
Isomorphic metal malonates with N-aminoguanidine: MCo2O4 (M = Ni & Zn) nanoparticle synthesis via a (AmgH)2[M1/3Co2/3(mal)2(H2O)2] precursor solid solution†
Rajendran
Selvakumar
a,
Steven J.
Geib
b,
Thathan
Premkumar
*c,
Sundararajan
Vairam
d and
Subbiah
Govindarajan
*a
aDepartment of Chemistry, Bharathiar University, Coimbatore – 641 046, India. E-mail: drsgovind@yahoo.co.in
bDepartment of Chemistry, University of Pittsburgh, Pittsburgh, PA 15260, USA
cThe University College, Department of Chemistry, Sungkyunkwan University, Suwon 440-746, South Korea. E-mail: thathanpremkumar@gmail.com
dDepartment of Chemistry, Government College of Technology, Coimbatore, India
First published on 30th October 2015
Abstract
Divalent metal complexes of malonate with aminoguanidine possessing (AmgH)2[M(mal)2(H2O)2] [M = Co (1), Ni (2) or Zn (3); mal = malonate anion and AmgH = aminoguanidinium cation] stoichiometry and their solid solutions, (AmgH)2[Ni0.5Co0.5(mal)2(H2O)2] (4) and (AmgH)2[M1/3Co2/3(mal)2(H2O)2] [where M = Ni (5) or Zn(6)], were prepared and characterized by analytical, thermal and powder X-ray diffraction studies. The crystal and molecular structures of both cobalt and nickel compounds were isomorphic, crystallizing in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The complexes exhibit similar modes of endo-followed by exothermic decomposition to produce respective metal oxides below 550 °C. Metal cobaltites, MCo2O4 where M = Ni and Zn, were obtained from the above solid solutions as decomposition residues by heating at 600, 700 and 800 °C in a silica crucible for 3 h. Spinel oxides nanoparticles were characterized by infrared spectra, powder X-ray diffraction patterns, scanning electron microscope coupled with energy dispersive X-ray analysis and transmission electron microscope studies.
. The complexes exhibit similar modes of endo-followed by exothermic decomposition to produce respective metal oxides below 550 °C. Metal cobaltites, MCo2O4 where M = Ni and Zn, were obtained from the above solid solutions as decomposition residues by heating at 600, 700 and 800 °C in a silica crucible for 3 h. Spinel oxides nanoparticles were characterized by infrared spectra, powder X-ray diffraction patterns, scanning electron microscope coupled with energy dispersive X-ray analysis and transmission electron microscope studies.
Introduction
Transition metal oxides are a well-established class of materials with diverse applications due to their useful properties such as magnetic, electrical, catalytic activities.1–10 Many researchers have worked on the synthesis of such metal oxides with uniform nanosized particles.11–15 The method employed for the preparation of nanosized metal oxide uses the decomposition of a precursor with hydrazine or its derivatives and carboxylate bonded to metal ions. The chemistry of hydrazine is interesting because it possesses a potent N–N bond along with two free electron pairs and four substitutable hydrogen atoms. Moreover, it is known to form various complexes with transition metals. The participation of the carboxylate ion as an important ligand in inorganic and bioinorganic chemistry has created increasing interest in the coordination chemistry of carboxylic acids. The versatility of carboxylate ions on the coordination is well known, and combined with hydrazine or hydrazine derivatives makes the coordination chemistry interesting. Further interest in their thermal chemistry is due to the fueling character of hydrazine.16 Therefore, isomorphic metal hydrazine compounds and their solid solutions have been used for the preparation of metal or mixed-metal (spinel) oxides. In the preparation of advanced ceramic powders, various chemical methods such as co-precipitation, sol–gel, spray–dry, etc., have been used. However, preparation of ceramic powders using a solid solution precursor method17,18 was reported to be more promising and advantageous than other methods as it achieves excellent stoichiometry and low impurity content.Solid solution precursors is a suitable method in order to achieve better homogeneity, controlled morphology, purity of desired phase, high specific surface and good physico chemical and electrocatalytic surface properties. Solid solution precursors containing hydrazine are all simple hydrazine complexes of mixed-metal hydrazidocarboxylate19,20 and oxalate.21,22 The corresponding malonate23 and malate24 analogs have been synthesized in our laboratory and we have studied their utility as starting materials for metal oxides and spinel cobaltites. Among the anions, only malonate (mal) forms both neutral hydrazine and hydrazinium metal malonates of similar compositions. Furthermore, guanylhydrazine, also referred to as aminoguanidine (Amg), is a hydrazine derivative with better chemical and thermal reactivity due to its asymmetric bifunctional nature and rich nitrogen content. Hence, for the first time, this study attempts to synthesize aminoguanidinium metal malonate, and the structurality among the complexes was examined by a single-crystal X-ray diffraction (XRD) study. Furthermore, since malonates of Co, Ni and Zn are isomorphous, solid solution malonates were prepared and used as precursors for nanosized spinel oxides (Scheme 1). Though, a number of methods have been employed to synthesize the oxide materials, the use of novel methods such as solid solution precursor method is very scant. Further, cobaltites are good anode materials for lithium ion batteries. Spinel NiCo2O4 and ZnCo2O4 nanostructures have shown great opportunities in energy related area. Hence, it is envisaged that the prepared cobaltites can be used in energy related materials and devices.
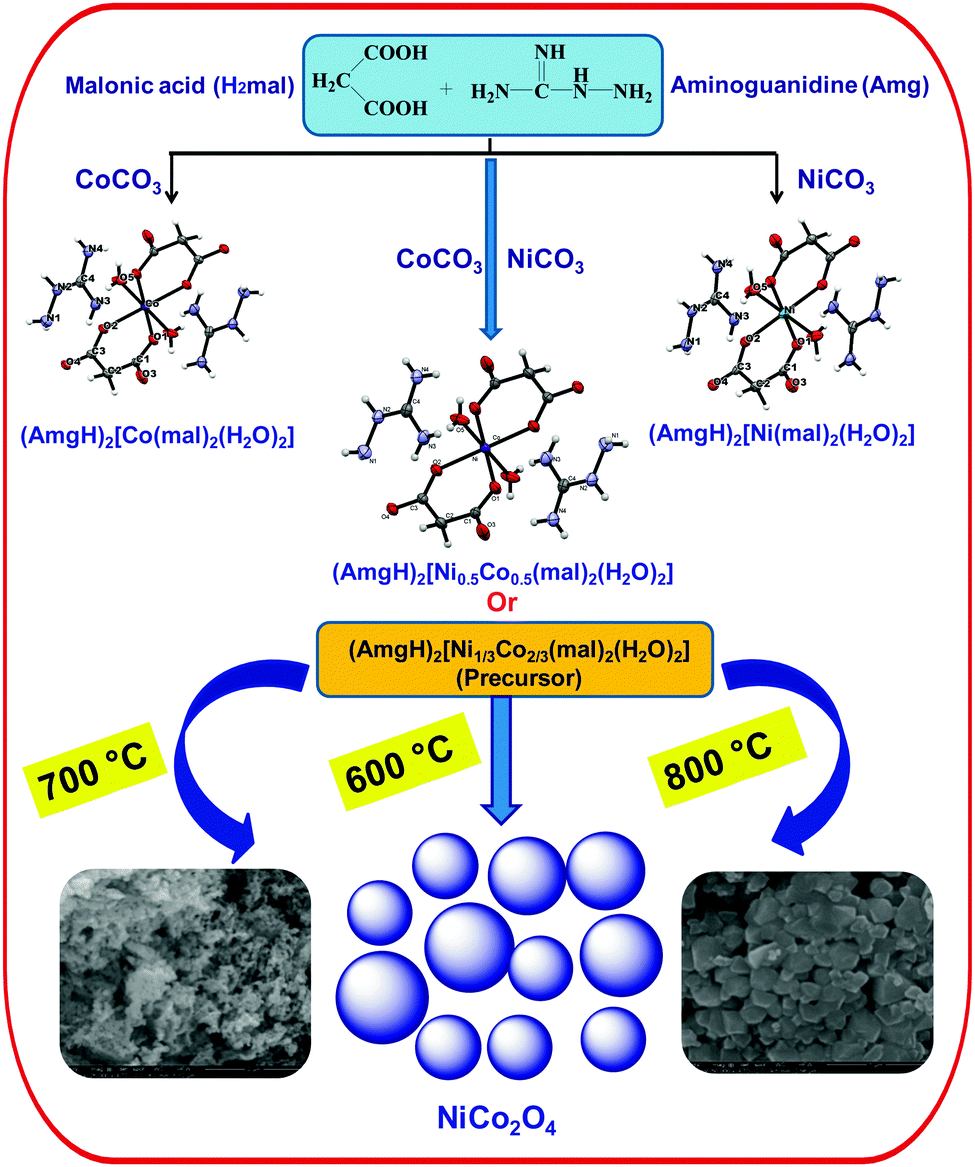 | ||
| Scheme 1 Scheme showing detailed experimental procedure and reaction conditions. | ||
Results and discussion
Isomorphic aminoguanidinium diaqua metal malonate, (AmgH)2[M(mal)2(H2O)2], where M = Co (1), Ni (2), Zn (3) and aminoguanidinium mixed-metal malonate dihydrate, (AmgH)2[Ni0.5Co0.5(mal)2(H2O)2] (4), and (AmgH)2[M1/3Co2/3(mal)2(H2O)2], where M = Ni (5), Zn (6), were prepared by the reaction of respective metal ion precursors with malonic acid followed by the addition of aminoguanidine bicarbonate in water medium. Nanocystalline metal cobaltites, MCo2O4, where M = Ni and Zn, were prepared from the above solid solutions 5 and 6, as decomposition residues by heating at 600, 700 and 800 °C in a silica crucible for 3 h.The metal complexes crystallize in the same triclinic space group, P (Table 1). The X-ray crystallographic study reveals that the metal coordination spheres in cobalt (1) and nickel (2) complexes (Fig. 1a–c) are isomorphic with a monomeric building block, [M(mal)2(H2O)2]2−, that is charge balanced by the cationic aminoguanidine counterpart. In each case, the divalent metal ion is six-coordinated and exhibits an octahedral geometry, in which four coplanar oxygen atoms from two chelated malonate ligand furnish the equatorial plane with a bite angle of 89.08(4)° in 1, 91.01(10)° in 2 and 89.80(4)° in 4 and the axial positions are occupied by two aqua ligands. The M–O bond distances in the equatorial planes are 2.0636(9) (O1) and 2.0466(9) Å (O2) for complex 1, 2.019(2) (O1) and 2.008(2) Å (O2) for complex 2 and 2.0480(9) (O1) and 2.0339(9) Å (O2) for complex 4. The bond length and angles of compounds 1 and 2 are depicted in Tables S1 and S2 (ESI†). These values are in good agreement with malonate-containing complexes having similar coordinating modes.25 The apical M–O bond length of 2.0972(12) Å for Co and 2.079(3) Å for Ni, are slightly longer than those of the equatorial M–O bonds. The bond distance and angle of the aminoguanidine moiety were also comparable to those observed for uncomplexed cations in the solid state.26–29
| 1 | 2 | 4 | |
|---|---|---|---|
| Empirical formula | C8H22CoN8O10 | C8H22N8NiO10 | C8H19Co0.5N8Ni0.5O10 |
| Formula weight | 449.27 | 449.05 | 446.13 |
| Temperature | 203(2) K | 203(2) K | 203(2) K |
| Wavelength | 0.71073 Å | 0.71073 Å | 0.71073 Å |
| Crystal system | Triclinic | Triclinic | Triclinic |
| Space group |
P |
P |
P |
| Unit cell dimensions | a = 6.9244(12) Å | a = 6.881(4) Å | a = 6.9096(10) Å |
| b = 7.3583(12) Å | b = 7.283(4) Å | b = 7.3355(10) Å | |
| c = 9.5353(16) Å | c = 9.541(5) Å | c = 9.5299(13) Å | |
| α = 105.537(3)° | α = 105.601(10)° | α = 105.577(2)° | |
| β = 110.219(3)° | β = 109.834(10)° | β = 110.156(2)° | |
| γ = 102.545(3)° | γ = 102.526(10)° | γ = 102.494(2)° | |
| Volume | 412.65(12) Å3 | 407.3(4) Å3 | 410.52(10) Å3 |
| Z | 1 | 1 | 1 |
| Density (calculated) | 1.808 Mg m−3 | 1.831 Mg m−3 | 1.805 Mg m−3 |
| Absorption coefficient | 1.114 mm−1 | 1.266 mm−1 | 1.188 mm−1 |
| F(000) | 233 | 234 | 231 |
| Crystal size | 0.25 × 0.22 × 0.16 mm3 | 0.21 × 0.18 × 0.12 mm3 | 0.24 × 0.21 × 0.13 mm3 |
| Theta range for data collection | 2.36 to 32.26° | 2.36 to 32.23° | 2.36 to 32.24° |
| Index ranges | −10 ≤ h ≤10, −10 ≤ k ≤ 10, −14 ≤ l ≤ 14 | −9 ≤ h ≤ 10, −10 ≤ k ≤ 10, −14 ≤ l ≤ 13 | −10 ≤ h ≤ 10, −10 ≤ k ≤ 10, −14 ≤ l ≤ 14 |
| Reflections collected | 5131 | 5014 | 5106 |
| Independent reflections | 2679 [R(int) = 0.0207] | 2604 [R(int) = 0.0533] | 2655 [R(int) = 0.0237] |
| Completeness to theta = 32.23° | 91.20% | 90.20% | 91.1% |
| Absorption correction | None | Multi-scan (Sadabs) | None |
| Max. and min. transmission | 0.8419 and 0.7681 | 0.8629 and 0.7769 | 0.8629 and 0.7745 |
| Refinement method | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 2679/0/168 | 2604/0/168 | 2655/0/168 |
| Goodness-of-fit on F2 | 0.885 | 1.088 | 0.944 |
| Final R indices [I > 2σ(I)] | R 1 = 0.0304, wR2 = 0.0891 | R 1 = 0.0667, wR2 = 0.1499 | R 1 = 0.0304, wR2 = 0.0888 |
| R indices (all data) | R 1 = 0.0333, wR2 = 0.0925 | R 1 = 0.0949, wR2 = 0.1637 | R 1 = 0.0318, wR2 = 0.0914 |
| Largest diff. peak and hole | 0.497 and −0.287 e Å−3 | 2.400 and −1.288 e Å−3 | 0.530 and −0.318 e Å−3 |
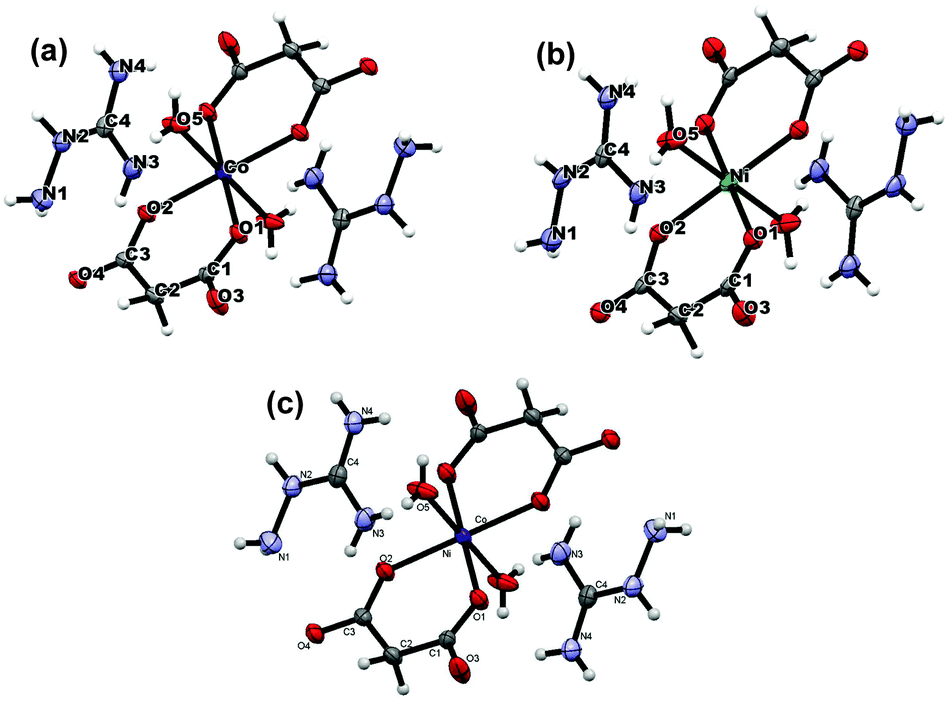 | ||
| Fig. 1 The molecular structure and atom labelling scheme for (AmgH)2[M(Mal)2(H2O)2] where M = (a) Co & (b) Ni, and (c) (AmgH)2[Ni0.5Co0.5(mal)2(H2O)2]. | ||
Further analysis of this structure indicates that the self-complementary hydrogen bonds of the metal-containing anionic moieties occur between the aqua ligand and the uncoordinated carbonyl oxygen atom donors (O5–H5O⋯O4 and O5–H5OB⋯O3) and a weak C–H⋯O interaction (see Tables 2 and 3 for a detailed bond parameter) consolidating the packing. This leads to the formation of a lamellar structure (Fig. 2) along the ab plane. As expected, the procumbent aminoguanidinium cationic components produce strong charge assistant hydrogen bonding (N–H⋯O) with adjacent anionic layers to fulfil the compact 3D pillar-layered architecture (Fig. 3).
| D–H | H⋯A | D–H⋯A | D–H⋯A | |
|---|---|---|---|---|
| Symmetry codes: (i) −x, 1 − y, 1 − z (ii) x, −1 + y, z (iii) 1 − x, 1 − y, −z, (iv) x, y, −1 + z. | ||||
| N1–H1⋯O1(iii) | 0.84(3) | 2.47(3) | 3.1701(19) | 143(3) |
| N2–H2N⋯O2(iv) | 0.86(3) | 2.10(3) | 2.9459(17) | 166(3) |
| N4–H4NA⋯O4(iv) | 0.783(18) | 2.174(19) | 2.9543(18) | 174(2) |
| O5–H5O⋯O4(i) | 0.73(3) | 1.98(3) | 2.6985(19) | 171(3) |
| O5–H5OB⋯O3(ii) | 0.76(3) | 1.98(3) | 2.715(2) | 165(3) |
| D–H | H⋯A | D–H⋯A | D–H⋯A | |
|---|---|---|---|---|
| Symmetry codes: (i) −x, 1 − y, 1 − z (ii) x, −1 + y, z (iii) 1 − x, 1 − y, −z, (iv) x, y, −1 + z. | ||||
| N1–HiNB⋯O1(iii) | 0.78(5) | 2.54(5) | 3.191(5) | 142(5) |
| N2–H2N⋯O2(iv) | 0.66(5) | 2.30(6) | 2.97(5) | 173(9) |
| N4–H4NB⋯O4(iv) | 0.86(5) | 2.12(6) | 2.969(5) | 172(6) |
| O5–H5OA⋯O3(ii) | 0.79(7) | 1.96(6) | 2.729(5) | 167(6) |
| O5–H5OB⋯O4(i) | 0.89(7) | 1.80(7) | 2.695(5) | 178(8) |
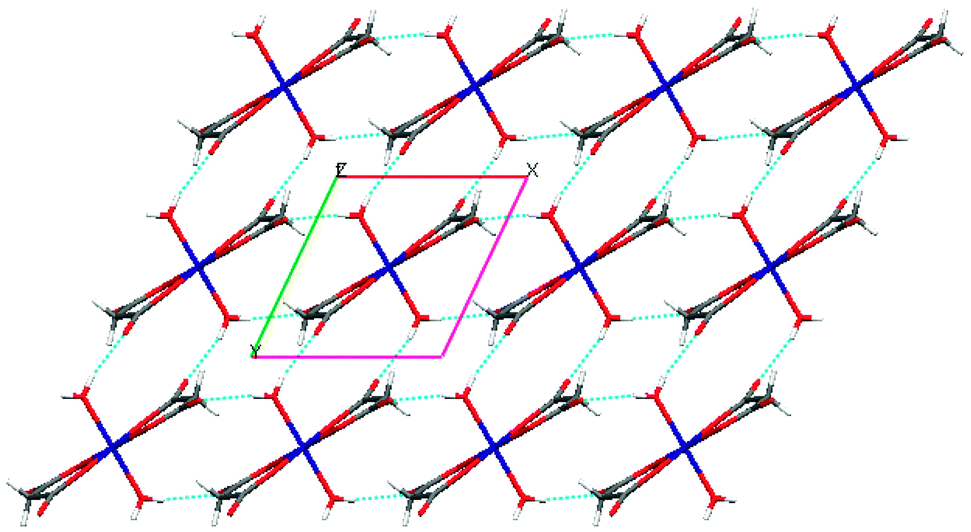 | ||
| Fig. 2 Perspective view of the 2D supramolecular layered array in the Co complex, showing the self-complementary hydrogen bonds between complex anions [Co(mal)2(H2O)2]2− (viewed down the c axis). | ||
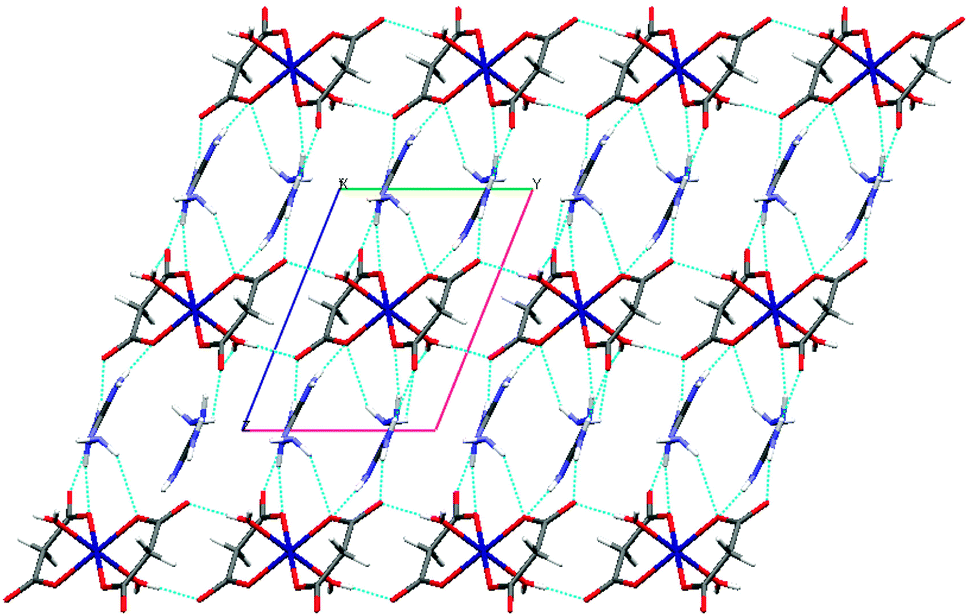 | ||
| Fig. 3 Supramolecular network in the Co complex, in which the top 2D layers are linked by the (AmgH)+ through charge-assistant N–H⋯O interactions (viewed down a). | ||
The infrared spectra of all complexes (1–6) were superimposable and show a band near 3420 cm−1 due to O–H stretching of the water molecules. The IR spectra of compounds 1, 4 and 6 are shown in Fig. S1 (ESI†) as representative examples. The deprotonation of both carboxyl groups present in the parent acid is evident from the disappearance of the band near 1730 cm−1. The asymmetric and symmetric stretching frequencies of the carboxylate ions were observed near 1580 and 1385 cm−1, respectively, with an average separation Δν (νasym − νsym) of 195 cm−1, documenting the monodentate coordination of both carboxylate groups in the dicarboxylate ion.30 The presence of the aminoguanidinium (+1) cation in the crystal structure is manifested by characteristic intense broad bands observed at 1680 and 1660 cm−1, which can be assigned to the overlapping of δNH2, νCN and δCNH vibrations.31,32 The peak near 1125 cm−1 can be attributed to the N–N stretching vibration of the hydrazinic moiety of the cation. The IR spectra of the nickel and zinc cobaltites register two absorption bands in the region of 667 and 587 cm−1 for metal oxygen stretching in the tetrahedral and octahedral sites,33 respectively.
The electronic spectrum (Fig. 4) of compound 1 shows a band at 512 nm, which is assigned to the 4T1g(F) → 4T1g(P) transition favouring octahedral geometry34 of the Co(II) complex. The bands at 661 and 392 nm for compound 2 that are assigned to the transitions 3A2g → 3T1g(F) and 3A2g → 3T1g(P), respectively, are in conformity with the octahedral arrangement35 of the Ni(II) ion. The mixed-metal complexes also reveal characteristic absorptions for the individual metal ions.
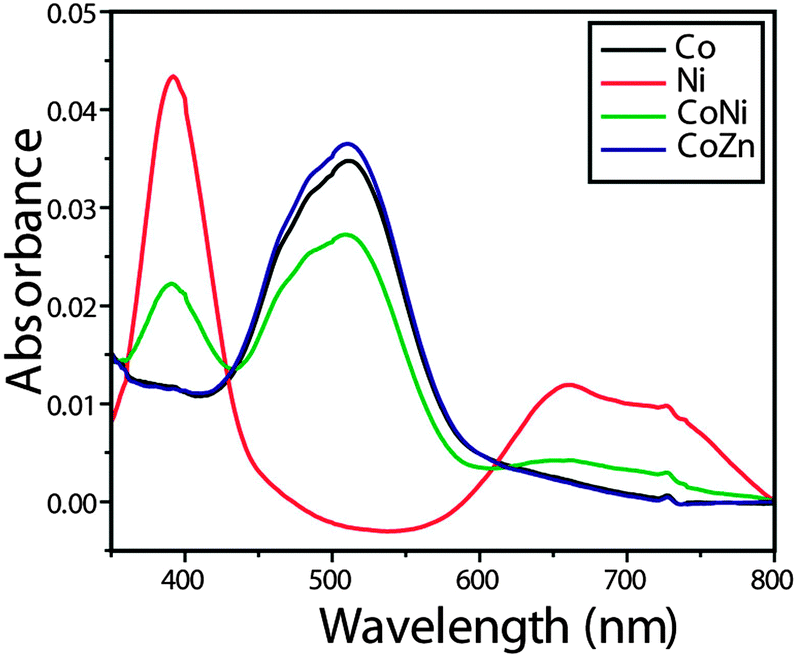 | ||
| Fig. 4 Electronic spectra of Co, Ni and mixed-metal complexes. | ||
Powder XRD analysis was used to characterize the new product. The powder XRD patterns of the single metal (1–3) and mixed-metal (5 and 6) complexes are shown in Fig. 5. Sharp line diffractograms were obtained in all cases, indicating good sample crystallinity. Comparisons of the diffraction patterns were used to confirm that the products were single-phase materials. The XRD patterns were indexed and the detailed lattice parameters of the single metal (1–3) and mixed-metal (4–6) complexes are given in Table 4. The results indicate that all the complexes crystallized in the triclinic space group P. Further, the values of h, k, l for each powder XRD peak have been calculated and are depicted in the Fig. 5. It was observed that the lattice parameters derived from power XRD patterns are in good agreement with lattice parameters obtained through single crystal X-ray analysis. It is evident that the powder XRD patterns of all the complexes exhibited identical patterns indicating their isomorphism. Furthermore, it was noticed that the powder XRD pattern of Co complex (1) and the powder X-ray pattern of the same derived from single crystal X-ray data (Fig. S2, ESI†) are showing almost similar patterns, which unambiguously confirm the accuracy of both the data.
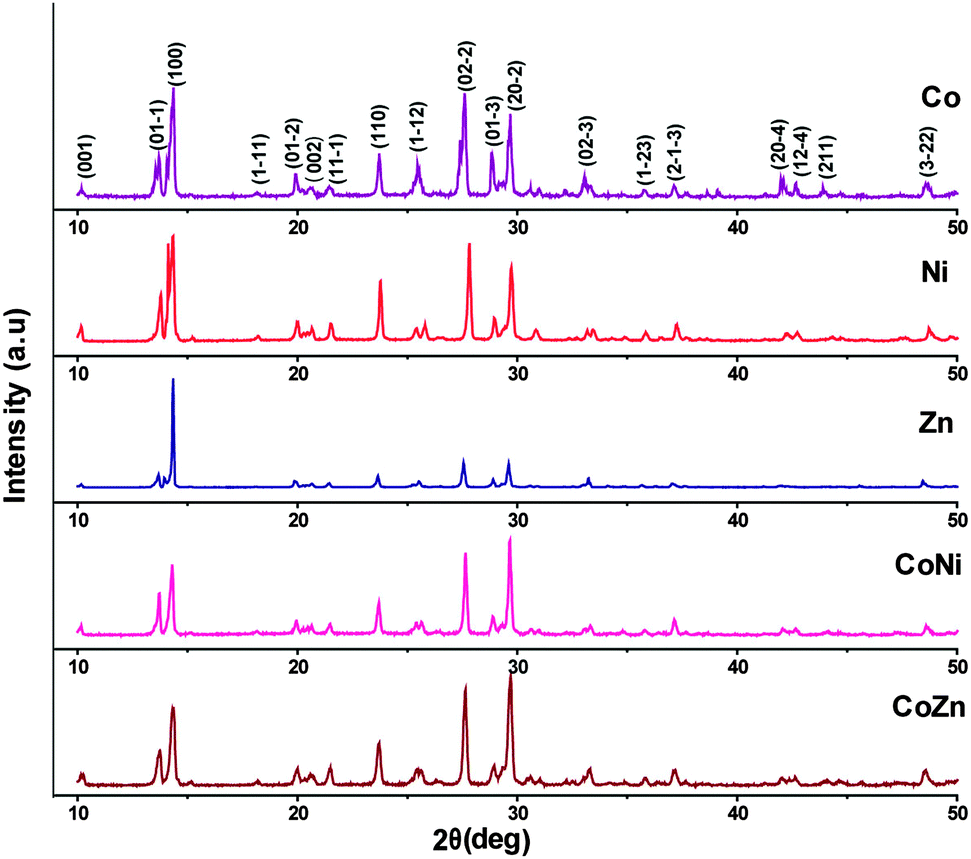 | ||
| Fig. 5 Powder X-ray patterns of metal and mixed-metal complexes. | ||
| Compound | Cell parameters from single crystal XRD method for 1, 2 & 4 | Cell parameters from powder XRD method for compounds 3, 5 & 6 | ||||
|---|---|---|---|---|---|---|
| 1 | 2 | 4 | 3 | 5 | 6 | |
| Crystal system | Triclinic | Triclinic | Triclinic | Triclinic | Triclinic | Triclinic |
| Space group |
P |
P |
P |
P |
P |
P |
| Unit cell dimensions | a = 6.9244(12) Å | a = 6.881(4) Å | a = 6.9096(10) Å | a = 6.892 Å | a = 6.879 Å | a = 6.903 Å |
| b = 7.3583(12) Å | b = 7.283(4) Å | b = 7.3355(10) Å | b = 7.507 Å | b = 7.393 Å | b = 7.349 Å | |
| c = 9.5353(16) Å | c = 9.541(5) Å | c = 9.5299(13) Å | c = 9.460 Å | c = 9.492 Å | c = 9.533 Å | |
| α = 105.537(3)° | α = 105.601(10)° | α = 105.577(2)° | α = 104.943° | α = 105.059° | α = 105.725° | |
| β = 110.219(3)° | β = 102.526(10)° | β = 102.494(2)° | β = 102.208° | β = 102.516° | β = 102.361° | |
| γ = 102.545(3)° | γ = 109.834(10)° | γ = 110.156(2)° | γ = 110.316° | γ = 109.978° | γ = 109.646° | |
| Volume | 412.65(12) Å3 | 407.3(4) Å3 | 410.52(10) Å3 | 418.302 Å3 | 412.516 Å3 | 412.7931 Å3 |
The typical XRD patterns of the cobaltite spinels obtained from the mixed-metal precursors are shown in Fig. 6. The XRD lines are very sharp and intense indicating the pure and highly crystalline nature of these oxide materials. The XRD lines were indexed and the lattice parameters for NiCo2O4 and ZnCo2O4 were calculated using sin2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ = (n2λ2/4a2)(h2 + k2 + l2), where a = 8.150 and 8.012 Å, respectively. These values are in good agreement with the literature values of the spinels36,37 (JCPDS File No. 20-781 for NiCo2O4 and 23-1390 for ZnCo2O4).
θ = (n2λ2/4a2)(h2 + k2 + l2), where a = 8.150 and 8.012 Å, respectively. These values are in good agreement with the literature values of the spinels36,37 (JCPDS File No. 20-781 for NiCo2O4 and 23-1390 for ZnCo2O4).
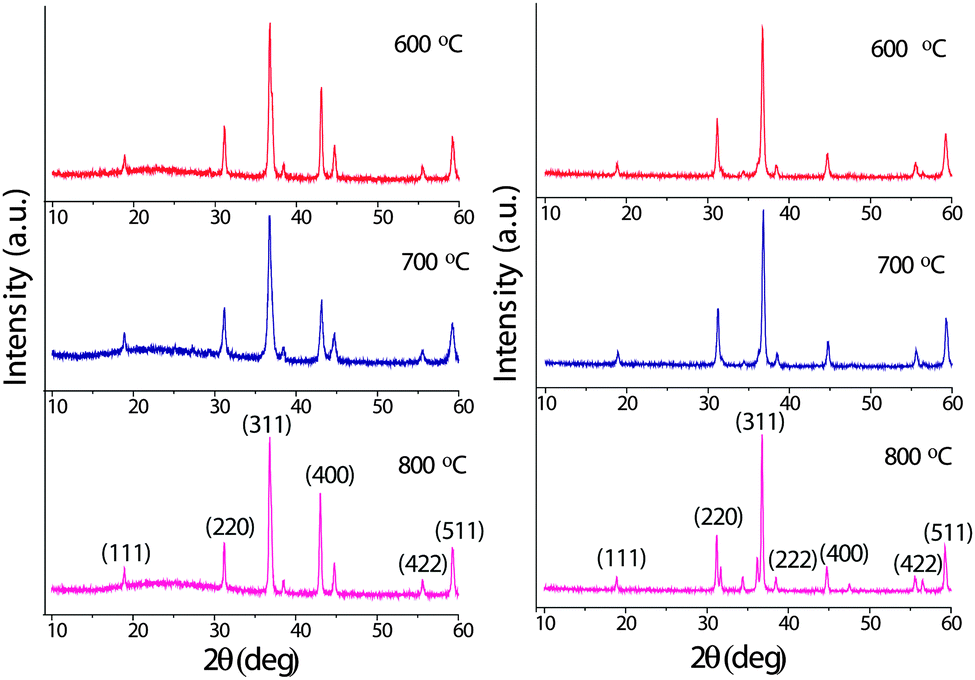 | ||
| Fig. 6 Powder XRD patterns of NiCo2O4 (left) and ZnCo2O4 (right) obtained at 600, 700 and 800 °C. | ||
All the complexes show similar thermal behavior upon heating. The DTA curve of compound 1 shows an endotherm at 150 °C corresponding to dehydration. Another endotherm can be seen at 230 °C, where the anhydrous compound decomposes to produce cobalt hydrogenmalonate, [Co(Hmal)2], as intermediate. This further decomposes with high exothermicity at 395 °C to produce Co3O4 as the end residue, which is shown in Fig. 7a. The TG also shows three steps of decomposition and the observed mass losses corresponding to the formation of intermediates and the end products are in good agreement with the calculated values. For compounds 2 and 3, the final step of decomposition was highly exothermic (Fig. 7b and c) and the end residue was invariably the metal oxide as detailed in the reaction scheme.
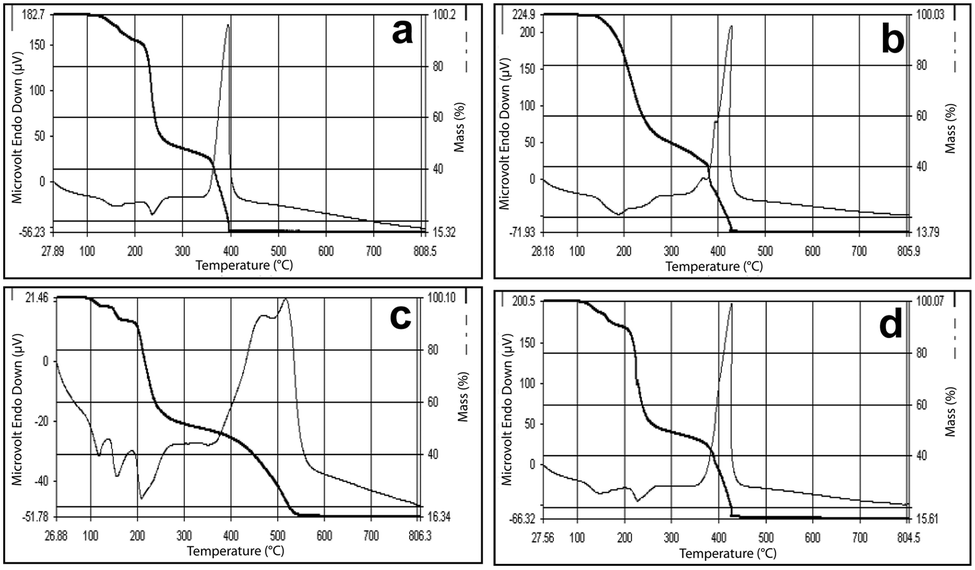 | ||
| Fig. 7 Simultaneous TG-DTA of (AmgH)2[M(mal)2(H2O)2], where M = (a) Co, (b) Ni and (c) Zn, and (d) (AmgH)2[Zn1/3Co2/3(mal)2(H2O)2]. | ||
This high exothermicity has been exploited for the preparation of mixed-metal oxides from the precursors. The mixed-metal (Zn–Co) complex (Fig. 7d) also shows a similar decomposition pattern resembling more of the simple cobalt compound, probably due to more cobalt content. It is interesting to note that the Ni and Ni–Co complexes upon decomposition produce a foamy material that comes out of the crucible.
The decomposition intermediate, [Co(Hmal)2], was further characterized by IR spectroscopic analysis. The peak near 3420 cm−1 due to O–H stretching of the water molecules and a strong peak at 1125 cm−1 attributed to the N–N stretching vibration of the aminoguanidinium cation, observed in the IR spectra (Fig. S1, ESI†) of the complexes, were completely disappeared in the IR spectrum (Fig. S3, ESI†) of the thermal decomposition intermediate, metal hydrogenmalonate, which confirms that the water molecules and aminoguanidinium cations were completely eliminated during the decomposition. Further, characteristic band in the region 1605 cm−1 and 1390 cm−1, which were ascribed to the asymmetric and stretching vibrations, respectively, of carboxylate groups.30 This corroborates the monodentate coordination of carboxylate groups and formation of metal hydrogenmalonate intermediate.
SEM studies were used to determine the size and morphology of the mixed-metal oxides (obtained at 800 °C) produced by thermal decomposition of the as-prepared solid solution precursors. Representative SEM micrographs are shown in Fig. 8a and b. The obtained particles were mostly irregular in shape and not significantly agglomerated. The reason for the irregular shape and agglomeration may be due to the absence of any protecting agents to minimize the interparticle attraction. Based on the SEM images, the average size of the mixed-metal oxide particles prepared by thermal decomposition of the precursor complexes was 40–50 nm for NiCo2O4 and 35–40 nm for the ZnCo2O4. The EDX analysis (Fig. 8c and d) of the calcined mixed-metal oxide confirmed the presence of Co and Ni/Zn cations at a 2:1 ratio without any other impurities. Thus, the calcination of the mixed-metal precursor in air at 800 °C for three hours resulted in the formation of respective spinel oxides in the crystalline phase. Additionally, the particle agglomeration was minimized without affecting the purity.
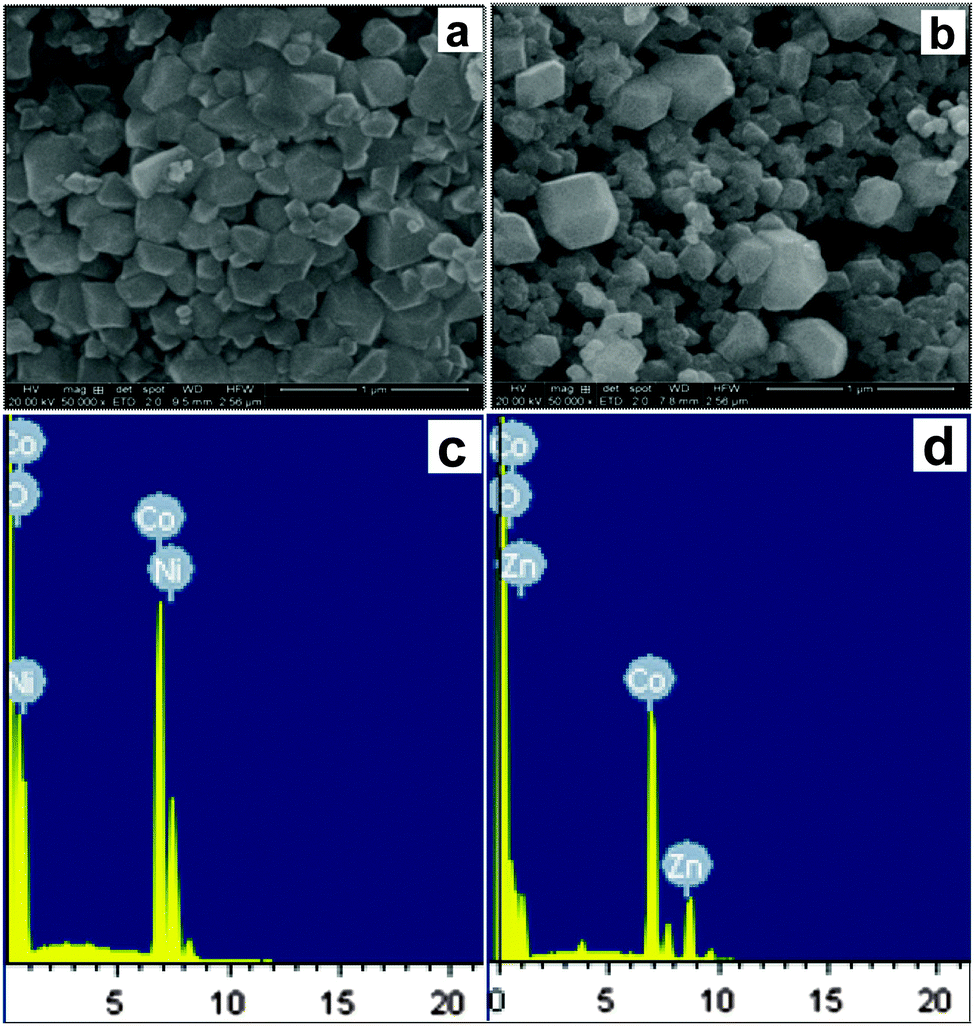 | ||
| Fig. 8 SEM image of (a) NiCo2O4 and (b) ZnCo2O4 obtained at 800 °C; EDAX analysis of (c) NiCo2O4 and (d) ZnCo2O4 obtained at 800 °C. | ||
The calcination of the mixed metal precursors such as 5 and 6 {(AmgH)2[(Ni or Zn)1/3Co2/3(mal)2(H2O)2]} at 800 °C under atmospheric air for 3 h resulted in the formation of corresponding mixed metal oxides such as NiCo2O4 and ZnCo2O4 nanoparticles. The size and nanocrystallinity of NiCo2O4 and ZnCo2O4 were further corroborated by TEM and high resolution TEM (HRTEM) analysis. The typical images are shown in Fig. 9a–f. The presence of NiCo2O4 and ZnCo2O4 nanoparticles with a size distribution of ∼50 nm was clearly defined and the morphology of the nanoparticles was characterized as mostly irregular bead-type oriental aggregation throughout the region. Furthermore, the selected area electron diffraction (SAED) pattern (Fig. 9b and d) clearly shows well-defined spots that are indicative of the presence of single-crystalline particles that correlate well with the powder XRD results. The HRTEM images (Fig. 9e and f) of nanoparticles show some obvious visible lattice fringes with the equal interplanar distances, which demonstrate a crystalline nature of the mixed metal oxides.
 | ||
| Fig. 9 TEM image (a) and SAED pattern (b) of NiCo2O4; TEM image (c) and SAED pattern (d) of ZnCo2O4. HRTEM images of (e) NiCo2O4 and (f) ZnCo2O4. | ||
Conclusions
Isomorphic aminoguanidinium diaqua metal malonate, (AmgH)2[M(mal)2(H2O)2], where M = Co(1), Ni(2), Zn(3) and aminoguanidinium mixed-metal malonate dihydrate, (AmgH)2[Ni0.5Co0.5(mal)2(H2O)2] (4), and (AmgH)2[M1/3Co2/3(mal)2(H2O)2], where M = Ni(5), Zn(6), were prepared and characterized by various analytical techniques. IR, elemental, powder XRD and single-crystal XRD studies reveal that the compounds 1, 2 and 3 are isomorphic. The compounds 5 and 6 were investigated as precursor solid solutions for nanosized spinel cobaltites. As compared to metal hydrazine carboxylate precursors,18,23,24 these precursors yielded spinel oxide with a homogeneous, high purity nanoparticle regime (50 nm) after corresponding thermal treatment. Recently, it is reported that the mesoporous ZnCo2O4 nanorods as efficient and high stable co-catalyst for the photochemical reduction of CO2 under mild reaction conditions.36 Also the porous nickel cobaltites are formed to have improved super capacitive properties.37 Hence, it is believed that the prepared cobaltites can be used in energy related materials, devices and as a good catalyst in low temperature reduction reactions. Further, the studies are in progress to prepare nanorods or 2D nanosheets38 of mixed metal oxides using solid solution precursors with different experimental conditions as well as different ligands and solvents. The synthesis of spinel ferrite nanoparticles using the same protocols is ongoing and will be reported in due course.Experimental
Synthesis of complexes 1, 2 and 3
Aminoguanidinium diaqua metal malonate complexes (AmgH)2[M(mal)2(H2O)2], where M = Co(1), Ni(2) and Zn(3), were prepared by the reaction of respective metal carbonates (Co – 0.118 g, 1 mmol; Ni – 0.125 g, 0.33 mmol; Zn – 0.125 g, 1 mmol) with malonic acid (0.208 g, 2 mmol) followed by the addition of aminoguanidine bicarbonate (0.272 g, 2 mmol in 1:2:2 mole ratio) in aqueous medium. The resulting solution upon evaporation at room temperature produced crystals suitable for X-ray study. The crystals were filtered, washed with alcohol and air dried. Analytical data for compound 1- found: Amg-14.30, Co-13.10, C-21.30, H-5.00, N-24.90%; calc.: Amg-14.25, Co-13.14, C-21.38, H-4.90, N-24.94%, compound 2- found: Amg-14.20, Ni-13.10, C-21.30, H-4.40, N-24.90%; calc.: Amg-14.25, Ni-13.07, C-21.38, H-4.45, N-24.96%, compound 3- found: Amg-14.10, Zn-14.30, C-21.00, H-4.80, N-23.60%; calc.: Amg-14.05, Zn-14.36, C-21.08, H-4.83, N-24.59%.
Synthesis of compounds 4, 5 and 6
Materials and physical measurements
All reagents for the synthesis were obtained commercially and used as received. Elemental analysis of C, H and N were performed on a Elementar Vario EL III analyzer. The FT-IR spectra were recorded using KBr pellets in the range of 4000–400 cm−1 on an AVATAR-320-FT-IR spectrometer. The powder X-ray diffraction was recorded on a Rikagu diffractometer with copper radiation (λ = 0.154 06 nm) at 40 kV and 40 mA. The simultaneous TG-DTA curves were recorded on a Perkin Elmer Pyris Diamond instrument at a heating rate of 5 °C min−1 under air. Absorption spectral measurements were performed using a JASCO V-530 UV-visible spectrophotometer in the range 200–800 nm. Quartz cuvettes with a 1 cm path length were used to record the absorption spectra. Scanning electron microscope (SEM) observation was conducted with an FEI QUANTA 200/EDAX microscope. Selected area electron diffraction (SAED) patterns and TEM image obtained using Philips CM20 transmission electron microscope operating at 200 kV.The hydrazine content in aminoguanidine was estimated by titrating against standard KIO3 under Andrews' condition.39 The metal content were determined by EDTA titration.40
Final Fourier syntheses showed only chemically insignificant differences electron density (with the largest difference peaks close to metal atoms). An inspection of Fovs. Fc values and trends based upon sinθ, the Miller index, or the parity group did not reveal any systematic errors in the data. The detailed crystallographic data for 1, 2 and 4 are listed in Table 1.
Acknowledgements
R. Selvakumar wishes to thank UGC for the award of a research fellowship in science for meritorious students under the Basic Science Research (BSR) program.Notes and references
- N. Kikukawa, M. Takemori, Y. Nagano, M. Sugasawa and S. Kobayashi, J. Magn. Magn. Mater., 2004, 284, 206–214 CrossRef CAS.
- A. S. Prakash, A. M. A. Khadar, K. C. Patil and M. S. Hegde, J. Mater. Synth. Process., 2002, 10, 135–141 CrossRef CAS.
- G. Vaidyanathan, S. Sendhilnathan and R. Arulmurugan, J. Magn. Magn. Mater., 2007, 313, 293–299 CrossRef CAS.
- J. Azadmanjiri, H. K. Salehani, M. R. Barati and F. Farzan, Mater. Lett., 2007, 61, 84–87 CrossRef CAS.
- M. A. Ahmed and M. M. El-Sayed, J. Magn. Magn. Mater., 2007, 308, 40–45 CrossRef CAS.
- C. K. Kim, L. H. Lee, S. Katoh, R. Murakami and M. Yoshimura, Mater. Res. Bull., 2001, 36, 2241–2250 CrossRef CAS.
- N. Moumen and M. P. Pileni, Chem. Mater., 1996, 8, 1128–1134 CrossRef CAS.
- Q. Song and Z. J. Zhang, J. Phys. Chem. B, 2006, 110, 11205–11209 CrossRef CAS PubMed.
- M. Han, C. R. Vestal and Z. J. Zhang, J. Phys. Chem. B, 2004, 108, 583–587 CrossRef CAS.
- J. Z. Jhang, G. F. Goya and H. R. Rechenberg, J. Phys.: Condens. Matter, 1999, 11, 4063–4078 CrossRef.
- A. Verma, T. C. Goel, R. G. Mendiratta and M. I. Alam, Mater. Sci. Eng., B, 1999, 60, 156–162 CrossRef.
- Q. Chen, A. J. Rondinone, B. C. Chakoumakos and Z. J. Zhang, J. Magn. Magn. Mater., 1999, 194, 1–7 CrossRef CAS.
- A. Lagashetty, V. Havanoor, S. Basavaraja and A. Venkataraman, Bull. Mater. Sci., 2005, 28, 477–481 CrossRef CAS.
- A. J. Rondinone, A. C. S. Samia and Z. J. Zhang, J. Phys. Chem. B, 1999, 103, 6876–6880 CrossRef CAS.
- A. B. Fuertes, T. Valdes-Solis and M. Sevilla, J. Phys. Chem. C, 2008, 112, 3648–3654 Search PubMed.
- R. A. Porob, S. Z. Khan, S. C. Mojumdar and V. M. S. Verenkar, J. Therm. Anal. Calorim., 2006, 86, 605–608 CrossRef CAS.
- K. C. Patil and M. M. A. Sekar, Int. J. Self-Propag. High-Temp. Synth., 1994, 3, 181–196 CAS.
- B. N. Sivasankar and S. Govindarajan, Z. Naturforsch., B: J. Chem. Sci., 1994, 49, 950–954 CAS.
- N. A. Dhas and K. C. Patil, J. Solid State Chem., 1993, 102, 440–445 CrossRef CAS.
- B. N. Sivasankar and S. Govindarajan, Synth. React. Inorg. Met.-Org. Chem., 1995, 25, 127–138 CrossRef CAS.
- D. Gajapathy, K. C. Patil and V. R. Paiverneker, Mater. Res. Bull., 1982, 17, 29–32 CrossRef.
- K. C. Patil, D. Gajapathi and V. R. Paiverneker, J. Mater. Sci. Lett., 1983, 2, 272–274 CrossRef CAS.
- B. N. Sivasankar and S. Govindarajan, Mater. Res. Bull., 1996, 31, 47–54 CrossRef CAS.
- S. Yasodhai and S. Govindarajan, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 1999, 38, 1244–1248 Search PubMed.
- X.-J. Zhao, Z.-H. Zhang, Y. Wang and M. Du, Inorg. Chim. Acta, 2007, 360, 1921–1928 CrossRef CAS.
- M. Koskinen, I. Mutikainen and H. Elo, Z. Naturforsch., B: J. Chem. Sci., 1994, 49, 556–560 CAS.
- J. T. Koskinen, M. Koskinen, I. Metikainen, B. Mannfors and H. Elo, Z. Naturforsch., B: J. Chem. Sci., 1996, 51, 1771–1778 CAS.
- M. Koskinen, I. Mutikainen, P. Tilus, E. Pelttari, M. Korvela and H. Elo, Monatsh. Chem., 1997, 128, 767–775 CrossRef CAS.
- R. L. Davidovich, V. B. Logviniva, V. V. Tkachev and L. O. Atovmyan, Russ. J. Coord. Chem., 1995, 21, 783–787 CAS.
- A. Braipandi, F. Dallavalle, M. A. Pellinghelli and E. Leporati, Inorg. Chem., 1968, 7, 1430–1433 CrossRef.
- I. Nemec, Z. Machackova, K. Teubner, I. Cisarova, P. Vanek and Z. Micka, J. Solid State Chem., 2004, 177, 4655–4664 CrossRef CAS.
- Z. Machackova, I. Nemec, K. Teubner P. Nemec, P. Vanek and Z. Micka, J. Mol. Struct., 2007, 832, 101–107 CrossRef CAS.
- K. B. Gudasi, S. A. Patil, R. S. Vadavi, R. V. Shenoy and M. Nethaji, Transition Met. Chem., 2006, 31, 586–592 CrossRef CAS.
- E. Canpolat, A. Yazici and M. Kaya, Transition Met. Chem., 2006, 31, 653–657 CrossRef CAS.
- B. Lefez, P. Nikeng, J. Lopitaux and G. Poillerat, Mater. Res. Bull., 1996, 31, 1263–1267 CrossRef CAS.
- S. Wang, Z. Ding and X. Wang, Chem. Commun., 2015, 51, 1517–1519 RSC.
- C. An, G. Liu, Y. Wang, L. Li, F. Qiu, Y. Xu, C. Xu, Y. Wang, L. Jiao and H. Yuan, RSC Adv., 2013, 3, 15382–15388 RSC.
- Y. Zhu, C. Cao, J. Zhang and X. Xu, J. Mater. Chem. A, 2015, 3, 9556–9564 CAS.
- G. H. Jeffery, J. Bassett, J. Mendham and R. C. Denny, Vogel's Textbook of Quantitative Chemical Analysis, 5th edn, 1986 Search PubMed.
- A. I. Vogel, A Textbook of Quantitative Inorganic Analysis Including Elementary Instrumental Analysis, Longman, 3rd edn, 1996, pp. 415–457 Search PubMed.
- G. M. Sheldrick, SHELXTL, version 6.10, Bruker Analytical X-ray Systems, Madison, WI, 2001 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Bond angle and bond length of compound 1 and 2 and IR spectra of the compounds 1, 4, and 6. CCDC 917228 (1), 917229 (2) and 1046628 (4). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj01134e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |