
Transition-metal free N-arylation of cyanamides by diaryliodonium triflates in aqueous media†
Jihui
Li
,
Xinyi
Zheng
,
Wei
Li
,
Wei
Zhou
,
Wen
Zhu
and
Yucang
Zhang
*
College of Materials and Chemical Engineering, Hainan University, Haikou, 570228, P. R. China. E-mail: zhangyucang88@163.com
First published on 11th November 2015
Abstract
An operationally simple protocol is established for the synthesis of disubstituted cyanamides through the transition-metal free N-arylation of cyanamides by diaryliodonium triflates in aqueous media. Both alkyl and aryl cyanamides are well compatible with the mild reaction conditions. The one-pot synthesis of ureas is also possible through sequential arylation and hydrolysis of cyanamides, diaryliodonium triflates and H2O with good yields.
Cyanamides have attracted considerable attention as a class of versatile organic molecules. They have a wide range of uses in organic synthetic chemistry1 and coordination chemistry;2 they can be used as building blocks for the construction of not only diverse N-containing compounds but also metal ligands with their unique reactivity and structure of the cyanamide unit. Additionally, it has been found that some cyanamide-based compounds show a diversity of interesting bioactivities,3 such as anti-cathepsins K and L,3a inhibition of spontaneous myogenic,3d and peptide activator activities.3e It has also been proved that cyanamide is a natural product present in higher plants even though the distribution is limited.4 As a valuable dinitrogen resource, there have been some synthetic methods developed for cyanamides, which can be divided into these two categories: (i) conventional electrophilic substitution and condensation of cyanamide, or nucleophilic substitution of cyanogen bromide5 and (ii) transition-metal catalyzed transformations of various starting materials.6 The latter method is an alternative approach to achieving a diversity of complex cyanamides by using easily available starting materials. For example, palladium-catalyzed three component reactions of aryl isocyanides, allyl methyl carbonate, and trimethylsilyl azide were developed for the synthesis of allyl cyanamides and N-cyanoindoles;6a,b a copper-catalyzed cascade reaction of 1-(2-iodoaryl)thioureas and aryl iodides was adopted to build 2-arylsulfanylarylcyanamides.6d The palladium-catalyzed N-arylation of alkyl cyanamides by arylhalides with bidentate phosphane as a ligand was developed for the synthesis of disubstituted cyanamides (Figure a).6e Moreover, an N-arylation of aryl cyanamides by diaryliodonium hexafluorophosphates was also developed more recently, and the reaction was needed to be performed under oxygen free conditions (Figure b).7
Diaryliodonium salts are popular arylating reagents used for the construction of C–C, C–O, C–N, C–S, C–Se and C–X (X = halides) bonds under mild reaction conditions in organic synthesis.8 For instance, the N-arylations of various N-nucleophiles by diaryliodoniums have been widely used for the construction of C–N bonds. In particular, the transition-metal free transformations, which are potentially environmentally benign synthetic methods, are highly pursued in the past decades.8c,f,g Moreover, diaryliodonium salts are easily available, air- and moisture-stable, thermostable, nontoxic and environmentally benign, and the reactions can generate one molecular aryliodide which is also valuable organic intermediate in arylation reactions.
Recently, we have reported a copper-catalyzed three-component reaction of cyanamides, boronic acids and amines for the synthesis of guanidines, where an N-arylation on the terminal nitrogen of cyanamides by arylboronic acids can be realized, and an N-arylation of the internal nitrogen of cyanamides was found to be a side reaction.9 As a part of our ongoing research in exploiting cyanamides as a dinitrogen resource, here, an N-arylation of the internal nitrogen of both alkyl and aryl cyanamides by diaryliodonium triflates in aqueous media is reported for making disubstituted cyanamides under transition-metal free conditions (Fig. 1c). The arylation of p-tolylcyanamide (1a) with diphenyliodonium triflate (2a) was initially screened in DMF with K2CO3 as a base at 80 °C without excluding oxygen, producing the cross-coupling product in 82% yield within 2 h (Table 1, entry 1). Then, higher and lower reaction temperatures were evaluated, and inferior yields were obtained (Table 1, entries 2–4). Additionally, the reaction only produced a trace amount of the desired product without the base (Table 1, entry 5). Other solvents including H2O, toluene, EtOH, and DMSO were further investigated (Table 1, entries 6–10). Pleasantly, a superior yield was produced in water (Table 1, entry 6), and the reaction in toluene also afforded a comparable yield (Table 1, entry 7). Different bases such as K3PO4, Cs2CO3, NaOH, CH3COONa and Et3N were finally explored in H2O; all the reactions provided inferior yields (Table 1, entries 11–15). Thus, the best reaction conditions were K2CO3, H2O, 80 °C, 2 h (Table 1, entry 6), and the exclusion of oxygen was not required comparing with the reaction between cyanamides and diaryliodonium triflates.7
 | ||
| Fig. 1 N-Arylation of cyanamides. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Base (2 eq.) | Solvent | T (°C) | t (h) | Yieldb (%) |
| a Reaction conditions: A mixture of p-tolylcyanamide 1a (0.2 mmol), diphenyliodonium triflate 2a (0.2 mmol), solvent (1 mL) and base (0.4 mmol) was stirred at an identified reaction temperature. b Isolated yields. | |||||
| 1 | K2CO3 | DMF | 80 | 2 | 82 |
| 2 | K2CO3 | DMF | 50 | 2 | 55 |
| 3 | K2CO3 | DMF | 130 | 2 | 55 |
| 4 | K2CO3 | DMF | rt | 24 | 15 |
| 5 | — | DMF | 50 | 2 | Trace |
| 6 | K2CO3 | H2O | 80 | 2 | 91 |
| 7 | K2CO3 | Toluene | 80 | 2 | 87 |
| 8 | K2CO3 | EtOH | 80 | 2 | 60 |
| 9 | K2CO3 | Dioxane | 80 | 2 | 84 |
| 10 | K2CO3 | DMSO | 80 | 2 | 77 |
| 11 | K3PO4 | H2O | 80 | 2 | 87 |
| 12 | Cs2CO3 | H2O | 80 | 2 | 58 |
| 13 | NaOH | H2O | 80 | 2 | 79 |
| 14 | Et3N | H2O | 80 | 2 | 55 |
| 15 | CH3COONa | H2O | 80 | 2 | 34 |

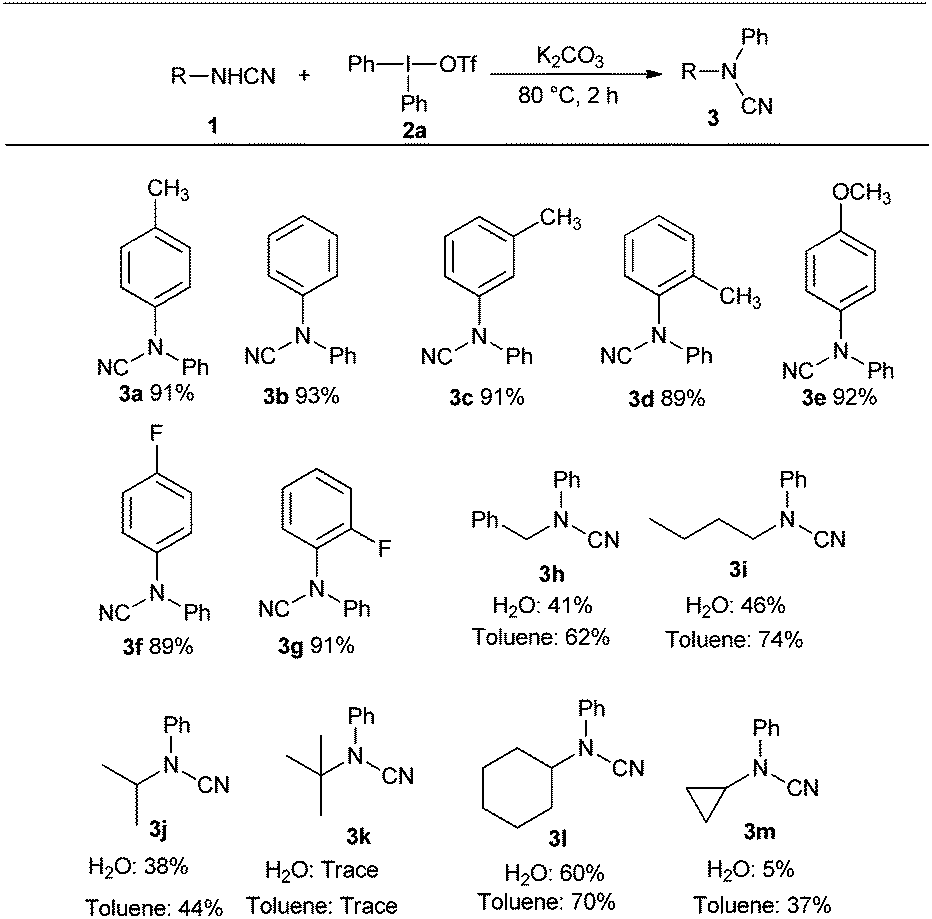
The optimized conditions were subsequently applied to various cyanamides as depicted in Scheme 1. p-, m-, o-Tolyl cyanamides all furnished coupling products with comparable yields (3a, 3b, 3c), indicating the steric hindrance of the aryl cyanamides was not a problem for the reactions. The aryl cyanamides with electron-withdrawing and -donating groups were both compatible with the reaction conditions with high yields. For example, p-methoxyphenyl and p-fluorophenyl cyanamides provided 91% (3e) and 88% (3f) yields, respectively. Gratifyingly, although the aliphatic cyanamides can be easily hydrolyzed, the reactions of alkyl cyanamides including linear and cyclic afforded the desired products in moderate yields in water (3h–3j, 3l, 3m); these have not been reported in the reaction with diaryliodonium hexafluorophosphates.7 Cyclopropyl cyanamide also produced the corresponding product, giving 5% yield (3l), yet t-butyl cyanamide did not produce any arylation product (3k). The reactions of alkyl cyanamides were also investigated in toluene for comparison; the results were similar to that in water but higher yields were obtained, and the cyclopropyl cyanamides are well compatible under the mild reaction conditions with moderate yields. It is worth noting that the reaction on a larger scale also provided the desired coupling product in high yield and aryliodide was recovered with good yield under the same reaction conditions.10
 | ||
Scheme 1 Phenylation of various cyanamides.a a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reaction conditions: a mixture of cyanamides 1 (0.2 mmol), diphenyliodonium triflate 2a (0.2 mmol) and K2CO3 (0.4 mmol) in H20 or toluene (1 mL) was stirred at 80 °C for 2 h. Reaction conditions: a mixture of cyanamides 1 (0.2 mmol), diphenyliodonium triflate 2a (0.2 mmol) and K2CO3 (0.4 mmol) in H20 or toluene (1 mL) was stirred at 80 °C for 2 h. | ||
The reaction scope was finally extended to other diaryliodonium salts in the aqueous media. It turned out that both symmetric and unsymmetric diaryliodonium triflates were fully tolerated under the reaction conditions, providing good yields (Table 2). For the symmetric substrates with electron-rich and -poor aryl groups, the arylation took place smoothly with similar yields (entries 1–5); and the halogen substituents, bromide and chloride, which can be further functionalized, survived under the gentle reaction conditions (entries 2 and 3). The diaryliodonium salts with an ortho-substituent (2f) also produced the corresponding product without reducing the yield (entry 4). In accordance with other N-nucleophiles,8b,c the aryl transfer of unsymmetric diaryliodonium triflates onto cyanamides generally preferred the electron-poor aryl groups to the electron-rich under transition-metal free conditions, and the selectivities are strongly dependent on the gap of the electron density between two aryl groups, which should become better as the gap grows.8a,11 For instance, the electron-poorer phenyl of phenyl(p-t-butylphenyl)iodonium (2g) and phenyl(p-acetylaminophenyl)iodonium (2h) is more easily transferred onto cyanamide rather than the other aryl groups (entries 6 and 7). Acetylamino was well compatible with the mild reaction conditions, as the product 4c and byproduct N-(4-iodophenyl)acetamide were formed. The treatment of p-tolylcyanamide by phenyl(p-nitrophenyl)iodonium triflate (2i) mainly produced p-tolyl(p-nitrophenyl)cyanamide (4d) in high yield (entry 8); similarly, phenyl(2-thiophenyl)iodonium triflate (2j) afforded an excellent selectivity (entry 9). Unexpectedly, the electron-poor p-iodophenyl of phenyl(p-iodophenyl)iodonium triflate (2k) is not a preferable transferred group in the reaction (entry 10); the reason for this discrepancy is unclear at this stage. This indicates that the iodo group is well accommodated with the reaction conditions. Considering that the steric hindrance of the substrates may be a factor affecting the aryl transfer selectivity,8b,12 the reactions of (2,5-dimethylphenyl)phenyliodonium triflate (2l) with both p-tolyl and o-tolyl cyanamides were evaluated. The treatment of 2l with p-tolylcyanamides provided a mixture of products with a 1.2/1 ratio of phenyl(p-tolyl)cyanamide/(2,5-dimethylphenyl)p-tolylcyanamides (3a/4a) (entry 11). However, the reaction of 2l with o-tolyl cyanamides produced more of the 2,5-dimethylphenylation product than the phenylation product with good yields, which shows an ortho-effect (3d/4g = 1/1.5) (entry 12).8c These results suggest that the steric hindrance of cyanamides plays a significant role on the chemoselectivities for unsymmetric diaryliodonium salts with the steric hindrance aryl group, which may due to the accelerated reductive elimination between both bulky cyanamide and bulky aryl groups via a T-shape intermediate.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ar1 | Ar2 | 2 | 3/4 (Ratio) | Yieldb (%) |
| a Reaction conditions: A mixture of p-tolylcyanamides 1a (0.2 mmol), diaryliodonium triflate 2 (0.2 mmol) and K2CO3 (0.4 mmol) in H2O (1 mL) was stirred at 80 °C for 2 h. b Isolated yields. c Ratios based on 1H NMR. d o-Tolylcyanamide was used. | |||||
| 1 | 4-CH3C6H4 | 4-CH3C6H4 | 2b | 3n/— (—) | 90 |
| 2 | 4-CIC6H4 | 4-CIC6H4 | 2c | 3o/— (—) | 97 |
| 3 | 4-BrC6H4 | 4-BrC6H4 | 2d | 3p/— (—) | 78 |
| 4 | 2,5-(CH3)2C6H3 | 2,5-(CH3)2C6H3 | 2f | —/4a (—) | 88 |
| 5 | 4-(t-C4H9)C6H4 | 4-(t-C4H9)C6H4 | 2e | —/4b (—) | 89 |
| 6 | C6H6 | 4-(t-C4H9)C6H4 | 2g | 3a/4b (2.4/1)c | 96 |
| 7 | C6H5 | 4-CH3CONHC6H4 | 2h | 3a/4c (6.8/1) | 86 |
| 8 | C6H5 | 4-NO2C6H4 | 2i | 3a/4d (<1/20) | 97 |
| 9 | C6H5 | 2-Thienyl | 2j | 3a/4e (>99/1) | 84 |
| 10 | C6H5 | 4-IC6H4 | 2k | 3a/4f (1.5/1)c | 97 |
| 11 | C6H5 | 2,5-(CH3)2C6H3 | 21 | 3a/4a (1.2/1)c | 89 |
| 12d | C6H5 | 2,5-(CH3)2C6H3 | 21 | 3d/4g (1/1.5)c | 85 |

As ureas are wildly used in pharmaceutical candidates13 and pesticides,14 the hydrolysis of cyanamides was explored to make ureas. The reaction was briefly investigated as shown in Scheme 2a; the treatment of the disubstituted cyanamides by water in toluene, which is promoted by trifluoroacetic acid, delivered the hydrolysis product ureas with good yields. The phenyl(p-tolyl) cyanamide and benzyl(phenyl) cyanamide both afforded the desired N,N-disubstituted ureas, which can be the precursors of bioactive benzimidazolones (Scheme 2a, 5a and 5b). The one-pot synthesis of ureas can also be easily realized by combining N-arylation and subsequent hydrolysis with good yield (Scheme 2b).
 | ||
| Scheme 2 The synthesis of ureas. | ||
In summary, a transition-metal free N-arylation of cyanamides with diaryliodonium triflates has been developed for the synthesis of disubstituted cyanamides with good yields. The reaction shows a general reaction scope; the aromatic and aliphatic cyanamides are both well compatible with the mild aqueous reaction conditions. Diaryliodonium triflates including symmetric and unsymmetric all work well, and generally the electron-poorer aryl groups are the preferentially transferred groups for the unsymmetric diaryliodonium triflates; the steric hindrance of both substrates is also an important factor for the reaction chemoselectivities. In addition, the disubstituted cyanamides can be easily hydrolyzed to valuable N,N-disubstituted ureas, and the N-arylation of cyanamides and subsequent hydrolysis can be combined into a one-pot procedure with good yields. The mild transition-metal free aqueous conditions and eco-friendly feature of diaryliodonium salts will make this method attractive. Further exploration of N,N-disubstituted ureas for the synthesis of bioactive heterocycles is in progress and will be detailed in the future.
Acknowledgements
The authors thank the Hainan innovation and entrepreneurship program for high-tech people, and Hainan international cooperation program (KJHZ 2014-02) for financial support. We also thank Xiaopeng Wu and Jiuhui Li [from the Institute of Tropical Bioscience and Biotechnology (ITBB), Chinese Academy of Tropical Agricultural Sciences (CATAS)] for their excellent technical and analytical support.Notes and references
- Selected examples: (a) A. Servais, M. Azzouz, D. Lopes, C. Courillon and M. Malacria, Angew. Chem., Int. Ed., 2007, 46, 576–579 CrossRef CAS PubMed; (b) M.-H. Larraufie, C. Ollivier, L. Fensterbank, M. Malacria and E. Laocte, Angew. Chem., Int. Ed., 2010, 49, 2178–2181 CrossRef CAS PubMed; (c) Z. Pan, S. M. Pound, N. R. Rondla and C. J. Douglas, Angew. Chem., Int. Ed., 2014, 53, 5170–5174 CAS; (d) H. Basavaprabhu and V. V. Sureshbabu, Org. Biomol. Chem., 2012, 10, 2528–2533 RSC; (e) V. Panduranga, Basavaprabhu and V. V. Sureshbabu, Tetrahedron Lett., 2013, 54, 975–979 CrossRef CAS; (f) S. Kamijo, T. Jin and Y. Yamamoto, Angew. Chem., Int. Ed., 2002, 41, 1780–1782 CrossRef CAS; (g) L. V. R. Boñaga, H.-C. Zhang and B. E. Maryanoff, Chem. Commun., 2004, 2394–2395 RSC; (h) K. Fukumoto, T. Oya, M. Itazaki and H. Nakazawa, J. Am. Chem. Soc., 2009, 131, 38–39 CrossRef CAS PubMed; (i) R. L. Giles, J. D. Sullivan, A. M. Steiner and R. E. Looper, Angew. Chem., Int. Ed., 2009, 48, 3116–3120 CrossRef CAS PubMed; (j) S. Guin, S. K. Rout, A. Gogoi, W. Ali and B. K. Patela, Adv. Synth. Catal., 2014, 356, 2559–2565 CrossRef CAS; (k) T. K. Lane, B. R. D'Souza and J. Louie, J. Org. Chem., 2012, 77, 7555–7563 CrossRef CAS PubMed; (l) V. Kumar, M. P. Kaushik and A. Mazumdar, Eur. J. Org. Chem., 2008, 1910–1916 CrossRef.
- Selected examples: (a) C. J. Adams, J. Chem. Soc., Dalton Trans., 1999, 2059–2064 RSC; (b) A. S. Smirnov, E. S. Butukhanova, N. A. Bokach, G. L. Starova, V. V. Gurzhiy, M. L. Kuznetsov and V. Yu. Kukushkin, Dalton Trans., 2014, 43, 15798–15811 RSC; (c) M. Yuan, S. Gao, H.-L. Sun and G. Su, Inorg. Chem., 2004, 43, 8221–8223 CrossRef CAS PubMed.
- (a) D. G. Barrett, D. N. Deaton, A. M. Hassell, R. B. McFadyen, A. B. Miller, L. R. Miller, J. A. Payne, L. M. Shewchuk, D. H. Willard, Jr. and L. L. Wright, Bioorg. Med. Chem. Lett., 2005, 15, 3039–3043 CrossRef CAS PubMed; (b) H.-W. K. Bischofsheim, H.-J. L. Hogheim, J.-R. S. Kelkheim, A. W. Egelsbach, S. F. Idstein, H.-W. J. Niedernhausen and S. P. Frankfurt, US 6369069B1, 2002; (c) K. S. Atwal, G. J. Grover, S. Z. Ahmed, P. G. Sleph, S. Dzwonczyk, A. J. Baird and D. E. Normandin, J. Med. Chem., 1995, 38, 3236–3245 CrossRef CAS PubMed; (d) P. W. Manley and U. Quast, J. Med. Chem., 1992, 35, 2327–2340 CrossRef CAS PubMed; (e) G. Danger, A. Michaut, M. Bucchi, L. Boiteau, J. Canal, R. Plasson and R. Pascal, Angew. Chem., Int. Ed., 2013, 52, 611–614 CrossRef CAS PubMed.
- T. Kamo, M. Endo, M. Sato, R. Kasahara, H. Yamaya, S. Hiradate, Y. Fujii, N. Hirai and M. Hirota, Phytochemistry, 2008, 69, 1166–1172 CrossRef CAS PubMed.
- (a) C. Grundmann, in Methoden der Organischen Chemie (Houben-Weyl), ed. J. Falbe, Georg Thieme Verlag, Stuttgart, 1985, vol. E5, pp. 1611–1658 Search PubMed; (b) S. R. Sandler and W. Karo, Organic Functional Group Preparations, Academic, New York, 1972, vol. 3, pp. 286–287 and vol. 2, pp. 174–178 Search PubMed.
- Selected transition-metal catalyzed synthesis of cyanamides: (a) S. Kamijo, T. Jin and Y. Yamamoto, J. Am. Chem. Soc., 2001, 123, 9453–9454 CrossRef CAS PubMed; (b) S. Kamijo and Y. Yamamoto, J. Am. Chem. Soc., 2002, 124, 11940–11945 CrossRef CAS PubMed; (c) S. Kamijo, T. Jin and Y. Yamamoto, Angew. Chem., Int. Ed., 2002, 41, 1780–1782 CrossRef CAS; (d) S. K. Sahoo, L. Jamir, S. Guin and B. K. Patel, Adv. Synth. Catal., 2010, 352, 2538–2548 CrossRef CAS; (e) R. M. Stolley, W. Guo and J. Louie, Org. Lett., 2012, 14, 322–325 CrossRef CAS PubMed; (f) P. Kumari, R. Nagpal, P. Chauhan, V. Yatindranath and S. M. Chauhan, J. Chem. Sci., 2015, 127, 13–18 CrossRef CAS; (g) S. Cerezo, J. Cortés, M. Moreno-Mañas, R. Pleixats and A. Roglans, Tetrahedron, 1998, 54, 14869–14884 CrossRef CAS.
- P. Li, G. Cheng, H. Zhang, X. Xu, J. Gao and X. Cui, J. Org. Chem., 2014, 79, 8156–8162 CrossRef CAS PubMed.
- (a) E. A. Merritt and B. Olofsson, Angew. Chem., Int. Ed., 2009, 48, 9052–9070 CrossRef CAS PubMed; (b) M. S. Yusubov, A. V. Maskaev and V. V. ZhdankinARKIVOC, 2011, 370–409 Search PubMed; (c) J. Malmgren, S. Santoro, N. Jalalian, F. Himo and B. Olofsson, Chem. – Eur. J., 2013, 19, 10334–10342 CrossRef CAS PubMed; (d) M. Bielawski, M. Zhu and B. Olofsson, Adv. Synth. Catal., 2007, 349, 2610–2618 CrossRef CAS; (e) Y. Kita, K. Morimoto, M. Ito, C. Ogawa, A. Goto and T. Dohi, J. Am. Chem. Soc., 2009, 131, 1668–1669 CrossRef CAS PubMed; (f) M. A. Carroll and R. A. Wood, Tetrahedron, 2007, 63, 11349–11353 CrossRef CAS; (g) F. Tinnis, E. Stridfeldt, H. Lundberg, H. Adolfsson and B. Olofsson, Org. Lett., 2015, 17, 2688–2691 CrossRef CAS PubMed; (h) I. P. Beletskaya, D. V. Davydov and M. Moreno-Manas, Tetrahedron Lett., 1998, 39, 5621–5622 CrossRef CAS; (i) J. Yan, M. Zhu and Z. Zhou, Eur. J. Org. Chem., 2006, 2060–2062 CrossRef CAS.
- J. Li and L. Neuville, Org. Lett., 2013, 15, 6124–6127 CrossRef CAS PubMed.
- The reaction of phenyl cyanamide with di(p-tolyl)iodonium triflate in 1.25 mmol produced 223 mg phenyl(p-tolyl)cyanamide (3a, 86% yield) and 196 mg p-tolyliodide (72% yield).
- M. Ochiai, Y. Kitagawa and M. Toyonari, ARKIVOC, 2003, 43–48.
- K. M. Lancer and G. H. Wiegand, J. Org. Chem., 1976, 41, 3360–3364 CrossRef CAS.
- (a) B. A.-K. Wulfrath, S. S. Goch and E. K. Hattingen, US 2011/0263657, 2011; (b) C. P Decicco, J. L. Seng, K. E. Kennedy, M. B. Covington, P. K. Welch, E. C. Arner, R. L. Magolda and D. J. Nelson, Bioorg. Med. Chem. Lett., 1997, 7, 2331–2336 CrossRef; (c) J. A. W. Kruijtzer, D. J. Lefeber and R. M. J. Liskamp, Tetrahedron Lett., 1997, 38, 5335–5338 CrossRef CAS; (d) A. R. Katritzky, N. Kirichenko and B. V. Rogovoy, ARKIVOC, 2003(VIII), 8–14.
- (a) A. Retnakaran and J. E. Wright, Chitin and Phenzoylphenyl Ureas, 1987, 38, 205–282 Search PubMed; (b) L. Chen, Q. Wand, R. Huang, C. Mao, J. Shang and F. Bi, J. Agric. Food Chem., 2005, 53, 38–41 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures as well as spectral data of all synthesized compounds. See DOI: 10.1039/c5nj02153g |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |