
Hypervalent-iodine(III) oxidation of hydrazones to diazo compounds and one-pot nickel(II)-catalyzed cyclopropanation†
Haixuan
Liu
,
Yunyang
Wei
and
Chun
Cai
*
School of Chemical Engineering, Nanjing University of Science and Technology, Xiaolingwei 200, Nanjing 210094, People's Republic of China. E-mail: c.cai@njust.edu.cn
First published on 16th November 2015
Abstract
A one-pot process for the catalytic cyclopropanation of various alkenes with unsubstituted hydrazones is described. Iodosobenzene (PhI![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) was found to be a competent oxidant of hydrazones to diazo compounds. Ni(OH)2 was chosen as an effective and cheap metal catalyst. The cyclopropane products can be generated efficiently (5 min–4 h) in moderate to good yields (42–91%) under mild (80 °C) and neat conditions.
O) was found to be a competent oxidant of hydrazones to diazo compounds. Ni(OH)2 was chosen as an effective and cheap metal catalyst. The cyclopropane products can be generated efficiently (5 min–4 h) in moderate to good yields (42–91%) under mild (80 °C) and neat conditions.
Introduction
The cyclopropyl moiety is ubiquitous in the structure of naturally occurring compounds.1 As the smallest strained cycloalkane, cyclopropane arouses chemists' interest in many aspects including the theoretical study of bonding,2 potential synthetic transformations3 and its structural importance in drugs and enzyme inhibitors.4 Consequently, a variety of synthetic routes to cyclopropanes have been developed.5 Among them, transition metal catalytic decomposition of diazo compounds in the presence of alkenes is a classic and common method.6 The active metal complexes include rhodium,7 ruthenium,8 copper,9 and cobalt,10 while only scattered reports concern nickel catalysts.11 And the investigations have been restricted to diazo esters or diazo ketones (Scheme 1a). Diazo compounds are toxic, irritating and sometimes explosive. In order to broaden substrate scopes and to avoid handling of diazo compounds, protocols that combine in situ generation of diazo compounds and carbene addition to alkenes are most desirable. Either base elimination of tosylhydrazones or the oxidation of hydrazones is a choice of generating diazo compounds in situ. The first strategy has been extensively scanned using aromatic aldehyde tosylhydrazones or substituted acetophenone tosylhydrazones as substrates with a wide range of olefins (Scheme 1b).12 However, this strategy involves using a strong base or high temperatures and a relatively long reaction time is often needed. The latter strategy faces more challenge since it is hard to tolerate alkenes under an oxidation environment. Conventionally, the oxidizing agents used to oxidize hydrazones to diazo compounds have been mercuric oxide,13 manganese dioxide,14 silver oxide15 and lead tetraacetate.16 Manganese dioxide was chosen to operate cyclopropanation from hydrazones in some reports, but an excess amount of the oxidant was used and the reaction had to be operated in a two-step manner, meaning long reaction times and tedious work-up procedures.17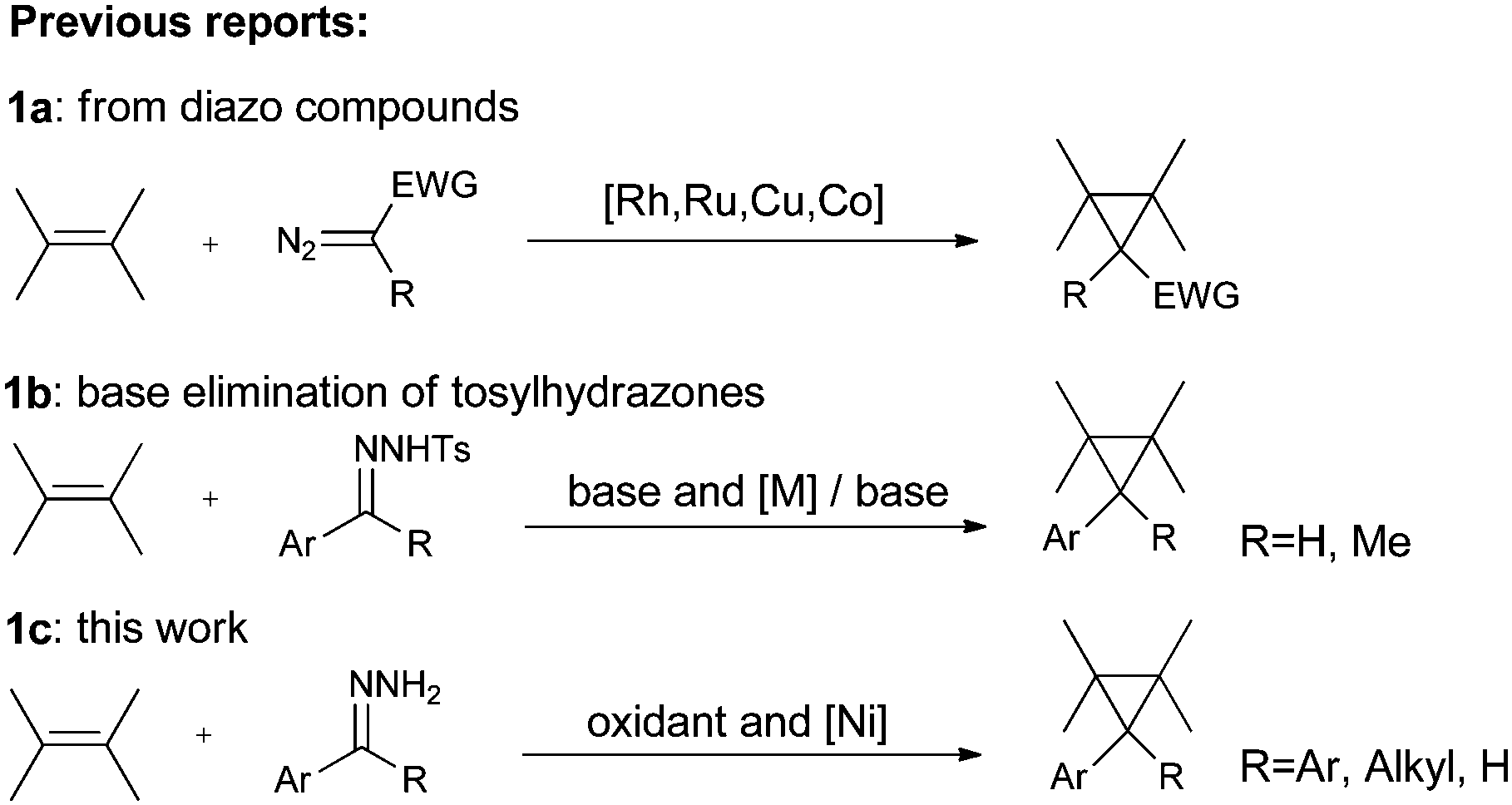 | ||
| Scheme 1 Cyclopropanation strategies. | ||
On the other hand, the wide application of hypervalent iodine reagents in modern synthesis has been well-documented.18 Cyclopropanation of alkenes can be achieved by formation of iodonium ylide derived from an active methylene compound and subsequent carbene transfer.19 However, the strategy of in situ generating diazo species mediated by a hypervalent iodine reagent was only applied in one-pot esterification reactions using benzophenone hydrazones or N-tert-butyldimethylsilyl hydrazones with carboxylic acid.20 In these esterification reactions, since both the active intermediate (diazo compound) and the substrate (hydrazone) tended to decomposition, low temperature (−78 °C or −10 °C) had to be used. We intended to broaden the application of this strategy by exploring new catalytic systems that would be compatible with new reaction types. Also, in order to continue our research on hypervalent iodine reagents21 and circumvent the existing shortcomings in cyclopropanation, we present here a one-pot cyclopropanation from hydrazones and alkenes, which is highly efficient and can be adapted to general hydrazones (Scheme 1c).
Results and discussion
We began our investigations by using benzophenone hydrazone (1a) as a model substrate. Upon treating the substrate with hypervalent iodine reagent such as (diacetoxyiodo)benzene (DIB), hydroxy(tosyloxy)iodobenzene (HTIB), [bis(trifluoroacetoxy)iodo]benzene (PIFA), and 2-iodoxybenzoic acid (IBX) in chloroform at room temperature, only decomposition products could be isolated which is consistent with the literature.22 When iodosobenzene was used as an oxidant, the accumulation of diphenyldiazomethane was visible by the outstanding red-violet color spot on the thin layer chromatography (TLC) plate. In order to confirm by GC-MS analysis, methanol was added to the reaction mixture to consume the unstable diazo compound and the corresponding (methoxymethylene) dibenzene could be examined in 35% yield. The parent ketone was found in 40% yield formed by decomposition of the starting hydrazone in solution and reaction with oxygen. The conversion of the starting material is 85% after 10 minutes. With the initial findings, we continued our study by using styrene to trap the in situ generated diphenyldiazomethane, the desired cyclopropane product (3a) was formed in 35% yield at 80 °C after 20 hours (Table 1, entry 1). Styrene was used in excess to depress side reactions of the starting hydrazone.|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Additive | Solvent | Time | Yieldb (%) |
| a Conditions: 1a (0.5 mmol), 2a (4.0 equiv.), catalyst (30 mol%), additive (20 mol%) in solvent (2 mL) at 80 °C. TBAC = tetrabutylammonium chloride, SDS = sodium dodecyl sulfate, TMEDA = tetramethylethylenediamine. b Isolated yield. c Under argon. d Reaction was performed at 60 °C. e Ni(OH)2 (20 mol%) was used. f Ni(OH)2 (10 mol%) was used. | |||||
| 1 | — | — | CHCl3 | 20 h | 35 |
| 2 | CuBr2 | — | CHCl3 | 20 h | — |
| 3 | VOSO4·xH2O | — | CHCl3 | 20 h | — |
| 4 | BiCl3 | — | CHCl3 | 20 h | — |
| 5 | Ni(OAc)2·4H2O | — | CHCl3 | 20 h | 46 |
| 6 | Ni(OAc)2·4H2O | TBAC | CHCl3 | 20 h | 26 |
| 7 | Ni(OAc)2·4H2O | SDS | CHCl3 | 20 h | 32 |
| 8 | Ni(OAc)2·4H2O | Ph3P | CHCl3 | 20 h | 28 |
| 9 | Ni(OAc)2·4H2O | TMEDA | CHCl3 | 20 h | 22 |
| 10 | Ni(OAc)2·4H2O | K2CO3 | CHCl3 | 20 h | 35 |
| 11 | Ni(acac)2 | — | CHCl3 | 20 h | 40 |
| 12 | NiF2·4H2O | — | CHCl3 | 4 h | 55 |
| 13 | Ni(OH)2 | — | CHCl3 | 1 h | 58 |
| 14 | Ni(OH)2 | — | — | 40 min | 72 |
| 15c | Ni(OH)2 | — | — | 1 h | 73 |
| 16d | Ni(OH)2 | — | — | 20 h | 25 |
| 17 | Ni(OH)2 | — | — | 1 h | 67 |
| 18 | Ni(OH)2 | — | — | 2 h | 62 |
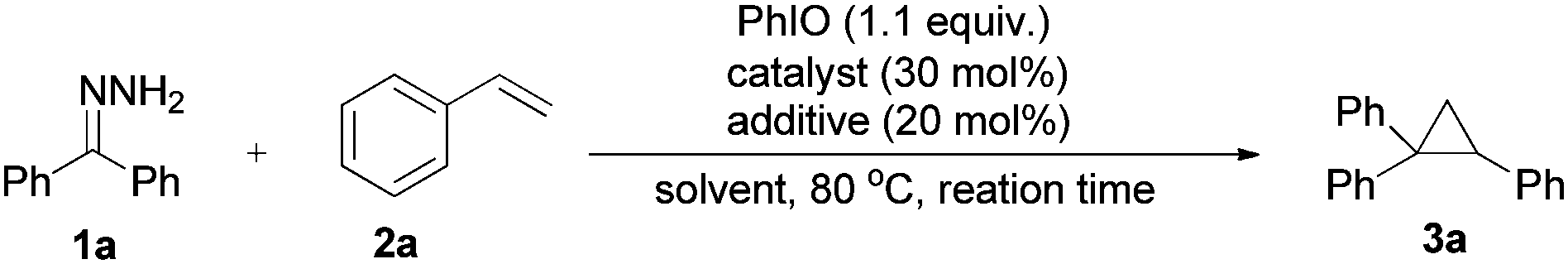
In order to accelerate the reaction, various metal catalysts were then scanned at similar reaction conditions, to our delight, Ni(OAc)2·4H2O exhibited catalytic activity and 3a could be obtained in 46% yield (Table 1, entries 2–5). The attempt to improve product yield using additives like surfactants, ligands and bases only gave inferior results (Table 1, entries 6–10). We then tended to survey Ni(II) salts with different counter ions, the reaction greatly speeded up when using NiF2·4H2O or Ni(OH)2 as a catalyst (Table 1, entries 11–13). As GC-MS analysis showed that byproducts result from the solvent, we ran the reaction under neat conditions and the product yield climbed to 72% (Table 1, entry 14). Careful screening of alkene equivalents showed that 4 equivalents of styrene (with regard to hydrazones) is optimal (Table S1 in the ESI†). The cumbersome nature of running the reaction under argon had little effect on the yield of 3a (Table 1, entry 15). But in this case, the in situ generated diazo compound was decomposed by disproportionation to give the symmetrical azine as the main byproduct. Lowering the reaction temperature to 60 °C led to a drop in reaction performance (Table 1, entry 16). It is worth noting that an acceptable yield (62%) could also be obtained when decreasing the catalyst loading to 10 mol%, only longer reaction time was needed (Table 1, entries 17 and 18).
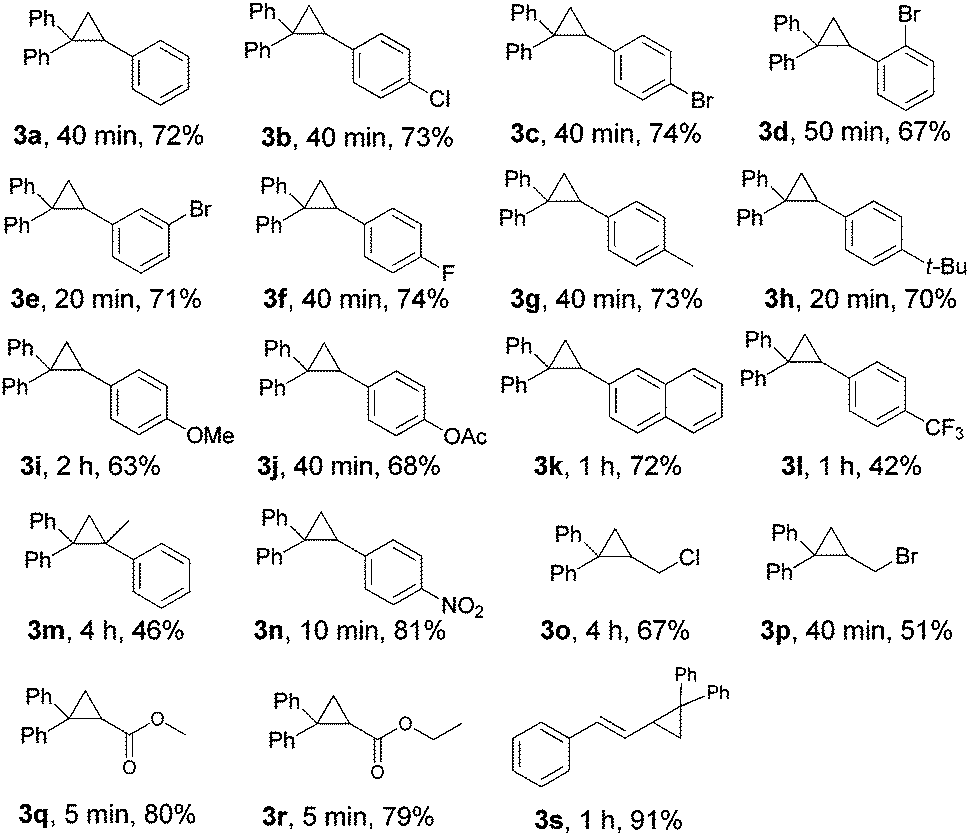
With the optimized conditions in hand (Table 1, entry 14), the scope of this protocol was examined for the reaction of benzophenone hydrazone (1a) with a series of substituted alkenes (Table 2). The reactions proceeded smoothly to afford the target cyclopropanes in moderate to good yields. For 4-substituted styrenes in which the substituent is a halogen and an alkyl group (2b, 2c, 2f–2h), the protocol was applicable without significant differences in product yields (3b, 3c, 3f–3h). When 2-bromostyrene was used as a substrate, the yield of the product was a bit reduced compared with 3- and 4-bromostyrene which shows an influence of steric effects (3c–3e). The presence of a moderate electron-donating group (–OAc) or a strong electron-donating group (–OMe) on the benzene ring of styrene gave the products (3j, 3i) in slightly lower yields. When we employed 4-nitrostyrene as the substrate, the corresponding product (3n) was achieved in 81% yield, suggesting that styrene with a strong electron-withdrawing group is favored in this transformation. But unexpectedly, the trifluoromethyl-substituted product (3l) was only isolated in 32% yield, which may be attributed to the high volatility of 4-(trifluoromethyl) styrene. The protocol was also applicable to α-methylstyrene and 2-vinylnaphthalene, affording the desired products (3m, 3k) in 46% and 72% yields, respectively. Aliphatic alkenes were also examined. To our delight, allyl chloride and allyl bromide could react and afford the desired cyclopropanes in moderate yields (3o, 3p). Halogen containing products were highly meaningful since the halogen enables further functional group manipulations to more complex structures. Meanwhile, this kind of product can't be synthesized from tosylhydrazones, because, under basic conditions, the known N-alkylation of tosylhydrazones with an alkyl halide will be a dominant side reaction.23 Alkyl acrylates such as methyl acrylate and ethyl acrylate can react with benzophenone hydrazone efficiently and generated the corresponding cyclopropanes in 80% and 79% yields, respectively (3q, 3r). Unfortunately, acyclic internal alkenes show no reactivity under the reaction conditions, only the decomposed products of benzophenone hydrazone (azine and ketone) were detected by GC-MS (for example 1-chloro-2-butene, (E)-stilbene etc. was tested). A cyclic internal alkene such as norbornene was used, but the 1,3-dipolar cycloaddition product was isolated in 89% yield.24 The product does not exclude molecular nitrogen when it came into contact with Ni(OH)2 or is heated at 120 °C. When cyclohexene was used, C–H insertion out competes cyclopropanation and the C–H insertion product was formed in 47% yield.24 The special reactivity toward the allylic C–H insertion product may contribute to both steric and electronic effects.25 For conjugated alkenes, (E)-1-phenyl-1,3-butadiene could also smoothly undergo the reaction and the desired product was obtained in 91% yield (3s).
|
|
|---|
|
a Conditions: 1a (0.5 mmol), 2 (4.0 equiv.), Ni(OH)2 (30 mol%), PhIO (1.1 equiv.), at 80 °C. For solid alkenes, chloroform (0.2 mL) was used as solvent. The isolated yields of the products were shown.
|
|
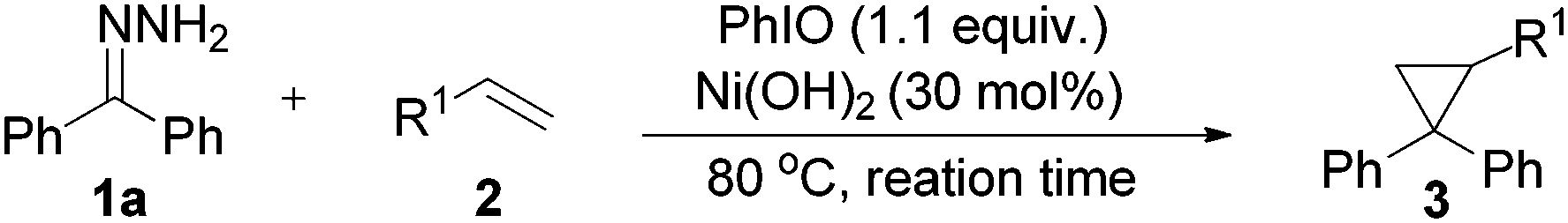
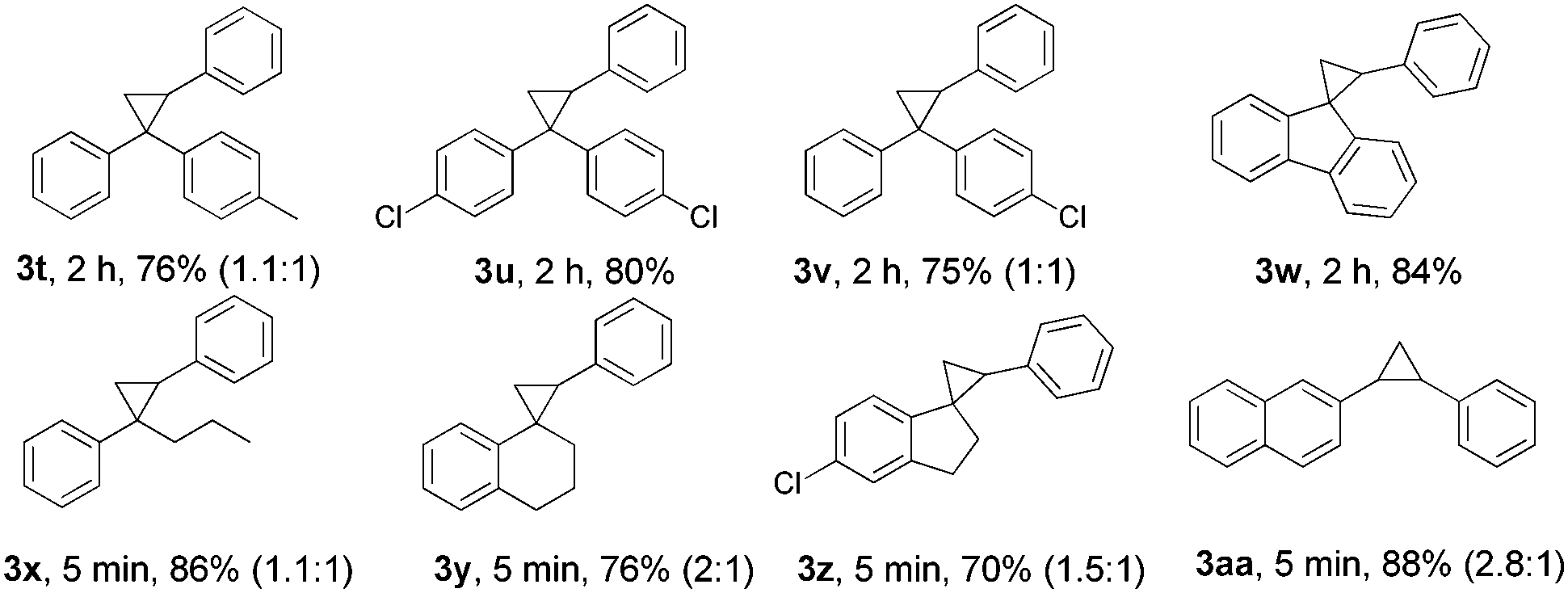
Then, the generality of this protocol concerning the hydrazone part was investigated (Table 3). Diaryl ketone hydrazones with chlorine or a methyl substituent and 9-fluorenone hydrazone all furnished the corresponding cyclopropanes (3t–3w) in satisfactory yields. Furthermore, aryl–alkyl ketone hydrazones exhibited high reactivity and completed the reactions in 5 min with good product yields (3x–3z). Notably, the thermally sensitive aldehyde hydrazone substrate (3aa) could undergo this transformation efficiently, and generated the desired product in 88% yield. Products 3t, 3v, and 3x–3aa were isolated as mixtures of diastereoisomers and the trans/cis ratios were calculated according to NMR spectra. Compared to the diaryl diazo compound, the aryl–alkyl diazo compound and the aryl diazo compound are even more unstable and difficult to handle for the lack of one stabilized benzene ring and the electron-donating effect of the alkyl- or the H-group.26 We believe that the efficiency applicable to 3x–3aa makes this protocol more practically useful and a better choice in this field. Next, when cyclohexanone hydrazone was used, again, only the 1,3-dipolar cycloaddition product was obtained in 43% yield.24
|
|
|---|
|
a Conditions: 1a (0.5 mmol), 2 (4.0 equiv.), Ni(OH)2 (30 mol%), PhIO (1.1 equiv.), at 80 °C. For solid alkenes, chloroform (0.2 mL) was used as solvent. The isolated yields of the products were shown. For 3t, 3v, and 3x–3aa, the trans/cis ratios of diastereoisomer were shown in brackets.
|
|
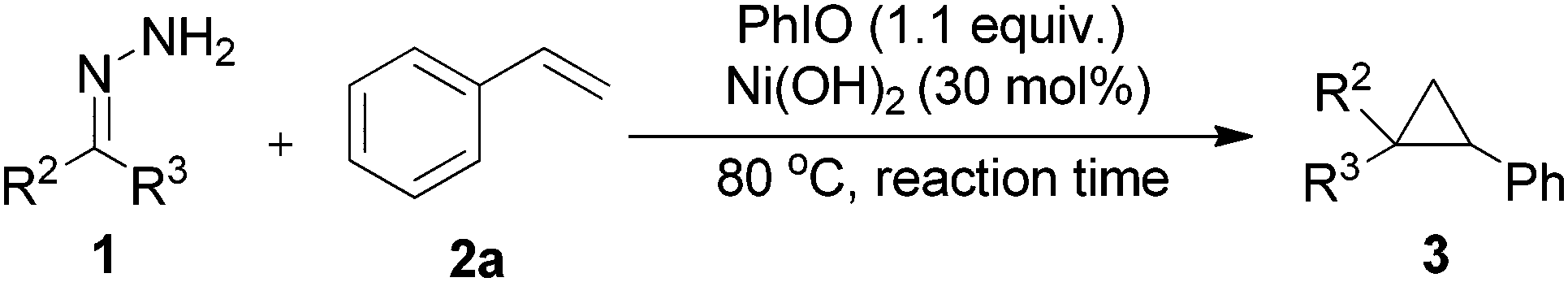
Based on the knowledge of transition metal-catalyzed cyclopropanation and the above experiments we explored, we proposed a reaction pathway for this one-pot nickel(II)-catalyzed cyclopropanation of alkenes with hydrazones (Scheme 2). Iodosobenzene functions as the oxidizing agent and in situ oxidize hydrazones to diazo compounds. The interaction of diazo compounds with the nickel salt generates metal–carbene intermediates and subsequent carbene addition leads to the desired product.
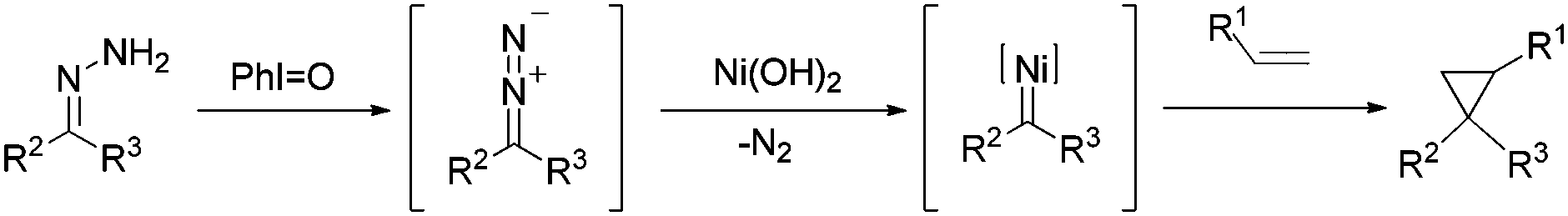 | ||
| Scheme 2 Proposed mechanisms. | ||
Conclusions
In summary, a new approach to cyclopropane compounds has been introduced in this paper. The one-pot and easy operation procedure utilizes iodosobenzene as an oxidant to generate non-stabilized diazo compounds in situ from unsubstituted hydrazones. There's no need to isolate the diazo compound or to run the reaction stepwise. With the aid of a nickel catalyst, the diazo compound decomposes readily and reacts with the alkene at 80 °C to furnish the cyclopropane efficiently (5 min–4 h) and in moderate to good yields (42–91%). The mild protocol is generally applicable to aryl alkenes with different electronic effect substituents (–OMe, –NO2, –t-Bu, –F, etc.), aliphatic alkenes with halogen functional groups, alkyl acrylates and hydrazones derived from diaryl ketones, aryl–alkyl ketones and aldehydes. If lower catalyst loading was used, an acceptable yield could also be obtained and for all reactions the termination can be clearly indicated by the vanishing of the special color of diazo compounds. We concentrated on the synthesis of 1,1-diaryl cyclopropanes because of its lack of systematic investigation and a good supplement to the existing methods. The enantioselective version of this protocol through adding chiral ligands or chiral auxiliaries of the substrate, and further exploration of this reliable way of generating diazo intermediates is now in progress in our laboratory.Experimental
A screwed tube was charged with the corresponding hydrazone (0.5 mmol), Ni(OH)2 (30 mol%, 0.014 g) and alkene (4 equiv.). For solid alkenes, chloroform (0.2 mL) was used as the solvent. A magnetic stirrer with an oil bath was used and pre-heated to 80 °C. The screwed tube was then put to the oil bath and at the same time iodosobenzene (1.1 equiv., 0.121 g) was added. The reaction was stirred at 80 °C until total disappearance of the red-violet color which indicated the completion of the reaction. The reaction mixture was purified by column chromatography using petroleum ether or petroleum ether/ethyl acetate as an eluent.Acknowledgements
We thank the Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD). We also thank Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences for testing HRMS.Notes and references
- (a) R. Faust, Angew. Chem., Int. Ed., 2001, 40, 2251 CrossRef CAS; (b) Y. Fujimoto, F. Irreverre, J. M. Karle, I. L. Karle and B. Witkop, J. Am. Chem. Soc., 1971, 93, 3471 CrossRef CAS PubMed; (c) W. A. Donaldson, Tetrahedron, 2001, 57, 8589 CrossRef CAS.
- A. De Meijere, Angew. Chem., Int. Ed., 1979, 18, 809 CrossRef.
- (a) A. Maercker, K. S. Oeffner and U. Girreser, Tetrahedron, 2004, 60, 8245 CrossRef CAS; (b) J. P. Dinnocenzo, H. Zuilhof, D. R. Lieberman, T. R. Simpson and M. W. McKechney, J. Am. Chem. Soc., 1997, 119, 994 CrossRef CAS.
- C. J. Suckling, Angew. Chem., Int. Ed., 1988, 27, 537 CrossRef.
- (a) G. Brunner, S. Elmer and F. Schröder, Eur. J. Org. Chem., 2011, 4623 CrossRef CAS; (b) M. G. Apenova, O. O. Semivrazhskaya, E. V. Borkovskaya, N. M. Belov, I. N. Loffe, V. Y. Markov, S. I. Troyanov, N. S. Lukonina, L. N. Sidorov and A. A. Goryunkov, Chem. – Asian J., 2015, 10, 1370 CrossRef CAS PubMed.
- (a) J. R. Denton and H. M. L. Davies, Org. Lett., 2009, 11, 787 CrossRef CAS PubMed; (b) M. Verdecchia, C. Tubaro and A. Biffis, Tetrahedron Lett., 2011, 52, 1136 CrossRef CAS; (c) T. M. Gregg, M. K. Farrugia and J. R. Frost, Org. Lett., 2009, 11, 4434 CrossRef CAS PubMed; (d) Z. H. Zhang and J. B. Wang, Tetrahedron, 2008, 64, 6577 CrossRef CAS . For some reviews, see: ; (e) H. Lebel, J. Marcoux, C. Molonaro and A. B. Charette, Chem. Rev., 2003, 103, 977 CrossRef CAS PubMed; (f) G. Maas, Chem. Soc. Rev., 2004, 33, 183 RSC; (g) T. Ye and M. A. Mckervey, Chem. Rev., 1994, 94, 1091 CrossRef CAS; (h) M. P. Doyle and D. C. Forbes, Chem. Rev., 1998, 98, 911 CrossRef CAS PubMed.
- (a) M. P. Doyle and R. J. Pieters, J. Am. Chem. Soc., 1991, 113, 1424 CrossRef; (b) H. M. L. Davies and S. A. Panaro, Tetrahedron, 2000, 56, 4871 CrossRef CAS; (c) T. Nishimura, Y. Maeda and T. Hayashi, Angew. Chem., Int. Ed., 2010, 49, 7324 CrossRef CAS PubMed; (d) K. P. Kornecki, J. F. Briones, V. Boyarskikh, F. Fullilove, J. Autschbach, K. E. Schrote, K. M. Lancaster, H. M. L. Davies and J. F. Berry, Science, 2013, 342, 351 CrossRef CAS PubMed; (e) P. Muller, G. Bernardinelli, Y. F. Allenbach, M. Ferri and H. D. Flack, Org. Lett., 2004, 6, 1725 CrossRef PubMed; (f) Y. Su, M. Bai, J. B. Qiao, X. J. Li, R. Li, Y. Q. Tu and P. Gu, Tetrahedron Lett., 2015, 56, 1805 CrossRef CAS.
- (a) H. Nishiyama, Y. Itoh, H. Matsumoto, S. B. Park and K. Itoh, J. Am. Chem. Soc., 1994, 116, 2223 CrossRef CAS; (b) C. M. Che, J. S. Huang, F. W. Lee, Y. Li, T. S. Lai, H. L. Kwong, P. F. Teng, W. S. Lee, W. C. Lo, S. M. Peng and Z. Y. Zhou, J. Am. Chem. Soc., 2001, 123, 4119 CrossRef CAS PubMed; (c) B. G. Kim and M. L. Snapper, J. Am. Chem. Soc., 2006, 128, 52 CrossRef CAS PubMed; (d) J. A. Miller, W. Jin and S. T. Nguyen, Angew. Chem., Int. Ed., 2002, 114, 3077 CrossRef; (e) S. Priya, M. S. Balakrishna, S. M. Mobin and R. McDonald, J. Organomet. Chem., 2003, 688, 227 CrossRef CAS.
- (a) R. E. Lowenthal, A. Abiko and S. Masamune, Tetrahedron Lett., 1990, 31, 6005 CrossRef CAS; (b) W. Zhang, F. Xie, S. Matsuo, Y. Imahori, T. Kida, Y. Nakatsuji and I. Ikeda, Tetrahedron: Asymmetry, 2006, 17, 767 CrossRef CAS; (c) X. Dai and T. H. Warren, J. Am. Chem. Soc., 2004, 126, 10085 CrossRef CAS PubMed.
- (a) S. F. Zhu, J. A. Perman and X. P. Zhang, Angew. Chem., Int. Ed., 2008, 47, 8460 CrossRef CAS PubMed; (b) S. F. Zhu, J. V. Ruppel, H. J. Lu, L. Wojtas and X. P. Zhang, J. Am. Chem. Soc., 2008, 130, 5042 CrossRef CAS PubMed; (c) L. Y. Huang, Y. Chen, G. Y. Gao and X. P. Zhang, J. Org. Chem., 2003, 68, 8179 CrossRef CAS PubMed; (d) M. Otte, P. F. Kuijpers, O. Troeppner, I. Ivanović-Burmazović, J. N. H. Reek and B. D. Bruin, Chem. – Eur. J., 2014, 20, 4880 CrossRef CAS PubMed; (e) S. Fantauzzi, E. Gallo, E. Rose, N. Raoul, A. Caselli, S. Issa, F. Ragaini and S. Cenini, Organometallics, 2008, 27, 6143 CrossRef CAS; (f) S. L. Marquad, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, J. Am. Chem. Soc., 2013, 135, 6018 CrossRef PubMed.
- (a) J. J. Eisch, A. A. Aradi, M. A. Lucarelli and Y. Qian, Tetrahedron, 1998, 54, 1169 CrossRef CAS; (b) A. Nakamura, T. Yoshida, M. Cowie and S. Otsuka, J. Am. Chem. Soc., 1977, 99, 2108 CrossRef CAS; (c) M. Ishikawa, A. Naka, S. Okazaki and H. Sakamoto, Organometallics, 1993, 12, 87 CrossRef CAS; (d) D. J. Mindiola and G. L. Hillhouse, J. Am. Chem. Soc., 2002, 124, 9976 CrossRef CAS PubMed; (e) R. Waterman and G. L. Hillhouse, J. Am. Chem. Soc., 2003, 125, 13350 CrossRef CAS PubMed; (f) B. Gabor, C. Kruger, B. Marczinke, R. Mynott and G. iEke, Angew. Chem., Int. Ed., 1991, 30, 1666 CrossRef; (g) B. Gabor, C. Kruger, B. Marczinke, R. Mynott and G. WiEke, Angew. Chem., Int. Ed., 2015, 54, 10670 CrossRef PubMed.
- (a) V. K. Aggarwal, J. de Vicente and R. V. Bonnert, Org. Lett., 2004, 3, 2785 CrossRef; (b) H. Jiang and H. Chen, Chem. – Eur. J., 2012, 18, 11884 CrossRef CAS PubMed; (c) J. Barluenga, N. Quiñones, M. Tomás-Gamasa and M. P. Cabal, Eur. J. Org. Chem., 2012, 2312 CrossRef CAS.
- L. I. Smith and K. L. Howard, Org. Synth., 1944, 24, 53 CrossRef CAS.
- S. Kumar and R. W. Murray, J. Am. Chem. Soc., 1984, 106, 1040 CrossRef CAS.
- W. Schroeder and L. Katz, J. Org. Chem., 1954, 19, 718 CrossRef CAS.
- T. L. Holton and H. Shechter, J. Org. Chem., 1995, 60, 4725 CrossRef CAS.
- (a) J. R. Denton, D. Sukumaran and H. M. L. Davies, Org. Lett., 2007, 9, 2625 CrossRef CAS PubMed; (b) N. M. Roda, D. N. Tran, C. Battilocchio, R. Labes, R. J. Ingham, J. M. Hawkins and S. V. Ley, Org. Biomol. Chem., 2015, 13, 2550 RSC.
- (a) V. V. Zhdankin, ARKIVOC, 2009, 1 CAS; (b) P. J. Stang, Chem. Rev., 1996, 96, 1123 CrossRef CAS PubMed; (c) G. Qian, B. Liu, Q. Tan, S. Zhang and B. Xu, Eur. J. Org. Chem., 2014, 4837 CrossRef CAS.
- (a) S. Lin, M. Li, Z. Dong, F. Liang and J. Zhang, Org. Biomol. Chem., 2014, 12, 1341 RSC; (b) H. T. Bonge and T. Hansen, Synlett, 2007, 55 CAS; (c) B. Moreau, D. Albrico, V. N. G. Lindsay and A. B. Charette, Tetrahedron, 2012, 68, 3487 CrossRef CAS.
- (a) M. E. Furrow and A. G. Myers, J. Am. Chem. Soc., 2004, 126, 12222 CrossRef CAS PubMed; (b) L. Lapatsanis, G. Milias and S. Paraskewas, Synthesis, 1985, 513 CrossRef CAS.
- H. Liu and Y. Wei, Tetrahedron Lett., 2013, 54, 4645 CrossRef CAS.
- K. C. Nicolaou, C. J. Mathison and T. Montagnon, J. Am. Chem. Soc., 2004, 126, 5192 CrossRef CAS PubMed.
- (a) S. Cacchi, F. La Torre and D. Misiti, Synthesis, 1977, 301 CrossRef CAS; (b) Y. Kong, W. Zhang, M. Tang and H. Wang, Tetrahedron, 2013, 69, 7487 CrossRef CAS.
- For the structure and spectroscopic data of the products, see the ESI†.
- (a) Y. Li, J. S. Huang, Z. Y. Zhou, C. M. Che and X. Z. You, J. Am. Chem. Soc., 2002, 124, 13185 CrossRef CAS PubMed; (b) P. Müller and S. Tohill, Tetrahedron, 2000, 56, 1725 CrossRef.
- (a) M. Regitz and G. Mess, Diazo compounds: Properties and Synthesis, Academic press, New York, 1986, p. 71 Search PubMed; (b) G. L. Closs and R. A. Moss, J. Am. Chem. Soc., 1964, 86, 4042 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nj02378e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |