
Efficient saturated red light-emitting polyfluorenes containing iridium complexes in side chains†
Ting
Guo
,
Wenkai
Zhong
,
Anqi
Zhang
,
Jianhua
Zou
,
Lei
Ying
*,
Wei
Yang
and
Junbiao
Peng
Institute of Polymer Optoelectronic Materials and Devices, State Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Guangzhou 510640, China. E-mail: msleiying@scut.edu.cn
First published on 21st October 2015
Abstract
A series of electrophosphorescent copolymers were synthesized by Suzuki polymerization. A diketone-ended alkyl chain which is grafted in the N-position of the carbazole unit is used as a ligand to form a pendant cyclometalated Ir complex with 1-(9,9-dioctyl-9H-fluoren-2-yl)isoquinoline and 1-(N,N-diphenyl-benzenamine-4-yl)isoquinoline. The electroluminescence from the backbone of the copolymers is completely quenched by tethered iridium complexes even though with the content of iridium complexes being as low as 1 mol%. Saturated red light-emitting diodes were fabricated on the basis of these two series of copolymers. The best device performance with a maximum external quantum efficiency of 7.8% on the basis of copolymer PF-IrNiq1 was achieved, which was among the highest efficiencies of the reported electrophosphorescent red light-emitting polymers comprising iridium ligands.
Introduction
Electrophosphorescent polymer light-emitting diodes (PLEDs) based on iridium complexes have attracted great attention due to their capabilities of harvesting both singlet and triplet excitons to realize quantitative internal quantum efficiency.1–3 Additional advantages of PLEDs are their great potential for the fabrication of large-area and flexible devices based on the low-cost solution processing technique.4–6 Although PLEDs based on blending phosphorescent dyes into polymeric hosts can realize high-efficiency, the distinct incompatibility of phosphorescent dyes with host materials may lead to serious phase separation during the solution blending procedure, which in turn results in fast efficiency roll-off.7–10 An effective strategy to avoid such a problem is chemically tethering phosphorescent dyes to polymer backbones to attain molecular-level dispersion of dyes, for example attaching organic complexes as polymer side chains, coordinating organic complexes in the main chain, or constructing hyperbranched architectures by using organic complexes as core units.11–14Early investigation on phosphorescent conjugated copolymers has been carried out by chemically grafting red light-emitting phosphorescent dye into the side chain of polyfluorenes, providing phosphorescent polymers with moderate efficiencies.15 The efficiency of such polymers tethering red light-emitting iridium complexes in the side chains can be significantly improved by using the poly(fluorene-alt-carbazole) backbone.16 An alternative strategy to fabricate red light-emitting phosphorescent conjugated copolymers is by covalently incorporating iridium complexes into the polymer backbone.17 Based on this strategy, red light-emitting phosphorescent polymers consisting of chelated iridium complexes in the main chain were developed, which have achieved a maximal external quantum efficiency of 7.0% and 2.93% by copolymerizing the primary ligand and the auxiliary ligand into the backbone, respectively.18,19 Another interesting case is the incorporation of iridium complexes into the amino-alkyl containing polyfluorenes, which results in a series of phosphorescent copolymers that can achieve efficient red light-emission based on high work-function Al or Au as the cathode.20 It is also worth mentioning that the hyperbranched red light-emitting phosphorescent polymers consisting of iridium complexes as core units, give efficient emission with a maximum luminous efficiency of 6.5 cd A−1.21 These investigations demonstrated that this type of covalently copolymerized dye exhibited slow efficiency roll-off relative to that of the physically blending dye/polymer matrix system, indicating the great potential of this strategy.
Device performances highly depend on the efficiencies of phosphorescent emitters. Thus, it is of particular interest to develop new red-light emitting polymers by covalently tethering more efficient phosphorescent iridium complexes as side chains. With this in mind, herein we try to introduce two iridium complexes 1-(9,9-dioctyl-9H-fluoren-2-yl)isoquinoline (Fiq) and 1-(N,N-diphenyl-benzenamine-4-yl)isoquinoline (Niq) into the side chain of polyfluorenes, and incorporate the β-diketone as the terminal group of the side chain tethering into the N-atom of the carbazole unit. Efficient red light-emitting polymers with a maximum quantum efficiency of over 7% with CIE coordinates of (0.67, 0.33) were obtained based on the higher efficiency Niq-based iridium complex.
Experimental section
Materials
All manipulations involving air-sensitive reagents were performed under an atmosphere of dry argon. All manipulations involving air-sensitive reagents were performed under an atmosphere of dry argon. All reagents, unless otherwise specified, were obtained from Aldrich, Acros, and TCI Co. and were used as received. 2,7-Bis(4,4,5,5-tetramethyl-1,3,2-dixaborolan-2-yl)-9,9-dioctylfluorene (4),22 2,7-dibromo-9,9-dioctyl-9H-fluorene (5),23 3,6-dibromo-9-(12,14-pentadecyl-diketone)carbazole24 and poly(fluorene-alt-carbazole) (PFCz)21 were synthesized according to similar reported procedures.Synthesis of monomers
Synthesis of the copolymers
General procedures of Suzuki polycondensation of copolymers, taking PF-IrFiq05 as an example.2,7-Bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-9,9-dioctyl-fluorene (4) (321.3 mg, 0.5 mmol), 2,7-dibromo-9,9-dioctylfluorene (5) (271.5 mg, 0.495 mmol), 3,6-Dibromo-9-(iridium(III)bis(1-(9,9-dioctyl-9H-fluoren-2-yl)isoquinoline-N,C2′)-12,14-pentadecyldiketone)carbazole (8.9 mg, 0.005 mmol), palladium(II) acetate (Pd(OAc)2, 1.5 mol% equivalent) and tricyclohexylphosphine (P(Cyh)3, 4 mol% equivalent) were dissolved in the mixture of toluene (8 mL), after being stirred for 0.5 h, deionized H2O (2 mL) and Et4NOH (35 wt%) aqueous solution (0.2 mL). The mixture was heated to 90 °C and stirred for 48 hours under an argon atmosphere. The reaction was then capped by adding phenyl boric acid (25 mg) and bromobenzene (1 mL) successively and stirred for another 12 h. The whole mixture was poured into methanol. The precipitated polymer was recovered by filtration and purified by silica column chromatography with toluene as the eluent to remove a small molecular fraction of catalyst residues (yield: 89%). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.85 (br, 2H), 7.78 (brs, 2H), 7.72 (br, 2H), 1.96 (m, 4H), 1.21–0.97 (m, 24H), 0.82 (br, 6H).
Measurements
1H and 13C NMR spectra were recorded on a Bruker DRX 500 spectrometer operating at 500 and 125 MHz, respectively, and a Bruker DRX 300 spectrometer operating at 300 and 75 MHz, respectively, with tetramethylsilane as a reference. Molecular weight was obtained using a Waters GPC 2410 in tetrahydrofuran via a calibration curve of polystyrene standards. Elemental analyses were performed on a Vario EL Elemental analysis instrument (Elementar Co.). Samples were pressed as homogeneous tablets (Φ = 30 mm) of compressed (375 MPa) powder of the copolymers. UV-vis absorption spectra were recorded on a HP 8453 spectrophotometer. The PL quantum yields were determined in an integrating sphere ISO80 (Labsphere) with 325 nm excitation of the HeCd laser (MellsGriot). Cyclic voltammetry was carried out on a CHI660A electrochemical workstation with platinum at a scan rate of 50 mV s−1 against a saturated calomel reference electrode with nitrogen-saturated solution of 0.1 M tetrabutyl ammonium hexafluoro-phosphate (Bu4NPF6) in acetonitrile (CH3CN). The iridium content analyses were determined by using a Philips (Magix PRO) sequential X-ray fluorescence (XRF) spectrometer, with a rhodium tube operated at 60 kV and 50 mA, a LiF 200 crystal and a scintillation counter. Tris(acetylacetonate)iridium(III) (Ir content of 38%, from Alfa Aesar Co.) was used as an internal reference specimen. Samples and specimens were pressed into homogeneous tablets (Φ = 30 mm) of compressed (375 MPa) powder of the copolymers.Device fabrication and characterization
Polymers were dissolved in p-xylene and filtered using a 0.45 μm PTFE filter. Patterned ITO coated glass substrates were cleaned with acetone, detergent, distilled water and 2-propanol in an ultrasonic bath. After treatment with oxygen plasma, 150 nm of poly(3,4-ethylenedioxythiophene) doped with poly(styrene sulfonic acid) (PEDOT:PSS, Batron-P 4083, Bayer AG) was spin-coated onto the ITO substrates followed by drying in a vacuum at 80 °C for 18 h. A thin film of polymers was coated onto the anode by spin-casting inside a dry box. The film thickness of the active layers was around 75–80 nm, measured using an Alfa Step 500 surface profiler (Tencor). Device performances were measured inside a dry box. Current–voltage (I–V) characteristics were recorded using a Keithley 236 sourcemeter. EL spectra were obtained by an Oriel Instaspec IV CCD spectrograph. Luminance was measured using a PR 705 photometer (Photo Research). The external quantum efficiencies were determined by a Si photodiode with calibration in an integrating sphere (ISO80, Labsphere).Results and discussion
Synthesis and characterization
Scheme 1 shows the synthetic route to iridium bis-chelate complexes and copolymers. The ligands 1a and 2a were synthesized in comparatively high yield. Monomers with iridium complexes 3a and 3b were synthesized from 3,6-dibromo-9-(12,14-pentadecyl-diketone) carbazole and the corresponding iridium chloride-bridged dimers in moderate yields, where the dimer intermediate can be easily synthesized from the starting materials of 1a, 1b and iridium trichloride hydrate. It has been reported that the utilization of long alkyl side chains can effectively suppress the energy back transfer from the phosphorescent iridium complex to the polymer backbone.25 The copolymers from fluorene and iridium complex-attached carbazole units were synthesized based on palladium-catalyzed Suzuki polymerization. The feed ratios of Ir complexes in polycondensation are 0.5, 1 and 2, and the corresponding copolymers are denoted as PF-IrFiq05, PF-IrFiq1, PF-IrFiq2, PF-IrNiq05, PF-IrNiq1 and PF-IrNiq2, respectively.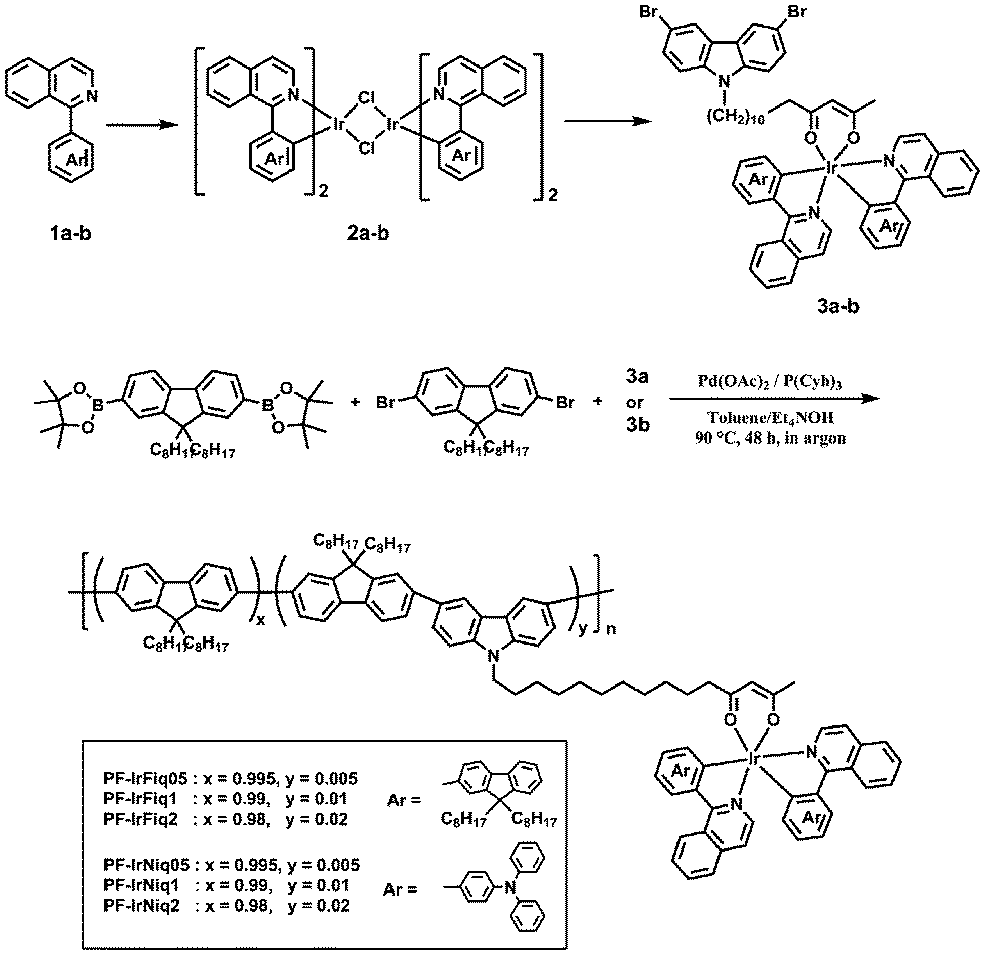 | ||
| Scheme 1 Synthesis of monomers and copolymers. | ||
All copolymerization was end-capped by adding 2-(4,4,5,5-tetramethyl-1,3,2-dioxabora-2-yl)-9,9-dioctylfluorene and 2-bromo-9,9-dioctylfluorene to remove bromide and dioxaborate end-groups, respectively, which have been proved to be relevant to reduce the effects of end-group traps. It was also noted that all copolymers can be readily dissolved in organic solvents including tetrahydrofuran (THF), toluene, chloroform and xylenes at room temperature. The number average molecular weight (Mn) as estimated by gel permeation chromatography (GPC) with THF as the eluent and polystyrene as the standard is in the range of 17.6–104.6 kDa, with a polydispersity index (PDI) of 1.94–2.31, which agrees with the traditional Suzuki polymerization (Table 1).
| Polymer | M n × 10−4 | PDI | Ir content in (mol%) | |
|---|---|---|---|---|
| Feed ratio | Polymera | |||
| a Calculated from the content of C, H, N and Ir complexes in copolymers. | ||||
| PF-IrFiq05 | 1.76 | 2.20 | 0.5 | 0.22 |
| PF-IrFiq1 | 4.58 | 1.94 | 1 | 0.40 |
| PF-IrFiq2 | 2.53 | 2.26 | 2 | 1.01 |
| PF-IrNiq05 | 6.25 | 1.99 | 0.5 | 0.33 |
| PF-IrNiq1 | 5.17 | 2.31 | 1 | 0.53 |
| PF-IrNiq2 | 10.46 | 2.24 | 2 | 0.99 |
The content of iridium in the copolymers was estimated by X-ray fluorescence (XRF) spectrometry, and the molar ratios of Ir complexes in the copolymers were calculated by combining elemental analysis with XRF data. It was noted that the ratios of Ir complexes afforded by XRF measurements were slightly lower than the theoretical values, which was understandable since the small molecular weight fraction has been removed during the work-up. Actual compositions of the copolymers and the feed ratios of the monomers are listed in Table 1.
Photophysical properties
Fig. 1a shows the UV-vis absorption spectra of Ir(Fiq)2(acac) and Ir(Niq)2(acac) complexes, and the photoluminescence (PL) spectra of the host PFO. From Fig. 1a one can clearly see that the iridium complexes exhibited a relatively broad absorbance ranging from 270 to 520 nm. The strong absorption peaks below 350 nm can be attributed to the spin-allowed singlet state 1π–*π transition of cyclometalated ligands, and the absorption peaks at 432 and 408 nm can be assigned to the spin-allowed singlet metal-to-ligand charge-transfer (1MLCT) transition.26 The absorption peaks of 493 nm for Ir(Fiq)2(acac) and 503 nm for Ir(Niq)2(acac) can be correlated with the triplet metal-to-ligand charge-transfer (3MLCT) transition.27,28 The spin forbidden 3MLCT can be ascribed to the strong orbital coupling of the heavy metal Ir and the ligand.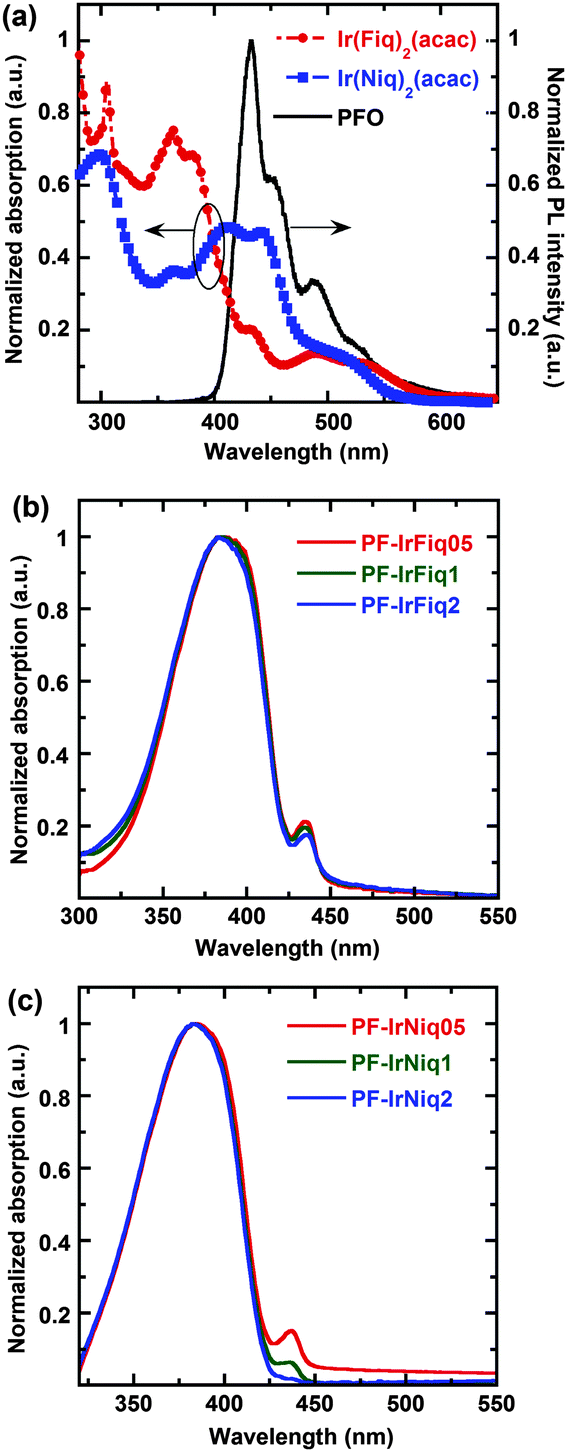 | ||
| Fig. 1 UV-vis absorption spectra of Ir(Fiq)2(acac) and Ir(Niq)2(acac) and PL of PFO (a); PL spectra of PF-IrFiq (b) and PF-IrNiq (c) as thin films. | ||
As shown in Fig. 1a, there is good overlap between the PL spectrum of the host PFO and the absorption spectra of iridium complexes Ir(Fiq)2(acac) and Ir(Niq)2(acac), indicating good Förster energy transfer can be occurred from PFO to the iridium complex.29 The comparison demonstrated more pronounced overlap between the absorbance of Ir(Niq)2(acac) and the PL spectrum of PFO than that of Ir(Fiq)2(acac), illustrating the higher Förster energy transfer efficiency for copolymers containing Ir(Niq)2(acac) as the side chain.
From Fig. 1b and c, we note that there is no distinct difference between the UV-vis absorption profiles of the two series of copolymers containing Ir complex side chains, which predominantly exhibited the absorption profiles of PFO with characteristic peak absorption at 380 nm. The relatively low absorbance can be attributed to the comparatively low molar content of iridium complexes Ir(Fiq)2(acac) and Ir(Niq)2(acac). It was also realized that all copolymers displayed obvious absorption characteristics corresponding to the β-phase of poly(9,9-diocylfluorene)s, which typically located at 437 nm as thin films.
Fig. 2 displays the photoluminescence (PL) spectra of copolymers as thin films. From Fig. 2, one can realize that the PL spectra of copolymer PF-IrFiqs are dominated at 439 and 467 nm, while the emission peaked at 644 nm corresponding to the relatively weak tethered iridium complex, and the emission peaked at 644 nm did not exhibit obvious enhancement with the increased iridium complex. In contrast, from Fig. 2b one can realize that the emission of the iridium complex at an iridium molar ratio of 0.5 mol% is nearly negligible; however, at a relatively high molar ratio of the iridium complex of 2 mol%, the characteristic emission of the iridium complex is dominated in the PL spectrum, and the emission corresponding to the host of PFO is significantly quenched, indicating the efficient Förster energy transfer from PFO to Ir(Niq)2(acac). It is also worth pointing out that the strong electron-donating N,N-diphenyl functional groups can effectively elongate the emission of the Ir(Niq)2(acac) complex with respect to the iridium complex based on phenyl-isoquinoline as the ligand. Although the emission peaks of copolymers PF-IrNiqs exhibited a blue shift of about 7 nm, the PL efficiency is higher in comparison to PF-IrFiqs, indicating that the introduction of N,N-diphenyl functional group Ir(Niq)2(acac) is beneficial to increase the emission efficiency. All polymers exhibited a relatively high PL quantum efficiency between 52 and 70%.
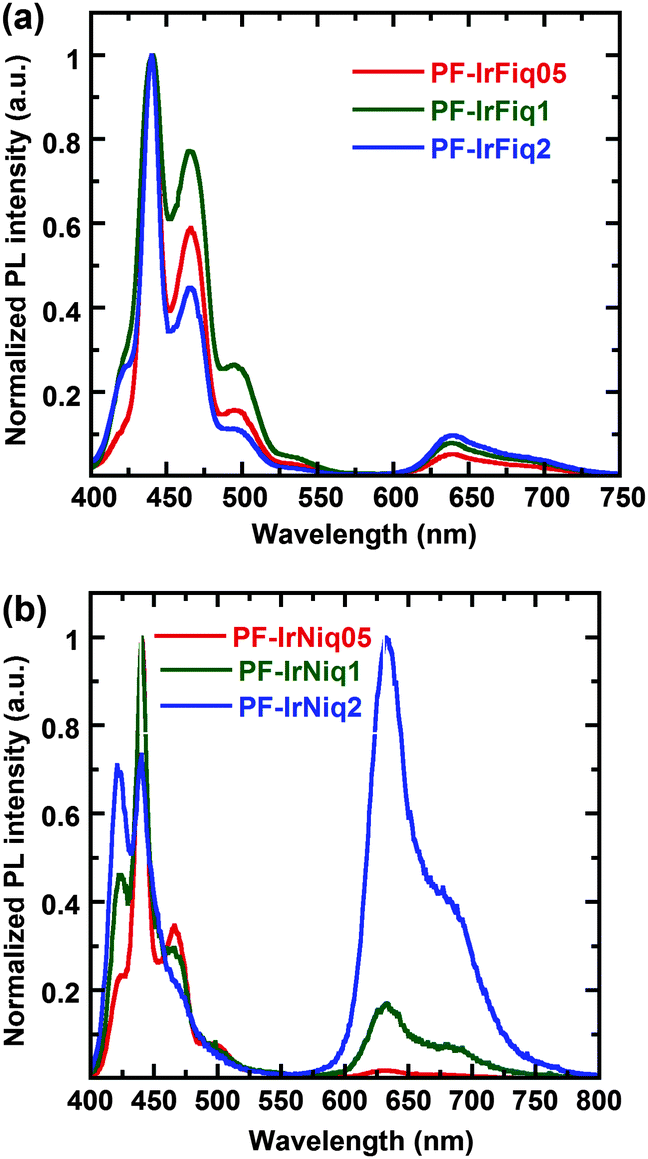 | ||
| Fig. 2 PL spectra of copolymers as thin films. | ||
We also note the characteristic PL emission of PF-IrFiq located at about 439 nm (0–0 band), 467 nm (0–1 band) and 496 nm (0–2 band) correlated well with the β-phase of poly(9,9-diocylfluorene)s, which agrees with the relatively high absorption characteristics at about 438 nm (Fig. 1b). In contrast, the characteristic PL emission of PF-IrNiq located at about 420 nm (0–0 band), 447 nm (0–1 band) and 470 nm (0–2 band), which can be correlated with the amorphous-phase of poly(9,9-diocylfluorene)s. This emission characteristic gradually decreased with the enhanced ratio of the iridium complex, which is in consistence with the decreased content of β-phase absorption characteristics at about 437 nm (Fig. 1c). The detailed photophysical properties are summarized in Table 2.
| Polymer | λ abs (nm) | PL |
E
optg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (eV) a (eV) |
E ox (V) | HOMO (eV) | LUMOb (eV) | |
|---|---|---|---|---|---|---|---|
| λ PL (nm) | Q PL (%) | ||||||
| a Estimated from the onset of the absorption edge of copolymers. b Calculated from the optical band gap (Eg) and HOMO. c Data in CH2Cl2 solution (10−5 mol L−1) of 0.1 mol L−1n-Bu4NPF6. | |||||||
| PF-IrFiq05 | 382 | 439, 467, 644 | 62 | 2.92 | 1.36 | −5.76 | −2.84 |
| PF-IrFiq1 | 382 | 439, 467, 644 | 50 | 2.96 | 1.32 | −5.72 | −2.86 |
| PF-IrFiq2 | 382 | 439, 467, 644 | 52 | 2.96 | 1.30 | −5.70 | −2.84 |
| PF-IrNiq05 | 383 | 420, 447, 637 | 70 | 2.93 | 1.32 | −5.72 | −2.79 |
| PF-IrNiq1 | 383 | 420, 447, 637 | 66 | 2.93 | 1.35 | −5.75 | −2.82 |
| PF-IrNiq2 | 383 | 439, 467, 637 | 65 | 2.95 | 1.37 | −5.77 | −2.82 |
| Ir(Fiq)2(acac)c | 348, 432, 493 | 644 | — | 2.08 | 0.85 | −5.25 | −3.17 |
| Ir(Niq)2(acac) | 290, 408, 503 | 637 | — | ||||
Electrochemical properties
The electrochemical behavior of these copolymers was investigated by cyclic votammetry (CV) in the acetonitrile solution of tetra-n-butylammonium hexafluorophosphate (n-Bu4NPF6, 0.1 M). The measurements were carried out in an inert atmosphere by using the ITO-coated glass as the working electrode, a platinum wire as the counter electrode, and a saturated calomel electrode as the reference electrode. The ferrocene/ferrocenium (Fc/Fc+) redox couple was used as the standard, which has a potential of −4.8 eV relative to the vacuum.30The onsets of oxidation potential (Eox) of the copolymers PF-IrFiq and PF-IrNiq are located in the range of 1.30–1.36 and 1.32–1.37 V, respectively. Under the same experimental conditions, the potential of Fc/Fc+ was measured to be 0.40 V relative to the saturated calomel electrode. Thus the highest occupied molecular orbital energy levels (EHOMO) of copolymers can be calculated through the semi-empirical equation EHOMO = −e(Eox − EFc/Fc+ + 4.80) (eV),21 which are determined to be ranged from −5.76 to −5.70 eV for PF-IrFiq and from −5.77 to −5.72 eV for PF-IrNiq. These results indicated that the EHOMO levels of copolymers were essentially insensitive to the molar ratio of the iridium complex. The optical band gaps (Eoptg) of the copolymers calculated from the absorption edge of their thin films are nearly identical around 2.9 eV. The lowest unoccupied molecular orbital energy levels (ELUMO) were estimated from the EHOMO and Eoptg, which ranged from −2.86 to −2.84 eV for PF-IrFiq, and from −2.82 to −2.79 eV for PF-IrNiq. The detailed electrochemical data are summarized in Table 2.
Electroluminescence and device performances
Electroluminescence (EL) performances of the resulted copolymers were investigated based on light-emitting devices with the configuration of ITO/PEDOT:PSS (50 nm)/poly(N-vinylcarbazole) (PVK) (40 nm)/copolymer + (PBD) (30 wt%) (80 nm)/Ba (4 nm)/Al (150 nm). Here, 2-(4-bisphenylyl)-5-(4-tert-butylphenyl)-1,3,4-oxadiazole (PBD) was employed to blend with the copolymer in the emissive layer to improve electron transportation.31 The resulted EL spectra are shown in Fig. 3. It was realized that the EL spectra of all devices are dominant with the red-light emission, which located at 644 and 637 nm, respectively, with CIE coordinates of (0.70, 0.31) and (0.68, 0.31) for PF-IrFiq and PF-IrNiq copolymers, respectively, which are very close to the saturated red light-emission.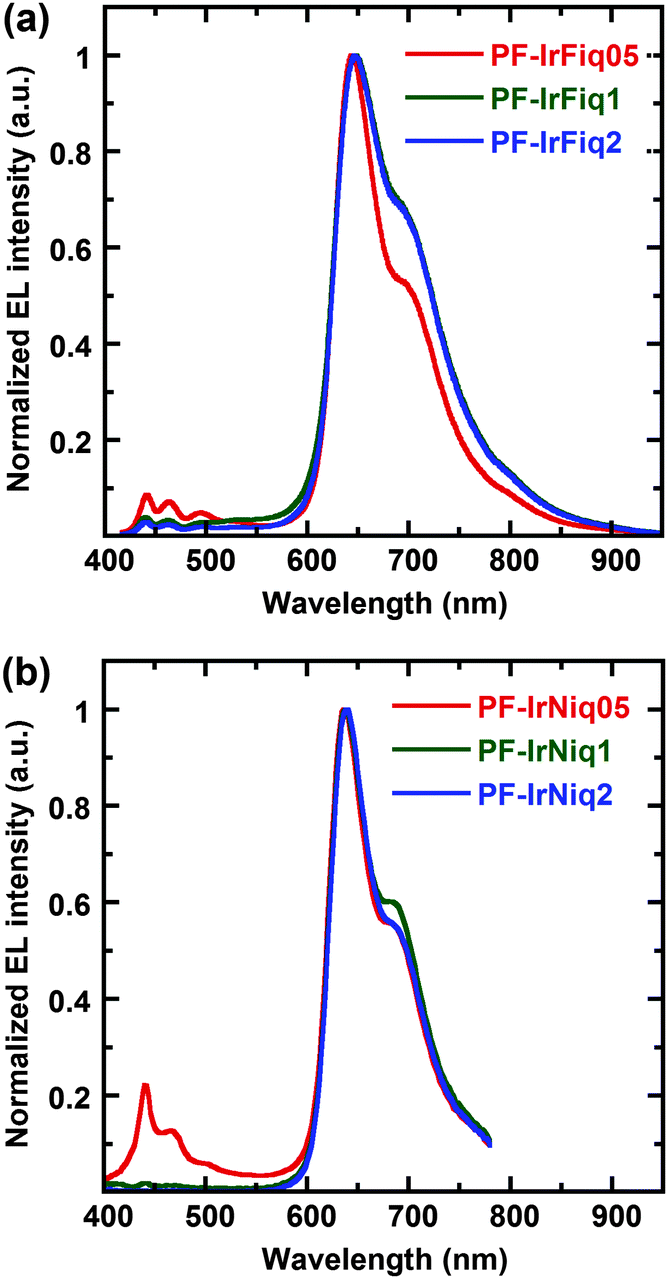 | ||
| Fig. 3 EL spectra of copolymers PF-IrFiq (a) and PF-IrNiq (b). | ||
The EL emission characteristics corresponding to PFO were nearly completely quenched with 1 mol% of the Ir complexes compared with the PL spectra, indicating that energy transfer in the EL procedure was more pronounced than the PL procedure. It was also recognized that the device performances varied with the ratio of the tethered iridium complexes. The EL spectra of copolymers revealed that copolymers PF-IrNiq exhibited a blue shift of about 7 nm blue shift relative to the copolymers PF-IrFiq incorporated fluorenyl-isoquinoline, which agreed with the PL spectra. On the other hand, devices based on the copolymer comprising PF-IrNiq as the emissive layer exhibited improved EL performances with respect to those based on PF-IrFiq. The best device performance was achieved for the device fabricated with copolymer PF-IrNiq containing 1 mol% Ir(Niq)2(acac), corresponding to an external quantum efficiency (EQE) of 7.80% and a luminous efficiency (LE) of 3.90 cd A−1, the efficiency of this device remained as high as an EQE of 5.18% and an LE of 2.59 cd A−1 at a current density of 100 mA cm−2, indicating that the favorable stability of the device. The maximum luminance (Lmax) achieved is 2803 cd cm−2, and the CIE coordinate of the device was (0.68, 0.29), which was very close to the NTSC standard red coordinates of (0.67, 0.33). To our knowledge, this device performance was among the most highly efficient electrophosphorescent pure red light-emitting polymers based on the Ir-complex as emitters. The detailed EL performance data are listed in Table 3. We noted that much higher device performances were attained based on copolymers comprising of Ir(Niq)2(acac) as the side chains. The fact can be understood since the spectra overlap of the absorbance of Ir(Niq)2(acac) with PL of host polymer PFO is more pronounced than that of Ir(Fiq)2(acac) (see Fig. 1a).
| Copolymer | V on (V) | At maximal QE | J = 100 mA cm−2 | L max (cd m−2) | λ EL (nm) | ||
|---|---|---|---|---|---|---|---|
| QEext (%) | LE (cd A−1) | QEext (%) | LE (cd A−1) | ||||
| Device structure: ITO/PEDOT/PVK/copolymer+PBD (30 wt%)/Ba/Al. | |||||||
| PF-IrFiq05 | 7.4 | 0.63 | 0.31 | 0.60 | 0.29 | 184 | 644 |
| PF-IrFiq1 | 5.8 | 2.71 | 1.35 | 1.45 | 0.72 | 850 | 644 |
| PF-IrFiq2 | 7.7 | 1.02 | 0.50 | 0.81 | 0.40 | 535 | 644 |
| PF-IrNiq05 | 9.2 | 1.71 | 0.86 | 0.72 | 0.36 | 376 | 637 |
| PF-IrNiq1 | 11.3 | 7.80 | 3.90 | 5.18 | 2.59 | 2803 | 637 |
| PF-IrNiq2 | 11.0 | 3.87 | 1.93 | 2.53 | 1.26 | 1625 | 637 |
The characteristics of external quantum efficiency (EQE) and luminance (L) as a function of current density (J) are shown in Fig. 4. The device based on copolymer PF-IrNiq1 achieved better EL efficiency than the device based on PF-IrFiq1. In addition, unlike a physically doping system comprising dyes and polymers,10 the EQE of these two devices exhibited slow roll-off regarding to current density. The device performances based on the resulted copolymers in terms of luminance and efficiency were much lower than the vacuum evaporated devices based on small molecules. Our current understanding is the lack of effective purification protocol for conjugated polymers with respect to the sublimation under high vacuum for small molecules, or the existence of non-conductive solubilizing alkyl side chains of the polymer backbone that is unfavorable for the charge transport and recombination. Nevertheless, these observations indicated that the strategy of preparing electrophosphorescent polymers through grafting of the iridium complexes onto the copolymer through a long alky side chain, not only can decrease concentration quenching by complexes stacking, but also increase the distance between the polymer host and Ir complexes, effectively suppressing the triplet state energy back transfer from the Ir complex guest to the polymer host.32
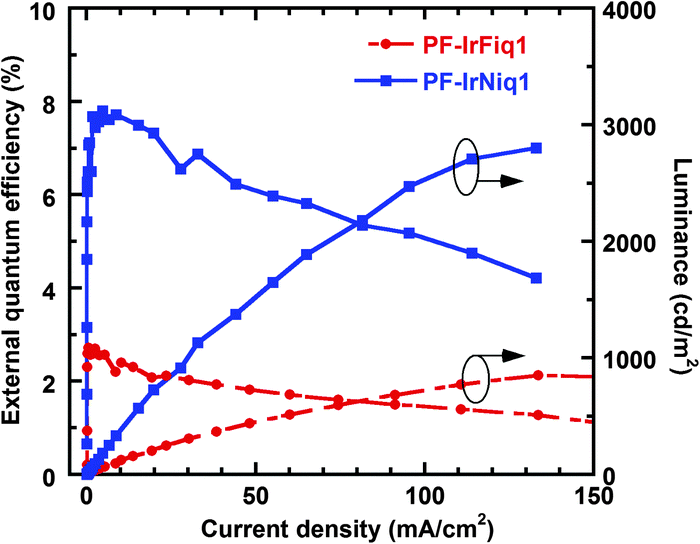 | ||
| Fig. 4 EQE – J – L characteristics of PF-IrFiq1 and PF-IrNiq1. | ||
Conclusion
In summary, two electrophosphorescent polymers PF-IrFiqs and PF-IrNiqs containing Ir complexes grafted on the side chain were synthesized, and the Ir complexes were Ir(Fiq)2(acac) and Ir(Niq)2(acac) which consists of fluoren-2-yl-isoquinoline and triphenylamine-isoquinoline as the primary ligand, respectively. The electroluminescent spectra of the resulting copolymers exclusively exhibited the grafted Ir complex characteristic emission, while the emission corresponding to the host polymer was nearly completely quenched by the tethered Ir complexes. For copolymer PF-IrFiq, the best device performance was based on the copolymer containing 1 mol% Ir complexes, in which the maximum external quantum efficiency of 2.71% was realized. For the devices based on copolymer PF-IrNiq, the maximum external quantum efficiency of 7.80% was obtained on the basis of 1 mol% Ir complexes, and the external quantum efficiency remained to be 5.18% at a current density of 100 mA cm−2, indicating the good stability of this polymer archetype. As the accessed external quantum efficiency is among the highest electrophosphorescent red light-emitting polymers based on Ir-complexes as the emitting centers, these observations highlighted the great advantage of tethering iridium complexes in the side chain of conjugated polymers.Acknowledgements
The authors are grateful for financial support from the Ministry of Science and Technology-China (2015AA033402 and 2015CB655004), the National Natural Science Foundation of China (Grants 51473054 and 51273069) and Fujian Key Laboratory of Polymer Materials (Fujian Normal University) (FJKL-POLY 201503).Notes and references
- A. H. Liang, L. Ying and F. Huang, J. Inorg. Organomet. Polym. Mater., 2014, 24, 905 CrossRef CAS.
- G. J. Zhou, W. Y. Wong and S. Suo, J. Photochem. Photobiol., C, 2010, 11, 133 CrossRef CAS.
- L. Ying, C.-L. Ho, H. B. Wu, Y. Cao and W. Y. Wong, Adv. Mater., 2014, 26, 2459 CrossRef CAS PubMed.
- S. Y. Shao, J. Q. Ding, L. X. Wang, X. F. Jing and F. S. Wang, J. Am. Chem. Soc., 2012, 134, 20290 CrossRef CAS PubMed.
- H. Xu, R. Chen, Q. Sun, W. Y. Lai, Q. Su, W. Huang and X. Liu, Chem. Soc. Rev., 2014, 43, 3259 RSC.
- L. Cui, Y. Xie, Y. Wang, C. Zhong, Y. Deng, X. Liu, Z. Jiang and L. Liao, Adv. Mater., 2015, 27, 4213 CrossRef CAS PubMed.
- Q. Mei, L. Wang, Y. Guo, J. Weng, F. Yan, B. Tian and B. Tong, J. Mater. Chem., 2012, 22, 6878 RSC.
- V. Jankus, K. Abdullah, G. C. Griffiths, A. A. Hameed, Y. Zheng, R. B. Martin and A. P. Monkman, Org. Electron., 2015, 20, 97 CrossRef CAS.
- C. Huang, F. Chang, Y. Chu, C. Lai, T. Lin, C. Zhu and S. Kuo, J. Mater. Chem. C, 2015, 3, 8142 RSC.
- F. Dumur, L. Beouch, S. Peralta, G. Wantz, F. Goubard and D. Gigmes, Org. Electron., 2015, 25, 21 CrossRef CAS.
- L. Ying, J. H. Zou, A. Q. Zhang, B. Chen, W. Yang and Y. Cao, J. Organomet. Chem., 2009, 694, 2727 CrossRef CAS.
- H. Y. Zhen, J. Luo, W. Yang, Q. L. Chen, L. Ying, J. H. Zou, H. B. Wu and Y. Cao, J. Mater. Chem., 2007, 17, 2824 RSC.
- T. Guo, L. Yu, Y. Yang, Y. H. Li, Y. Tao, Q. Hou, L. Ying, W. Yang, H. B. Wu and Y. Cao, J. Lumin., 2015, 167, 179 CrossRef CAS.
- Z. Huang, B. Liu, Y. He, X. Yan, X. Yang, X. Xu, G. Zhou, Y. Ren and Z. Wu, J. Org. Chem., 2015, 794, 1 CrossRef CAS.
- X. Chen, J. L. Liao, Y. Liang, M. O. Ahmed, H. E. Tseng and S. A. Chen, J. Am. Chem. Soc., 2003, 125, 636 CrossRef CAS PubMed.
- J. X. Jiang, C. Y. Jiang, W. Yang, H. Y. Zhen, F. Huang and Y. Cao, Macromolecules, 2005, 38, 4072 CrossRef CAS.
- A. J. Sandee, C. K. Williams, N. R. Evans, J. E. Davies, C. E. Boothby, A. Koehler, R. H. Friend and A. B. Holmes, J. Am. Chem. Soc., 2004, 126, 7041 CrossRef CAS PubMed.
- H. Zhen, C. Luo, W. Yang, W. Y. Song, B. Du, J. X. Jiang, C. Y. Jiang, Y. Zhang and Y. Cao, Macromolecules, 2006, 39, 1693 CrossRef CAS.
- K. Zhang, Z. Chen, Y. Zou, S. L. Gong, C. L. Yang, J. G. Qin and Y. Cao, Chem. Mater., 2009, 21, 3306 CrossRef CAS.
- L. Ying, Y. H. Xu, W. Yang, L. Wang, H. B. Wu and Y. Cao, Org. Electron., 2009, 10, 42 CrossRef CAS.
- T. Guo, R. Guan, J. H. Zou, J. Liu, L. Ying, W. Yang, H. B. Wu and Y. Cao, Polym. Chem., 2011, 2, 2193 RSC.
- T. Guo, L. Yu, B. F. Zhao, L. Ying, H. B. Wu, W. Yang and Y. Cao, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1043 CrossRef CAS.
- Y. Yang, L. Yu, Y. Xue, Q. H. Zou, B. Zhang, L. Ying, W. Yang, J. B. Peng and Y. Cao, Polymer, 2014, 55, 1698 CrossRef CAS.
- Z. H. Ma, J. Q. Ding, Y. X. Cheng, Z. Y. Xie, L. X. Wang, X. B. Jing and F. S. Wang, Polymer, 2011, 52, 2189 CrossRef CAS.
- N. R. Evans, L. S. Devi, C. S. K. Mak, S. E. Watkins, S. I. Pascu, A. Köhler, R. H. Friend, C. K. Willianms and A. B. Holmes, J. Am. Chem. Soc., 2006, 128, 6647 CrossRef CAS PubMed.
- M. Lian, Y. Yu, J. Zhao, Z. Huang, X. Yang, G. Zhou, Z. Wu and D. Wang, J. Mater. Chem. C, 2014, 2, 9523 RSC.
- Z. Ma, L. Chen, J. Q. Ding, L. X. Wang, X. F. Jing and F. S. Wang, Adv. Mater., 2011, 23, 3726 CrossRef CAS PubMed.
- Z. Niu, T. Zheng, Y. Su, P. Wang, X. Li, F. Cui, J. Liang and G. Li, New J. Chem., 2015, 39, 6025 RSC.
- S. H. Rhee, S. H. Kim, H. S. Kim, J. Y. Shin, J. Bastola and S. Y. Ryu, ACS Appl. Mater. Interfaces, 2015, 7, 16750 CAS.
- L. Ying, Y. H. Li, C. H. Wei, M. Q. Wang, W. Yang, H. B. Wu and Y. Cao, Chin. J. Polym. Sci., 2013, 31, 88 CrossRef.
- S. Lamansky, P. I. Djurovich, R. F. Abdel, S. Garon, D. L. Murphy and M. E. Thompson, J. Appl. Phys., 2002, 92, 1570 CrossRef CAS.
- C. H. Chang, R. Griniene, Y. D. Su, C. C. Yeh, H. C. Kao, J. V. Grazulevicius, D. Volyniuk and S. Grigalevicius, Dyes Pigm., 2015, 122, 257 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nj02185e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |