
One-pot, efficient and green synthesis of acridinedione derivatives using highly monodisperse platinum nanoparticles supported with reduced graphene oxide†
Burak
Aday‡
a,
Yunus
Yıldız‡
b,
Ramazan
Ulus
a,
Sinan
Eris
b,
Fatih
Sen
*b and
Muharrem
Kaya
*b
aChemistry Department, Faculty of Arts and Science, Dumlupınar University, Evliya Çelebi Campus, 43100 Kütahya, Turkey
bSen Research Group, Biochemistry Department, Faculty of Arts and Science, Dumlupınar University, Evliya Çelebi Campus, 43100 Kütahya, Turkey. E-mail: fatih.sen@dpu.edu.tr; Fax: +90 274 265 20 56; Tel: +90 274 265 20 31 ext. 3702
First published on 17th November 2015
Abstract
An efficient, high yielding, quick method has been developed for the synthesis of acridinedione derivatives via a one-pot, multi-component condensation of dimedone (1), various aromatic aldehydes (2a–i), and various aromatic amines (3a,b) using highly monodisperse platinum nanoparticles supported with reduced graphene oxide (Pt NPs@rGO) as a recyclable heterogeneous catalyst. This method takes advantage of the fact that water/ethanol (a green solvent) is used in combination with Pt NPs@rGO nanoparticles as a catalyst which can be easily recovered and reused for further runs. This highly monodisperse catalyst has been used for the first time as a highly stable, exceptionally reusable, isolable, bottleable, long-lived and highly efficient catalyst for the synthesis of acridinedione derivatives from dimedone, aromatic aldehydes and various amines with great catalytic performance. The synthesized catalyst was characterized using transmission electron microscopy, high resolution electron microscopy, X-ray diffraction, Raman and X-ray photoelectron spectroscopy techniques. The results of these analyses show the formation of highly crystalline and colloidally stable highly monodisperse Pt NPs@rGO. This highly monodisperse catalyst is a most efficient catalyst which gives the high yield of products in a short time.
Introduction
Acridinediones are heterocyclic compounds containing nitrogen that are the core structure of a number of biologically interesting compounds because they contain a 1,4-dihydropyridine ring similar to nicotinamide adenine dinucleotide (NADH).1 Furthermore, acridinedione derivatives are known to be biological agents with anti-fungal,2 anti-tumor,3 anti-cancer,4 and anti-glaucoma5 properties. So far, a great number of acridinedione compounds have also been synthesized and used clinically as anti-microbial6 and anti-bacterial agents.7 There are many reported methods for the synthesis of acridinedione including multi-component condensation (MCR) of various aromatic aldehydes, cyclic diketones, and various aromatic amines using different catalysts such as Amberlyst-15,8 silica functionalised propyl sulfonic acid (SiO2–Pr-SO3H),9 dodecylbenzenesulfonic acid (DBSA),5a,10 cetyltrimethylammonium bromide,11 proline,12 and sulfuric acid (H2SO4).5c Because these homogeneous catalysts pose difficulties for reusability, recovery, separation and synthesis, the heterogeneous alternatives have already aroused much attention. Recently, the use of several heterogeneous catalytic systems such as iron(II,III) oxide (magnetite, Fe3O4),13 silica coated magnetite (Fe3O4@SiO2),14 12-tungstophosphoric acid supported on silica coated magnetite (Fe2O3@SiO2–PW),15 cadmium oxide (CdO),16 and zinc iron oxide (ZnFe2O4)17 have been reported because of their easy recovery and reusability. However, the problem in most of those catalytic systems is unsatisfactory yield, long reaction times, laborious work-up procedures, the requirement for special apparatus and harsh reaction conditions. To avoid these limitations and to improve the reaction conditions available for the synthesis of acridinedione, the discovery of new methodologies using new heterogeneous and reusable catalysts is still needed. So to improve the long-term durability and activity of catalysts, carbon nanotubes (CNTs) and graphene containing robust graphitic structures have been widely studied as supporting materials.18a–e Graphene oxide (GO) has abundant surface functional groups and defects, which are chemically active sites for use in catalytic reactions and also act as the anchoring sites for metal nanoparticles (NPs), making it a promising supporting material for catalysts for organic reactions. However, an excessive amount of oxygen-containing functional groups can reduce the electrical conductivity, activity and chemical stability of CNTs and graphene, which can make them weak to chemical oxidation and thus the long-term durability of the supported electrocatalyst deteriorates. All these reasons make the use of reduced graphene oxide (rGO) even more attractive in organic and engineering applications. In this paper an efficient, green and novel method is reported for the first time, for the synthesis of acridinedione derivatives via one-pot MCRs of cyclic diketones (1), various aromatic aldehyde (2a–i), and various aromatic amines (3a,b) with high yields and short reaction times using ethanol–water (EtOH–H2O) as a green solvent and highly monodisperse platinum (Pt) NPs@rGO as a highly efficient, robust, reusable and easily recoverable catalyst for the synthesis of acridinedione derivatives. Target molecules were characterized using Fourier-transform infrared (FT-IR) spectroscopy, proton nuclear magnetic resonance spectroscopy (1H-NMR) and carbon-13 (13C)-NMR data and high resolution mass spectral (HRMS) analyses.Results and discussion
The characterization of highly monodisperse Pt NPs@rGO
Pt NPs@rGO was easily and reproducibly prepared using the sonochemical double solvent reduction method at room temperature. The Pt(0) NPs which were formed initially, quickly agglomerated and precipitated out of the tetrahydrofuran (THF) solution without stabilizing and supporting materials such as 1-aminooctane and rGO, respectively. In the presence of 1-aminooctane and rGO, the Pt NPs@rGO has strong resistance to agglomeration over months. This indicates that the 1-aminooctane ligand and rGO are very strong stabilizing agents for the Pt NPs@rGO. The solid NPs can be isolated by evaporating the solvent under vacuum after washing the Pt NPs@rGO with dry EtOH to eliminate the excess ligand. Structural and chemical characterizations of Pt NPs@rGO were performed using transmission electron microscopy (TEM), high resolution TEM (HRTEM), Raman spectroscopy, X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS).TEM, HRTEM and the particle size histogram of the Pt NPs@rGO show the highly monodisperse morphology of the catalyst as shown in Fig. 1. The average particle size of Pt NPs@rGO is found to be 2.54 ± 0.27 nm and a uniform distribution of the catalyst particles with a very narrow range on rGO was detected. The TEM results also indicated that most of the particles were spherical and no agglomeration was observed in the prepared catalyst as shown in Fig. S1 [(ESI†)]. Fig. 1 also shows the atomic lattice fringes obtained using HRTEM for Pt NPs@rGO and it is found that Pt(111) plane spacing is 0.228 nm which is very close to the figure of 0.227 nm which is found in the literature.20
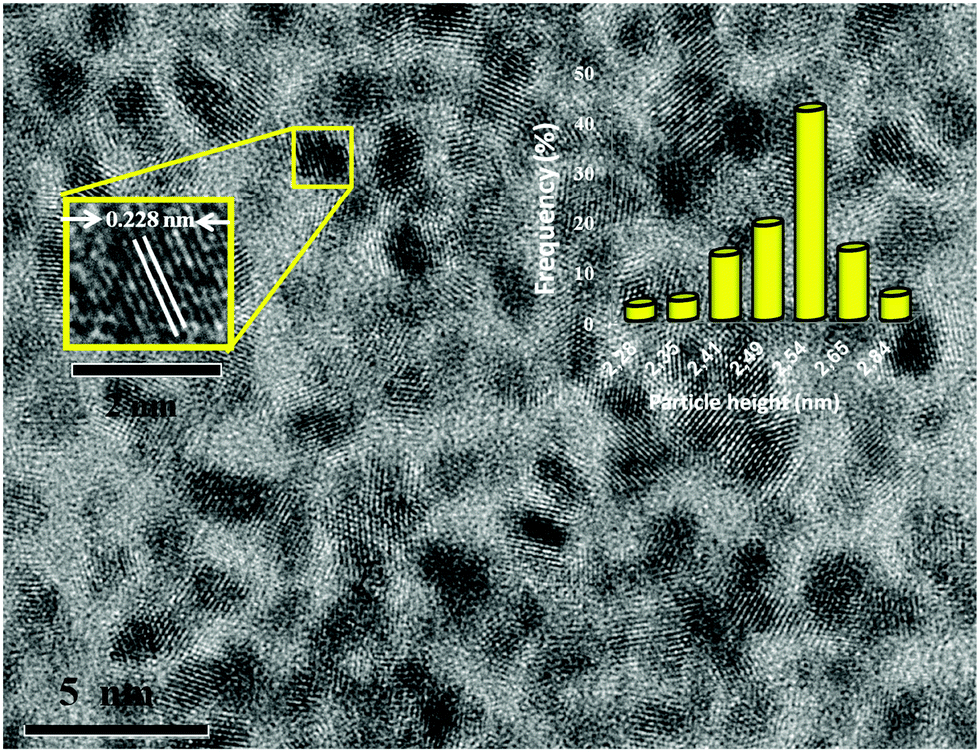 | ||
| Fig. 1 High resolution transition electron micrograph and particle size histogram of a Pt NPs@rGO. | ||
X-ray diffraction patterns of Pt NPs@rGO are illustrated in Fig. S2 (ESI†). XRD measurements indicate that Pt NPs@rGO have diffraction peaks of a face-centered cubic (fcc) crystal lattice at around 2θ = 25.4, 41.3, 49.1, 70.8, 81.4 and 85.4 which are as a result of C(002), Pt(111), (200), (220), (311), and (320), respectively, (JCPDS-ICDD, Card No. 04-802). The Pt(220) diffraction peaks of the prepared catalyst are used to calculate the lattice parameters (αPt) and average crystallite sizes of the metal particles. The lattice constant was found to be 3.932 Å, which is in good agreement with 3.923 Å for pure Pt, by using the following equation:21
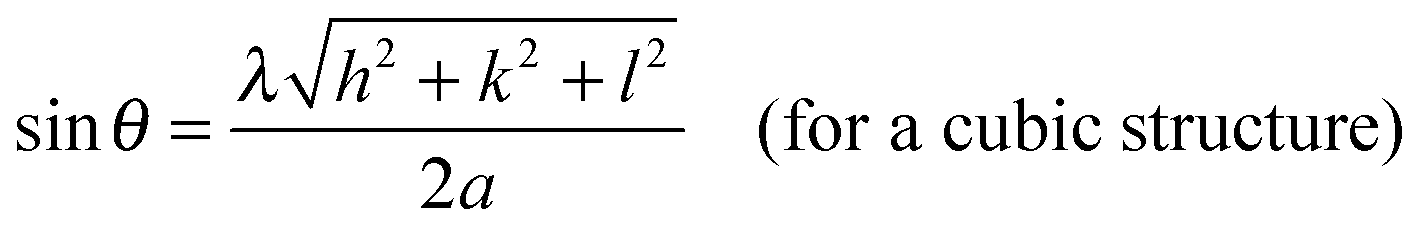
where k = a coefficient (0.9); λ = the wavelength of the X-rays used (1.54056 Å); β = the full width half-maximum of the respective diffraction peak (rad); and θ = the angle at the position of the peak maximum (rad).
XPS was used to designate the surface composition and the oxidation state of Pt in the highly monodisperse Pt NPs@rGO. For this purpose, the Pt 4f region of the spectrum was analyzed and fitted by using a Gaussian–Lorentzian method and the relative intensity of the species was calculated by integrating each peak. Accurate binding energies (±0.3 eV) were confirmed by comparing them to the energies of the C 1s peak at 284.6 eV in the XPS spectrum. The doublet consists of two double Pt 4f photoelectron spectrums for the prepared catalyst as shown in Fig. S3 (ESI†). The ratio of the 4f7/2/4f5/2 signals for Pt NPs was found to be 4/3, which agrees with the values found in the literature.23 The line at 71.0–74.1 eV is undoubtedly because of Pt(0), whereas the higher binding energy component, 74.3–77.6 eV, is most probably because of the unreduced Pt precursor or PtOx species such as PtO2 or Pt(OH)4. This result indicates that Pt is mostly in the zero oxidation state (85.6%) in our catalyst. As shown in Fig. S4 (ESI†), the C 1s XPS spectrum of the pristine GO exhibits three different peaks centered at 285.0, 287.0, and 288.6 eV, corresponding to the C–C (sp2), C–OH and HO–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups in GO, respectively. However, Pt NPs@rGO shows highly reduced functional groups over the pristine GO, indicating the co-reduction of Pt and GO during the Pt loading step.
O groups in GO, respectively. However, Pt NPs@rGO shows highly reduced functional groups over the pristine GO, indicating the co-reduction of Pt and GO during the Pt loading step.
Raman spectroscopy is also an effective method to distinguish the ordered and disordered structure of carbon in carbonaceous materials. The Raman spectra of rGO and Pt NPs@rGO are shown in Fig. S5 (ESI†). Two prominent scattering peaks located at 1350 and 1595 cm−1 can be assigned to the E2g phonon of the sp2 carbon atoms (D-band) and the breathing mode of κ-point phonons of A1g symmetry (G-band), respectively. The intensity ratio of the D to G band (ID/IG) could be used to evaluate the degree of modification or defects of rGO. In this work, the ID/IG values of rGO and Pt NPs@rGO are 1.12 and 1.34, respectively, which indicate that the Pt NPs have been functionalized with the rGO.
The synthesis of acridinedione derivatives in the presence of highly monodisperse Pt NPs@rGO
Pt NPs@rGO is often used as a catalyst in alcohol oxidation, batteries, hydrogen storage, sensors, and super-capacitors24–28 but its use as a catalyst for synthesizing acridinedione derivatives from different aromatic aldehydes and various amines has not been reported before. The synthesis of acridinedione compounds has been performed in EtOH–H2O (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) using dimedone (1), substitute aromatic aldehydes (2a–i) and various amines (3a,b) in a single process using MCR. These compounds were prepared using a one-pot reaction in an effective yield process, and with a simple work-up procedure using highly monodisperse Pt NPs@rGO. With the assistance of this catalyst, a new, easy, rapid, green, one-pot and efficient procedure has been designed for synthesizing acridinedione derivatives from different aromatic aldehydes and various amines in the presence of highly stable, exceptional reusable, isolable, bottleable, long lived and highly efficient monodisperse Pt NPs@rGO as a catalyst (Scheme 1).
:1) using dimedone (1), substitute aromatic aldehydes (2a–i) and various amines (3a,b) in a single process using MCR. These compounds were prepared using a one-pot reaction in an effective yield process, and with a simple work-up procedure using highly monodisperse Pt NPs@rGO. With the assistance of this catalyst, a new, easy, rapid, green, one-pot and efficient procedure has been designed for synthesizing acridinedione derivatives from different aromatic aldehydes and various amines in the presence of highly stable, exceptional reusable, isolable, bottleable, long lived and highly efficient monodisperse Pt NPs@rGO as a catalyst (Scheme 1).
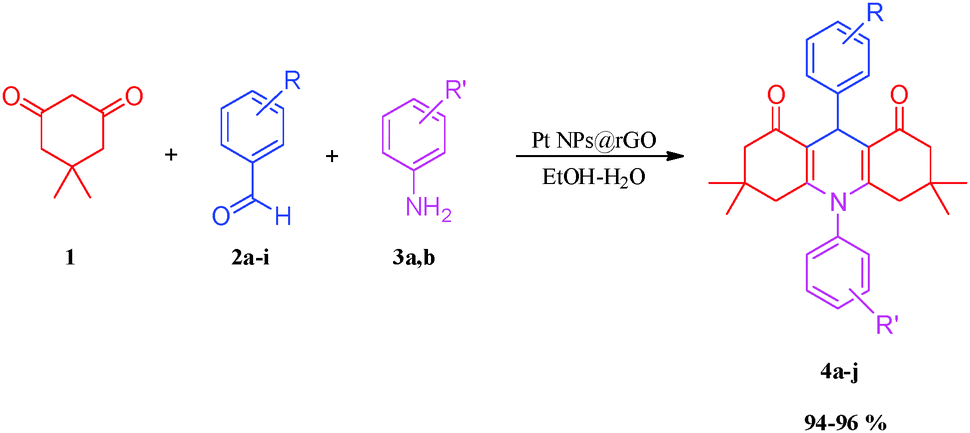 | ||
| Scheme 1 Synthesis of acridinedione derivatives in the presence of Pt NPs@rGO. | ||
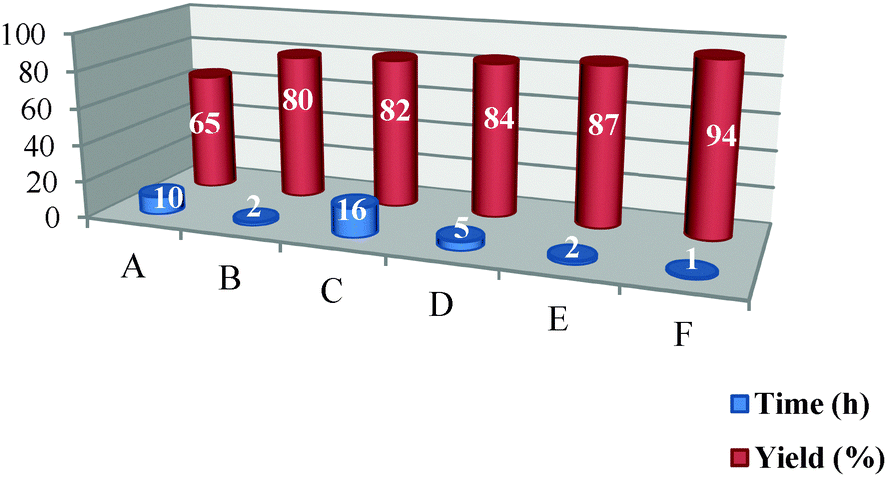
In this research, the results obtained using Pt NPs@rGO in the synthesis of target molecules were compared with those obtained using other catalysts such as Pt NPs (A), glacial acetic acid29 (B), DBSA (C), H2SO4 (D), hydrochloric acid (E), Pt NPs@rGO (F). As shown in Table 1, Pt NPs@rGO was the most efficient catalyst which gives the highest yield in model reactions.
|
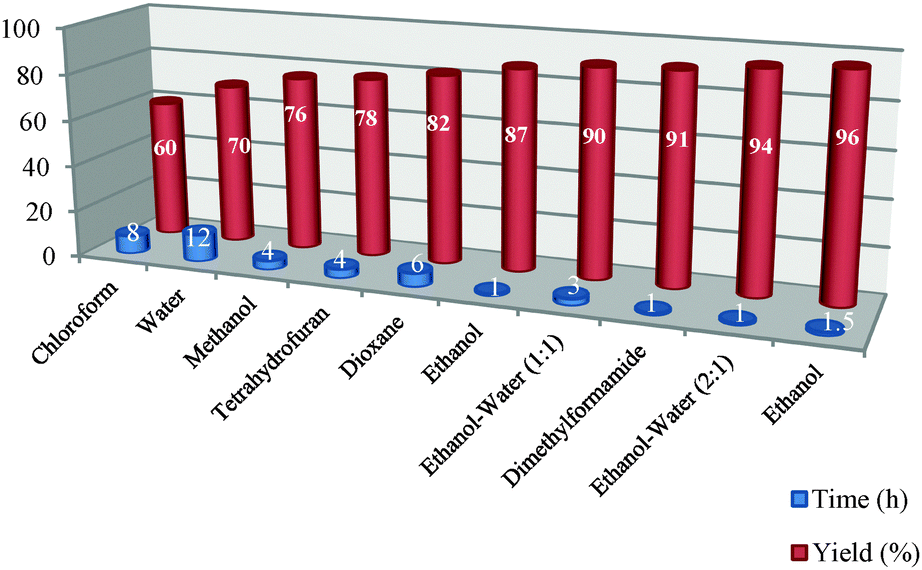
This model reaction has been standardized by using various solvents, amounts of catalyst and reaction conditions based on different temperatures. Firstly, the effect of the solvent has been investigated on the model reaction as shown in Table 2. The model reaction was performed in different solvents such as chloroform, water, methanol, THF, dioxane, EtOH, EtOH–H2O (1:1), dimethylformamide, EtOH–H2O (2:1) and EtOH under the same reaction conditions. Because the reaction with the highest yield and shortest reaction time was obtained using EtOH rather than the other solvents used, EtOH–H2O (2:1) was chosen as the optimum solvent for the model reaction, because this solvent’s conditions were more environmentally friendly than using EtOH alone. Therefore, it can be thought that EtOH–H2O (2:1) mixture is a green and superior solvent compared to the others shown in Table 2.
|
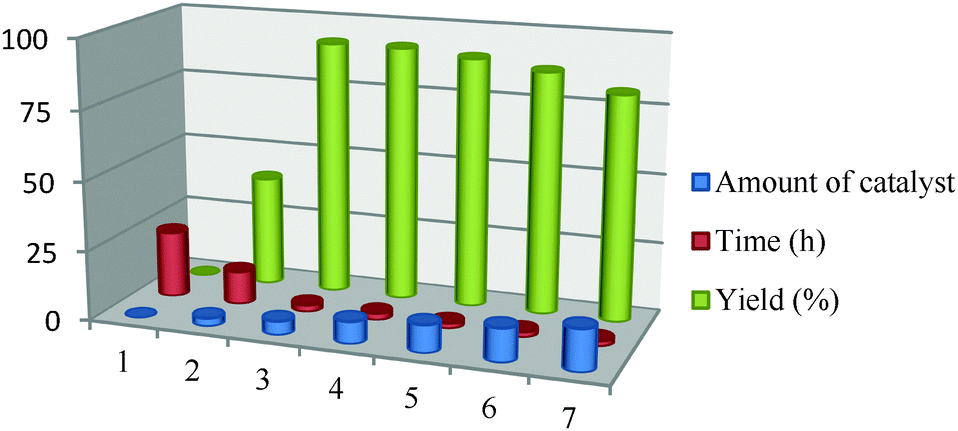
Because the amount of catalyst is another important parameter in terms of the reaction efficiency, seven sets of model reactions were carried out to determine the optimum catalyst concentration for the best possible yield. The first set of reactions where no catalyst was used led to no formation of product but as the amount of catalyst was increased gradually from 3 mg to 8 mg a steady increase was observed in the product yield (Table 3). Furthermore, an increase in the catalyst amount caused a decrease in the product yield which was possibly because of the protonation of the amine groups in the aromatic ring from the carboxylic acid groups of the catalyst.
:1) at 90 °C
|
|
|||||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
| Amount of catalyst | 0 | 3 | 5 | 8 | 12 | 15 | 20 |
| Time (h) | 24 | 12 | 2.5 | 1 | 2 | 2 | 2 |
| Yield (%) | 0 | 40 | 92 | 94 | 90 | 87 | 81 |
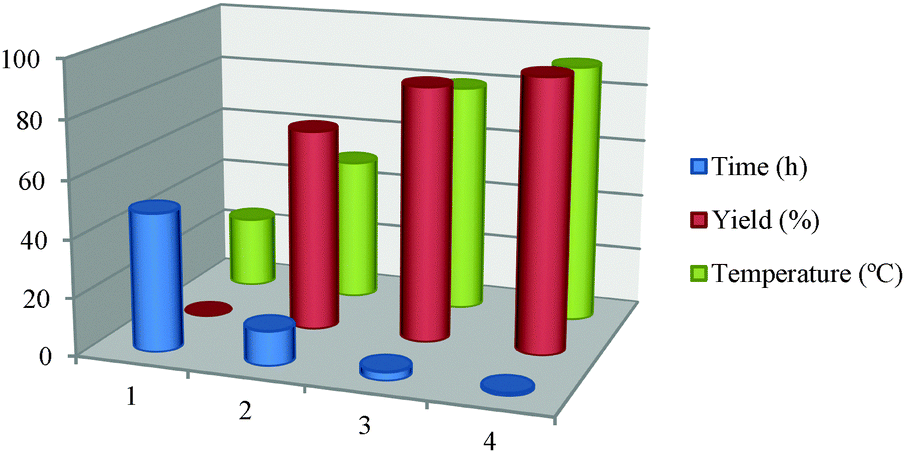
After standardizing the solvent and catalyst amount, the temperature effect on the model reaction was examined. It was observed that at room temperatures no desired product was formed even after 48 h of stirring. The yield of the desired product gradually increased with temperature from room temperature to 90 °C as shown in Table 4. The effect of the molar ratio between Pt and rGO was also determined and it was observed that increasing the ratio of Pt:rGO increases the yield of the model reaction as shown in Table S1 (ESI†).
:1) at 90 °C
|
|
||||
|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |
| Time (h) | 48 | 12 | 3 | 1 |
| Yield (%) | 0 | 70 | 88 | 94 |
| Temperature (°C) | 25 | 50 | 80 | 90 |
In summary, the highest efficiency and fastest reaction time for the model reaction was observed at 90 °C by using 8 mg of Pt NPs@rGO in EtOH–H2O. These various substituents subsumed cyano, nitro, halogen (–F, –Br), methyl, methoxy, and hydroxyl moieties. The results of all reactions performed under these conditions are shown in Table 5.
| Derivative number | R | R′ | Time (min) | Yieldb (%) | Melting point (mp, °C) | |
|---|---|---|---|---|---|---|
| Found | Reported | |||||
|
a Reactions were carried out with dimedone, aromatic aldehyde and various aromatic amines, in a 2:1:1 molar ratio.
b Yields refer to isolated pure products.
|
||||||
| 4a | –H | 4-Cl | 60 | 94 | 300–302 | 309–31130 |
| 4b | 4-NO2 | 4-Cl | 45 | 95 | 315–317 | >30031 |
| 4c | 3-NO2 | 4-Cl | 45 | 96 | 285–287 | 285–28632 |
| 4d | 4-Br | 4-Cl | 75 | 95 | 304–305 | >30031 |
| 4e | 4-F | 4-Cl | 45 | 94 | 280–282 | >30033 |
| 4f | 4-CH3 | 4-Cl | 60 | 94 | 262–265 | 268–26932 |
| 4g | 4-OCH3 | 4-Cl | 75 | 95 | 255–257 | 269–27034 |
| 4h | –H | 4-Br | 60 | 96 | 303–305 | 269–27235 |
| 4i | 4-CN | 4-Cl | 60 | 95 | 317–319 | — |
| 4j | 4-OH | 4-Cl | 30 | 94 | 352 | — |
All the products were characterized using melting point, FT-IR, NMR, and HRMS analyses (Fig. S6–S15, ESI†).
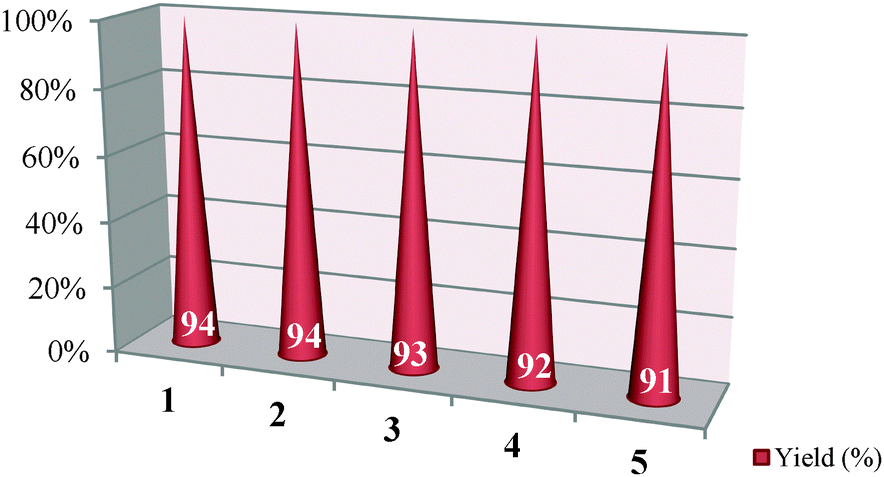
Finally, the reusability performances of the highly monodisperse Pt NPs@rGO were also investigated. For this purpose, the used catalyst was isolated from the reaction media, washed with acetone, dried and reused in another synthesis reaction. It was observed that the Pt NPs@rGO can be reused effectively for five times and gives satisfactory yields (Table 6). It should be pointed out that no extra care needs to be taken to store or handle the catalyst because it is not air or moisture sensitive. As far as is known, Pt NPs@rGO shows the best reusability performance found in the literature.13–18
|
A plausible mechanism for the synthesis acridinedione derivatives in the presence of Pt NPs@rGO is shown in Scheme 2. Firstly, rGO binds with the oxygen of the carbonyl group, and thus it increases the carbonyl activity which in turn makes the alpha hydrogen very acidic, thereby facilitating the enolization and nucleophilic attack to the aromatic aldehydes leading to the formation of a knoevenagel product. Then the knoevenagel product was reacted in a Michael reaction with another molecule of the C–H active molecule and produced an intermediate. Then the aniline derivatives induced cyclization with the help of Pt NPs@rGO and produced one of the highest yields for the desired products.
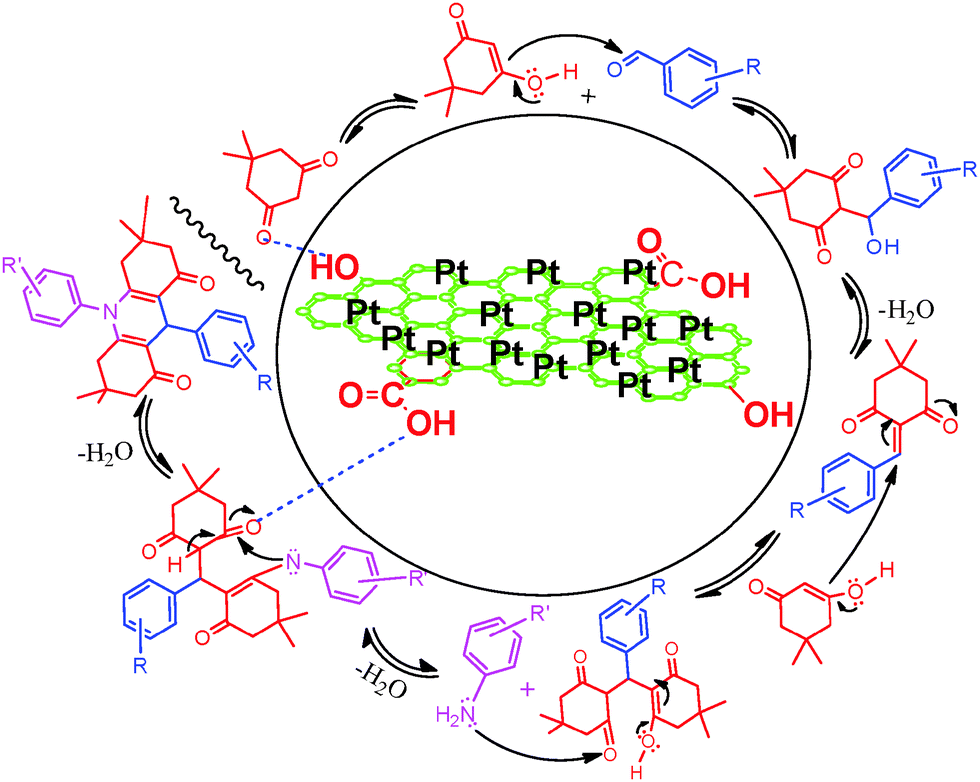 | ||
| Scheme 2 Recommended mechanism pathway for the Pt NPs supported with rGO catalyzed synthesis of acridinedione derivatives. | ||
Conclusions
An easy, effective and practical synthetic method has been developed for the synthesis of acridinedione cycloaddition reactions of the various amine, aromatic aldehyde compounds and dimedone in the presence of a highly stable, exceptionally reusable, isolable, bottleable, long-lived and highly efficient monodisperse rGO furnished with Pt NPs as the catalyst. This highly monodisperse catalyst is one of the most efficient catalysts which gives the highest yields of products in the shortest time and this is most likely because of the highly monodispersed, low crystallite particle size, high chemical surface area and high Pt(0) contents (%) of rGO furnished Pt NPs. Furthermore, the methodology described is efficient and environmentally benign and rGO furnished Pt NPs offer advantages such as a simple work-up and high yields are possible because the catalyst shows no air or moisture sensitivity.Experimental
Synthesis of graphene oxide
Firstly, GO was synthesized using the modified Hummers’ method.19 Typically, 1.0 g of graphite powder was mixed with 1.0 g of sodium nitrate (NaNO3) and 33 mL H2SO4 (98%) in an iced water bath. Next, 6.0 g of potassium permanganate (KMnO4) was slowly added to the previously obtained mixture at 35 °C for 1.5 h. Then, 40 mL of H2O was gradually added into the mixture, followed by maintaining the temperature at 95 °C for 35 min. Next, the mixture was diluted with 100 mL H2O, and then 6 mL of hydrogen peroxide (H2O2; 30%) was added to the mixture. The mixture was purified using centrifugation and then thoroughly washed with water.Hydrothermal reduction of GO with hydrazine hydrate
In a typical procedure, GO (100 mg) was loaded into a 250 mL round bottomed flask and H2O (100 mL) was then added, yielding an inhomogeneous yellow-brown dispersion. This dispersion was ultrasonically mixed using a Kudos KS3310HP ultrasonic bath cleaner (150 W) until it became clear with no visible particulate matter. Hydrazine monohydrate (1.00 mL) was then added and the solution heated in an oil bath at 100 °C under a water-cooled condenser for 24 h over which the reduced GO gradually precipitated out as a black solid. This product was isolated by filtration over a small fritted glass funnel, washed copiously with H2O and methanol, and dried on the funnel under a continuous air flow through the solid product cake.Synthesis of Pt NPs@rGO
Pt NPs@rGO was prepared using a sonochemical double solvent reduction method. Briefly, superhydride and EtOH were used to reduce the mixture of 0.25 mmol (0.081 g) of platinum(IV) chloride (PtCl4) dissolved in a small amount of anhydrous THF and 0.25 mmol of 1-aminooctane ligands for 1 h with ultrasonic mixing. The observation of a brown-black color in the solution indicated the formation of Pt NPs. The prepared Pt NPs were mixed in a 1:1 ratio with rGO using a tip sonicator. Finally, the solid Pt NPs@rGO NPs were dried under vacuum at room temperature.
General procedure for preparation of acridinedione derivatives (4a–j) using Pt NPs@rGO as the catalyst
A mixture of 2 mmol dimedone 1, 1 mmol 4-methyl benzaldehyde 2f, 1 mmol 4-chloroaniline 3a and Pt NPs@rGO (8 mg) in 9 mL EtOH–H2O solvent mixture (2:1) was continuously heated to 90 °C for 60 min. The reaction progress was monitored using thin layer chromatography. The solid product was filtered, washed with 500 mL of H2O, and then recrystallized from EtOH (94–96%).
O), 1605 s (CC); HRMS (quadrupole time-of-flight with electrospray ionisation; QTOF-ESI): m/z calculated for C30H29ClN2O2: 484.1918; found: 485.2006 [M + H]+.
O), 1589 s (CC); HRMS (QTOF-ESI): m/z calculated for C29H30ClNO3: 475.1914; found: 476.1997 [M + H]+.
Acknowledgements
This research was financed by the Dumlupinar University Research Fund (Grant No. 2014-05 and 2015-35). The partial support from the Science Academy and Fevzi Akkaya Research Funding (FABED) are gratefully acknowledged.Notes and references
- S. Singh, S. Chhina, V. K. Sharma and S. S. Sachev, J. Chem. Soc., Chem. Commun., 1982, 8, 453–454 RSC.
- A. Srivastava and C. Nizamuddin, Indian J. Heterocycl. Chem., 2004, 13, 261–264 CAS.
- Y. Mikata, M. Yokoyama, K. Mogami, M. Kato, I. Okura, M. Chikira and S. Yano, Inorg. Chim. Acta, 1998, 279, 51–57 CrossRef CAS.
- (a) S. A. Gamage, J. A. Spicer, G. J. Atwell, G. J. Finlay, B. C. Baguley and W. A. Denny, J. Med. Chem., 1999, 42, 2383–2393 CrossRef CAS PubMed; (b) M. G. R. Pitta, E. S. Souza, F. W. A. Barros, M. O. M. Filho, O. Pessoa, M. Z. Hernandes, M. D. C. A. D. Lima, S. L. Galdino and I. D. R. Pitta, Med. Chem. Res., 2013, 22, 2421–2429 CrossRef CAS; (c) K. Venkatesan, S. S. Pujari and K. V. Srinivasan, Synth. Commun., 2008, 39, 228–241 CrossRef.
- (a) M. Kaya, E. Basar, E. Çakir, E. Tunca and M. Bulbul, J. Enzyme Inhib. Med. Chem., 2012, 27, 509–514 CrossRef CAS PubMed; (b) R. Ulus, İ. Yeşildağ, M. Tanc, M. Bülbül, M. Kaya and C. T. Supuran, Bioorg. Med. Chem., 2013, 21, 5799–5805 CrossRef CAS PubMed; (c) İ. Yeşildağ, R. Ulus, E. Basar, M. Aslan, M. Kaya and M. Bülbül, Monatsh. Chem., 2014, 145, 1027–1034 CrossRef.
- (a) L. Ngadi, A. M. Galy, J. P. Galy, J. Barbe, A. Cremieux and D. Sharples, Eur. J. Med. Chem., 1990, 25, 67–70 CrossRef CAS; (b) M. Wainwright, J. Antimicrob. Chemother., 2001, 47, 1–13 CrossRef CAS PubMed.
- K. Palani, D. Thirumalai, P. Ambalavanan, M. N. Ponnuswamy and V. T. Ramakrishnan, J. Chem. Crystallogr., 2005, 35, 751–760 CrossRef CAS.
- (a) B. Das, P. Thirupathi, I. Mahender, V. S. Reddy and Y. K. Rao, J. Mol. Catal. A: Chem., 2006, 247, 233–239 CrossRef CAS; (b) M. Kaya, Y. Yıldırır and G. Y. Çelik, Med. Chem. Res., 2011, 20, 293–299 CrossRef CAS.
- G. M. Ziarani, A. Badiei, M. Hassanzadeh and S. Mousavi, Arab J. Chem., 2014, 7, 335–339 CrossRef CAS.
- T. S. Jin, J. S. Zhang, T. T. Guo, A. Q. Wang and T. S. Li, Synlett, 2004, 1425–1427 Search PubMed.
- J. J. Xia and K. H. Zhang, Molecules, 2012, 17, 5339–5345 CrossRef CAS PubMed.
- K. Venkatesan, S. S. Pujari and K. V. Srinivasan, Synth. Commun., 2009, 39, 228–241 CrossRef CAS.
- A. G. Mohammad, S. G. Javad and M. Halimeh, C. R. Chim., 2012, 15, 969–974 CrossRef.
- D. Binoyargha, N. Sibaji and K. P. Amarta, Tetrahedron Lett., 2014, 55, 5236–5240 CrossRef.
- R. Ezzat, E. Sara and K. Maryam, Chin. J. Catal., 2013, 34, 1513–1518 CrossRef.
- V. B. Ashok, U. K. Bhagwat and G. G. Anil, Res. Chem. Intermed., 2015, 41, 1447–1458 CrossRef.
- D. Pamarida, D. Arghya, B. Asim and M. Chhanda, Green Chem., 2014, 16, 1426–1435 RSC.
- (a) H. Pamuk, B. Aday, F. Sen and M. Kaya, RSC Adv., 2015, 5, 49295–49300 RSC; (b) M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132–145 CrossRef CAS PubMed; (c) A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed; (d) S. Rana, S. Maddila and S. B. Jonnalagadda, Catal. Sci. Technol., 2015, 5, 3235–3241 RSC; (e) A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS PubMed; (f) Y. Lia, W. Gaob, L. Cib, C. Wanga and P. M. Ajayan, Carbon, 2010, 48, 1124–1130 CrossRef; (g) D. Wang, W. Niu, M. Tan, M. Wu, X. Zheng, Y. Li and N. Tsubaki, ChemSusChem, 2014, 7, 1398–1406 CrossRef CAS PubMed; (h) Z. Xing, J. Tian, Q. Liu, A. M. Asiri, P. Jiang and X. Sun, Nanoscale, 2014, 6, 11659–11663 RSC; (i) Y. Zhou, Q. Bao, L. A. L. Tang, Y. Zhong and K. P. Loh, Chem. Mater., 2009, 21, 2950–2956 CrossRef CAS; (j) J. Tian, S. Liu, Y. Zhang, H. Li, L. Wang, Y. Luo, A. M. Asiri, A. O. Al-Youbi and X. Sun, Inorg. Chem., 2012, 51, 4742–4746 CrossRef CAS PubMed; (k) J. Shen, M. Shi, B. Yan, H. Ma, N. Li and M. Ye, J. Mater. Chem., 2011, 21, 7795–7801 RSC.
- S. H. William and E. O. Richard, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- (a) T. Yonezawa, N. Toshima, C. Wakai, M. Nakahara, M. Nishinaka, T. Tominaga and H. Nomura, Colloids. Surf., A, 2000, 169, 35–45 CrossRef CAS; (b) F. Sen, S. Sen and G. Gökağaç, Phys. Chem. Chem. Phys., 2011, 13, 1676 RSC; (c) F. Sen and G. Gökağaç, J. Appl. Electrochem., 2014, 44, 199 CrossRef CAS; (d) S. Sen, F. Sen and G. Gökağaç, Phys. Chem. Chem. Phys., 2011, 13, 6784 RSC; (e) F. Sen and G. Gökağaç, J. Phys. Chem. C, 2007, 111, 1467 CrossRef CAS.
- (a) Z. Liu, X. Y. Ling, X. Su and J. Y. Lee, J. Phys. Chem. B, 2004, 108, 8234–8240 CrossRef CAS; (b) F. Şen and G. Gökağaç, Energy Fuels, 2008, 22, 1858–1864 CrossRef; (c) Z. Ozturk, F. Sen, S. Sen and G. Gokagac, J. Mater. Sci., 2012, 47, 8134–8144 CrossRef CAS; (d) E. Erken, H. Pamuk, Ö. Karatepe, G. Başkaya, H. Sert, O. M. Kalfa and F. Şen, J. Cluster Sci., 2015 DOI:10.1007/s10876-015-0892-8.
- (a) H. Klug and L. Alexander, X-Ray Diffraction Procedures: For Polycrystalline and Amorphous Materials, Wiley, New York, 2nd edn, 1954 Search PubMed; (b) B. Celik, E. Erken, S. Eris, Y. Yıldız, B. Sahin, H. Pamuk and F. Sen, Catal. Sci. Technol., 2015 10.1039/c5cy01371b.
- (a) F. Sen and G. Gökagac, J. Phys. Chem. C, 2007, 111, 5715–5720 CrossRef CAS; (b) P. T. A. Sumodjo, E. J. Silva and T. Rabochai, J. Electroanal. Chem., 1989, 271, 305 CrossRef CAS; (c) S. Ertan, F. Sen, S. Sen and G. Gokagac, J. Nanopart. Res., 2012, 14, 922–926 CrossRef; (d) F. Sen, G. Gokagac and S. Sen, J. Nanopart. Res., 2013, 15, 1979 CrossRef; (e) F. Sen, Y. Karatas, M. Gulcan and M. Zahmakiran, RSC Adv., 2014, 4, 1526–1531 RSC; (f) T. C. Deivaraj, W. X. Chen and J. Y. Lee, J. Mater. Chem., 2003, 13, 2555 RSC.
- Y. Li, W. Gao, L. Ci, C. Wang and P. M. Ajayan, Carbon, 2010, 48, 1124–1130 CrossRef CAS.
- E. Yoo, T. Okata, T. Akita, K. Kohyama, J. Nakamura and I. Honma, Nano Lett., 2009, 9, 2255–2259 CrossRef CAS PubMed.
- C. Xu, X. Wang and J. Zhu, J. Phys. Chem. C, 2008, 112, 19841–19845 CAS.
- R. Kou, Y. Shao, D. Wang, M. H. Engelhard, J. H. Kwak, J. Wang, V. V. Viswanathan, C. Wang, Y. Lin, Y. Wang, I. A. Aksay and J. Liu, Electrochem. Commun., 2009, 11, 954–957 CrossRef CAS.
- S. Hrapovic, Y. Liu and K. B. Erkek, Anal. Chem., 2004, 76, 1083–1088 CrossRef CAS PubMed.
- I. O. El-Sabbagh, A. Mohamed, H. Hanan and E. S. Al-Din, Arch. Pharm., 2010, 343, 519–527 CrossRef PubMed.
- R. Rezaei, R. Khalifeh, M. Rajabzadeh, L. Dorosty and M. M. Doroodmand, Heterocycl. Chem., 2013, 19, 57–63 CAS.
- Z. Q. Tang, Y. Chen, C. C. Liu, K. Y. Cai and S. J. Tu, J. Heterocycl. Chem., 2010, 47, 363–367 CAS.
- O. I. El-Sabbagh, M. A. Shabaan, H. H. Kadry and E. S. Al-Din, Arch. Pharm., 2010, 9, 519–527 CrossRef PubMed.
- L. L. Zhao and D. Teng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, 1772–1773 Search PubMed.
- Y. Chen, W. J. Hao, Z. Q. Tang, B. Jiang and C. M. Li, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, 3934–3940 Search PubMed.
- M. Kidwai and D. Bhatnagar, Tetrahedron Lett., 2010, 50, 2700–2703 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nj02098k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |