
Biosourced 1,2,3-triazolium ionic liquids derived from isosorbide†
Malek
M'sahel
abc,
Mona Marie
Obadia
a,
Raouf
Medimagh
*b,
Anatoli
Serghei
a,
Mongia
Said Zina
c and
Eric
Drockenmuller
*a
aLaboratoire d'Ingénierie des Matériaux Polymères (UMR CNRS 5223), Université Claude Bernard Lyon 1, 15 Boulevard Latarjet, 69622 Villeurbanne Cedex, France. E-mail: eric.drockenmuller@univ-lyon1.fr
bLaboratoire des Substances Naturelles (LR10INRAP02), Institut National de Recherche et d'Analyse Physico-chimique, Sidi Thabet Biotechpole, 2020 Ariana, Tunisia. E-mail: raouf.medimagh@inrap.rnrt.tn
cFaculté des Sciences de Tunis El Manar, Université de Tunis, El Manar, 2092 Tunis, Tunisia
First published on 16th November 2015
Abstract
Two series of 1,2,3-triazolium chiral ionic liquid (CIL) stereoisomers are synthesized from isosorbide by combining the robust attributes of the copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC), alkylation of the resulting 1,2,3-triazoles by methyl iodide and further anion metathesis with different fluorinated salts. These novel biosourced CILs are characterized in details by 1H and 13C NMR spectroscopy, high-resolution mass spectrometry (ESI-HRMS), thermogravimetric analysis (TGA), differential scanning calorimetry (DSC) and broadband dielectric spectroscopy (BDS). The effect of stereochemistry on their physical, thermal and ion conducting properties is discussed. Markedly, it is demonstrated for the first time that stereochemistry can drastically influence ionic conductivity by several orders of magnitude.
Introduction
Ionic liquids (ILs), i.e. organic salts with melting temperature (Tm) below 100 °C, exhibit valuable physical properties such as low vapour pressure, enhanced chemical, electrochemical or thermal stabilities as well as recyclability which make them a good alternative to conventional volatile organic solvents.1–3 They have been widely applied in the chemical industry,4,5 catalysis,6–9 extraction phase,10 biomass conversion,11 anion receptors,12 synthesis and stabilization of nanostructured materials,13–15 energy storage,16 or polymerization media.17–19 Chiral ionic liquids (CILs), i.e. ILs having either a chiral cation and/or anion, have been applied in chiral recognition and asymmetric synthesis.20–22 The design of novel CILs using the broad palette of available chiral ions, particularly from renewable resources, is an emerging field of research.23,24Isosorbide (1), the most studied and readily available stereoisomer of 1,4:3,6-dianhydrohexitols (DAHs), is an appealing biosourced building block due to its rigid structure, difunctional nature and asymmetric reactivity.25,26 DAHs and their functionalized derivatives are versatile renewable monomers for the synthesis of polymers by step27–29 and chain30,31 growth polymerization techniques. Several CILs have been obtained by chemical modification of DAHs (Scheme 1). These include ammonium CILs synthesized from isosorbide32 (2–5) or isomannide33 (6, 7). Besides, imidazolium CILs were obtained from isosorbide (8)34 and isomannide (9, 10).35 Although the influence of the DAH stereochemistry on physical properties of the resulting CILs has been pointed out,32,33 it has a rather limited impact on the isolated yields and enantiomeric excess in the aza-Diels Alder reaction of Danishefsky's diene with various imines.32,34 1,3,4-Trisubstituted-1,2,3-triazolium ILs (TILs) have recently gained an increasing interest due to their modular synthesis through copper-catalyzed azide–alkyne cycloaddition (CuAAC), efficient alkylation of 1,2,3-triazoles and anion metathesis.36–38 TILs have been previously applied as chiral organocatalysts,39–41 gas separation,42 and counter cations in anionic polyionic liquids (PILs).43 Besides, a new class of PILs having 1,2,3-triazolium groups in their repeating unit has been recently developed by merging different polymerization methods with CuAAC, 1,2,3-triazoles alkylation and anion metathesis reactions.44–46 These materials are an extremely promising recent addition to the broad palette of ILs and PILs.47–49
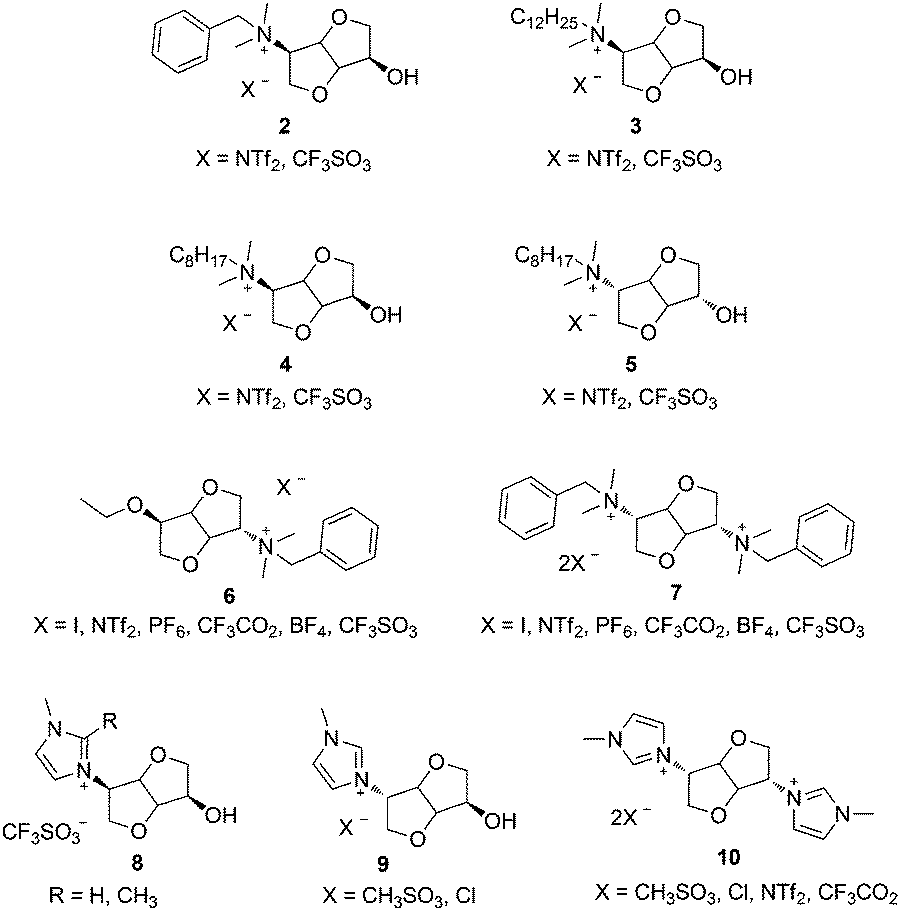 | ||
| Scheme 1 Chiral ionic liquids derived from 1,4:3,6-dianhydrohexitols. | ||
We present herein the synthesis and the characterization of two series of biosourced CILs issued from selective chemical modifications of either the endo or the exo hydroxyl groups of 1 using an original synthetic approach not requiring any protection/deprotection steps. The influences of the DAH stereochemistry and the chemical nature of the counter anion are discussed based on the measurements of physical and ion conducting properties.
Results and discussion
Synthesis and characterization
Biosourced TILs 17–24 were synthesized in 4–5 steps starting from 1 (Scheme 2). Initially, pure mono-tosyl isosorbide stereoisomers 11 and 12 were obtained by reacting 1 with tosyl chloride (TsCl) in pyridine and further separation by column chromatography to eliminate the di-tosyl isosorbide, mostly precipitated in cold ethanol. Azide-functionalized stereoisomers 13 and 14 were then obtained almost quantitatively by treating mono-tosylates 11 and 12 with NaN3.50,51 Consequently to the SN2 mechanism of the azidation reaction, a configuration inversion occurs at the C-b or C-e carbon atoms. Therefore, azides 13 and 14 as well as each of their derivatives exhibit isoidide and isomannide configurations, respectively. Further CuAAC between 13 and 14 with pent-1-yne afforded after purification by column chromatography pure 1,2,3-triazoles 15 and 16 in 64% and 48% yields, respectively. The obtained 1,2,3-triazoles are readily soluble in both protic and aprotic polar solvents such as water, methanol, acetone, dimethylformamide (DMF), dimethylsulfoxide (DMSO), chloroform (CHCl3), dichloromethane (CH2Cl2) and tetrahydrofuran (THF), but insoluble in hexane, diethyl ether (Et2O), ethyl acetate (EtOAc) and toluene (Table S1 in the ESI†). 1H and 13C NMR spectroscopy (Fig. 1 and 2) and electrospray ionization high-resolution mass spectrometry (ESI-HRMS, Table S2 in the ESI†) demonstrate the high purity of 1,2,3-triazole stereoisomers 15 and 16. Chemical shifts, multiplicity, and splitting constants of the proton signals of the DAH moiety are significantly impacted by the difference in stereochemistry between 15 and 16. However, an identical single peak is obtained in the ESI-HRMS spectra, confirming that pure stereoisomers are obtained.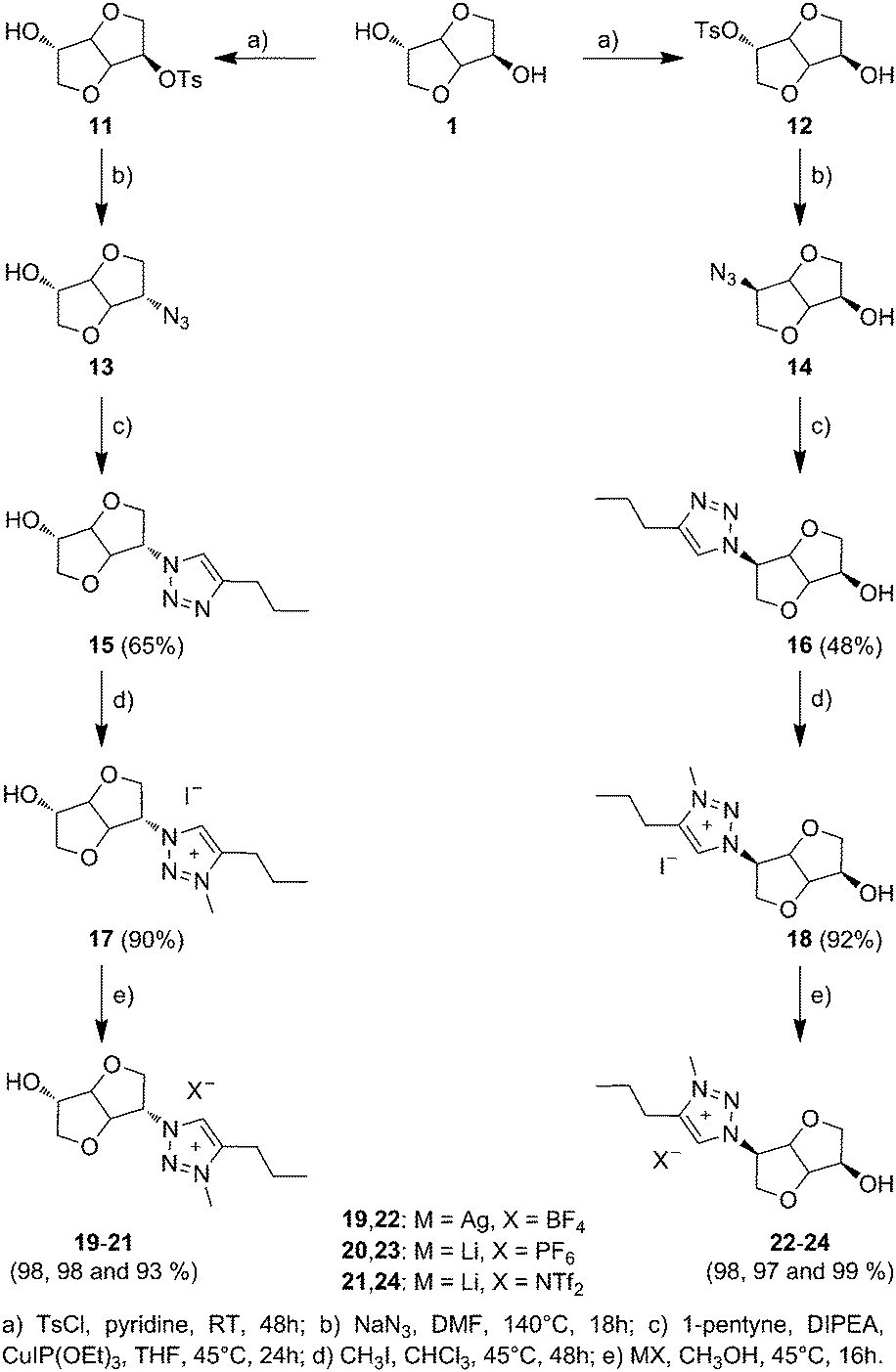 | ||
| Scheme 2 Synthesis of biosourced 1,2,3-triazolium chiral ionic liquids derived from isosorbide. | ||
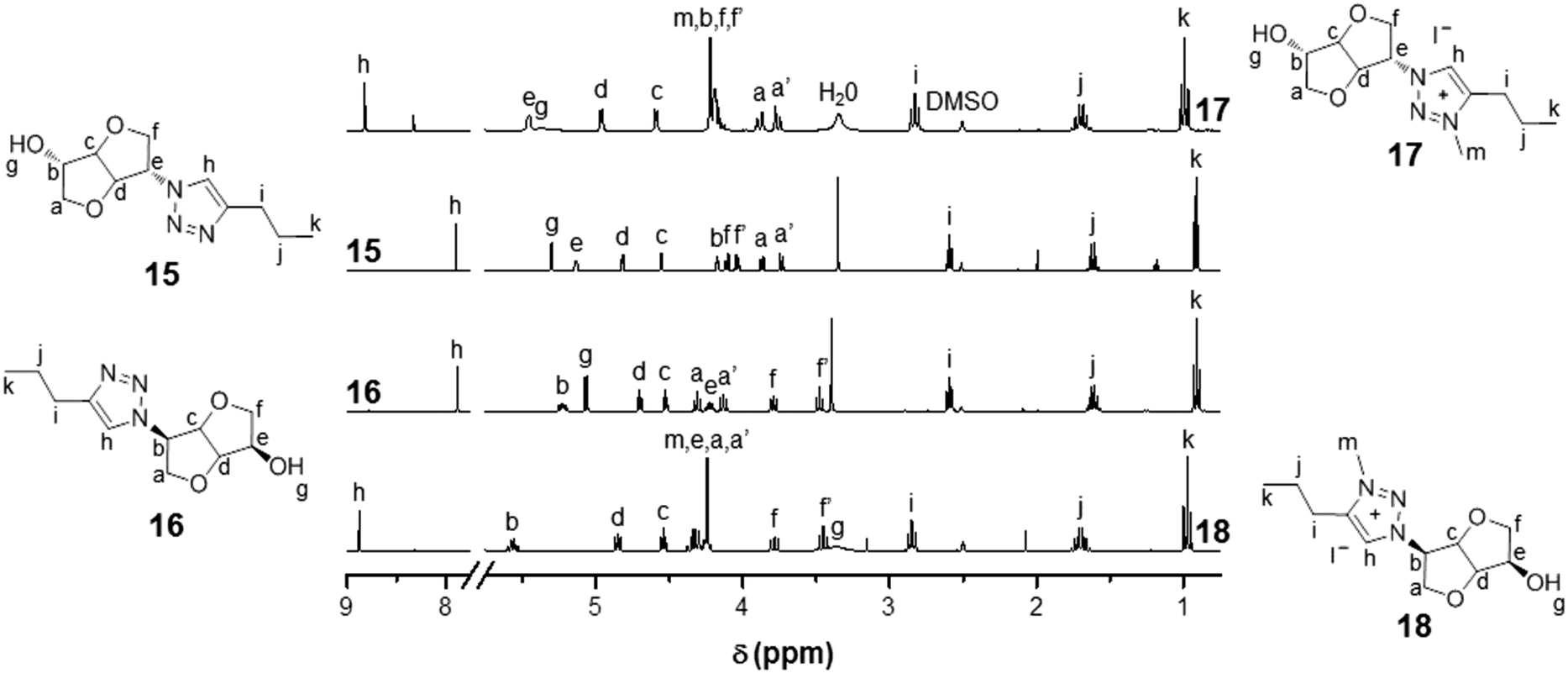 | ||
| Fig. 1 1H NMR (DMSO-d6, 25 °C) of biosourced 1,2,3-triazoles 15 and 16 and the corresponding 3-methyl-1,2,3-triazolium iodides 17 and 18. | ||
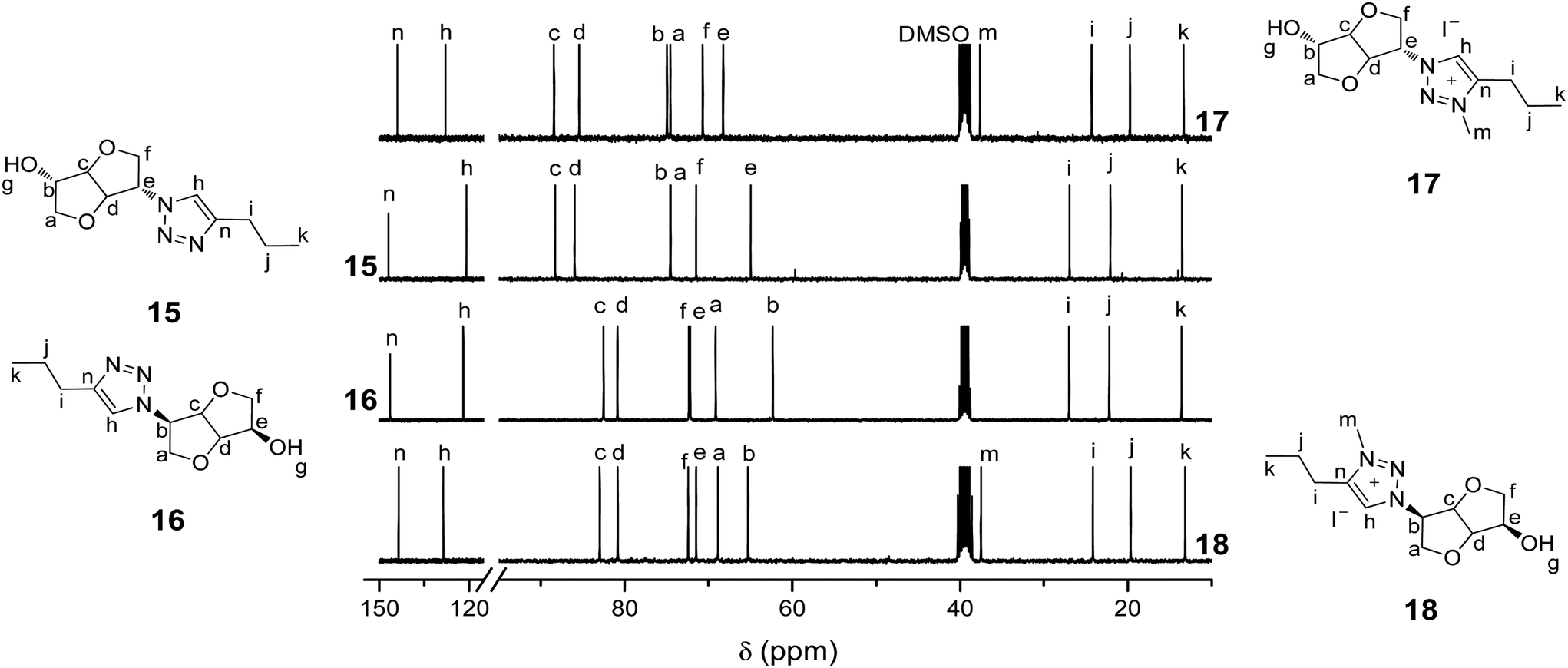 | ||
| Fig. 2 13C NMR (DMSO-d6, 25 °C) of biosourced 1,2,3-triazoles 15 and 16 and the corresponding 3-methyl-1,2,3-triazolium iodides 17 and 18. | ||
Further alkylation of 1,2,3-triazoles 15 and 16 by iodomethane (CH3I) in CHCl3 afforded the corresponding 3-methyl-1,2,3-triazolium iodides 17 and 18 as glassy orange materials in 90% and 92% yields, respectively. 1H NMR of CILs 17 and 18 confirms the quantitative alkylation of the 1,2,3-triazole groups with the exclusive appearance of a single peak for 1,2,3-triazolium protons at respectively 8.81 and 8.88 ppm as well as the appearance of the signals of methyl protons at 4.20 and 4.24 ppm with an expected 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 integration ratio. The absence of 1,2,3-triazole signals at 7.89 and 7.87 ppm corroborates the quantitative alkylation of the 1,2,3-triazole groups. Besides, most of the signals adjacent to the 1,2,3-triazolium group are shifted after alkylation due to the difference in electron density between 1,2,3-triazole and 1,2,3-triazolium groups.
:3 integration ratio. The absence of 1,2,3-triazole signals at 7.89 and 7.87 ppm corroborates the quantitative alkylation of the 1,2,3-triazole groups. Besides, most of the signals adjacent to the 1,2,3-triazolium group are shifted after alkylation due to the difference in electron density between 1,2,3-triazole and 1,2,3-triazolium groups.
13C NMR of 17 and 18 (Fig. 2) also corroborate the quantitative alkylation since signals corresponding to 1,2,3-triazolium carbons at 144.10/143.16 and 128.00/128.17 ppm and the methyl group at 37.69/37.08 ppm are observed together with the total disappearance of the 1,2,3-triazole carbon signals initially located at 146.96/146.44 and 120.97/122.03 ppm. Most other carbon signals are shifted after alkylation of the 1,2,3-triazole groups. As for 15 and 16, ESI-HRMS revealed the presence of a single peak for 17 and 18, indicative of the recovery of pure CIL stereoisomers. The alkylation of the 1,2,3-triazole groups has little effect on the solubility of CILs 17 and 18. Indeed, the only differences with their neutral precursors 15 and 16 are that 17 and 18 are insoluble in EtOAc and partially soluble in THF (Table S1 in the ESI†).
3-Methyl-1,2,3-triazolium iodides 17 and 18 were then subjected to anion metathesis with silver tetrafluoroborate (AgBF4), lithium hexafluorophosphate (LiPF6) and lithium bis(trifluoromethylsulfonyl)imide (LiNTf2, Scheme 2). To ensure the absence of any salt residuals, all CILs were dissolved in acetonitrile, centrifuged and filtrated through a thin layer of neutral alumina. This general procedure allows obtaining two series of CILs having isoidide (19–21) or isomannide (22–24) configurations with yields above 90%.
The high purity of CILs 19–24 is confirmed by 1H and13C NMR spectroscopy (Fig. S1–S4 in the ESI†) as well as ESI-HRMS (Table S2 in the ESI†). For each series, i.e. having isoidide or isomannide configuration, the change in the counter anion has little to no effect on the chemical displacement of almost every proton and carbon signals. However, different chemical displacements are observed for the two stereoisomeric series: e.g. while 1,2,3-triazolium and methyl signals of 21 appear at respectively 8.77 and 4.18 ppm, those of 24 are detected at 8.86 and 4.23 ppm. 13C NMR confirms the fidelity of the anion metathesis reaction as well as the effect of stereochemistry on the chemical displacements of CILs. For the isoidide series, 1,2,3-triazolium carbon signals exhibit a minor shift going from 144.10/128.00 ppm for 17 to 144.07/127.95, 144.09/128.00 and 144.22/128.07 ppm for 19, 20 and 21, respectively. This difference is however more pronounced for the isomannide series, i.e. 1,2,3-triazolium carbon signals are located at 143.16/128.17 ppm for 18 and 143.75/128.75 ppm for 24. The most significant changes though are associated to the stereochemistry of the DAH unit. A combination of the positive and negative modes of ESI-HRMS demonstrates the high purity of CIL stereoisomers and confirms the chemical structures of both the cations and counter anions. It is worth mentioning that anion metathesis reactions do not induce any change in the solubility of CILs 19–24 compared to their iodide analogues 17 and 18 (Table S1 in the ESI†).
Thermal properties
The thermal properties of CILs 17–24 were investigated by DSC and TGA (Table 1). Alkylation of 1,2,3-triazole groups changes the physical state of 1,2,3-triazolium iodides compared to their neutral 1,2,3-triazole precursors. Indeed, 15 is a crystalline solid (Tg = −12 °C and Tm = 85 °C), whereas 17 is an orange viscous liquid (Tg = −8 °C) while Tg of 16 (−28 °C) is 7 °C lower than Tg of 18 (−21 °C). Besides, while Tg values of CILs having an isoidide configuration increase in the following order: 21 (−58 °C, NTf2−) < 20 (−37 °C, PF6−) < and 19 (−27 °C, BF4−), Tg values of CILs having an isomannide configuration are lower and increase in the following order: 22 (−68 °C, BF4−) < 24 (−60 °C, NTf2−) < and 23 (−51 °C, PF6−). The differences between stereoisomers having isoidide (17, 19–20) or isomannide (18, 22–23) configuration and I−, BF4− and PF6− counter anions could somehow be explained by the presence of strong intramolecular hydrogen bonding between the 1,2,3-triazolium proton and the C3–O–C6 ether group (Scheme S1 in ESI†). The absence of intermolecular hydrogen bonding for the isomannide series induces lower Tg values. Conversely, there is almost no difference between the values of stereoisomers 21 and 24 (Tg = −58 and −60 °C, respectively). In this case, the bulky and weakly coordinating characters of NTf2− which usually confers low Tg and high conductivity materials exceed the impact of the DAH stereochemistry.Thermal stabilities under inert atmosphere are closely structure related (Table 1 and Fig. 3). Temperatures at 10% weight loss (Td10) are influenced by the DAH stereochemistry and increase in the following orders: I− < BF4− ∼ PF6− < NTf2− for the isoidide series and I− ∼ BF4− ∼ PF6− < NTf2− for the isomannide series. Higher Td10 values are observed for the isoidide series and in both series the highest values were obtained for the NTf2− counter anions (Td10 = 269 and 245 °C for CILs 21 and 24, respectively). Unexpectedly, 1,2,3-triazolium CILs are more thermally stable than their neutral 1,2,3-triazole precursors and the increase in Td10 is more significant for the isoidide series, i.e. Td10 = 237 and 269 °C for 15 and 21, respectively.
 | ||
| Fig. 3 Thermogravimetric analysis of biosourced 1,2,3-triazoles 15 and 16 and 1,2,3-triazoliums 17–24. | ||
Ionic conductivity
NTf2− containing CILs 21 and 24 were then studied by broadband dielectric spectroscopy (BDS) in order to pinpoint the impact of stereochemistry on the temperature dependence of ionic conductivity (σDC) under anhydrous conditions. Only NTf2− containing CILs were studied by BDS because (1) they generally provide higher σDC than other anions, and (2) since both stereoisomers have almost identical Tg values, any difference would be solely due to the influence of stereochemistry and not molecular relaxation. Coughlin et al. reported σDC of different TILs with tosylate or triflate counter anions which ranges between 1 × 10−6 and 1 × 10−3 S cm−1.52 However, to the best of our knowledge, the impact of stereochemistry on σDC of ILs has not been addressed so far. 200 μm thick samples sandwiched between platinum electrodes were annealed at 110 °C for 24 h under vacuum to eliminate solvent and water residuals. Their frequency-dependent evolution of the electrical response was studied at temperatures ranging from −70 to 110 °C. σDC was derived from the plateau value observed in the spectral dependence of the conductivity function (σ′ = ωε′′ε0, where ω is the frequency, ε0 the vacuum permittivity and ε′′ the dielectric loss), and plotted as a function of reciprocal temperature (Fig. 4). The evolution of σDC follows a classical Vogel–Fulcher–Tammann (VFT) dependence with temperature due to the correlation between the charge transport of the ionic species and molecular mobility. They were thus fitted using the VFT eqn (1):| σDC = σ∞ × exp(−B/(T − T0)) | (1) |
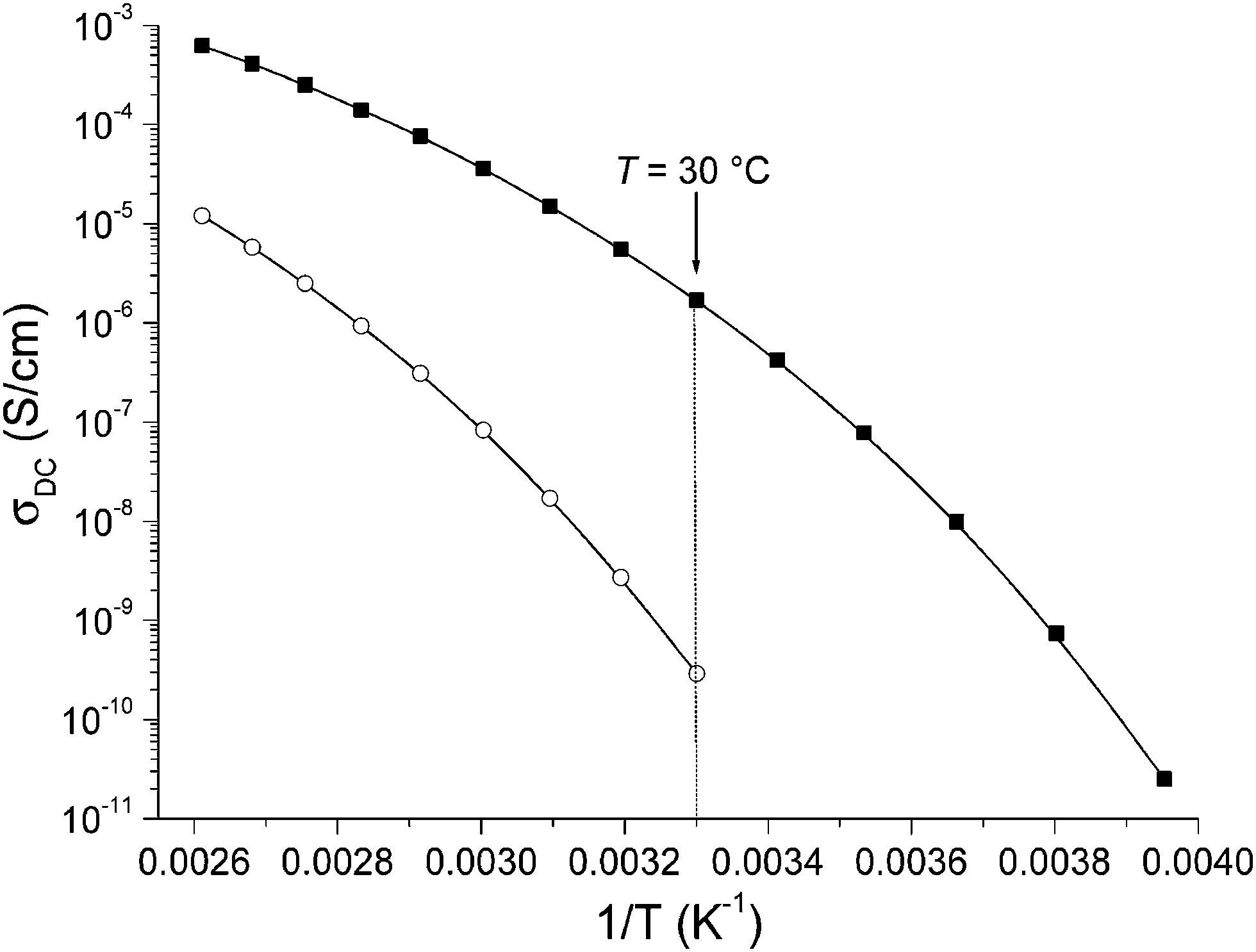 | ||
| Fig. 4 Direct current conductivity (σDC) versus reciprocal temperature for TILs 21 (solid squares) and 24 (open circles). The solid line represents the best VFT fit of experimental data using eqn (1) with σ∞ = 1.40 S cm−1, B = 1451 K and T0 = 195 K for 21 and σ∞ = 1.03 S cm−1, B = 1874 K and T0 = 218 K for 24. | ||
Conclusions
We have developed an efficient route for the synthesis of two series of biosourced CILs derived from isosorbide by CuAAC, N-alkylation of 1,2,3-triazoles and anion metathesis reactions. These novel stereoisomers illustrate the robustness of the developed approach and the capacity to broaden the field of ionic liquids and the understanding of their structure/properties relationship. All synthesized TILs are amorphous liquids with Tg values ranging from −68 to −8 °C and remain thermally stable above 200 °C. Most importantly, we have demonstrated the significant impact of the DAH stereochemistry on ion conducting properties. The application of this new class of CILs as biosourced electrolytes in electrochemical storage technologies is under investigation.Experimental section
General methods
Pent-1-yne (99%), diisopropylethylamine (DIPEA, 99%), copper(I)iodide triethylphosphite (CuIP(OEt)3, 97%), iodomethane (CH3I, 99%), silver tetrafluoroborate (AgBF4, ≥99.99%), lithium hexafluorophosphate (LiPF6, ≥99.99%), bis(trifluoromethane)sulfonimide lithium salt (LiNTf2, 99.95%) and all solvents were purchased from Aldrich and used as received. Mono-tosyl 11–1250 and mono-azide 13–1451 1,4:3,6-dianhydrohexitols were synthesized as described earlier. 1H NMR (400 MHz) spectra were recorded on a Bruker DRX400 spectrometer in DMSO-d6. Peak multiplicities are designated as m = multiplet, s = singlet, d = doublet, t = triplet, q = quartet, and dd = doublet of doublets. High-resolution mass spectrometry (HRMS) was performed on a MicroTOFQ-II (Bruker Daltonics, Bremen) equipped with an electrospray ionization (ESI) ion source in both positive and negative modes. The sample was infused at 150 μL h−1 in a mixture of water, methanol and dichloromethane with 0.1% of formic acid. The gas flow of the sprayer was 0.6 bar, the spray voltage was 3.5 kV, and the capillary temperature was 200 °C. The mass range of the time-of-flight mass spectrometer (TOF) was 50–1000 m/z. For calibration, a solution of formate was used. Differential scanning calorimetry (DSC) measurements were performed under helium using a DSC Q200 (TA Instruments) at a heating rate of 20 °C min−1. Tg values were measured during the second heating cycle. Thermogravimetric analysis (TGA) was performed under nitrogen using a TGA Q500 (TA Instruments) at a heating rate of 20 °C min−1.:1 mixture of petroleum ether and ethyl acetate as the eluent, affording the adduct 15 as a white crystalline solid (900 mg, 64.4%). M.p. 85–87 °C; 1H NMR (DMSO-d6): δ = 7.89 (s, 1H, Hh), 5.30 (d, 1H, Hg, J = 3.8 Hz), 5.13 (m, 1H, He), 4.81 (d, 1H, Hd, J = 3.8 Hz), 4.55 (d, 1H, Hc, J = 3.8 Hz), 4.17 (t, 1H, Hb, J = 3.5 Hz), 4.10 (dd, 1H, Hf, J = 5.4 Hz, J′ = 10.1 Hz), 4.03 (dd, 1H, Hf, J = 2.5 Hz, J′ = 10.1 Hz), 3.86 (dd, 1H, Ha, J = 3.5 Hz, J′ = 9.8 Hz), 3.73 (d, 1H, Ha, J = 9.8 Hz), 2.59 (t, 2H, Hi, J = 7.6 Hz), 1.61 (m, 2H, Hj), 0.91 ppm (t, 3H, Hk, J = 7.2 Hz); 13C NMR (DMSO-d6): δ = 146.96 (C-n), 120.97 (C-h), 88.26 (C-c), 85.94 (C-d), 74.57 (C-b), 74.50 (C-a), 71.46 (C-f), 64.95 (C-e), 26.93 (C-i), 22.08 (C-j), 13.53 ppm (C-k); HRMS (ESI-TOF) m/z: [M + H]+ calculated for C11H18N3O3 240.1343 g mol−1; found 240.1335 g mol−1.
:1 mixture of petroleum ether and ethyl acetate as the eluent, affording the adduct 16 as a transparent viscous liquid (600 mg, 47.7%). 1H NMR (DMSO-d6), δ = 7.87 (s, 1H, Hh), 5.22 (m, 1H, Hb), 5.06 (d, 1H, Hg, J = 6.6 Hz), 4.69 (dd, 1H, Hd, J = 4.5 Hz, J′ = 4.7 Hz), 4.51 (dd, 1H, Hc, J = 4.5 Hz, J′ = 4.7 Hz), 4.30 (dd, 1H, Ha, J = 8.0 Hz, J′ = 8.2 Hz), 4.21 (m, 1H, He), 4.12 (dd, 1H, Ha, J = 8.7 Hz, J′ = 9.8 Hz), 3.78 (dd, 1H, Hf, J = 6.6 Hz; J′ = 8.4 Hz), 3.47 (dd, 1H, Hf, J = 8.3 Hz, J′ = 8.5 Hz), 2.58 (t, 2H, Hi, J = 7.4 Hz), 1.60 (m, 2H, Hj), 0.90 ppm (t, 3H, Hk, J = 7.4 Hz); 13C NMR (DMSO-d6) δ = 146.44 (C-n), 122.03 (C-h), 82.59 (C-c), 80.92 (C-d), 72.40 (C-f), 72.25 (C-e), 69.21 (C-a), 62.40 (C-b), 27.09 (C-i), 22.30 (C-j), 13.67 ppm (C-k); HRMS (ESI-TOF) m/z: [M + H]+ calculated for C11H18N3O3 240.1343 g mol−1; found 240.1340 g mol−1.
General procedure for anion exchange
Acknowledgements
E.D. gratefully acknowledges the financial support from the Institut Universitaire de France (IUF). R.M. and M.M. are thankful to the Tunisian Ministry of Higher Education and Research for financial support.Notes and references
- T. Welton, Chem. Rev., 1999, 99, 2071 CrossRef CAS PubMed.
- J. H. Davis, Chem. Lett., 2004, 33, 1072 CrossRef CAS.
- P. N. Reddy, P. Padmaja, B. V. S. Reddy and G. Rambabu, RSC Adv., 2015, 5, 51035 RSC.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123 RSC.
- R. Gusain, R. Singh, K. L. N. Sivakumar and O. P. Khatri, RSC Adv., 2014, 4, 1293 RSC.
- T. Welton, Coord. Chem. Rev., 2004, 248, 2459 CrossRef CAS.
- M. A. P. Martins, C. P. Frizzo, D. N. Moreira, N. Zanatta and H. G. Bonacorso, Chem. Rev., 2008, 108, 2015 CrossRef CAS PubMed.
- P. Dominguez de Maria, Angew. Chem., Int. Ed., 2008, 47, 6960 CrossRef CAS PubMed.
- H. Li, P. S. Bhadury, B. Song and S. Yang, RSC Adv., 2012, 2, 12525 RSC.
- N. Papaiconomou, I. Billard and E. Chainet, RSC Adv., 2014, 4, 48260 RSC.
- K. Wilpiszewska and T. Spychaj, Carbohydr. Polym., 2011, 86, 424 CrossRef CAS.
- R. Tepper, B. Schulze, M. Jäger, C. Friebe, D. H. Scharf, H. Görls and U. S. Schubert, J. Org. Chem., 2015, 80, 3139–3150 CrossRef CAS PubMed.
- J. Dupont and J. D. Scholten, Chem. Soc. Rev., 2010, 39, 1780 RSC.
- S. Livi, J.-F. Gerard and J. Duchet-Rumeau, Chem. Commun., 2011, 47, 3589 RSC.
- T.-P. Fellinger, A. Thomas, J. Yuan and M. Antonietti, Adv. Mater., 2013, 25, 5838 CrossRef CAS PubMed.
- M. H. Chakrabarti, F. S. Mjalli, I. M. AlNashef, M. A. Hashim, M. A. Hussain, L. Bahadori and C. T. J. Low, Renewable Sustainable Energy Rev., 2014, 30, 254 CrossRef CAS.
- P. Kubisa, Prog. Polym. Sci., 2009, 34, 1333 CrossRef CAS.
- T. Erdmenger, C. Guerrero-Sanchez, J. Vitz, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2010, 39, 3317 RSC.
- D. Carriazo, M. C. Serrano, M. C. Gutierrez, M. L. Ferrer and F. del Monte, Chem. Soc. Rev., 2012, 41, 4996 RSC.
- C. Baudequin, D. Bregeon, J. Levillain, F. Guillen, J.-C. Plaquevent and A.-C. Gaumont, Tetrahedron: Asymmetry, 2005, 16, 3921 CrossRef CAS.
- T. Payagala and D. W. Armstrong, Chirality, 2012, 24, 17 CrossRef CAS PubMed.
- K. Bica and P. Gaertner, Eur. J. Org. Chem., 2008, 3235 CrossRef CAS.
- G. Imperato, B. Konig and C. Chiappe, Eur. J. Org. Chem., 2007, 1049 CrossRef CAS.
- X. Chen, X. Li, A. Hu and F. Wang, Tetrahedron: Asymmetry, 2008, 19, 1 CrossRef CAS.
- J. Wu, P. Eduard, S. Thiyagarajan, J. van Haveren, D. S. van Es, C. E. Koning, M. Lutz and C. F. Guerra, ChemSusChem, 2011, 4, 599 CrossRef CAS PubMed.
- M. Rose and R. Palkovits, ChemSusChem, 2012, 5, 167 CrossRef CAS PubMed.
- F. Fenouillot, A. Rousseau, G. Colomines, R. Saint-Loup and J.-P. Pascault, Prog. Polym. Sci., 2010, 35, 578 CrossRef CAS.
- P. Dimitrov-Raytchev, C. Besset, E. Fleury, J.-P. Pascault, J. Bernard and E. Drockenmuller, Pure Appl. Chem., 2013, 85, 511 Search PubMed.
- H. Bennour, A. Fildier, S. Chatti, H. R. Kricheldorf, M. Said Zina and R. Medimagh, Macromol. Chem. Phys., 2015, 216, 1081–1090 CrossRef CAS.
- S. Beghdadi, I. AbdelhediMiladi, H. BenRomdhane, J. Bernard and E. Drockenmuller, Biomacromolecules, 2012, 13, 4138 CrossRef CAS PubMed.
- J. J. Gallagher, M. A. Hillmyer and T. M. Reineke, ACS Sustainable Chem. Eng., 2015, 3, 662 CrossRef.
- O. N. Van Buu, A. Aupoix, N. D. T. Hong and G. Vo-Thanh, New J. Chem., 2009, 33, 2060 RSC.
- V. Kumar, C. E. Olsen, S. J. C. Schaffer, V. S. Parmar and S. V. Malhotra, Org. Lett., 2007, 9, 3905 CrossRef CAS PubMed.
- O. N. Van Buu, A. Aupoix and G. Vo-Thanh, Tetrahedron, 2009, 65, 2260 CrossRef.
- M. D. R. Gomes da Silva and M. M. A. Pereira, Carbohydr. Res., 2011, 346, 197 CrossRef CAS PubMed.
- H. B. Nulwala, C. N. Tang, B. W. Kail, K. Damodaran, P. Kaur, S. Wickramanayake, W. Shi and D. R. Luebke, Green Chem., 2011, 13, 3345 RSC.
- D. Mendoza-Espinosa, R. González-Olvera, C. Osornio, G. E. Negrón-Silva and R. Santillan, New J. Chem., 2015, 39, 1587 RSC.
- J. D. Watkins, E. A. Roth, M. Lartey, E. Albenze, M. Zhong, D. R. Luebke and H. B. Nulwala, New J. Chem., 2015, 39, 1563 RSC.
- S. S. Khan, J. Shah and J. Liebscher, Tetrahedron, 2010, 66, 5082 CrossRef CAS.
- Y. Jeong and J.-S. Ryu, J. Org. Chem., 2010, 75, 4183 CrossRef CAS PubMed.
- J. M. Aizpurua, R. M. Fratila, Z. Monasterio, N. Perez-Esnaola, E. Andreieff, A. Irastorza and M. Sagartzazu-Aizpurua, New J. Chem., 2014, 38, 474 RSC.
- F. Yan, M. Lartey, K. Jariwala, S. Bowser, K. Damodaran, E. Albenze, D. R. Luebke, H. B. Nulwala, B. Smit and M. Haranczyk, J. Phys. Chem. B, 2014, 118, 13609 CrossRef CAS PubMed.
- M. M. Obadia, B. P. Mudraboyina, A. Serghei, T. N. T. Phan, D. Gigmes and E. Drockenmuller, ACS Macro Lett., 2014, 3, 658 CrossRef CAS.
- P. Dimitrov-Raytchev, S. Beghdadi, A. Serghei and E. Drockenmuller, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 34 CrossRef CAS.
- B. Mudraboyina, M. M. Obadia, I. Allaoua, R. Sood, A. Serghei and E. Drockenmuller, Chem. Mater., 2014, 26, 1720 CrossRef CAS.
- B. J. Adzima, S. C. Taylor, H. He, D. R. Luebke, K. Matyjaszewski and H. B. Nulwala, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 417 CrossRef CAS.
- J. Yuan, D. Mecerreyes and M. Antonietti, Prog. Polym. Sci., 2013, 38, 1009 CrossRef CAS.
- A. S. Shaplov, R. Marcilla and D. Mecerreyes, Electrochim. Acta, 2015, 175, 18–34 CrossRef CAS.
- K. Stappert and A.-V. Mudring, RSC Adv., 2015, 5, 16886 RSC.
- F. Bachmann, J. Reimer, M. Ruppenstein and J. Thiem, Macromol. Rapid Commun., 1998, 19, 21 CrossRef CAS.
- C. Besset, S. Binauld, M. Ibert, P. Fuertes, J.-P. Pascault, E. Fleury, J. Bernard and E. Drockenmuller, Macromolecules, 2010, 43, 17 CrossRef CAS.
- S. Sanghi, E. Willett, C. Versek, M. Tuominen and E. B. Coughlin, RSC Adv., 2012, 2, 848 RSC.
- J. Sun, M. Forsyth and D. R. Macfarlane, J. Phys. Chem. B, 1998, 102, 8858 CrossRef CAS.
- Z. B. Zhou, H. Matsumoto and K. Tatsumi, Chem. – Eur. J., 2005, 11, 752 CrossRef CAS PubMed.
- J. Vila, L. M. Varela and O. Cabeza, Electrochim. Acta, 2007, 52, 7413 CrossRef.
- K. Tsunashima, A. Kawabata, M. Matsumiya, S. Kodama, R. Enomoto, M. Sugiya and Y. Kunugi, Electrochem. Commun., 2011, 13, 178 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR and HRMS characterization, solubility data and hydrogen-bonding scheme. See DOI: 10.1039/c5nj02660a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |