DOI:
10.1039/C5NJ02058A
(Paper)
New J. Chem., 2016,
40, 475-484
Solvent specific synthesis of nano corpse flowery lithiated iron oxide as an energy storage and gas sensing material†
Received
(in Montpellier, France)
4th August 2015
, Accepted 23rd October 2015
First published on 30th October 2015
Abstract
Iron oxide based materials are one of the most appealing matrices and promising futuristic materials for energy conversion/storage devices. The solvent dependent synthesis and growth of lithiated iron oxide/LiFeO2 nano flowers was established using a simple sol–gel method at low temperature. Herein, for the first time the development of flowery (corpse flower) shaped iron oxide based nano materials is reported. The effect of type of solvent on phase formation, shape and sizes of the as-synthesiszed samples was determined using charaterisation techniques such as X-ray diffraction, FTIR, Raman spectroscopy, surface area, TEM, UV, and XPS. Li was incorporated into the iron oxide matrix in ethylene glycol medium and developed a unique and uniform corpse flowery shape, depending on various reaction parameters, whereas in the presence of ethylene glycol monomethyl ether, the shape of the nano materials completely changed. The supercapacitive and gas sensing properties of some selected synthesiszed materials are evalutaed. The specific capacitance values of the materials depend on the nature of the solvent and lithium content of the as-prepared samples. The lithiated iron oxide samples exhibit a supercapacitance value of 241 F g−1 in 0.1 M Na2SO4 between −0.4 and 1 V versus Ag/AgCl. The gas sensing behaviour and optical properties are also included to open up the multidimensional applications of the samples.
1. Introduction
Iron oxide based materials are expected to be excellent candidates for numerous futuristic advance applications due to their favourable physicochemical properties, chemical stability and non-toxicity.1–6 Their inherent properties can be enhanced by carefully controlled synthetic procedures with the introduction of a definite amount of various secondary metal ions. The insertion of alkali metal ions, particularly lithium in iron oxides, through the aqueous route is currently a major interest for researchers. An iron oxide-based electrode, wherein only intercalation of Li ions occurs, has been recently found to be promising for cathode design.7 Li insertion resulted in the reduction of particle size and expansion of the d-spacing between the crystal layers of iron oxide.8 Therefore, lithiated iron oxide compounds, particularly those with a rock-salt structure, are of great interest for unique ionic conduction, electrical and electrochemical properties for use in lithium-ion batteries for the construction of second-generation galvanic cells.9 Various synthetic methods have been reported for the preparation and structural modification of Li containing iron oxides.10 Among all the solid-state reaction methods, the soft-chemistry method is preferred, wherein the H+/Li+ ion-exchange reaction occurs in an alcoholic medium at a relatively low reacting temperature.11 Low temperature synthetic processes are advantageous for obtaining a smaller particle size of lithiated iron oxide, which is also found to be a good candidate as an electroactive material.12–14 The various applications of iron oxides, such as electrochemical sensors and gas sensors, can be enhanced by adopting solvent mediation precipitation routes, which results in a modified structure, morphology and phase.
The present study emphasizes the development of synthetic protocols for lithium based iron oxide, in which a solvent mediated synthetic approach was chosen. The effect of both the nature of the solvent and lithium ion on phase formation, crystal structure, electrochemical and gas sensing properties are discussed.
2. Experimental
The chemicals used in the syntheses are iron nitrate (Fe(NO3)3·9H2O), ethylene glycol monomethyl ether (CH3OCH2CH2OH) (EGME), ethylene glycol (EG) and lithium hydroxide (LiOH) (E-Merck, India). All other chemicals were of Analar grade. Samples were synthesized by adopting our previous synthesis method.15 Iron oxides materials were synthesized by following solvent mediated precipitation routes in the presence of secondary metal ions, namely, Li. In this case, the desired solution of mixed metal ion (secondary metal ions + iron) was added to a calculated amount of solvent to obtain a fixed ratio. The desired amount of 0.1 M iron and Li ion solutions were vigorously stirred for 15 min in a three neck flask attached with a condenser, thermometer and pH meter (electrode). A stock solution of EGME or EG was added to the above mentioned solution (the ratio of solvent to total metal ion concentration was maintained at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with continuous stirring for another 10 min to form a homogeneous solution. After that the solution was heated to the desired temperature for 5 h, and then the flask was allowed to cool naturally to room temperature. The pH of the suspension was also monitored on completion of precipitation. The yellow slurry obtained after cooling was centrifuged and washed several times with deionized water and absolute alcohol and finally dried at 70 °C for 24 hours in an air oven. An iron analysis was done via atomic absorption spectroscopy (AAS) on a Perkin Elmer instrument, whereas the Li analysis was done via ICP-OES (inductively coupled plasma-optical emission spectroscopy) on an Optima-2100DV, PerkinElmer.
:1) with continuous stirring for another 10 min to form a homogeneous solution. After that the solution was heated to the desired temperature for 5 h, and then the flask was allowed to cool naturally to room temperature. The pH of the suspension was also monitored on completion of precipitation. The yellow slurry obtained after cooling was centrifuged and washed several times with deionized water and absolute alcohol and finally dried at 70 °C for 24 hours in an air oven. An iron analysis was done via atomic absorption spectroscopy (AAS) on a Perkin Elmer instrument, whereas the Li analysis was done via ICP-OES (inductively coupled plasma-optical emission spectroscopy) on an Optima-2100DV, PerkinElmer.
Crystal structures were determined by X-ray diffraction on a PAnalytical model X’Pert PRO PW-3040/60 with Mo-Kα radiation (λ = 0.709) at a scan speed of 1.2° min−1 over the range of 5–40°. FTIR spectra were obtained on a Nicolet-670. Raman spectra were obtained using a Renishaw (Renishaw plc, Gloucesteshire, UK) inVia Micro Raman Spectrophotometer equipped with a 514 nm green laser, which have a 1 cm−1 Raman shift spectra resolution, X–Y step resolution of 0.1 μm, and confocal resolution of 2.5 μm. The laser output power on the sample was set at 5.1 mW through a spectrum curve employing the Lorentzian FWHM. About 0.001 g of sample was used for the Raman analysis. TEM micrographs were obtained using an electron microscope (FEI, TECNAI G2 20, TWIN) operating at 200 kV, equipped with a GATAN CCD camera. Samples for transmission electron microscopy (TEM) were prepared by first diluting a small amount of suspension with ethanol, sonicating the suspension and then placing a single drop of the resulting suspension onto a 200 mesh carbon-coated copper grid, which was then allowed to dry in air. Selected area electron diffraction (SAED) patterns were obtained with the smallest area aperture available (10 μm). BET surface area and pore size was analysed in Autosorb-iQ and ASiQ-WiN device from Quanta chrome. The solid samples were sonicated for 5 min with deionized water for complete dispersion and then the analysis was done using a UV-Visible Perkin Elmer spectrophotometer. Cyclic voltammograms (CV) were obtained on a three-electrode cell workstation (CHI Instruments 660A). A standard Ag/AgCl electrode was used as the reference, whereas a platinum (Pt) wire was used as the counter electrode. Cyclic-voltammograms were recorded using a computer-controlled CHI-660C electrochemical analyser (CHI, USA). Cyclic voltammetry studies were performed using a two compartment, three-electrode cell, which had a glassy carbon working electrode (area 1/4 0.07 cm2), a platinum electrode and Ag/AgCl (0.1 M Na2SO4) as the reference electrode in the potential range of −0.4–1.0 V at a sweep rate of 5–100 mV s−1. All the electrochemical experiments were carried out in an argon atmosphere. The synthesized materials were dispersed over a glassy carbon electrode with 0.1 mL of 2% Nafion and dried prior to the electrochemical experiments.
The synthesized iron oxide was dispersed in isopropyl alcohol containing 50% of hydroxypropyl cellulose (HPC) of the material weight using an ultrasonic disperser for 20 min. This synthesized paste was deposited on a commercial Al2O3 substrate (15 mm × 15 mm) with previously screen-printed gold electrodes (10 mm × 10 mm) using the doctor blade technique, and then dried at 200 °C for 1 h followed by baking in a furnace at 400 °C for 1 h. The gas sensing activity of the device was investigated using a home-made gas-sensing evaluation system equipped with digital mass flow controllers, a rapid heating system with programmed temperature controllers, and a quartz reactor to carry out the sensing reactions. The change in resistance of the device due to the presence of the target gas was obtained using a high resistance meter (Keithley source meter 2400). The device was tested in the temperature range of 25–400 °C at various concentrations of the target gas (100–500 ppm) in a temperature-controlled environment. The background gas was N2, mixed with atmospheric air to give 10% oxygen content. The flow of gas (100 mL min−1) over the sensor device was altered with the sample gas and dry air to record the sensor response in terms of electric resistance. The gas sensitivity is defined as the resistance ratio:
where
Rair and
Rgas are the electrical resistances for the sensors in dry air and in gas, respectively.
3. Results and discussion
3.1 Chemical analysis
The synthesized samples were named as FeEGLi-1, FeEGLi-2, FeEGLi-3, FeEGLi-4 and FeEGMELi-1, FeEGMELi-2, FeEGMELi-3, FeEGMELi-4, which were obtained from EG and EGME medium. Samples were synthesized by varying the concentration of lithium ion solution to iron solution at ratios of 1:9, 1:4, 1:2.33 and 1:1 during the precipitation of iron oxide using EG and EGME as the solvent. The dark wine colour precursor solutions (mixture of iron and lithium) became visibly dark brown in colour after precipitation. Fig. S1a and b (ESI†) shows the percentage of iron and lithium in the lithiated iron oxide samples via EG and EGME synthesis media. The % of iron and lithium precipitation from the precursor is shown in Fig. S1c and d (ESI†), respectively.
As shown in Fig. 1a, the percentage of iron precipitated was more in the presence of lithium for the EG mediated precipitation routes in comparison to the EGME route. There was a slight variation in percentage of iron precipitated with Li:Fe ratio. As observed from Fig. 1b, the lithium content in the precipitated products obtained in an EG medium is slightly more as compared to that in the EGME medium and in both the cases an opposite trend is observed between lithium content in the solid product and Li ion used in the solution. Around 90% of iron is precipitated irrespective of lithium concentration and nature of solvent, as shown in Fig. S1c (ESI†); whereas, the percentage of lithium precipitation decreased with an increase in Li content in the solution.
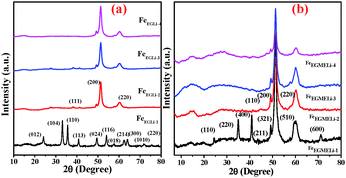 |
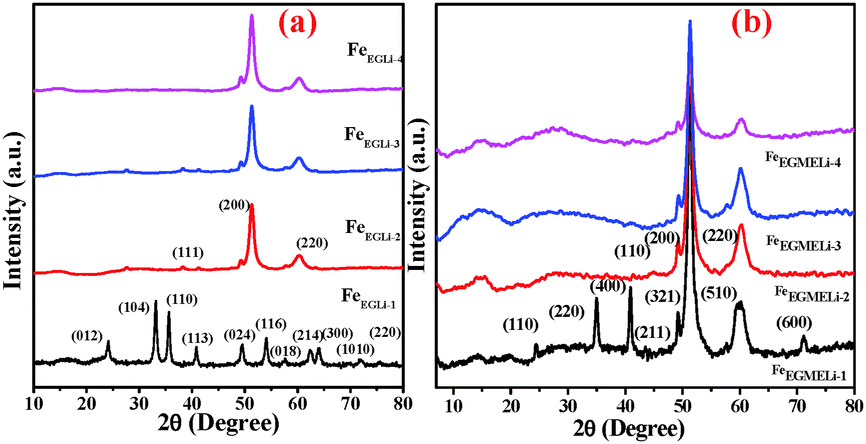
| | Fig. 1 XRD patterns of the as-synthesised samples in the presence of Li (a) EG and (b) EGME solvent mediated precipitation routes. | |
3.2 XRD Study
The XRD patterns of the different samples are given in Fig. 1. The different planes observed for FeEGLi-1 were assigned to (012), (104), (110), (113), (024), (116), (018), (214), (300), (1010) and (220) of the α-Fe2O3 phase (ICDD: 01-084-0310). In addition to the above mentioned peaks, there were no other peaks found, which could be assigned to the crystalline phases of lithium containing compounds.
However, the presence of lithium in the chemical analysis indicated that in the FeEGLi-1 sample, Li may be present either as an amorphous phase or a phase that was XRD transparent. The XRD patterns of the samples FeEGLi-2 to FeEGLi-4 exhibited several peaks, which could be ascribed to the α-LiFeO2 phase and were indexed to the corresponding planes (111), (200) and (220) of this phase (JCPDS file No. 01-074-2284), thus confirming that these samples were purely crystalline α-LiFeO2. This result is quite consistent with the results reported by several authors.16,17 The approximate crystallite sizes of the samples were calculated using the Debye–Scherer equation using the peak corresponding to the (220) plane. The crystal sizes were found to be 10.04, 7.12 and 5.4 nm for the FeEGLi-2, FeEGLi-3, and FeEGLi-4 sample. Lithium ferrite is an inverse spinel compound wherein Li+ and Fe3+ ions occupy the octahedral B-sites. Its synthesis is facilitated when the replacement of 2Fe2+ with Li+ Fe3+ in the iron oxide complex is easier. In this case, the reducing nature of the solvent helps in the formation of LiFeO2. Moreover, the occupation of Li+ in interstitial positions causes the reduction of Fe3+. All the peaks showed shifts that increased with an increase in Li concentration due to the topotactic incorporation of surface Li ions in the hematite matrix. This may also increase the crystallinity of the sample.10,18 Herein, it has been noted that the percentage of metal ion precipitated along with iron is very less, therefore the Li:Fe ratio in the synthesised samples is smaller in stoichiometry ratio as compared to the theoretical Li:Fe ratio in LiFeO2. Thus, the signature peak for α-LiFeO2 in the XRD pattern, i.e. the peak corresponding to the (220) and (111) planes, shifted. However, there is hardly any effect on the (200) plane. This observation is in contrast to the previous reported results wherein it was mentioned that doping in the above mentioned compound did not affect its (111) plane.19–21 The lattice parameter, a, was calculated to be 4.149 Å, which agrees with the literature data for the pure phase formation.20 However, the diffraction peaks were comparatively broader, which indicated the smaller size of the crystallites.21Fig. 3 shows the change in relative intensities (RI) of the (111) and (220) planes and their ratios in the various samples obtained in the EG medium. It was observed that their RI as well as their ratios increased with an increase in the percentage of Li for the EG mediated samples.
In the case of EGME mediated samples, the XRD patterns of FeEGMELi-1 were well matched with LiFeO2 in contrast to the EG mediated sample with same Li:Fe ratio. The different planes (110), (220), (400), (211), (321), (510) and (600) were well matched with the JCPDS file No. 01-070-2711, whereas for the FeEGMELi-2 to FeEGMELi-4 samples the (110), (200) and (220) planes were well matched with JCPDS file No. 01-074-2284, as shown in Fig. S2 (ESI†). It is noted that one broad peak was observed at around the 2θ value of 14–15°, which might be due to existence of reduced phases of iron oxides in these samples. Further characterisation was done to study the purity of the synthesised phases. EGME creates a favourable environment for the possible formation of α-LiFeO2. The crystal sizes were found to be 12.38, 11.54, 8.31 and 4.8 nm for FeEGLi-1, FeEGLi-2, FeEGLi-3 and FeEGLi-4, respectively. A reverse trend in RI of the (111) and (220) planes were observed for the EGME mediated samples. The RI of the (220) plane decreased with an increase in the % of Li. Therefore, crystal growth was favourable in the (200) direction. The crystallinity of the samples decreased as compared to the EG mediated samples.
3.3 FTIR study
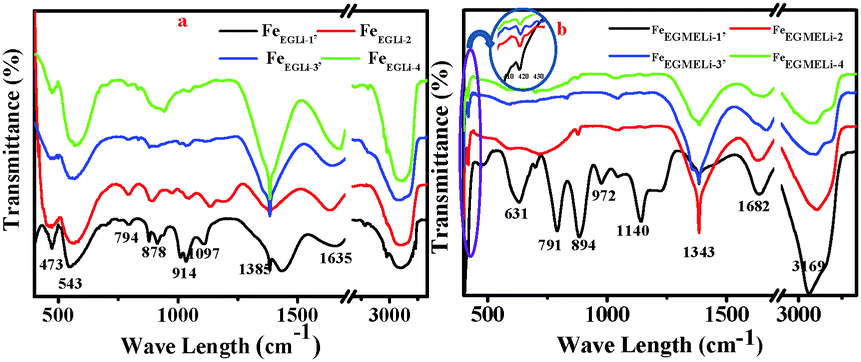
The FTIR spectra of the synthesized samples in both media are shown in Fig. 2. The presence of a band at 1343 cm−1 confirmed the presence of metal-solvent bonding in FeEGLi-1.13 This might be due to the adsorption of the complex solution over the hematite surface. The band at 1642 cm−1 was assigned to the bending mode of –OH (due to adsorbed water). The appearance of a peak for the –OH bending mode at 894 cm−1 was due to the transition moment lying in the a–b plane. The other peak at 791 cm−1 corresponded to the transition moment parallel to the c axis.9,21 For the present samples, both the peaks mentioned above were shifted to 791 and 894 cm−1. These two peaks gradually diminished and completely vanished in the samples prepared at higher Li:Fe ratios in both media. The absorption band at 543 cm−1, which was present in the FeEG series samples, shifted to a higher wavelength with the increase in lithium content. This band falls in the range of primary bands for pure Li ferrite in the range of 585–546 cm−1.22 The positions of the infrared bands were gradually shifted due to the shortening of the bond length of the tetrahedral site by the greater intercalation of Li ions. Surprisingly, this particular band was not observed in the IR spectra of the FeEGME series samples, instead a band at 631 cm−1 is visibly observed, which was assigned to the longitudinal absorptions (Au) of hematite. The band at 631 cm−1 completely vanished at a higher concentration of Li, which is perhaps due to the synergistic effect of the incorporation of lithium and variation in shape and sizes of the particles. The high frequency band at around 420 cm−1 (encircled in Fig. 4b) was recorded only for the FeEGME series samples. This band was observed for Li2CO3 on the basis of 7Li–6Li isotopic shifts,23 which is assigned to the LiO4 tetrahedron (Li+–O2−complexes at the tetrahedral site). The intensity of this band continues to decrease with an increase in lithium content in the samples. The variation in the frequency range of the IR bands is dependent on the microstructural properties of the sample.
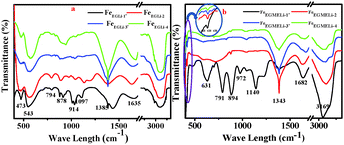 |
| | Fig. 2 IR spectra of the as synthesised samples in the presence of Li (a) in EG and (b) EGME solvent mediated precipitation routes. | |
3.4 Raman study
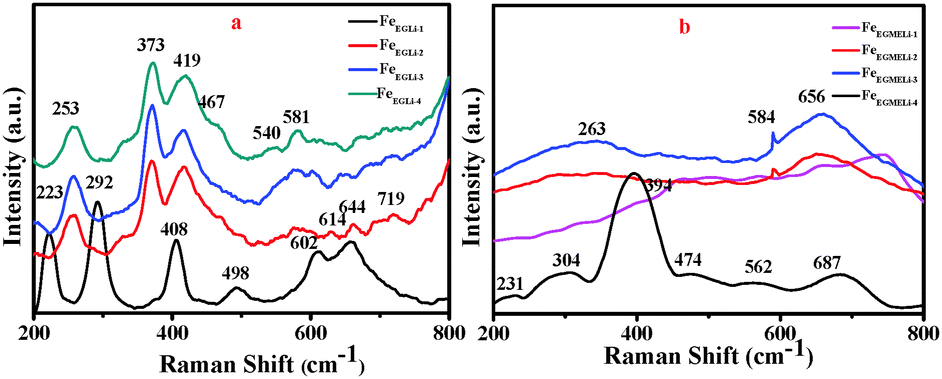
To understand the phase formation of Fe2O3 and LiFeO2 and arrangement of Li ion in these phases, a Raman analysis was carried out. The Raman spectra of all the synthesized samples are shown in Fig. 3. The Raman bands observed for FeEGLi-1 at 223 (A1g), 292 (Eg), 408 (Eg), 498 (A1g) and 614 (Eg) cm−1 were assigned to the hematite phase.24,25 These were attributed to the Fe–O stretching mode of the FeO6 octahedral linked to α-Fe2O3.26 However, these peaks were slightly shifted due to the presence of Li. The bands at 223 and 292 cm−1 experienced greater broadening as compared to the remaining oxygen-related bands. Therefore, it could be clearly observed that the % of Li doping plays a significant role in phase formation as discussed in the XRD data. The peaks observed for the other samples in EG medium at 253 and 581 cm−1 were assigned to the Li-cage in an octahedral environment, and these results were in good agreement with bulk α-LiFeO2. The Raman bands at 419 and 464 cm−1 were assigned to the LiO4 tetrahedron.26 The vibrational bands that appeared over 390–500 cm−1 were assigned as internal modes of LiO4.26 The Raman band at 720 cm−1 was assigned to the strong Fe–O stretching mode of lithium ferrite.4
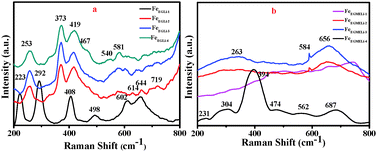 |
| | Fig. 3 Raman bands (200–800 cm−1) for synthesised LiFeO2 (a) in EG and (b) EGME solvent mediated precipitation routes. | |
The Raman spectra for the EGME series samples are shown in Fig. 3b. In case of FeEGMELi-1, five bands at 231, 304, 394, 474 and 687 cm−1 were observed. These peaks could be attributed to the O–Fe–O bending and Fe–O stretching mode of the FeO6 octahedra.27–30 Only three broad peaks were observed at 263, 584 and 656 cm−1, which correspond to the Li peaks for the rest of the EGME series.31 The broadness in Raman band observed in the 600–700 cm−1 region might be due to the overlapping of vibrations of the two types of cations at the same site.
3.5 Microstructure study: insight mechanism
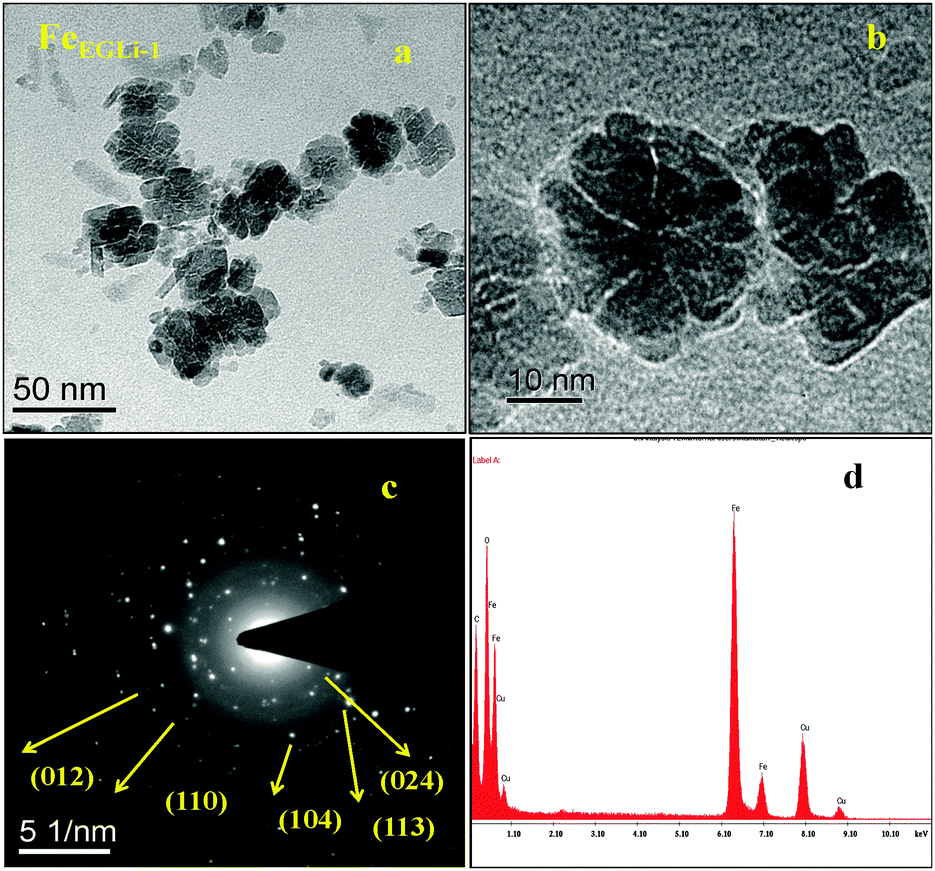
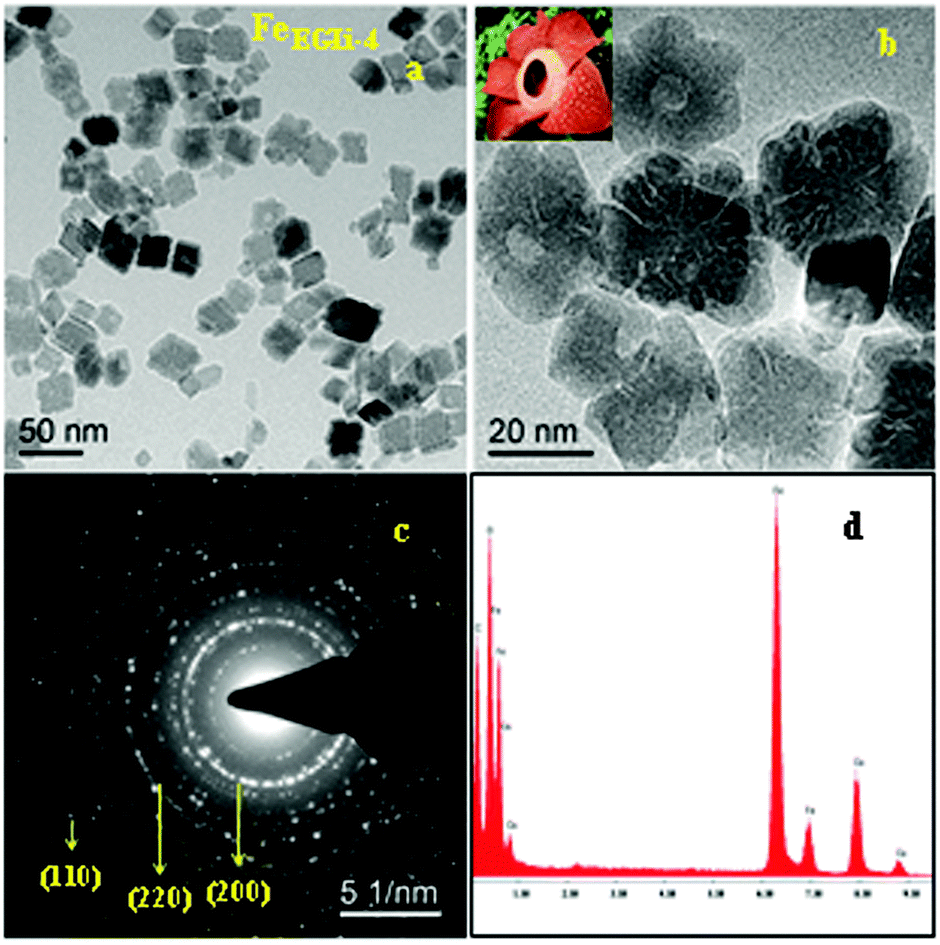
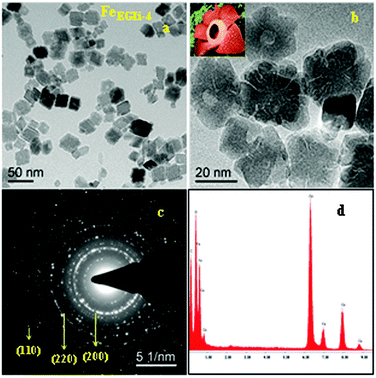
The TEM image and selected area electron diffraction (SAED) pattern of the EG mediated Li doped iron oxide samples FeEGLi-1 and FeEGLi-4 are shown in Fig. 4 and 5, respectively.
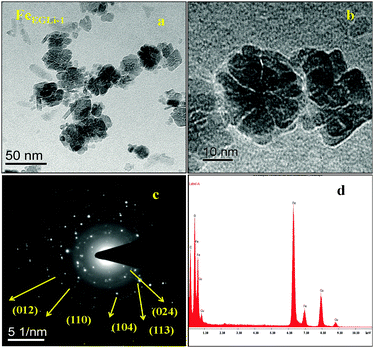 |
| | Fig. 4 (a) and (b) TEM images, (c) SAED pattern and (d) EDAX of the FeEGLi-1 sample. | |
 |
| | Fig. 5 (a) and (b) TEM images, (c) SAED pattern and (d) EDAX of the FeEGLi-4 sample. | |
A hematite phase with 20–30 nm nano rods and nano flowers were clearly observed. The selected area electron diffraction (SAED) patterns in Fig. 4c and 5c suggest that the sheet is a single-crystal, and the crystal structure can be indexed to a hexagonal type of α-Fe2O3. Close-packed Fe2O3 flowers were also observed. A porous structure was observed either between the granules or the flowers petals. The sheets are randomly assembled and regular-shaped, but they grew from the same root and resembled Plumeria or Frangipani. The petal has a shape of a trapezoid and has a smooth surface. However, with the increase in % of Li the complete formation of nano flowers appeared, as shown in Fig. 4b and 5b. They both looked similar to the Corpse flower, which is scientifically named “Rafflesia arnoldii”, which in shown as the inset Fig. 5b. The petals have an average length of about 50 nm. Interestingly, a self-assembly ordered array of LiFeO2 was observed due to the functionalized surface by EG. The self-assembly of EG-iron oxide crystals occurred via the slow evaporation of solvent in the solution of iron oxide particles. This phenomenon arose from the van der Waals interaction among the OH groups of EG at the surface. The hole in the central part of the flower is formed due to the overall etching reaction. A cationic exchange reaction occurs during the transformation of hematite to LiFeO2 and the dissolution of Fe2O3 in alkali condition occurred due to the low release of ionic precursors such as Fe2+ and Li+ ions, which are the driving force for the reductive etching reaction of Fe3+ ions due to their higher Lewis acidity. In the presence of Li+ ions on the surface of hematite, the reduction of iron (II) and inclusion of Li occur at the same time through the transportation of oxygen from iron (II) to Li ion. During this reaction time, a hollow structure was formed, which had the crystalline state of iron oxide. The SAED pattern is shown in Fig. 6c, wherein all the ring spots were evaluated to represent the d-spacings of 0.24, 0.20, and 0.14 nm, which could be referred to the crystallographic directions of (111), (200) and (220), respectively. These results are also consistent with the standard information provided by the XRD patterns. The corresponding EDAX is shown in Fig. 4d and 5d. They show that the ratio of O to Fe in the sample matched the theoretical ratio of O to Fe in the LiFeO2 material. Only Fe and O are shown, and due to its low atomic number Li could not be detected by EDAX.
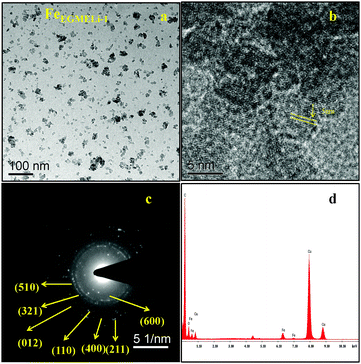 |
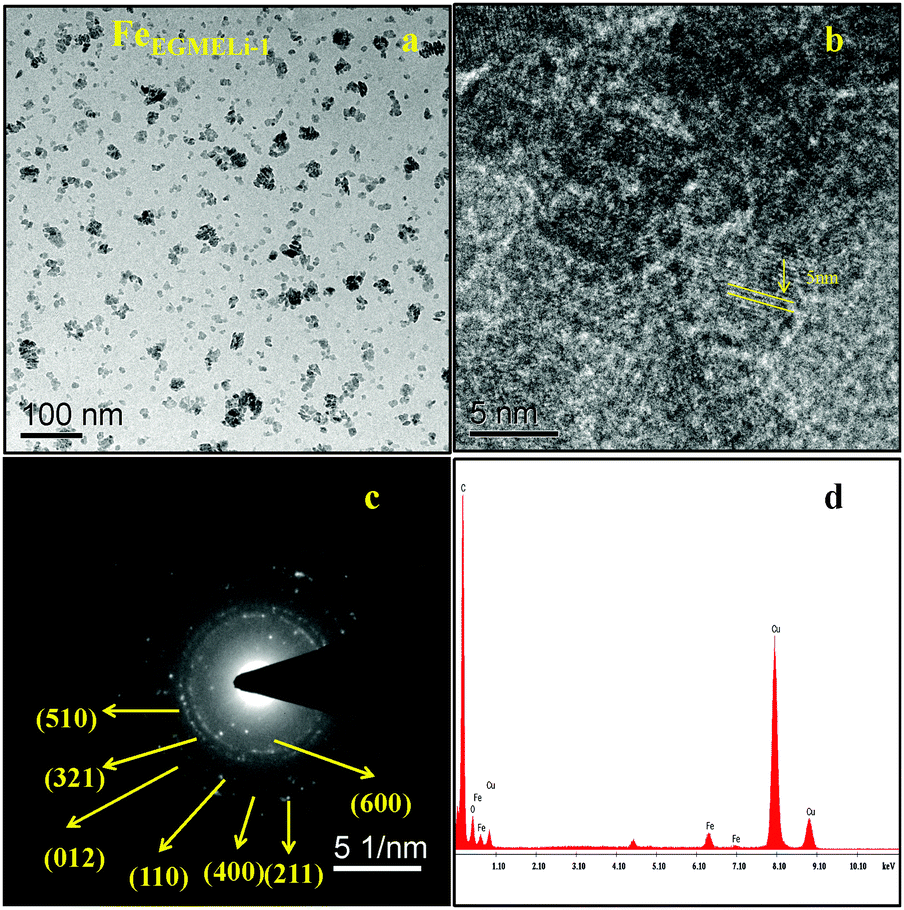
| | Fig. 6 (a) TEM, (b) HRTEM (c) SAED pattern and (d) EDAX of the FeEGMELi-1 sample. | |
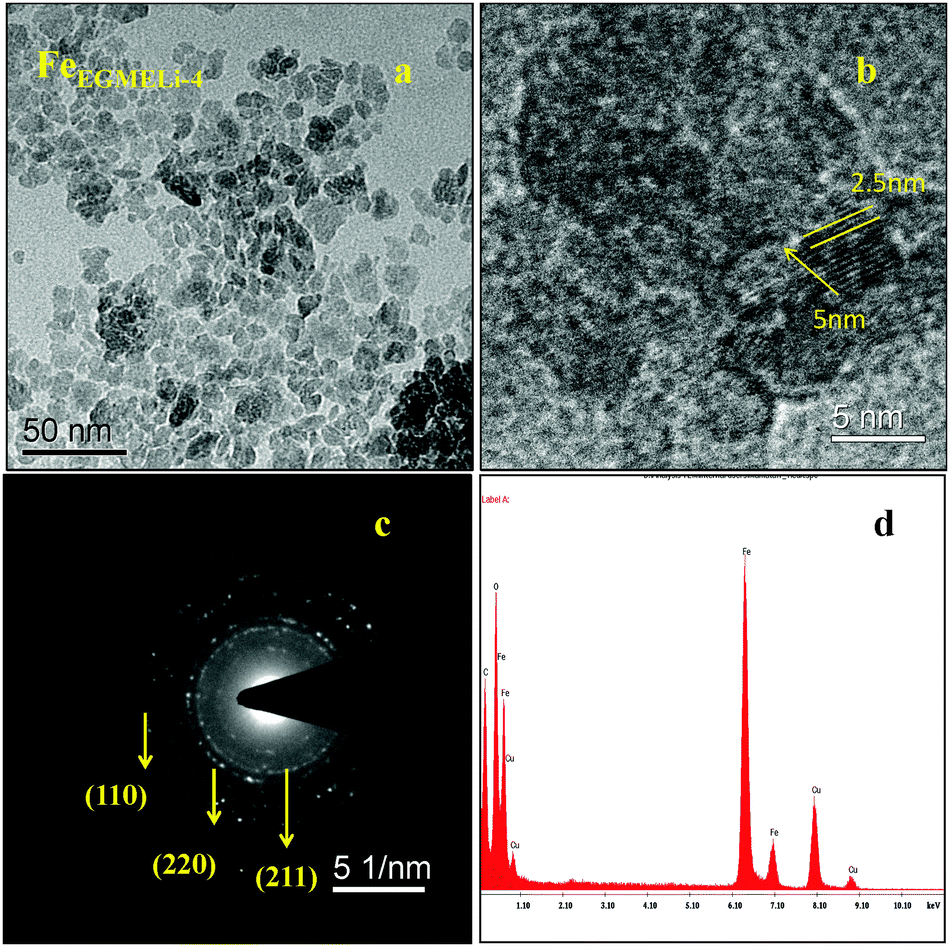
In the case of the EGME mediated Li containing iron oxide samples, tiny nanoparticles upto 5 nm were observed for FeEGMELi-1, as shown in Fig. 6a. Moreover, it is clearly observed that the resulting product has an average diameter of 2.5 nm and average length of 5 nm and a fairly narrow particle size distribution range, which supports the XRD results. The nanoparticles also have a spheroidal shape. The SAED pattern shown in Fig. 6c well matched with the crystal planes shown in the corresponding XRD patterns; whereas with the increase in % of Li, the sample showed a decreased particle size with a narrow size distribution, as shown in Fig. 7. The particles showed the development of small random tiny particles over bigger particles as a base. The nanoparticles have spheroidal shape. The formation of nanoparticles can be generally attributed the interfacial energy and magnetic dipole–dipole interaction.32 To get insight into the growth and self-assembly of the nanoparticles, the HRTEM fringes are shown in Fig. 7b, which shows that the average sizes of the d spacing are 5 nm and 2.5 nm and these are well matched with the crystal planes of the corresponding phase confirmed in the XRD study. Moreover, the EDAX patterns in Fig. 6d and 7d indicate that the observed atomic ratio of Fe and oxygen match that of the theoretical value for the LiFeO2 compound, which confirms the pure phase formation.
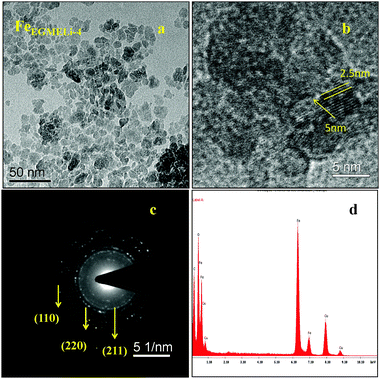 |
| | Fig. 7 (a) TEM, (b) HRTEM, (c) SAED pattern and (d) EDAX of the FeEGMELi-4 sample. | |
3.6 Surface area study
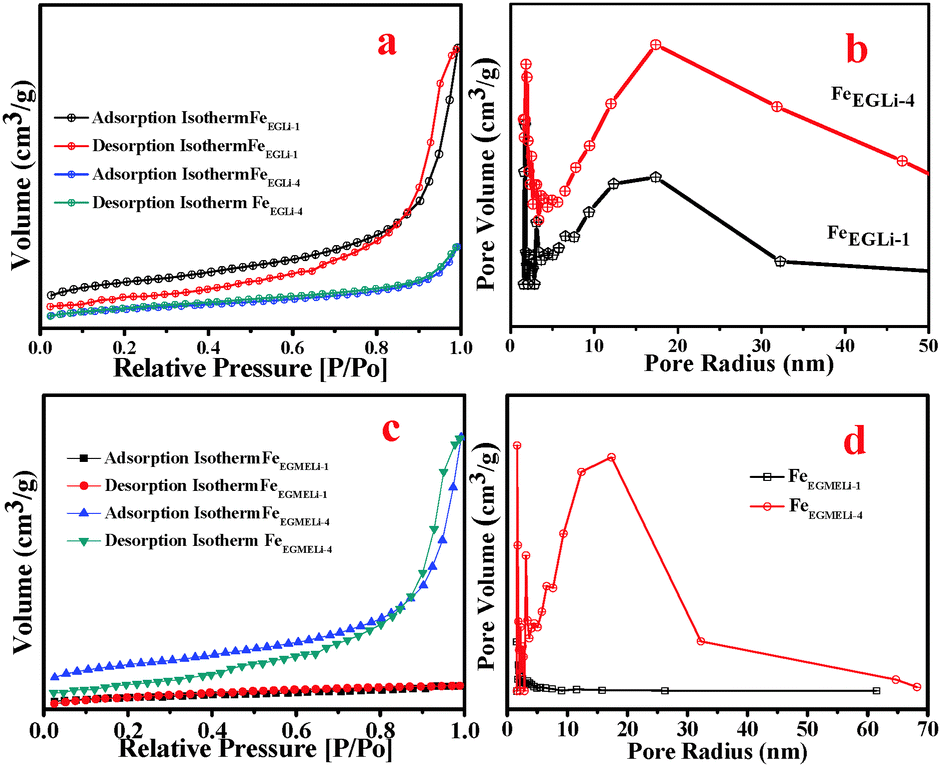
The surface area and pore size distribution of the α-Fe2O3-based nanostructures and the LiFeO2 materials were calculated through N2 adsorption–desorption measurements. The Brunauer–Emmett–Teller (BET) surface areas of the samples in both solvent media were estimated and are given in Table S1 (ESI†). In the case of FeEGLi-1, as shown in Fig. 8, a characteristic type IV isotherm was observed with a type H3 distinct and broad hysteresis loop at a relative pressure P/Po of 0.9–1. A surface area value of 123.9 m2 g−1 was estimated. The corresponding hysteresis loop may be associated with aggregates of thorny particles forming slit-like pores. Based on the BJH calculation, the pore size distribution (Fig. 8b) of FeEGLi-1 was 12.13 nm, which indicates the homogeneity of the pores. These pores arise from the void space generated during the oriented organization of nanocrystals.
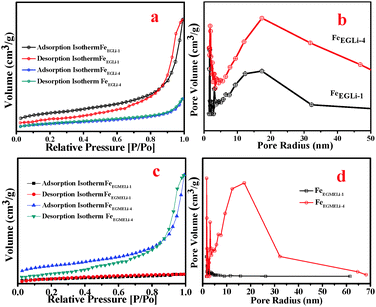 |
| | Fig. 8 (a) Surface area and (b) pore size distribution plots of FeEGLi-1 and FeEGLi-4. (c) Surface area and (d) pore size distribution plots of FeEGMELi-1 and FeEGMELi-4. | |
In contrast, the more flowery structure-like nano architecture of FeEGFeLi-4 showed the relatively low surface area of 40.86 m2 g−1 and small absorbance of N2, as shown in Fig. 8c, with a typical H3-type hysteresis loop at a relative pressure P/Po of 0.9–1.0. The shifting of the relative pressure towards a higher value suggests that the pore distribution might originate from the holes between compactly packed primary nanocrystals and the large mesoporous distribution or it might originate from some vacancies caused by the absence of subunits.31 However, almost the same pore-size distribution of around 10 nm was observed from the BJH analysis (Fig. 8d), which indicates that small pores exist in both nanostructures. The characteristic N2 adsorption–desorption isotherms and pore size distributions revealed that both samples have mesoporous structures. FeEGMELi-1 showed surface area 105.1 m2 g−1 and has a broad H3-type hysteresis loop as compared to that of FeEGMELi-4 (surface area value of 38.33 m2 g−1) at a relative pressure P/Po of 0.85–1.0. The BJH analyses showed that the sample possessed a bimodal (small and large) mesoporous distribution, with a mean pore size of 5–8 nm.
3.7 XPS study
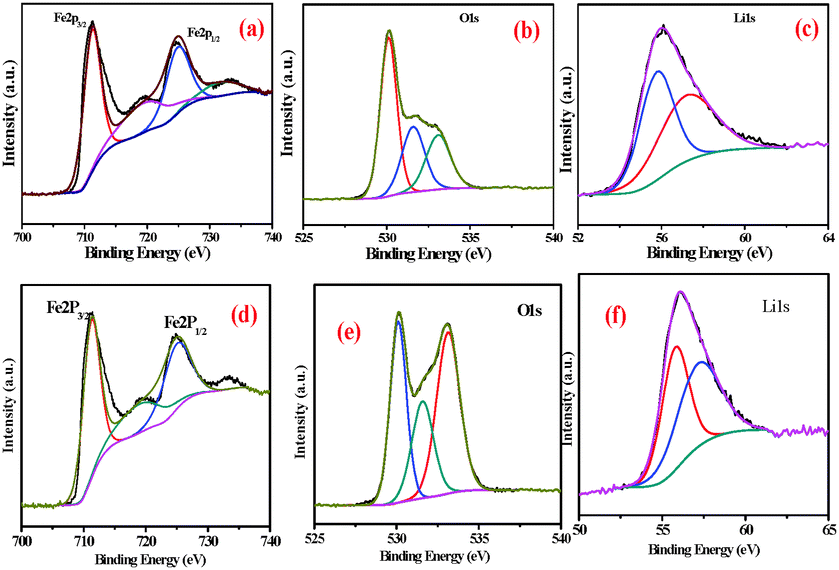
X-ray photoelectron spectroscopy (XPS) was used to further confirm the surface phenomena of the synthesized samples. The XPS spectra of FeEGLi-4 are shown in Fig. 9. The XPS spectrum for iron is split by the Fe 2p spin–orbit effect into 2p3/2 and 2p1/2 components. The 2p3/2 and 2p1/2 signals were accompanied by typical satellite peaks at 719 eV. The binding energy for Fe 2p3/2 was estimated to be 711.3 eV, and this value is comparable with the binding energy (BE) value of the 2p3/2 peak in Fe2O3 for Fe(III),32–35 which confirms the presence of iron as the hematite phase in the sample. Furthermore, the BE difference between O1s also agreed with the expected value. The obtained O/Fe atomic ratio is 1:4. The asymmetry of the O1s peak occurs at higher BE (Fig. 9b suggests the presence of surface hydroxyl groups). In principle, these OH groups can be classified according to three different types: free or isolated OH groups, H-bonded OH groups and OH groups interacting with chemisorbed water molecules. They could be assigned to different chemical states such as hydroxyl, and two peaks were also observed in the O1s region. The broad and lower intensity peak near 531.0 eV is complex and could be assigned to different chemical states, such as hydroxyl, water species or precursor impurities, for LiFeO2.36 The Li 1s signal was observed at 56 eV, which is the typical value for Li+ species. Therefore, LiFeO2 oxide formation was confirmed.37–39 Similar observations were obtained for the EGME mediated samples. There was no significant change as compared to the EG mediated samples. Fig. 11d shows the binding energies of Fe 2p3/2 and Fe 2p1/2 as 711.3 and 725.3 eV, respectively. Due to the presence of Li, a slight shift in binding energy was observed. Two O1s peaks were obtained at a higher BE due to the presence of surface hydroxyl groups in the FeEGMELi-4 sample. The Li 1s signal was observed at 56 eV.
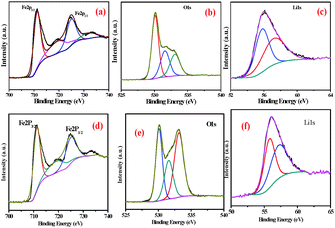 |
| | Fig. 9 XPS spectra of FeEGLi-4 (a) Fe2p, (b) O1s and (c) Li1s and XPS spectra of FeEGMELi-4 (d) Fe2p, (e) O1s and (f) Li1s. | |
3.8 Optical properties
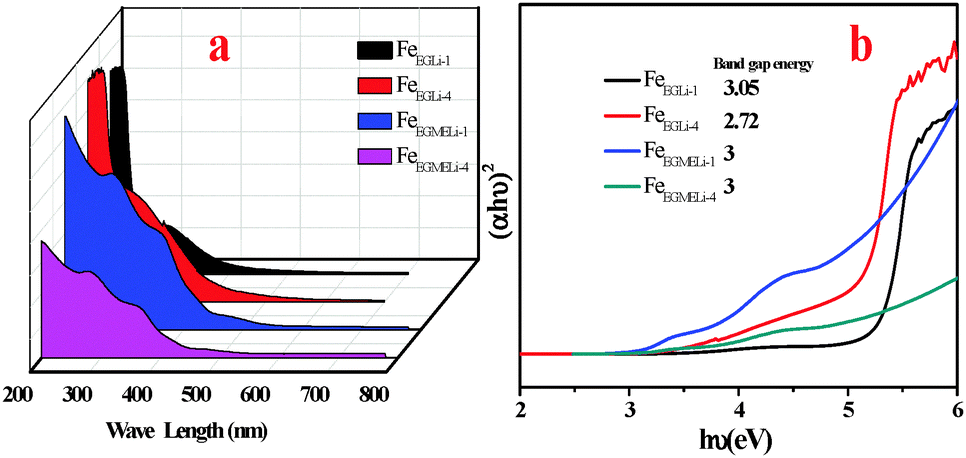
The optical absorption properties of the as synthesized samples were investigated at room temperature via UV-Vis spectroscopy, and the spectra are displayed in Fig. 10. The UV-Vis spectra were obtained in the range of 200–700 nm. The two types of optical absorptions in the UV and visible regions are mainly attributed to the direct charge transition of O2−2p → Fe3+3d (UV absorption), and the latter originates from the indirect charge transition of Fe3+3d → 3d (visible absorption).40,41
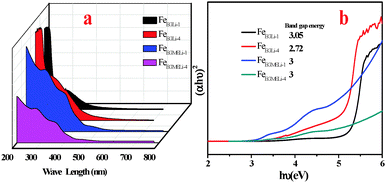 |
| | Fig. 10 (a) UV-Visible absorption spectra of the synthesized samples, (b) plot of (αhν)2versus photon energy for direct transition. | |
For sample FeEGLi-1, two absorption edges were observed with a sharp edge at 256 nm and 394 nm. The band gap energy 3.05 eV was estimated for the FeEGLi-1 sample, whereas the band gap energy of FeEGLi-4 slightly decreased as compared to FeEGLi-1 due to the higher percentage doping of lithium. However, the band gaps were found to be same for the samples obtained in EGME medium.
3.9 Electrochemical properties
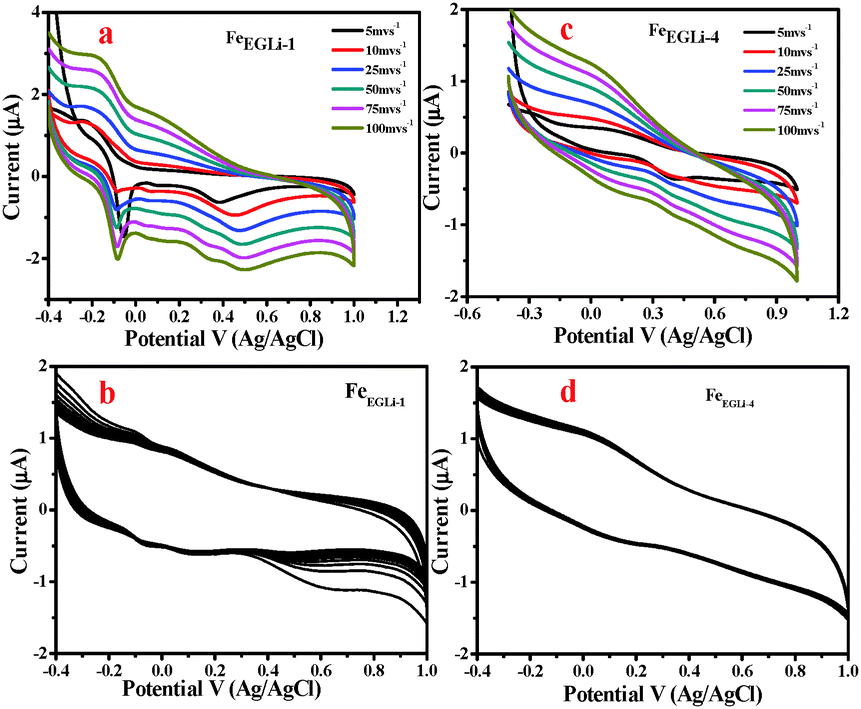
To evaluate rate capability, tests at different discharge currents were conducted for the Li doped synthesized nanomaterial. The electrochemical performances of the samples were evaluated by following the procedure described in the Experimental Section. Cyclic voltametric tests for the samples prepared in the presence of Li in EG and EGME media were carried out with the neutral electrolyte 0.1 M Na2SO4 in the range of −0.4–1 V and the results are shown in Fig. 11 and 12, respectively. The specific capacitance values were calculated using eqn (2).where I is the discharge current, Δt is the discharging time period in seconds, m is the mass of the electroactive materials and ΔV is the potential difference. 1D nanomaterials normally show excellent supercapacitance behaviour because possible electron transfer may occur through the 1D material. However, in the presence of lithium, both the phase and shape of the samples obtained in the studied solvent mediated precipitation routes are completely different. Herein EG medium, the as-synthesised LiFeO2 phase has a flowery shape without and with a prominent hole in the centre of the flower, which developed in low and high lithium concentration, respectively. This unusual shape of the materials along with their high surface area value may affect the electrochemical properties of the materials. Again, the alkali metal ion plays a vital role in the specific capacitance value. The redox coupling of iron oxide was developed with a lower % of Li in the matrix, whereas a higher % of Li lowered the specific capacitance value was observed. Two redox peaks were observed due to the reducing nature of the EG environment. Fe+2 to Fe+3 electron transfers occurred in the presence of Li+. The peak at −0.089 V corresponded to the Li redox coupling, whereas the peak at 0.4 V corresponded to the Fe redox. The pseudo capacitance behavior is due to the combined effect of the redox reactions. The pseudo capacitive behavior could also be due to the diffusion of electrolyte ions in the different channels present in the porous nanostructures.39 As given in Fig. 11, the pseudo capacitor nature of the CV curve changed to electric double layer with an increase in doping percentage. The FeEGLi-1 sample shows a specific capacitance of 241 F g−1. The peak observed at 0.5 V in the CV curves indicates Li ion intercalation in the iron matrix.40 Possible electrochemical reactions occurred in the α-LiFeO2 cathode due to the charge–discharge of Fe3+/Fe2+.40–42| | | Li1+x FeO2 → LiFeO2 + xLi+ + xe− | (3) |
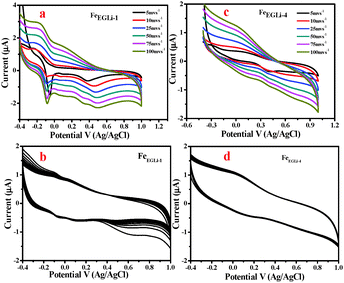 |
| | Fig. 11 (a) CV curve of FeEGLi-1 using 0.1 M Na2SO4 as the electrolyte, (b) CV of FeEGLi-1 for 500 cycles at 100 mV s−1, (c) CV curve of FeEGLi-4 using 0.1 M Na2SO4 as the electrolyte and (d) CV of FeEGLi-4 for 500 cycles at 100 mV s−1. | |
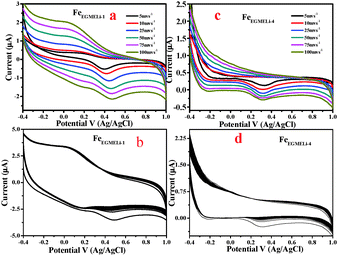 |
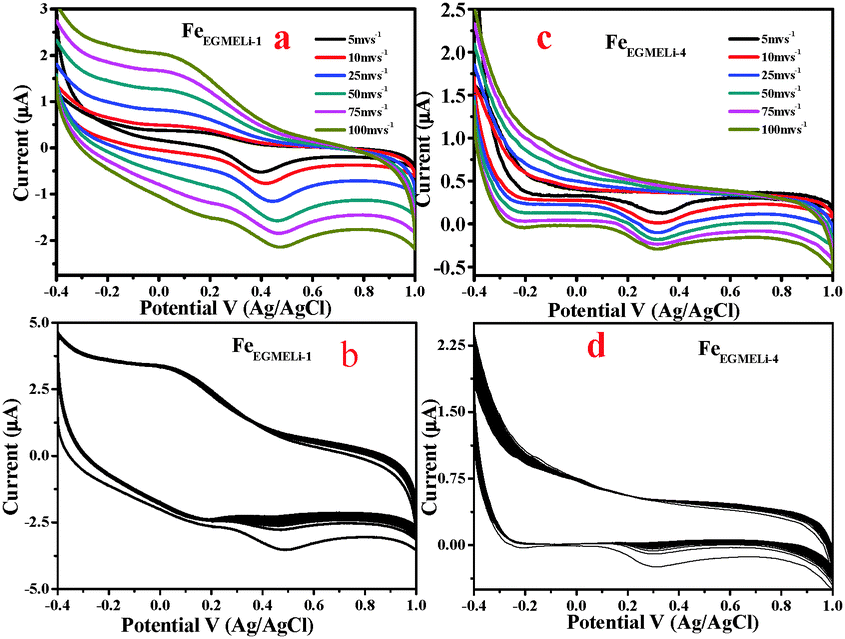
| | Fig. 12 (a) CV curve of FeEGMELi-1 using 0.1 M Na2SO4 as the electrolyte, (b) CV of FeEGMELi-1 for 500 cycles at 100 mV s−1 (c) CV curve of FeEGMELi-4 using 0.1 M Na2SO4 as the electrolyte, (d) CV of FeEGMELi-4 for 500 cycles at 100 mV s−1. | |
It is likely that due to this reason the α-LiFeO2 sample shows a higher specific capacitance value. However, the sample with more Li incorporation showed a lower specific capacitance value. A significant change in the CV curve was observed with the increase in % of Li, wherein the pseudo-capacitance nature of the CV curve changed to an electrical double layer nature CV curve. The capacitance behaviors of different materials are conditional to their cation characteristics and crystalline states,43–45i.e. the ionic electronegativity of constituent cations and closely related to the BET the surface area value. The specific capacitance values are mentioned in Fig. S3 (ESI†).
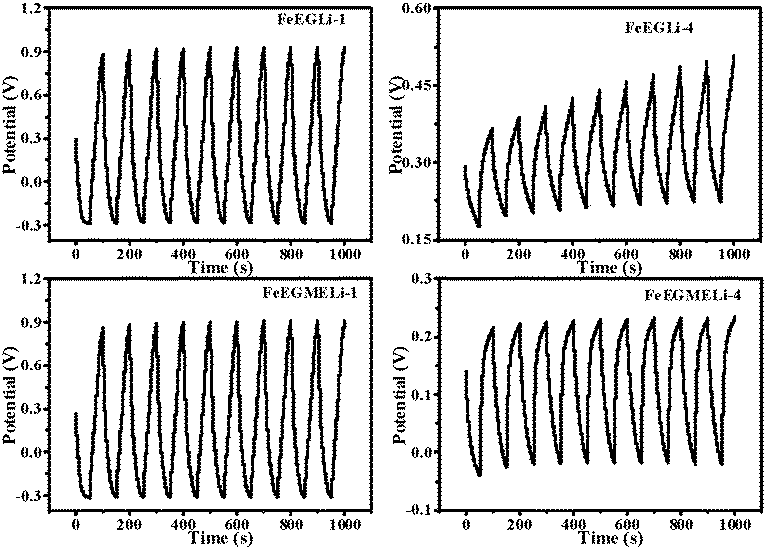
The cycling stability of the FeEGLi-1, FeEGLi-4, FeEGMELi-1 and FeEGMELi-4 electrodes were examined for 200 cycles at the specific current density of 5 mA g−1, as shown in Fig. 11(b and d) and 12(b and d). The FeEGLi-4 sample showed better super capacitance behavior with the retention percentage of 98% up to 200 cycles. The specific capacitance slightly decreased for the FeEGLi-4 sample, whereas for the FeEGLi-1 sample this value remained almost constant. Similar observations were made for the EGME sample (Fig. 12). In the EGME series, the FeEGMELi-1 sample showed better super capacitance behavior with a retention percentage of 98% up to 200 cycles. From the abovementioned observation, it can be concluded that the samples are affected by the % of lithium in the synthesized LiFeO2, irrespective of the solvent medium. The charge–discharge plots for the FeEGLi-1, FeEGLi-4, FeEGMELi-1 and FeEGMELi-4 samples at the current density of 5 mA g−1 are shown in Fig. 13. The discharge capacity remains constant for all the samples.
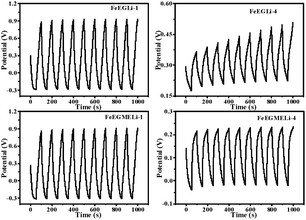 |
| | Fig. 13 Typical charge–discharge plots for the FeEGLi-1, FeEGLi-4, FeEGMELi-1 and FeEGMELi-4 samples at the current density of 5 mA g−1. The nature of the curve shows a linear variation of the voltage during the charging–discharging process. | |
3.10 Gas sensor study
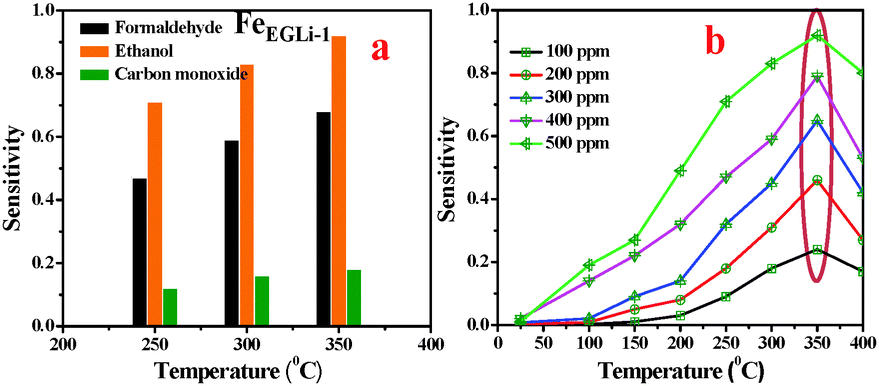
The gas sensing property of LiFeO2 may be developed due to electron jumping between the two valence states of iron, Fe2+ and Fe3+, on the octahedral sites in the spinel lattice structure.37 Considering the high surface area and inherent porosity gas sensing ability of FeEGLi-1 and FeEGMELi-1 samples, different toxic gases (HCHO, C2H5OH and CO) were studied at a concentration of 500 ppm at 250, 300, and 350 °C, and the results are shown in Fig. 14 and 15, respectively. It was observed that the selected samples exhibited the highest responses to ethanol, a good response towards formaldehyde and weak response for carbon monoxide. At 350 °C, the samples showed the sensitivity of 0.73 and 0.16 for 500 ppm, HCHO and CO, respectively, whereas for C2H5OH, the sensitivity was 0.92 for the FeEGLi-1 sample. Gas sensitivity was calculated using eqn (1). The response curve shown in Fig. 15 showed a parabolic-shape in the entire test range with the highest sensitivity (for 500 ppm) observed at 350 °C in the linear range. The excellent sensing properties for ethanol shown could be due to its porous structure associated with the small grain size, which enables C2H5OH gas to access more surfaces of the porous flowery material contained in the sensing unit. Therefore, the higher surface area of the nanostructure sample provides more chances to adsorb and desorb C2H5OH gas molecules, which lead to higher sensitivity at high temperatures.25,39 However, the FeEGLi-4 sample showed very negligible sensitivity towards toxic gases, which might be due to its smaller surface area.
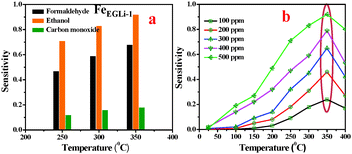 |
| | Fig. 14 Sensitivity of the sensors based on the FeEGLi-1 sample. (a) Comparative sensor performance for HCHO, C2H5OH and CO and (b) as a function of C2H5OH concentration at different temperatures. | |
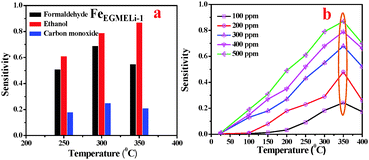 |
| | Fig. 15 Sensitivity of the sensors based on FeEGMELi-1. (a) Comparative sensor performance for HCHO, C2H5OH and CO, and (b) effect of C2H5OH concentration at different temperatures. | |
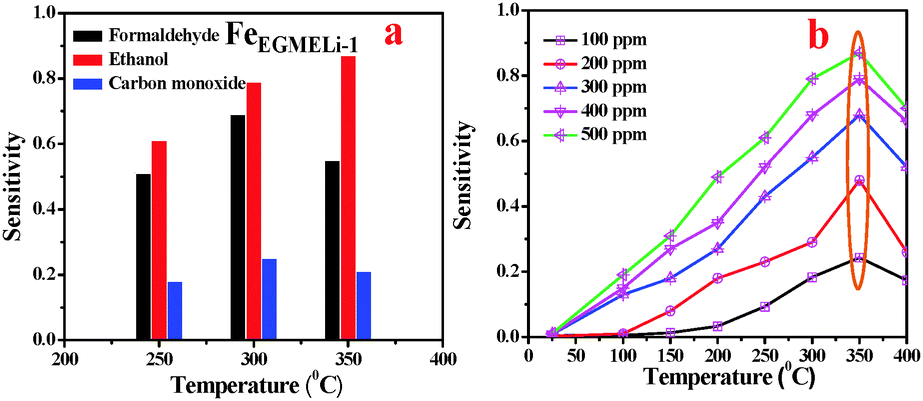
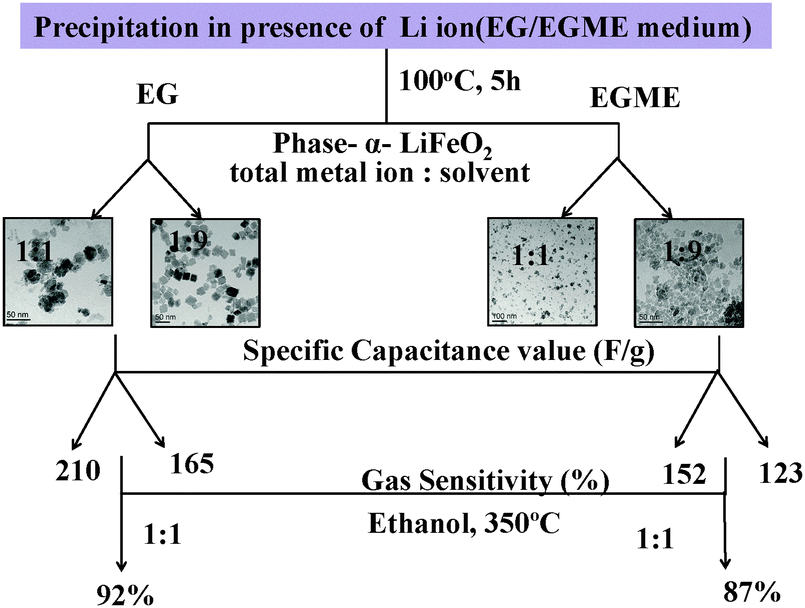
Similarly, the FeEGMELi-1 sample gas sensing behavior is shown in Fig. 15. It showed the sensitivity of 0.56 and 0.18 for 500 ppm at 350 °C for C2H5OH and CO, respectively, whereas for HCHO, the sensitivity was found to be 0.87. FeEGMELi-1 exhibited a maximum response to ethanol at 350 °C. According to the gas sensor, the principle diffusion of gases into the porous sample was independent of the working temperature, but dependant on the porosity within the sample. Similarly to FeEGLi-4, FeEGMELi-4 did not show better sensitivity towards any toxic gases. The present method offers a simple solvent mediated synthetic route for the preparation of the LiFeO2 phase, which can be utilized for multi-applications, as shown in Fig. 16. A comparison study of the synthetic routes and the application of LiFeO2 (Table S2, ESI†) was made with the reported literature synthesis methods to evaluate the novelty of our method.
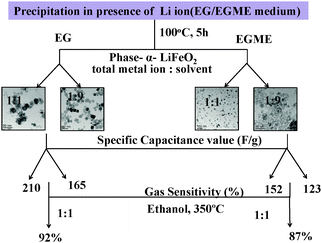 |
| | Fig. 16 Schematic of the results obtained from the EG and EGME mediated synthesis of iron oxide in the presence of Li. | |
4. Conclusions
In conclusion, α-LiFeO2 are synthesized for the first time-in EG and EGME solvent media at ambient temperature by varying the amount of LiOH. H2O and iron nitrate solution series of samples are prepared by varying the Li/Fe molar ratio from 1:9 to 1:1. Around 90% of the iron precipitated, irrespective of the lithium concentration and solvent, whereas the percentage of lithium precipitation decreased with an increase in Li content in the solution. α-LiFeO2 phases were developed irrespective of the Li:Fe ratio. The relative intensities (RI) of the (111) and (220) planes increased with the increase in % of Li for the EG mediated samples and the reverse effect was observed for the EGME mediated samples. In EG medium, depending on the concentration of Li, nano flowers with and without a hole at the central part of the flower were formed; whereas in the case of EGME, tiny nanoparticles up to 5 nm in size for FeEGMELi-1 were observed with a narrow size distribution. The charge–discharge behavior investigation of the prepared α-LiFeO2 nanoparticles as cathode materials showed that the sample obtained in EG medium has a higher specific capacitance value than the sample obtained EGME medium. The nature of the CV curve changed from pseudo capacitive to electrical double layer as the lithium content increased in the Li:Fe precursor solution. The FeEGLi-1 sample showed a sensitivity of 0.73 at 500 ppm for HCHO, whereas for C2H5OH, its sensitivity was 0.92. Similarly for the FeEGMELi-1 sample, the gas sensitivity of 0.56 and 0.18 was obtained for C2H5OH and CO (500 ppm), respectively, whereas for HCHO, its sensitivity was found to be 0.87. In both solvent media, the lithium containing samples showed better sensitivity for ethanol. Therefore, the present study provides the potentials for some of the synthesised samples as electrode and sensing materials for high performance devices.
Acknowledgements
The authors are thankful to the Prof. B. K. Mishra, Director, Institute of Minerals and Materials Technology, Bhubaneswar, for his kind permission to do the research. The authors wish to thank Dr I. N. Bhattacharya, Head Hydrometallurgy Department. One of the authors (Rasmita Barik) is thankful to DST Inspire Division (Govt. of India) for their financial support.
References
- K. Li, H. Chen, F. Shua, K. Chen and D. Xue, Electrochim. Acta, 2014, 136, 10–18 CrossRef CAS.
- K. Chen, S. Yin and D. Xue, Nanoscale, 2015, 7, 1161–1166 RSC.
- X. Chen, K. Chen, H. Wang and D. Xue, Electrochim. Acta, 2014, 147, 216–224 CrossRef CAS.
- X. Chen, K. Chen, H. Wang, S. Song and D. Xue, CrystEngComm, 2014, 16, 6707–6715 RSC.
- K. Y. Li, J. J. Shao and D. F. Xue, Mater. Res. Innovations, 2013, 17, 218–223 CrossRef CAS.
- K. Li, H. Chen, F. Shua, D. Xue and X. Guo, RSC Adv., 2014, 4, 36507–36512 RSC.
- B. Koo, H. Xiong, M. D. Slater, V. B. Prakapenka, M. Balasubramanian, P. Podsiadlo, C. S. Johnson, T. Rajh and E. V. Shevchenko, Nano Lett., 2012, 12, 2429–2435 CrossRef CAS PubMed.
- D. W. Junga, S. W. Hana, B. S. Kong and E. S. Oha, J. Power Sources, 2013, 242, 357–364 CrossRef.
- Y. Han, Y. J. Wang, L. Li, Y. P. Wang, L. F. Jiao, H. T. Yuan and S. X. Liu, Electrochim. Acta, 2011, 56, 3175–3181 CrossRef CAS.
- A. E. Abdel-Ghany, A. Mauger, H. Zaghib and C. M. Julien, J. Power Sources, 2012, 197, 285–291 CrossRef CAS.
- Y. Sakurai, H. Arai, S. Okada and J. I. Yamaki, J. Power Sources, 1997, 68, 711–715 CrossRef CAS.
- L. Croguennec, P. Deniard and R. Brec, J. Electrochem. Soc., 1997, 144, 3323–3330 CrossRef CAS.
- S. Zeng, K. Tang, T. Li, Z. Liang, D. Wang, Y. Wang, Y. Qi and W. Zhou, J. Phys. Chem. C, 2008, 112, 4836–4843 CrossRef CAS.
- J. Wang, Y. Zhou, Y. Hu, R. O'Hayre and Z. Shao, J. Mater. Sci., 2013, 48, 2733–2742 CrossRef CAS.
- R. Barik, B. Pandey, S. Anand and M. Mohapatra, J. Mater. Chem. A, 2014, 2, 12380–12389 RSC.
- Y. Ma, Y. Zhu, Y. Yu, T. Mei, Z. Xing, X. Zhang and Y. Qian, Int. J. Electrochem. Sci., 2012, 7, 4657–4662 CAS.
- X. Wang, L. Gao, F. Zhou, Z. Zhang, M. Ji, C. Tang, T. Shen and H. Zheng, J. Cryst. Growth, 2004, 265, 220–223 CrossRef CAS.
- R. Collongues and G. Chaudron, C. R. Acad. Sci., 1950, 124, 143 Search PubMed.
- Y. Wang, J. Wang, H. Liao, X. Qian, M. Wang, G. Song and S. Cheng, RSC Adv., 2014, 4, 3753–3757 CAS.
- A. M. Rahman, A. M. Glushenkov, Z. Chen, X. J. Dai, T. Ramireddy and Y. Chen, Phys. Chem. Chem. Phys., 2013, 15, 20371–20378 RSC.
- Y. Wang, J. Wang, H. Liao, X. Qian, Y. Zhu and S. Cheng, Int. J. Electrochem. Sci., 2013, 8, 8730–8739 CAS.
- S. A. Mazen, M. H. Abdallah, R. I. Nakhla, F. Metawe and H. M. Zaki, Mater. Chem. Phys., 1993, 34, 35–40 CrossRef CAS.
- K. S. Lee, J. H. Ko and V. H. Schmidt, J. Korean Phys. Soc., 2005, 46, 198–205 Search PubMed.
- I. V. Chernyshova and A. S. Madden, Phys. Chem. Chem. Phys., 2007, 9, 1736–1750 RSC.
- P. Makie, G. Westin, P. Persson and L. Osterlund, J. Phys. Chem. A, 2011, 115, 8948–8959 CrossRef PubMed.
- S. H. Shim and T. S. Duffy, Am. Mineral., 2002, 87, 318–326 CrossRef CAS.
- A. M. Jub and H. C. Allen, ACS Appl. Mater. Interfaces, 2010, 2, 2804–2812 Search PubMed.
- J. A. Glasscock, P. R. F. Barnes, I. C. Plumb, A. Bendavid and P. J. Martin, Thin Solid Films, 2008, 516, 1716–1724 CrossRef CAS.
- R. Barik, S. K. Tripathy and M. Mohapatra, J. Mater. Sci., 2014, 49, 5345–5354 CrossRef CAS.
- D. Bersani, P. P. Lottici and A. Montenero, J. Raman Spectrosc., 1999, 30, 355–360 CrossRef CAS.
- V. I. Tyutyunnik, J. Raman Spectrosc., 2000, 31, 559–563 CrossRef CAS.
- X. Fei, Z. Z. Sao and X. Chen, J. Mater. Chem. B, 2013, 1, 213–220 RSC.
- C. Z. Wu, Y. Xie, L. Y. Lei, S. Q. Hu and C. O. Yang, Adv. Mater., 2006, 18, 1727–1732 CrossRef CAS.
- G. Carraro, D. Barreca, E. Comini, A. Gasparotto, C. Maccato, C. Sadad and G. Sberveglieri, CrystEngComm, 2014, 14, 6469–6476 RSC.
- T. Fujii, F. M. F. de Groot and G. A. Sawatzky, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59(4), 3195 CrossRef CAS.
- L. Ferretto and A. Glisenti, J. Mol. Catal. A: Chem., 2002, 187, 119–128 CrossRef CAS.
- F. Martín, E. Navarrete, J. Morales, C. Roldan, J. R. Ramos-Barradob and L. Sanchez, J. Mater. Chem., 2010, 20, 2847–2852 RSC.
- I. V. Chernyshova, S. Ponnurangam and P. Somasundaran, Phys. Chem. Chem. Phys., 2010, 12, 14045–14056 RSC.
- D. K. Padhi and K. M. Parida, J. Mater. Chem. A, 2014, 2, 10300–10312 RSC.
- X. Zhang, W. Shi, J. Zhu, W. Zhao, J. Ma, S. Mhaisalkar, T. L. Maria, Y. Yang, H. Zhang, H. H. Hng and Q. Yan, Nano Res., 2010, 3, 643–652 CrossRef CAS.
- J. Chen, L. Xu, W. Li and X. Gou, Adv. Mater., 2005, 17, 582–586 CrossRef CAS.
- R. Kanno, T. Shirane, Y. Kawamoto, Y. Takeda, M. Takano, M. Ohashi and Y. Yamaguchi, J. Electrochem. Soc., 1996, 143, 2435–2442 CrossRef CAS.
- K. F. Chen and D. Xue, Sci. China: Technol. Sci., 2015, 58, 1768–1778 CAS.
- K. F. Chen and D. Xue, Journal of Materiomics, 2015, 1, 170–187 CrossRef.
- K. F. Chen, S. Song and D. Xue, RSC Adv., 2014, 4, 23338–23343 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nj02058a |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.