz, 1H of a, syn of C3H5), 2.16 (d, 3J(1H,1H) = 6.0 Hz, 1H of a, syn of C3H5), 1.03 (d, 3J(1H,1H) = 10.8 Hz, 1H of a, anti of C3H5), 0.83 (d, 3J(1H,1H) = 10.8 Hz, 1H of a, anti of C3H5), 0.72 (m, 1H of a, meso of C3H5), −0.11 (d, 3J(1H,1H) = 8.4 Hz, 1H of b, anti of C3H5), −1.11 (d, 3J(1H,1H) = 8.4 Hz, 1H of b, anti of C3H5). IR (ATR, cm−1): 1934vs [νa(CO)CO], 1866vs [νs(CO)CO], 1714s [ν(CO)CO]. Raman (capillary, cm−1): 1944(3) [νa(CO)CO], 1861(10) [νs(CO)CO], 1715(9) [ν(CO)CO]. Single crystals suitable for X-ray diffraction analysis were obtained by recrystallization of the product from hexane.
Synthesis of [(η3-C3H5){η5-1-(MeOCH2CH2OCO)C9H6}Mo(CO)2] (8)
The steps of synthesis followed the procedure for compound 7. Reagents: 3-(MeOCH2CH2OCO)C9H7 (3; 0.44 g, 2 mmol), 1.3 mL of nBuLi (1.6 mol L−1), [(η3-C3H5)Mo(CO)2(NCMe)2Cl] (6; 0.62 g, 2 mmol). Yield: 0.36 g (0.88 mmol, 44%). Yellow viscous oil. Anal. Calc. for C18H18MoO5: C: 52.69; H: 4.42. Found: C: 52.65; H: 4.48. 1H NMR (CDCl3, 400 MHz, δ ppm; 3:1 mixture of 8a (exo-C3H5) and 8b (endo-C3H5)): 7.86–7.78 (m, 1H of a and 1H of b, H4,7 of C9H6), 7.40–7.00 (m, 3H of a and 3H of b, H4–7 of C9H6), 6.26 (s-br, 1H of a, H2,3 of C9H6), 6.16 (s-br, 1H of b, H2,3 of C9H6), 6.05 (s-br, 1H of a, H2,3 of C9H6), 5.98 (s-br, 1H of b, H2,3 of C9H6), 4.51 (m, 2H of a and 2H of b, CH2), 3.73 (m, 2H of a and 2H of b, CH2), 3.71 (s-br, 1H of b, C3H5), 3.59 (s-br, 2H of b, C3H5), 3.43 (s, 3H of a and 3H of b, CH3), 2.36 (d, 3J(1H,1H) = 5.7 Hz, 1H of a, syn of C3H5), 2.15 (d, 3J(1H,1H) = 5.7 Hz, 1H of a, syn of C3H5), 1.03 (d, 3J(1H,1H) = 10.8 Hz, 1H of a, anti of C3H5), 0.83 (d, 3J(1H,1H) = 10.8 Hz, 1H of a, anti of C3H5), 0.75 (m, 1H of a, meso of C3H5), −0.03 (d, 3J(1H,1H) = 8.8 Hz, 1H of b, anti of C3H5), −1.17 (d, 3J(1H,1H) = 8.8 Hz, 1H of b, anti of C3H5). IR (ATR, cm−1): 1947vs [νa(CO)CO], 1869vs [νs(CO)CO], 1712s [ν(CO)CO].
Synthesis of [(η3-C3H5){η5-1-(PhCH2OCO)C9H6}Mo(CO)2] (9)
The steps of synthesis followed the procedure for compound 7. Reagents: 3-(PhCH2OCO)C9H7 (4; 0.50 g, 2 mmol), 1.3 mL of nBuLi (1.6 mol L−1), [(η3-C3H5)Mo(CO)2(NCMe)2Cl] (6; 0.62 g, 2 mmol). Yield: 0.60 g (1.36 mmol, 68%). Yellow crystals. Mp = 101 °C. Anal. Calc. for C22H18MoO4: C: 59.74; H: 4.10. Found: C: 59.79; H: 4.16. 1H NMR (CDCl3, 400 MHz, δ ppm; 3:1 mixture of 9a (exo-C3H5) and 9b (endo-C3H5)): 7.86–7.75 (m, 1H of a and 1H of b, H4,7 of C9H6), 7.52–7.00 (m, 9H of a and 9H of b, H4–7 of C9H6, C6H5), 6.25 (s-br, 1H of a, H2,3 of C9H6), 6.16 (s-br, 1H of b, H2,3 of C9H6), 6.03 (s-br, 1H of a, H2,3 of C9H6), 5.99 (s-br, 1H of b, H2,3 of C9H6), 5.40 (m, 2H of a and 2H of b, CH2), 3.44 (s-br, 1H of b, C3H5), 3.29 (s-br, 2H of b, C3H5), 2.26 (s-br, 1H of a, syn of C3H5), 2.12 (s-br, 1H of a, syn of C3H5), 0.99 (s-br, 1H of a, C3H5), 0.81 (s-br, 2H of a, C3H5), −0.03 (s-br, 1H of b, anti of C3H5), −1.19 (s-br, 1H of b, anti of C3H5). IR (ATR, cm−1): 1932vs [νa(CO)CO], 1865vs [νs(CO)CO], 1702s [ν(CO)CO]. Raman (capillary, cm−1): 1942(6) [νa(CO)CO], 1856(10) [νs(CO)CO], 1705(5) [ν(CO)CO].
Synthesis of [(η3-C3H5){η5-1-(4-MeOC6H4OCO)C9H6}Mo(CO)2] (10)
The steps of synthesis followed the procedure for compound 7. Reagents: 3-(4-MeOC6H4OCO)C9H7 (5; 0.53 g, 2 mmol), 1.3 mL of nBuLi (1.6 mol L−1), [(η3-C3H5)Mo(CO)2(NCMe)2Cl] (6; 0.62 g, 2 mmol). Yield: 0.25 g (0.55 mmol, 27%). Orange crystals. Mp = 124 °C. Anal. Calc. for C22H18MoO5: C: 57.65; H: 3.96. Found: C: 57.52; H: 3.80. 1H NMR (CDCl3, 400 MHz, δ ppm; 3:1 mixture of 10a (exo-C3H5) and 10b (endo-C3H5)): 7.90–7.80 (m, 1H of a and 1H of b, H4,7 of C9H6), 7.30–6.90 (m, 7H of a and 7H of b, H4–7 of C9H6 and C6H4), 6.37 (s-br, 1H of a, H2,3 of C9H6), 6.29 (s-br, 1H of b, H2,3 of C9H6), 6.13 (s-br, 1H of a, H2,3 of C9H6), 6.06 (s-br, 1H of b, H2,3 of C9H6), 3.82 (s, 3H of a and 3H of b, CH3), 3.58 (s-br, 1H of b, C3H5), 3.35 (s-br, 2H of b, C3H5), 2.40 (d, 3J(1H,1H) = 6.0 Hz, 1H of a, syn of C3H5), 2.24 (d, 3J(1H,1H) = 6.1 Hz, 1H of a, syn of C3H5), 1.13 (d, 3J(1H,1H) = 11.0 Hz, 1H of a, anti of C3H5), 0.89 (d, 3J(1H,1H) = 11.0 Hz, 1H of a, anti of C3H5), 0.63 (m, 1H of a, meso of C3H5), 0.01 (d, 3J(1H,1H) = 9.5 Hz, 1H of b, anti of C3H5), − 1.13 (d, 3J(1H,1H) = 9.6 Hz, 1H of b, anti of C3H5). IR (ATR, cm−1): 1944vs [νa(CO)CO], 1865vs [νs(CO)CO], 1722s [ν(CO)CO]. Raman (capillary, cm−1): 1946(3) [νa(CO)CO], 1862(7) [νs(CO)CO], 1714(9) [ν(CO)CO].
Synthesis of [{η3-1-(MeOCO)C9H6}(η5-Ind)Mo(CO)2] (12)
3-(MeOCO)C9H7 (2; 0.17 g, 1 mmol) was dissolved with 20 mL of THF, cooled at 0 °C and treated dropwise with 0.6 mL of nBuLi (1.6 mol L−1). The reaction mixture was stirred for 1 h and then added dropwise to the THF solution of [{(η5-Ind)Mo(CO)2(μ-Cl)}2] (11; 0.30 g, 0.5 mmol) precooled to −80 °C. The reaction mixture was stirred at room temperature overnight and then vacuum evaporated to dryness. The solid residue was washed with cold hexane (10 mL) and extracted several times with diethyl ether. The volatiles were vacuum evaporated and product was vacuum dried. Yield: 0.19 g (0.43 mmol, 43%). Port red solid. Mp = 120 °C (dec). Anal. Calc. for C22H18MoO4: C: 59.74; H: 4.10. Found: C: 59.87; H: 4.03. 1H NMR (CDCl3; 400 MHz; δ ppm): 7.52–6.65 (m, 9H, C9H6 and C9H7), 5.60 (m, 3H, C9H6 and C9H7), 4.73 (s-br, 1H, C9H7), 3.84 (s, 3H, (s, 3H, COOCH3)). IR (ATR, cm−1): 1943vs [νa(CO)CO], 1876vs [νs(CO)CO], 1828s [νs(CO)CO], 1694s [ν(CO)CO]. Single crystals suitable for X-ray diffraction analysis were obtained by recrystallization of the product from hot hexane.
Synthesis of 3-(MeCO)C9H7 (13)
The steps of synthesis followed the procedure for compound 2. Reagents: indene (1; 11.7 mL, 0.10 mol), sodium hydride (2.4 g; 0.10 mol), ethyl acetate (9.8 mL, 0.10 mol). The crude product was vacuum distilled (90 °C, 660 Pa). Yield: 6.0 g (38 mmol, 38%). Pale yellow solid. Mp = 58 °C. Anal. Calc. for C11H10O: C: 83.52; H: 6.37. Found: C: 83.44; H: 6.31. 1H NMR (CDCl3; 400 MHz; δ ppm): 8.19 (d, 3J(1H,1H) = 7.7 Hz, 1H, C9H7), 7.41 (d, 3J(1H,1H) = 7.4 Hz, 1H, C9H7), 7.30 (t, 3J(1H,1H) = 7.4 Hz, 1H, C9H7), 7.27 (t, 3J(1H,1H) = 1.8 Hz, 1H, C9H7), 7.22 (t, 3J(1H,1H) = 7.2 Hz, 1H, C9H7), 3.47 (d, 3J(1H,1H) = 1.8 Hz, 2H, C9H7), 2.46 (s, 3H, COCH3). 13C{1H} NMR (CDCl3; 101 MHz; δ ppm): 196.2 (1C, Cq, COMe), 145.0 (1C, CH, C9H7), 143.5, 143.2, 140.8 (3 × 1C, Cq, C9H7), 126.7, 125.8, 123.7, 123.5 (4 × 1C, CH, C9H7), 38.6 (1C, CH2, C9H7), 27.7 (1C, CH3, COCH3). IR (ATR, cm−1): 1669s [ν(CO)CO]. Raman (capillary, cm−1): 1666(8) [ν(CO)CO]. Single crystals suitable for X-ray diffraction analysis were obtained by recrystallization of the product from hexane.
Synthesis of [(η3-C3H5){η5-1-(MeCO)C9H6}Mo(CO)2] (14)
The steps of synthesis followed the procedure for compound 7. Reagents: 3-(MeCO)C9H7 (13; 0.79 g, 5 mmol), 3.1 mL of nBuLi (1.6 mol L−1), [(η3-C3H5)Mo(CO)2(NCMe)2Cl] (6; 1.55 g, 5 mmol). Yield: 1.1 g (3.1 mmol, 63%). Yellow powder. Mp = 124 °C. Anal. Calc. for C16H14MoO3: C: 54.78; H: 4.03. Found: C: 54.91; H: 4.16. 1H NMR (CDCl3, 400 MHz, δ ppm; 3:1 mixture of 14a (exo-C3H5) and 14b (endo-C3H5)): 8.05–7.98 (m, 1H of a and 1H of b, H4,7 of C9H6), 7.32–7.05 (m, 3H of a and 3H of b, H4–7 of C9H6), 6.05 (s-br, 2H of a and 2H of b, H2,3 of C9H6), 3.50 (s-br, 1H of b, C3H5), 3.29 (s-br, 2H of b, C3H5), 2.54 (s, 3H of a and 3H of b, CH3), 2.25 (s-br, 1H of a, syn of C3H5), 2.14 (s-br, 1H of a, syn of C3H5), 1.09 (d, 3J(1H,1H) = 9.3 Hz, 1H of a, anti of C3H5), 0.85 (d, 3J(1H,1H) = 9.5 Hz, 1H of a, anti of C3H5), 0.64 (s-br, 1H of a, meso of C3H5), 0.06 (s-br, 1H of b, anti of C3H5), −1.33 (s-br, 1H of b, anti of C3H5). IR (ATR, cm−1): 1936vs [νa(CO)CO], 1859vs [νs(CO)CO], 1660s [ν(CO)CO]. Raman (capillary, cm−1): 1932(5) [νa(CO)CO], 1836(10) [νs(CO)CO], 1666(7) [ν(CO)CO]. Single crystals suitable for X-ray diffraction analysis were obtained by recrystallization of the product from hexane.
Synthesis of 3-(tBuNHCO)C9H7 (15)
Lithium indenide (1-Li), freshly prepared from indene (1; 11.7 mL, 0.10 mol) and 62.5 mL of nBuLi (1.6 mol L−1) in THF (150 mL), was cooled at −80 °C, treated with tBuNCO (11.4 mL, 0.10 mol), slowly warmed at room temperature and stirred overnight. The reaction mixture was poured on an ice/water mixture. The pale yellow precipitate was filtered on a glass frit and washed with hexane. The crude product was dissolved in CH2Cl2 and water. The organic phase was separated, dried with magnesium sulfate and volatiles were evaporated on a rotavapor. The final product was washed with hexane on a glass frit and vacuum dried. Yield: 14.0 g (65 mmol, 65%). Colorless crystals. Mp = 122 °C. Anal. Calc. for C14H17NO: C: 78.10; H: 7.96; N: 6.51. Found: C: 78.22; H: 8.04; N: 6.67. 1H NMR (CDCl3; 400 MHz; δ ppm): 7.89 (d, 3J(1H,1H) = 7.7 Hz, 1H, C9H7), 7.49 (d, 3J(1H,1H) = 7.4 Hz, 1H, C9H7), 7.36 (t, 3J(1H,1H) = 7.5 Hz, 1H, C9H7), 7.28 (t, 3J(1H,1H) = 7.4 Hz, 1H, C9H7), 6.90 (s, 1H, C9H7), 5.89 (s, 1H, NHtBu), 3.48 (s, 2H, C9H7), 1.51 (s, 9H, C(CH3)3). 13C{1H} NMR (CDCl3; 101 MHz; δ ppm): 164.7 (1C, Cq, CONH), 143.9, 141.6 (2 × 1C, Cq, C9H7), 136.7, 126.7, 125.7, 124.1, 121.9 (5 × 1C, CH, C9H7), 51.7 (1C, Cq, C(CH3)3), 38.2 (1C, CH2, C9H7), 29.1 (3C, CH3, Cq, C(CH3)3). IR (ATR, cm−1): 3286m [ν(NH)], 1637s [ν(CO)CO]. Raman (capillary, cm−1): 1638(8) [ν(CO)CO].
Synthesis of 3-(PhNHCO)C9H7 (16)
3-(HOCO)C9H7 (0.80 g; 5.0 mmol) was dissolved in the excess of thionyl chloride (5 mL) and stirred at room temperature for 1 h and then heated at 60 °C for 10 min. The volatiles were vacuum evaporated. The sticky solid was dissolved in CH2Cl2, cooled at −80 °C, treated with the excess of PhNH2 (2.7 mL, 30 mmol) and stirred at room temperature for 2 h. The volatiles were vacuum evaporated and the solid product was washed with diethyl ether. The crude product was purified by column chromatography on silica (hexane/ethyl acetate = 7:3). Yield: 0.62 g (2.6 mmol, 53%). Colorless crystals. Mp = 155 °C. Rf(TLC; hexane/ethyl acetate = 7:3) = 0.44. Anal. Calc. for C16H13NO: C: 81.68; H: 5.57; N: 5.95. Found: 81.52; H: 5.55; N: 5.84. 1H NMR (CDCl3; 400 MHz; δ ppm): 7.94 (d, 3J(1H,1H) = 7.7 Hz, 1H, C9H7), 7.81 (s, 1H, NHPh), 7.63 (d, 3J(1H,1H) = 8.6 Hz, 2H, C6H5), 7.48 (d, 3J(1H,1H) = 7.4 Hz, 1H, C9H7), 7.37–7.32 (m, 1H of C9H7 and 2H of C6H5), 7.27 (td, 3J(1H,1H) = 7.4 Hz, 4J(1H,1H) = 1.2 Hz, 1H, C9H7), 7.13 (tt, 3J(1H,1H) = 7.5 Hz, 4J(1H,1H) = 1.0 Hz, 1H, C6H5), 7.06 (t, 3J(1H,1H) = 2.0 Hz, 1H, C9H7), 3.50 (d, 3J(1H,1H) = 2.0 Hz, 2H, C9H7). 13C{1H} NMR (CDCl3; 101 MHz; δ ppm): 163.3 (1C, Cq, CONH), 143.7, 141.2, 140.9, 137.9 (4 × 1C, Cq, C9H7 and C6H5), 137.0 (1C, CH, C9H7), 129.3 (2C, CH, C6H5), 127.0, 126.0 (2 × 1C, CH, C9H7), 124.7 (2C, CH, C6H5), 124.1, 122.2 (2 × 1C, CH, C9H7), 120.3 (2C, CH, C6H5), 38.6 (1C, CH2, C9H7). IR (ATR, cm−1): 3284s [ν(NH)], 1648s [ν(CO)CO]. Raman (capillary, cm−1): 1650(9) [ν(CO)CO].
Reaction of 1-Na with PhNCO
Lithium indenide (1-Li), freshly prepared from indene (1; 11.7 mL, 0.10 mol) and 62.5 mL of nBuLi (1.6 mol L−1) in THF (150 mL), was cooled at −80 °C, treated with PhNCO (10.9 mL, 0.10 mol), slowly warmed at room temperature and stirred overnight. The reaction mixture was poured on an ice/water mixture, treated with CH2Cl2 and neutralized with hydrochloric acid. The aqueous phase was separated and disposed. The organic phase was filtered on a glass frit. The collected solid was recrystallized from acetone to give colorless crystals of 18 (yield: 2.9 g, 8.2 mmol, 8%). The volatiles from the filtrate were vacuum evaporated and the product was washed with diethyl ether to give 9 g of a mixture of 16 and 17 in a molar ratio of 1:5 (according to 1H NMR). Analytically pure samples of 16 and 17 were obtained by column chromatography on silica (hexane/ethyl acetate = 7:3). 1,3-(PhNHCO)2C9H6 (18): white solid. Mp = 240 °C (dec.). Anal. Calc. for C23H18N2O2: C: 77.95; H: 5.12; N: 7.90. Found: C: 77.90; H: 5.04; N: 7.96. 1H NMR (acetone-d6; 400 MHz; δ ppm): 9.66 (s, 1H, NHPh), 9.14 (s, 1H, NHPh), 8.03 (d, 3J(1H,1H) = 7.6 Hz, 1H, C9H6), 7.85 (d, 3J(1H,1H) = 8.2 Hz, 2H, C6H5), 7.68–7.63 (m, 1H of C9H6 and 2H of C6H5), 7.42–7.27 (m, 3H of C9H6 and 4H of C6H5), 7.12 (tt, 3J(1H,1H) = 7.3 Hz, 4J(1H,1H) = 1.1 Hz, 1H, C6H5), 7.08 (tt, 3J(1H,1H) = 7.3 Hz, 4J(1H,1H) = 1.1 Hz, 1H, C6H5), 4.71 (d, 3J(1H,1H) = 2.1 Hz, H, C9H6). IR (ATR, cm−1): 3236m [ν(NH)], 1662s [ν(CO)CO], 1646s [ν(CO)CO]. Raman (capillary, cm−1): 1647(9) [ν(CO)CO]. 3-(PhNHCONPhCO)C9H7 (17): colorless crystals. Mp = 116 °C. Rf(TLC; hexane/ethyl acetate = 7:3) = 0.53. Anal. Calc. for C23H18N2O2: C: 77.95; H: 5.12; N: 7.90. Found: C: 77.84; H: 5.18; N: 7.79. 1H NMR (CDCl3; 400 MHz; δ ppm): 11.60 (s, 1H, NHPh), 7.75 (d, 3J(1H,1H) = 7.7 Hz, 1H, C9H7), 7.66 (d, 3J(1H,1H) = 8.2 Hz, 2H, C6H5), 7.43–7.25 (m, 3H of C9H7 and 7H of C6H5), 7.16 (tt, 3J(1H,1H) = 7.4 Hz, 4J(1H,1H) = 1.1 Hz, 1H, C6H5), 6.07 (t, 3J(1H,1H) = 2.1 Hz, 1H, C9H7), 3.25 (d, 3J(1H,1H) = 2.0 Hz, 2H, C9H7). 13C{1H} NMR (CDCl3; 101 MHz; δ ppm): 170.0, 152.2 (2 × 1C, Cq, CO), 142.5, 141.9, 138.9, 138.7, 137.9 (5 × 1C, Cq, C9H7 and C6H5), 139.0 (1C, CH, C9H7), 129.8, 129.2, 129.1 (3 × 2C, CH, C6H5), 128.9 (1C, CH, C6H5), 126.9, 126.0 (2 × 1C, CH, C9H7), 124.5, 124.0 (2 × 1C, CH, C9H7 and C6H5), 121.5 (1C, CH, C9H7), 120.6 (2C, CH, C6H5), 39.2 (1C, CH2, C9H7). IR (ATR, cm−1): 3214m [ν(NH)], 1646s [ν(CO)CO]. Raman (capillary, cm−1): 1723(1) [ν(CO)CO], 1648(9) [ν(CO)CO].
Synthesis of 3-(4-ClC6H4NHCO)C9H7 (19)
The steps of synthesis followed the procedure for compound 16. Reagents: 3-(HOCO)C9H7 (0.80 g, 5.0 mmol), 4-ClC6H4NH2 (2.6 g, 20 mmol). The crude product was washed with ether and purified by column chromatography on silica (hexane/ethyl acetate = 7:3). Yield: 0.60 g (2.2 mmol, 44%). Colorless crystals. Mp = 172 °C. Rf(TLC; hexane/ethyl acetate = 7:3) = 0.42. Anal. Calc. for C16H12ClNO: C: 71.25; H: 4.48; N: 5.19. Found: C: 71.35; H: 4.46; N: 5.12. 1H NMR (CDCl3; 400 MHz; δ ppm): 7.91 (d, 3J(1H,1H) = 7.5 Hz, 1H, C9H7), 7.80 (s, 1H, NHPh), 7.57 (d, 3J(1H,1H) = 8.8 Hz, 2H, C6H4), 7.48 (d, 3J(1H,1H) = 7.4 Hz, 1H, C9H7), 7.34 (t, 3J(1H,1H) = 7.5 Hz, 1H, C9H7), 7.29 (d, 3J(1H,1H) = 8.8 Hz, 2H, C6H4), 7.27 (td, 3J(1H,1H) = 7.4 Hz, 4J(1H,1H) = 1.1 Hz, 1H, C9H7), 7.05 (t, 3J(1H,1H) = 2.0 Hz, 1H, C9H7), 3.50 (d, 3J(1H,1H) = 2.0 Hz, 2H, C9H7). 13C{1H} NMR (CDCl3; 101 MHz; δ ppm): 163.2 (1C, Cq, CONH), 143.7, 141.1, 140.7, 136.5, 127.9 (5 × 1C, Cq, C9H7 and C6H4), 137.3 (1C, CH, C9H7), 129.3 (2C, CH, C6H5), 127.0, 126.2, 124.2, 122.1, (4 × 1C, CH, C9H7), 121.6 (2C, CH, C6H5), 38.6 (1C, CH2, C9H7). IR (ATR, cm−1): 3284m [ν(NH)], 1655s [ν(CO)CO]. Raman (capillary, cm−1): 1657(10) [ν(CO)CO].
Synthesis of [(η3-C3H5){η5-1-(tBuNHCO)C9H6}Mo(CO)2] (20)
The steps of synthesis followed the procedure for compound 7. Reagents: 3-(tBuNHCO)C9H7 (15; 0.43 g, 2 mmol), 1.3 mL of nBuLi (1.6 mol L−1), [(η3-C3H5)Mo(CO)2(NCMe)2Cl] (6; 0.62 g, 2 mmol). Yield: 0.50 g (1.23 mmol, 61%). Yellow powder. Mp = 190 °C (dec.). Anal. Calc. for C19H21MoNO3: C: 56.03; H: 5.20; N: 3.44. Found: C: 56.18; H: 5.33; N: 3.51. 1H NMR (CDCl3, 400 MHz, δ ppm; 3:1 mixture of 20a (exo-C3H5) and 20b (endo-C3H5)): 8.00–7.91 (m, 1H of a and 1H of b, H4,7 of C9H6), 7.21–6.98 (m, 3H of a and 3H of b, H4–7 of C9H6), 5.98 (d, 3J(1H,1H) = 3.0 Hz, 1H of a, H2,3 of C9H6), 5.91 (d, 3J(1H,1H) = 2.5 Hz, 1H of b, H2,3 of C9H6), 5.86 (d, 3J(1H,1H) = 3.0 Hz, 1H of a, H2,3 of C9H6), 5.79 (d, 3J(1H,1H) = 2.5 Hz, 1H of b, H2,3 of C9H6), 5.68 (s, 1H of a, NH), 5.62 (s, 1H of b, NH), 3.54 (s-br, 1H of b, C3H5), 3.33 (s-br, 2H of b, C3H5), 2.44 (d, 3J(1H,1H) = 6.7 Hz, 1H of a, syn of C3H5), 2.11 (d, 3J(1H,1H) = 6.6 Hz, 1H of a, syn of C3H5), 1.48 (s, 9H of a and 9H of b, C(CH3)3), 1.06 (d, 3J(1H,1H) = 10.7 Hz, 1H of a, anti of C3H5), 0.85 (d, 3J(1H,1H) = 10.8 Hz, 1H of a, anti of C3H5), 0.78 (m, 1H of a, meso of C3H5), −0.06 (d, 3J(1H,1H) = 9.6 Hz, 1H of b, anti of C3H5), −1.09 (d, 3J(1H,1H) = 9.7 Hz, 1H of b, anti of C3H5). IR (ATR, cm−1): 3372m [ν(NH)], 1932vs [νa(CO)CO], 1848vs [νs(CO)CO], 1635s [ν(CO)CO]. Raman (capillary, cm−1): 1931(6) [νa(CO)CO], 1850(10) [νs(CO)CO], 1637(4) [ν(CO)CO]. Single crystals of 20 suitable to X-ray diffraction analysis were prepared by slow evaporation of MeOH solution.
Synthesis of [(η3-C3H5){η5-1-(PhNHCO)C9H6}Mo(CO)2] (21)
The steps of synthesis followed the procedure for compound 7. Reagents: 3-(PhNHCO)C9H7 (16; 0.47 g, 2 mmol), 1.3 mL of nBuLi (1.6 mol L−1), [(η3-C3H5)Mo(CO)2(NCMe)2Cl] (6; 0.62 g, 2 mmol). Yield: 0.45 g (1.05 mmol, 53%). Yellow powder. Mp = 180 °C (dec.). Anal. Calc. for C21H17MoNO3: C: 59.03; H: 4.01; H: 3.28. Found: C: 58.91; H: 4.08; H: 3.21. 1H NMR (acetone-d6, 400 MHz, δ ppm; 4:1 mixture of 21a (exo-C3H5) and 21b (endo-C3H5)): 9.24 (1H of a, NH), 9.13 (1H of b, NH), 8.16–6.95 (m, 9H of a and 9H of b, H4–7 of C9H6, C6H5), 6.71 (d, 3J(1H,1H) = 3.2 Hz, 1H of a, H2,3 of C9H6), 6.61 (s-br, 1H of b, H2,3 of C9H6), 6.32 (d, 3J(1H,1H) = 3.2 Hz, 1H of a, H2,3 of C9H6), 6.28 (s-br, 1H of b, H2,3 of C9H6), 3.50 (s-br, 1H of b, C3H5), 3.36 (s-br, 2H of b, C3H5), 2.36 (d, 3J(1H,1H) = 7.2 Hz, 1H of a, syn of C3H5), 2.14 (d, 3J(1H,1H) = 7.0 Hz, 1H of a, syn of C3H5), 1.15 (m, 1H of a, meso of C3H5), 0.99 (d, 3J(1H,1H) = 11.3 Hz, 1H of a, anti of C3H5), 0.82 (d, 3J(1H,1H) = 11.4 Hz, 1H of a, anti of C3H5), −0.09 (d, 3J(1H,1H) = 10.7 Hz, 1H of b, anti of C3H5), −0.94 (d, 3J(1H,1H) = 10.8 Hz, 1H of b, anti of C3H5). IR (ATR, cm−1): 3307m [ν(NH)], 1932vs [νa(CO)CO], 1859vs [νs(CO)CO], 1633s [ν(CO)CO]. Raman (capillary, cm−1): 1933(5) [νa(CO)CO], 1866(9) [νs(CO)CO], 1635(8) [ν(CO)CO].
Synthesis of [(η3-C3H5){η5-1-(4-ClC6H4NHCO)C9H6}Mo(CO)2] (22)
The steps of synthesis followed the procedure for compound 7. Reagents: 3-(4-ClC6H4NHCO)C9H7 (19; 0.54 g, 2 mmol), 1.3 mL of nBuLi (1.6 mol L−1), [(η3-C3H5)Mo(CO)2(NCMe)2Cl] (6; 0.62 g, 2 mmol). Yield: 0.55 g (1.19 mmol, 60%). Yellow powder. Mp = 185 °C (dec.). Anal. Calc. for C21H16ClMoNO3: C: 54.62; H: 3.49; N: 3.03. Found: C: 54.70; H: 3.52; N: 3.08. 1H NMR (CDCl3, 400 MHz, δ ppm; 3:1 mixture of 23a (exo-C3H5) and 23b (endo-C3H5)): 7.98 (d, 3J(1H,1H) = 8.8 Hz, 1H of a and 1H of b, H4,7 of C9H6), 7.60–7.05 (m, 8H of a and 8H of b, H4–7 of C9H6, C6H4, NH), 6.10 (d, 3J(1H,1H) = 2.9 Hz, 1H of a, H2,3 of C9H6), 6.05 (d, 3J(1H,1H) = 2.9 Hz, 1H of a, H2,3 of C9H6), 6.03 (d, 3J(1H,1H) = 2.8 Hz, 1H of b, H2,3 of C9H6), 5.98 (d, 3J(1H,1H) = 2.8 Hz, 1H of b, H2,3 of C9H6), 3.62 (s-br, 1H of b, C3H5), 3.39 (s-br, 2H of b, C3H5), 2.47 (d, 3J(1H,1H) = 6.7 Hz, 1H of a, syn of C3H5), 2.21 (d, 3J(1H,1H) = 6.7 Hz, 1H of a, syn of C3H5), 1.14 (d, 3J(1H,1H) = 11.1 Hz, 1H of a, anti of C3H5), 0.90 (d, 3J(1H,1H) = 11.6 Hz, 1H of a, anti of C3H5), 0.64 (m, 1H of a, meso of C3H5), −0.06 (d, 3J(1H,1H) = 9.8 Hz, 1H of b, anti of C3H5), −1.00 (d, 3J(1H,1H) = 9.6 Hz, 1H of b, anti of C3H5). IR (ATR, cm−1): 3424m [ν(NH)], 1940vs [νa(CO)CO], 1852vs [νs(CO)CO], 1667s [ν(CO)CO]. Raman (capillary, cm−1): 1948(3) [νa(CO)CO], 1848(8) [νs(CO)CO], 1664(10) [ν(CO)CO]. Single crystals of 23·MeOH suitable to X-ray diffraction analysis were prepared by slow evaporation of MeOH solution.
X-ray crystallography
The X-ray data for the crystals of compounds 7, 12, 13, 14, 20 and 22·MeOH were obtained at 150 K using an Oxford Cryostream low-temperature device on a Nonius KappaCCD diffractometer with Mo Kα radiation (λ = 0.71073 Å) and a graphite monochromator. Data reductions were performed using DENZO-SMN.26 The structures were solved by direct methods (Sir92)27 and refined by full-matrix least squares based on F2 (SHELXL).28 Hydrogen atoms were mostly localized on a difference Fourier map. However, to ensure uniformity of the treatment of the crystals, all hydrogen atoms were recalculated into idealized positions (riding model) and assigned temperature factors Uiso(H) = 1.2[Ueq(pivot atom)] or 1.5Ueq for the methyl moiety with C–H = 0.96, 0.97, and 0.93 Å for methyl, methylene, and hydrogen atoms in aromatic rings or the allyl moiety, respectively. The structure of 7 contains a disorder of the allyl group which, is positionaly disordered on one of the carbon atoms (C13) and splits into two positions with an occupancy of about 7:3; this disorder was treated using SHELXL software.28
Acknowledgements
The NMR spectrometers at ITQB are part of the National NMR Facility, supported by Fundação para a Ciência e a Tecnologia (RECI/BBB-BQB/0230/2012). We thank University of Pardubice for the Erasmus mobility fellowship (Erasmus+ 2014/2015).References
- (a) T. Liu, X. Wang, C. Hoffmann, D. L. DuBois and M. R. Bullock, Angew. Chem., Int. Ed., 2014, 53, 5300–5304 CrossRef CAS PubMed; (b) M. Fang, E. S. Wiedner, W. G. Dougherty, W. S. Kassel, T. Liu, D. L. DuBois and M. R. Bullock, Organometallics, 2014, 33, 5820–5833 CrossRef CAS; (c) X. Wang, D. Li, W. Deuther-Conrad, J. Lu, Y. Xie, B. Jia, M. Cui, J. Steinbach, P. Brust, B. Liu and H. Jia, J. Med. Chem., 2014, 57, 7113–7125 CrossRef CAS PubMed; (d) S. Sulieman, D. Can, J. Mertens, H. W. P. N'Dongo, Y. Liu, P. Schmutz, M. Bauwens, B. Spingler and R. Alberto, Organometallics, 2012, 31, 6880–6886 CrossRef CAS; (e) D. Can, B. Spingler, P. Schmutz, F. Mendes, P. Raposinho, C. Fernandes, F. Carta, A. Innocenti, I. Santos, C. T. Supuran and R. Alberto, Angew. Chem., Int. Ed., 2012, 51, 3354–3357 CrossRef CAS PubMed; (f) E. Hillard, A. Vessieres, L. Thouin, G. Jaouen and C. Amatore, Angew. Chem., Int. Ed., 2006, 45, 285–290 CrossRef CAS PubMed; (g) C. Ornelas, New J. Chem., 2011, 35, 1973–1985 RSC; (h) J. H. Scarborough, K. Brusoski, S. Brewer, S. Rodich, K. S. Chatley, T. Nguyen and K. N. Green, Organometallics, 2015, 34, 918–925 CrossRef CAS; (i) Y. Yan, T. M. Deaton, J. Zhang, H. He, J. Hayat, P. Pageni, K. Matyjaszewski and C. Tang, Macromolecules, 2015, 48, 1644–1650 CrossRef CAS; (j) K. Strohfeldt and M. Tacke, Chem. Soc. Rev., 2008, 37, 1174–1187 RSC; (k) G. Erker, Dalton Trans., 2011, 40, 7475–7483 RSC; (l) S. S. Braga and A. M. S. Silva, Organometallics, 2013, 32, 5626–5639 CrossRef CAS; (m) S. Ursillo, D. Can, H. W. P. N'Dongo, P. Schmutz, B. Spingler and R. Alberto, Organometallics, 2014, 33, 6945–6952 CrossRef CAS.
- P. J. Chirik, Organometallics, 2010, 29, 1500–1517 CrossRef CAS.
- S. Manzini, J. A. Fernández-Salas and S. P. Nolan, Acc. Chem. Res., 2014, 47, 3089–3101 CrossRef CAS PubMed.
- (a) J. M. O'Connor and C. P. Casey, Chem. Rev., 1987, 87, 307–318 CrossRef; (b) H. Bang, T. J. Lynch and F. Basolo, Organometallics, 1992, 11, 40–48 CrossRef CAS; (c) C. C. Romão, Appl. Organomet. Chem., 2000, 14, 539–548 CrossRef.
- (a) M. J. Calhorda, C. C. Romão and L. F. Veiros, Chem. – Eur. J., 2002, 8, 868–875 CrossRef CAS PubMed; (b) M. J. Calhorda and L. F. Veiros, Coord. Chem. Rev., 1999, 185–186, 37–51 CrossRef CAS.
- G. P. McGovern, F. Hung-Low, J. W. Tye and C. A. Bradley, Organometallics, 2012, 31, 3865–3879 CrossRef CAS.
- J. Honzíček, C. C. Romão, M. J. Calhorda, A. Mukhopadhyay, J. Vinklárek and Z. Padělková, Organometallics, 2011, 30, 717–725 CrossRef.
- P. A. Deck, W. F. Jackson and F. R. Fronczek, Organometallics, 1996, 15, 5287–5291 CrossRef CAS.
- P. A. Deck and F. R. Fronczek, Organometallics, 2000, 19, 327–333 CrossRef CAS.
- K. Sünkel, S. Weigand, A. Hoffmann, S. Blomeyer, C. G. Reuter, Y. V. Vishnevskiy and N. W. Mitzel, J. Am. Chem. Soc., 2015, 137, 126–129 CrossRef PubMed.
- P. J. Graham, V. Lindsey, G. W. Parshall, M. L. Peterson and G. M. Whitman, J. Am. Chem. Soc., 1957, 79, 3416–3419 CrossRef CAS.
- W. P. Hart, D. W. Macomber and M. D. Rausch, J. Am. Chem. Soc., 1980, 102, 1196–1198 CrossRef CAS.
- (a) L. Busetto, C. Cassani, V. Zanotti, V. G. Albano and P. Sabatino, Organometallics, 2001, 20, 282–288 CrossRef CAS; (b) J. Honzíček, A. Mukhopadhyay, T. S. Silva, M. J. Romão and C. C. Romão, Organometallics, 2009, 28, 2871–2879 CrossRef; (c) A. R. Petrov, K. Jess, M. Freytag, P. G. Jones and M. Tamm, Organometallics, 2013, 32, 5946–5954 CrossRef CAS.
- (a) M. Oberhoff, L. Duda, J. Karl, R. Mohr, G. Erker, R. Fröhlich and M. Grehl, Organometallics, 1996, 15, 4005–4011 CrossRef CAS; (b) K. Klaß, R. Fröhlich and G. Erker, J. Chem. Soc., Dalton Trans., 1999, 4457–4461 RSC; (c) H. Goitia, Y. Nieto, M. D. Villacampa, C. Kasper, A. Laguna and M. C. Gimeno, Organometallics, 2013, 32, 6069–6078 CrossRef CAS.
- J. Honzíček, P. Kratochvíl, J. Vinklárek, A. Eisner and Z. Padělková, Organometallics, 2012, 31, 2193–2202 CrossRef.
- J. Schejbal, J. Honzíček, J. Vinklárek, M. Erben and Z. Růžičková, Eur. J. Inorg. Chem., 2014, 5895–5907 CrossRef CAS.
- (a) W. E. Noland, L. L. Landucci and V. Kameswaran, J. Org. Chem., 1980, 45, 3456–3461 CrossRef CAS; (b) T. Takahashi, H. Kameda, T. Kamei and M. Ishizaki, J. Fluorine Chem., 2006, 127, 760–768 CrossRef CAS; (c) G. R. Clemo, L. H. Groves, L. Munday and G. A. Swan, J. Chem. Soc., 1951, 863–867 RSC.
- J. W. Faller, C. C. Chen, M. J. Mattina and A. Jakubowski, J. Organomet. Chem., 1973, 52, 361–386 CrossRef CAS.
- (a) P. Neves, C. C. L. Pereira, F. A. A. Paz, S. Gago, M. Pillinger, C. M. Silva, A. A. Valente, C. C. Romão and I. S. Gonçalves, J. Organomet. Chem., 2010, 695, 2311–2319 CrossRef CAS; (b) F. Pammer, Y. Sun and W. R. Thiel, Organometallics, 2008, 27, 1015–1018 CrossRef CAS; (c) J. Honzíček, J. Vinklárek, M. Erben, Z. Padělková, L. Šebestová and M. Řezáčová, J. Organomet. Chem., 2014, 749, 387–393 CrossRef; (d) J. Honzíček, J. Vinklárek, Z. Padělková, L. Šebestová, K. Foltánová and M. Řezáčová, J. Organomet. Chem., 2012, 716, 258–268 CrossRef.
- J. Honzíček, J. Vinklárek, M. Erben, J. Lodinský, L. Dostál and Z. Padělková, Organometallics, 2013, 32, 3502–3511 CrossRef.
- J. Pietruszka, R. C. Simon, F. Kruska and M. Braun, Eur. J. Org. Chem., 2009, 6217–6224 CrossRef CAS.
- J. W. Faller, R. H. Crabtree and A. Habib, Organometallics, 1985, 4, 929–935 CrossRef CAS.
- M. G. B. Drew, V. Félix, I. S. Gonçalves, C. C. Romão and B. Royo, Organometallics, 1998, 17, 5782–5788 CrossRef CAS.
- J. R. Ascenso, C. G. de Azevedo, I. S. Gonçalves, E. Herdtweck, D. S. Moreno, M. Pessanha and C. C. Romão, Organometallics, 1995, 14, 3901–3919 CrossRef CAS.
- W. L. F. Armarego and D. D. Perrin, Purification of Laboratory Chemicals, Oxford, 1996 Search PubMed.
- Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 307–326 CAS.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435–436 Search PubMed.
- G. M. Sheldrick, SHELXL97, University of Göttingen, Germany, 2008 Search PubMed.
Footnote |
| † CCDC 1416908–1416913. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj02406d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |