DOI:
10.1039/C5NJ02322J
(Paper)
New J. Chem., 2016,
40, 464-474
Gel properties of T-shaped tetrathiafulvalene–pyridazine conjugates and F4TCNQ-induced morphological transformation†
Received
(in Montpellier, France)
31st August 2015
, Accepted 31st October 2015
First published on 6th November 2015
Abstract
A new series of T-shaped organic π-conjugate organogelators, monopyridazine–TTFs, were synthesized and characterized. All of the gelators could gelate the aromatic solvents, such as benzene, toluene and chlorobenzene, and the length of the alkyl chain had a certain influence on the gelation behavior. Interestingly, the gelators could react with 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4TCNQ) to form the charge-transfer complex organogels. FT-IR, UV-Vis, 1H NMR and CV experiments revealed that the cooperation of hydrogen bonding, π–π, and CT interactions was the main driving force for the formation of the native and CT gels. The FE-SEM images of the native gels reveal the characteristic gelation morphologies of fibrous structures, whereas the morphologies of CT complex gels show the amorphous block aggregates. XRD study of the native gel and the CT complex gel of 1b in DMF suggests that the molecules maintain lamellar and hexagonal columnar molecular packing models in the gel phase, respectively. Furthermore, the DMF gel of 1b could be destroyed only by the addition of the fluorine ions, while the toluene gel could be transformed into solution or suspension after adding halogen ions.
Introduction
Organic π-conjugated molecules, such as phthalocyanine,1 acene,2 anthracene,3 perylene,4 hexabenzocoronene,5 bisphenazine,6 tetrathiafulvalene,7 and so on, have been the focus of functional materials research in various application fields such as light-harvesting systems,8 photo-induced electron-transfer systems,9 organic light-emitting diodes,10 organic thin-film transistors,11 and solar cells.12 Self-assembly of small molecules has been extensively utilized as an efficient strategy to construct and modulate nanostructures in the past few decades.13 Amongst the soft assemblies, gels play an important role as materials for application in a number of areas including tissue engineering,14 drug delivery,15 modification agents for paints,16 the template synthesis of nanomaterials,17 catalysis of organic synthesis,18 separation,19 biological applications,20 controlling of crystal growth,21 and so on. Supramolecular gels, especially low-molecular-mass organogels (LMOGs), are a new class of materials in supramolecular chemistry, in which organogelators spontaneously self-assemble through noncovalent interactions, such as hydrogen bonding, π–π stacking, hydrophobic interactions, van der Waals forces, electrostatic interactions, charge transfer interactions, metal ion to ligand coordination, and so forth,22 to create three-dimensional networks capable of entrapping solvents therein. Due to the reversible nature of interactions, these LMOGs are responsive and tunable. In addition to thermal reversibility, these gels are responsive to stimuli such as light,23 ultrasound,24 pH,25 enzymes,26 mechanical stress,27 ionic strength,28 changes in the redox state,29 and so forth. Despite a large number of reports, creating new π-conjugated LMOGs is nontrivial by itself, and the generation of new functional supramolecular structures through the self-assembly of small organic molecules continues to be a significant and challenging scientific endeavor.
Tetrathiafulvalene (TTF), a strong electron-donor, has been widely studied in the research field of field-effect transistors,7b,30 switches,31 conductors,32 sensors,33 liquid crystalline materials,34 and so on. The strongly π-donating tetrathiafulvalene (TTF) core has recently appeared at the forefront of these topics, as a result of its ability to allow sequential formation of two stable oxidized states (TTF˙+ and TTF2+). The TTFs have been frequently used in many self-assembly aggregates with a close contact in between the π surfaces by using hydrogen bonds, and to form nanoscale fibers in a controlled way,35 one example being that of amide bonds which aid the formation of stacks of molecules.36 On the other hand, fibers have been formed in Langmuir films and gels with a random orientation,37 and gels of TTFs have shown some alignment of the fibers.38 Recently, electroactive fibrillar nanowires prepared from the functionalization of the TTF organogel were shown to have potential for conductivity at room temperature after doping with iodine.38a,39 Zhang et al. utilized the redox properties of the TTF moiety to tune the gel–sol transition behaviors.40 Results indicate that the use of intermolecular noncovalent interactions via a self-assembly process to build TTF-based supramolecular organizations is beneficial. More interestingly, the combination of TTF and electron acceptor charge transfer (CT) salt is recognized as an advantageous strategy of molecular design, and its conductivity has been studied extensively.41 For example, Shirai et al. have demonstrated that the amphiphilic TTFs can form conductive helical nanofibers by the formation of CT complexes with F4TCNQ and intermolecular hydrogen bonding among the chiral amide end groups.42 Recently, our group have explored the multiple stimulus responsive organogels based on monopyrrolotetrathiafulvalene (MPTTF) with different numbers of amide units and its CT complex.43 Taking a broad view of the previous reports, however, most of the TTF-based organogelators are linear rod molecules. Recently, we introduced successfully a pyridazine ring to the TTF skeleton to synthesize a series of monopyridazine–TTF conjugates. Their HOMO energy levels suggest that the conjugates can be considered as candidates for hole transporting organic semi-conductor materials.44 Annulation of the pyrazine ring to the TTF skeleton was effective in further reducing the electron-donating property and enhancing the stability in the air. To the best of our knowledge, however, pyridazine-annulated TTF derivatives have not yet been tested as gelators.
In the present work, we have reported the successful synthesis of a novel series of T-shaped organic π-conjugates LMOGs 1a–c with three aryl rings containing different lengths of alkyl chains in the TTF terminal (Scheme 1). These compounds showed the typical redox chemistry for this family of compounds and formed thermoreversible and stable organogels in aromatic solvents. Furthermore, due to the presence of alkyl chains of different length in the TTF terminal they showed obvious differences in the gelling ability. For example, the gelator 1b which contains n-dodecyl could gelatinize DMF and had the lowest critical gelation concentration (CGC) of 1.2 mg mL−1. Also, the organogels exhibited a quick gel–sol transition according to the stimulus of the fluorine ion. More interestingly, 1a–c could form stable binary gels with an electron acceptor (F4TCNQ) through charge-transfer and hydrogen-bonding interactions.
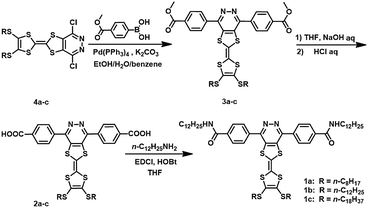 |
| | Scheme 1 The synthetic route and molecular structures of gelators 1a–c. | |
Experimental section
Synthesis
All solvents were purified by the standard procedures before use. p-(Methoxycarbonyl)phenylboronic acid, Pd(PPh3)4, 1-(3-(dimethylamino)propyl)-3-ethyl-carbodiimide hydrochloride (EDCI) and 1H-benzo[d][1,2,3]triazol-1-ol (HOBT) were purchased from Adamas, and were used as received without further purification. Compounds 4a–c were synthesized according to the literature method reported by our group.44
Typical procedure for compounds 3
A mixture of 4 (0.21 mmol), p-(methoxycarbonyl)phenyl-boronic acid (0.82 mmol), Pd(PPh3)4 (0.10 mmol) and K2CO3 (1.48 mmol) in a mixture of EtOH–H2O–C6H6 at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2 (10 mL) was stirred under N2 at 90 °C for 12 h. After cooling, the resulting mixture was dissolved in CH2Cl2, washed with water and dried over anhydrous Na2SO4. After evaporation of the solvent, the crude product was purified by column chromatography on silica gel using CH2Cl2:ethyl acetate at 30:1 to give a yellow solid.
:1:2 (10 mL) was stirred under N2 at 90 °C for 12 h. After cooling, the resulting mixture was dissolved in CH2Cl2, washed with water and dried over anhydrous Na2SO4. After evaporation of the solvent, the crude product was purified by column chromatography on silica gel using CH2Cl2:ethyl acetate at 30:1 to give a yellow solid.
Compound 3a.
Recrystallization from ethyl acetate gave a yellow flake solid, yield 58.6%. m.p.: 136–138 °C. 1H NMR (300 MHz, CDCl3): δ 8.26 (d, J = 8.16 Hz, 4H, Ar), 8.01 (d, J = 8.01 Hz, 4H, Ar), 3.99 (s, 6H, CH3), 3.99 (t, J = 7.41 Hz, 4H, CH2), 1.27–1.64 (m, 24H, CH2), 0.87 (t, J = 6.84 Hz, 6H, CH3); 13C NMR (75 MHz, CDCl3) δ 166.43, 151.41, 140.90, 140.38, 131.79, 130.25, 128.11, 128.00, 127.84, 52.50, 36.41, 31.88, 29.74, 29.17, 28.57, 22.69, 14.19; MALDI-TOF MS m/z calcd for C40H48N2O4S6: 813.21. Found: 813.2 (M+, 100).
Compound 3b.
Recrystallization from ethyl acetate gave a yellow flake solid, yield 54.8%. m.p.: 107–108 °C. 1H NMR (300 MHz, CDCl3): δ 8.25 (d, J = 8.4 Hz, 4H, Ar), 8.00 (d, J = 8.4 Hz, 4H, Ar), 3.98 (s, 6H, CH3), 2.81 (t, J = 7.5 Hz, 4H, CH2), 1.24–1.72 (m, 40H, CH2), 0.87 (t, J = 6.57 Hz, 6H, CH3); 13C NMR (75 MHz, CDCl3) δ 166.38, 151.44, 140.95, 140.38, 131.76, 130.24, 128.12, 127.96, 127.85, 52.44, 36.41, 31.92, 29.66, 29.36, 29.13, 28.48, 22.70, 14.21; MALDI-TOF MS m/z calcd for C48H64N2O4S6: 925.42. Found: 925.2 (M+, 100).
Compound 3c.
Recrystallization from ethyl acetate gave a yellow flake solid, yield 40.4%. m.p.: 94–95 °C. 1H NMR (300 MHz, CDCl3): δ 8.27 (d, J = 6.72 Hz, 4H, Ar), 8.02 (d, J = 6.72 Hz, 4H, Ar), 4.00 (s, 6H, CH3), 2.83 (t, J = 7.2 Hz, 4H, CH2), 1.26–1.65 (m, 66H, CH2), 0.90 (t, J = 6.42 Hz, 6H, CH3);13C NMR (75 MHz, CDCl3) δ 166.38, 151.46, 140.92, 140.45, 131.74, 130.24, 128.11, 128.06, 127.86, 52.44, 36.41, 31.94, 29.61, 29.38, 29.14, 28.48, 22.71, 14.15; MALDI-TOF MS m/z calcd for C60H88N2O4S6: 1093.74. Found: 1093.5 (M+, 100).
Typical procedure for compounds 2
A mixture of 3 (0.039 mmol) and NaOH (4 M, 8 mL) aqueous solution in THF (8 mL) was stirred at 50 °C overnight. After cooling and evaporation of the solvent, the pH was adjusted to 2–3. Thereafter, the mixture was filtered and washed with water. The filter cake was dried in vacuum to obtain a brownish yellow solid which was used in the next step without purification because of the low solubility.
Typical procedure for compounds 1
The mixture of 2 (0.06 mmol), EDCI (0.13 mmol), HOBt (0.13 mmol) and n-dodecylamine (0.12 mmol) was suspended in THF (10 mL). The reaction mixture was stirred at room temperature for one day. The solvent was eliminated and the residue was taken up in CH2Cl2. The solution was successively washed with aqueous HCl (1 M), aqueous NaOH (1 M) and brine. After drying and filtering, the solvent was eliminated and the residue was purified by column chromatography on silica gel using CH2Cl2:ethyl acetate at 5:1 to give a yellow solid.
Compound 1a.
Recrystallization from ethyl acetate gave a yellow powder, yield 71.4%, m.p.: 169–170 °C. 1H NMR (300 MHz, CDCl3): δ 7.94 (s, 8H, Ar), 6.31 (t, J = 5.28 Hz, 2H, NH), 3.50 (q, J = 6.57 Hz, 4H, CH2), 2.80 (s, 4H, CH2), 1.58–1.68 (m, 8H, CH2), 1.27–1.38 (m, 58H, CH2), 0.87 (t, J = 6.33 Hz, 12H, CH3); 13C NMR (75 MHz, CDCl3) δ 166.78, 151.34, 140.98, 138.77, 136.60, 128.32, 127.79, 127.59, 117.14, 40.33, 36.41, 31.93, 31.79, 29.68, 29.38, 29.08, 28.49, 26.09, 22.71, 14.15; MALDI-TOF MS m/z calcd for C62H94N4O2S6: 1119.83. Found: 1119.7 (M+, 100); elemental analysis calcd for C62H94N4O2S6: C 66.50, H 8.46, N 5.00; found: C 66.72, H 8.81, N 4.85.
Compound 1b.
Recrystallization from ethyl acetate gave a yellow powder, yield 56.0%, m.p.: 160–161 °C. 1H NMR (300 MHz, CDCl3): δ 7.96 (s, 8H, Ar), 6.22 (t, J = 5.49 Hz, 2H, NH), 3.50 (q, J = 6.48 Hz, 4H, CH2), 2.80 (t, J = 7.14 Hz, 4H, CH2), 1.27–1.58 (m, 16H, CH2), 1.24–1.27 (m, 64H, CH2), 0.89 (t, J = 6.39 Hz, 12H, CH3); 13C NMR (75 MHz, CDCl3) δ 166.88, 151.30, 140.73, 138.81, 136.57, 128.22, 127.78, 127.57, 116.77, 40.35, 36.39, 31.93, 29.64, 29.53, 29.37, 29.15, 28.51, 27.10, 22.70, 14.13; MALDI-TOF MS m/z calcd for C70H110N4O2S6: 1232.04. Found: 1231.9 (M+, 100); elemental analysis calcd for C70H110N4O2S6: C 68.24, H 9.00, N 4.55; found: C 68.45, H 9.26, N 4.29.
Compound 1c.
Recrystallization from ethyl acetate gave a yellow powder, yield 43.0%, m.p.: 162–163 °C. 1H NMR (300 MHz, CDCl3): δ 7.95 (s, 8H, Ar), 6.27 (t, J = 5.01 Hz, 2H, NH), 3.50 (q, J = 6.15 Hz, 4H, CH2), 2.80 (t, J = 6.81 Hz, 4H, CH2), 1.58–1.68 (m, 8H, CH2), 1.25–1.27 (m, 96H, CH2), 0.88 (t, J = 4.86 Hz, 12H, CH3); 13C NMR (75 MHz, CDCl3)δ 166.57, 151.20, 140.96, 138.64, 136.47, 128.13, 127.43, 108.46, 40.17, 36.28, 31.80, 29.59, 29.55, 29.24, 29.02, 28.36, 26.93, 22.57, 14.02; MALDI-TOF MS m/z calcd for C82H134N4O2S6: 1400.36. Found: 1400.0 (M+, 100). Elemental analysis calcd for C82H134N4O2S6: C 70.33, H 9.64, N 4.00; found: C 70.52, H 9.85, N 3.89.
Gelation study
A measured amount of gelator 1 with or without F4TCNQ and a measured volume of the solvent were placed in a sealed test tube and heated to obtain a clear solution. And then, the system was left at room temperature. The transition temperatures (Tgel) were determined by the ball-drop method. An inverted gel was immersed in a water bath initially at or below room temperature, and then was heated (about 2 °C min−1) slowly up to the point at which the gel fell due to the force of gravity, i.e. the Tgel.
NMR experiments
All solution state NMR studies were carried out on a Bruker AV-300 Spectrometer (300 MHz for 1H and 75 MHz for 13C) in deuterated solvents and chemical shifts were referenced relative to tetramethylsilane (δHδC = 0). All chemical shifts (δ) are quoted in ppm, and coupling constants (J) are given in Hz.
FT-IR spectroscopy
IR spectra were recorded on a Shimadzu FT-IR Prestige-21 instrument with the KBr disk technique. The CHCl3 solutions of gelators were loaded between two KBr windows and xerogels of native or CT complex were ground with KBr mixed together.
MALDI-TOF-MS spectrometry
Mass spectra were performed on a Shimadzu Axima CFRTM Plus using a 1,8,9-anthracenetriol (DITH) and β-phenylacrylic acid (CHCA) matrix.
Cyclic voltammetry
Cyclic voltammetry was performed on a CHI660D instrument in benzonitrile (10−3 M) with 0.1 M Bu4NPF6 as the supporting electrolyte and at a scan rate of 100 mV s−1. Counter and working electrodes were made of Pt and glass carbon, respectively, and Ag/AgCl was used as the reference electrode. For xerogel measurements, a small amount of the gel or CT gel was carefully put on the glass carbon electrode, which was left in air for 24 h. This modified glass carbon electrode (as working electrode), together with Pt as the counter electrode and Ag/AgCl as the reference electrode, was put into the benzonitrile solution containing 0.1 M Bu4NPF6, and the cyclic voltammograms were recorded at a scanning rate of 100 mV s−1.
UV-Vis spectroscopy
UV-vis spectra were recorded on a Hitachi U-3010 spectrophotometer. The measurable wavelength range was 220–1000 nm. Samples were prepared in quartz cuvettes with 1 cm path length.
Field emission scanning electron microscopy
The gel samples were placed on silicon wafer, and dried for 24 h under room temperature before imaging. A layer of gold was sputtered on the top to form a conducting surface and finally the specimen was transferred into a field emission scanning electron microscope (FE-SEM, Joel Scanning Microscope-JSM-6700F).
X-ray diffraction
Small-angle X-ray diffraction (SAXRD) measurements were carried out at 298 K on a beamline 1W2A synchrotron radiation X-ray small angle system at Beijing Synchrotron Radiation Facility (λ = 1.54 Å). Wide-angle X-ray diffraction (WAXRD) measurements were carried out at 298 K on the glass-sustained xerogel films and recorded on a Bruker D8/ADVANCE X-ray diffractometer (Germany) with radiation (λ = 1.54 Å).
TGA experiments
Thermogravimetric analysis (TGA) was performed by using a DTG60 at a heating rate of 10 °C min−1 under a N2 atmosphere. The solid samples were put in the TGA aluminium pan and heated from room temperature to 400 °C.
Results and discussion
Gelation behaviors
The gelation ability of T-shaped organic conjugates 1a–c was examined in 15 different organic solvents through a standard heating and cooling method. As summarized in Table 1, the gelation test indicated that these compounds could all immobilize aromatic solvents, such as benzene, toluene and chlorobenzene, and form transparent organogels. By contrast, these compounds could not dissolve in acetonitrile and formed a precipitate in ethyl acetate and DMSO, as well as formed homogeneous solutions in haloalkanes (CH2Cl2, CHCl3 and CCl4). From the results, we also found that the gelation capability of these conjugates was dependent on the length of the alkyl chain in the TTF terminal. For instance, 1a containing n-octyl could effectively gelatinize cyclohexane to form a yellow transparent gel, 1b containing n-dodecyl could form an opaque gel in DMF at the lowest CGC of 1.2 mg mL−1, and 1c containing n-octadecyl formed a transparent gel in n-hexane (Fig. 1). What is more, these gels were thermo-reversible and once formed the gel was stable at ambient temperature for at least a few weeks without crystallization or melting.
Table 1 Gelation properties of 1a–c in various solventsa
| Solvent |
1a
|
1b
|
1c
|
|
TG, transparent gel; OG, opaque gel; P, precipitate; I, insoluble; S, solution. The number is the CGC in mg mL−1.
|
| Cyclohexane |
TG, 11.5 |
S |
S |
|
n-Hexane |
S |
I |
TG, 2.4 |
| Benzene |
TG, 9.2 |
TG, 8.5 |
TG, 9.8 |
| Toluene |
TG, 7.7 |
TG, 21.5 |
TG, 9.8 |
| Chlorobenzene |
TG, 16.5 |
TG, 17 |
TG, 4.9 |
| Ethyl acetate |
P |
P |
P |
| Ethanol |
S |
P |
I |
| Acetone |
P |
P |
P |
| DMF |
P |
OG, 1.2 |
S |
| Acetonitrile |
I |
I |
I |
| Tetrahydrofuran |
S |
S |
S |
| DMSO |
P |
P |
P |
| Dichloromethane |
S |
S |
S |
| Chloroform |
S |
S |
S |
| Tetrachloromethane |
S |
S |
S |
 |
| | Fig. 1 The gel photographs of gelators (a) 1a–c in toluene and (b) 1a in cyclohexane, 1b in DMF and 1c in n-hexane. | |
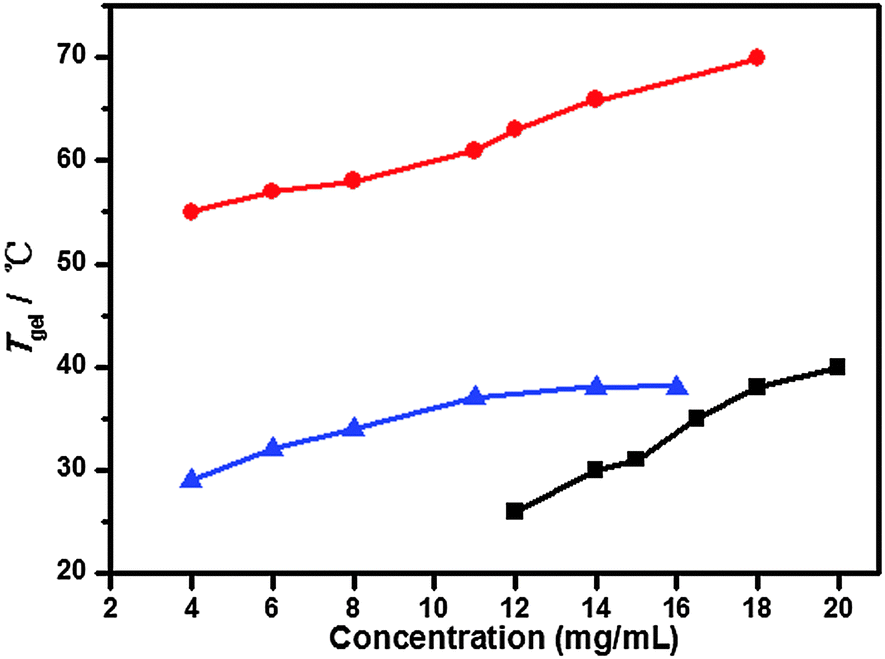
To investigate the thermal stability of the resulting gel phase material, the gel–sol phase transition temperature (Tgel) was determined through a conventional ball-drop method. As shown in Fig. 2, the Tgel values of gelators having different chain length 1a–c at different concentrations in toluene, DMF and n-hexane, respectively, were determined to examine their thermostability. The results showed that the Tgel values increased with increasing gelator concentration in three systems, indicating that the gels had more stable supramolecular structures at higher concentrations.
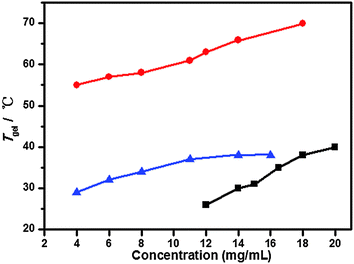 |
| | Fig. 2 Plots of Tgelversus the organogel concentration of gelators 1a in toluene (■), 1b in DMF (●) and 1c in n-hexane (▲). | |
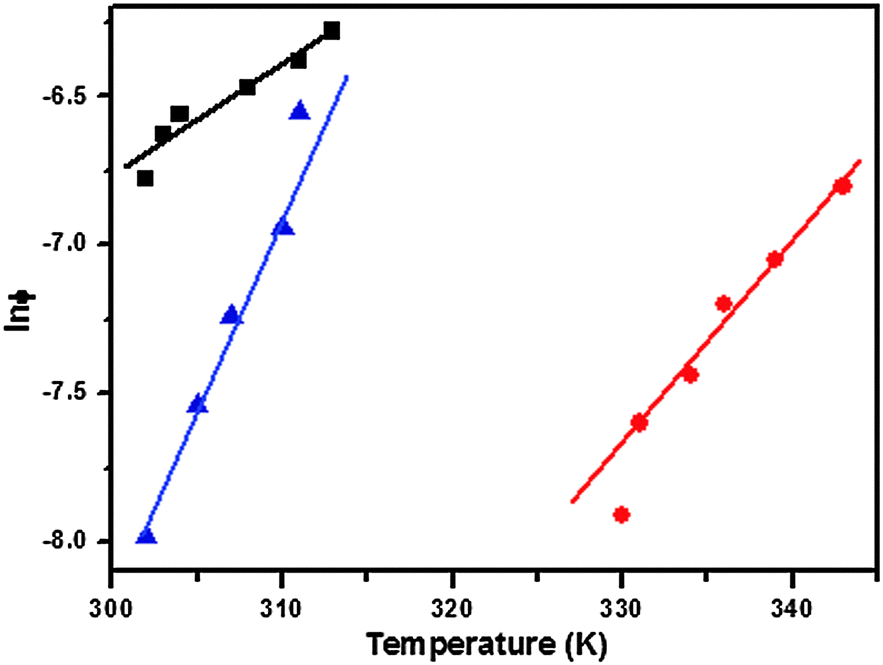
To determine further whether the formation of gels was a spontaneous process, a linear dependence of the melting temperature on the natural logarithm of the mole fraction of the gelator was observed (Fig. 3). The thermodynamic parameters of gelation were calculated using eqn (1)–(3) as follows:45
| | | ΔG° = RTln(ϕ) | (1) |
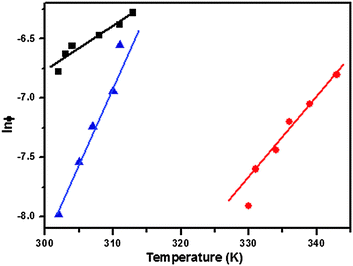 |
| | Fig. 3 ln ϕ vs. T plots of organogels of gelators 1a in toluene (■), 1b in DMF (●) and 1c in n-hexane (▲). | |
| | |
ΔH° = −RT2 [δln(ϕ)/δT]
| (2) |
| | |
ΔS° = −Rln(ϕ) − RT [δln(ϕ)/δT]
| (3) |
Here, ϕ (mole fraction of gelator) = a/(a + b), where a is the number of moles of the gelator, b is the number of moles of solvent, and T is the corresponding gel melting temperature. The value of the slope [δln(ϕ)/δT] in these equations was calculated and is shown in Fig. 3. The calculated enthalpy (ΔH°) and entropy (ΔS°) for the sol–gel transformation are shown in Table 2. The negative values of the Gibbs free energy (ΔG°) showed that gel formation was a spontaneous process.
Table 2 The calculated thermodynamic parameters of gelation
| Parameters |
1a/toluene |
1b/DMF |
1c/n-hexane |
|
δln(ϕ)/δT |
0.035 |
0.078 |
0.12 |
| ΔG° (kJ mol−1 K−1) |
−16.702 |
−19.870 |
−20.306 |
| ΔH° (kJ mol−1) |
−26.716 |
−69.767 |
−90.992 |
| ΔS° (J mol−1 K−1) |
−33.048 |
−143.728 |
−234.953 |
An advantageous property of the donor–acceptor system is its ability to assemble and form a stable CT complex gel without forming a precipitate, which enhances the dimensionality of the electronic structure by noncovalent interactions.46 With the electron-active gelators 1a–c in hand, we first explored the gelation behaviour of their CT complexes with TCNQ in the tested solvents. But unfortunately, no CT complexes were formed, indicating that introduction of a pyrazine ring to the TTF skeleton was effective in further reducing the electron-donating property compared with the previous reports of MPTTF.34 To our delight, the CT complex gels were formed when a stronger oxidant F4TCNQ was added (Fig. 4). Furthermore, as seen in the solution system, we also measured the UV-vis spectra upon addition of 1 equiv. of TCNQ and F4TCNQ to the 1b solution in CHCl3 (1 mM), respectively. Only addition of F4TCNQ exhibited three new absorption bands, centred at λmax 676 nm, corresponding to the TTF radical cation, and at 759 and 865 nm, corresponding to the progressive formation of the F4TCNQ radical anion (Fig. S1, ESI†).47 In fact, when 1 equivalent of F4TCNQ was directly added on the top of a DMF gel of 1b, the gel was gradually destroyed after several minutes, resulting in a brown solution under heating. Upon cooling, the solution changed to a brown CT complex gel. We also adopted the most effective gelation by using a 1:1 stoichiometry of the two components.34a However, the CGC of the CT complex gel (2.0 mg mL−1) was higher than that of the native gel of 1b in DMF (1.2 mg mL−1), and the Tgel was slightly elevated (from 29 to 31 °C). This finding implied that the stability of CT gel was higher than that of the native gel. The formation of the CT complex gel indicated that some driving forces for gelation were changed from S⋯S and π–π interactions between gelators to the CT interaction of TTF with F4TCNQ. Similar experimental results were also found in the toluene gels (Fig. S2, ESI†).
 |
| | Fig. 4 Tuning the gel formation of gelator 1b by the addition of 1.0 equiv. of F4TCNQ in DMF. | |
Next, the thermal stability of the conjugates was measured through thermogravimetric analysis (TGA) to evaluate the potential of the conjugates to act as soft materials. The results are shown in Fig. S3 (ESI†). The temperatures were higher than 300 °C when 5% weight loss occurred, which revealed that the target T-shaped conjugates had high thermal stability.
Morphological characterization
To investigate the morphology of organogels, dried samples of transparent gel 1a from toluene, opaque gel 1b from DMF and transparent gel 1c from n-hexane were subjected to field emission scanning electron microscopy (FE-SEM) measurements. As shown in Fig. 5, the SEM image of the dried gel of 1a from toluene appears much like an amorphous-like surface at the micrometer level and shows a wrinkled matrix morphology.48Fig. 5b shows that the native xerogel of 1b from DMF formed three-dimensional cross-linking networks of 100–600 nm width. When checking the fibers in detail, it was found that the thick fibers were actually composed of thinner wires. It is suggested that fiber–fiber interactions facilitated this hierarchical self-assembly. Similarly, the xerogel of 1c from n-hexane also formed fibrous aggregates which cross-linked into three-dimensional networks, and the average width of the single fibers was about 1 μm. In all these morphological experiments, the formation of uniform supramolecular aggregates suggests that the T-shaped pyrazine–TTF molecules are self-assembled in a hierarchical fashion to form a one-dimensional supramolecular polymer and further to form larger superstructures in organogel systems.49
 |
| | Fig. 5 FE-SEM images of native xerogels of 1a from toluene (a), 1b from DMF (b) and 1c from n-hexane (c), and CT complex xerogels of 1a from toluene (d), 1b from DMF (e) and 1c from n-hexane (f). | |
More interestingly, an evident change took place in the morphologies of the CT complex gels of gelators with F4TCNQ compared with those of the native gels, as shown in Fig. 5d–f. The supramolecular structures of native gels were changed into amorphous block aggregates. These changes in morphology showed that CT interaction played a key role in the formation of the CT complex gels.
XRD studies
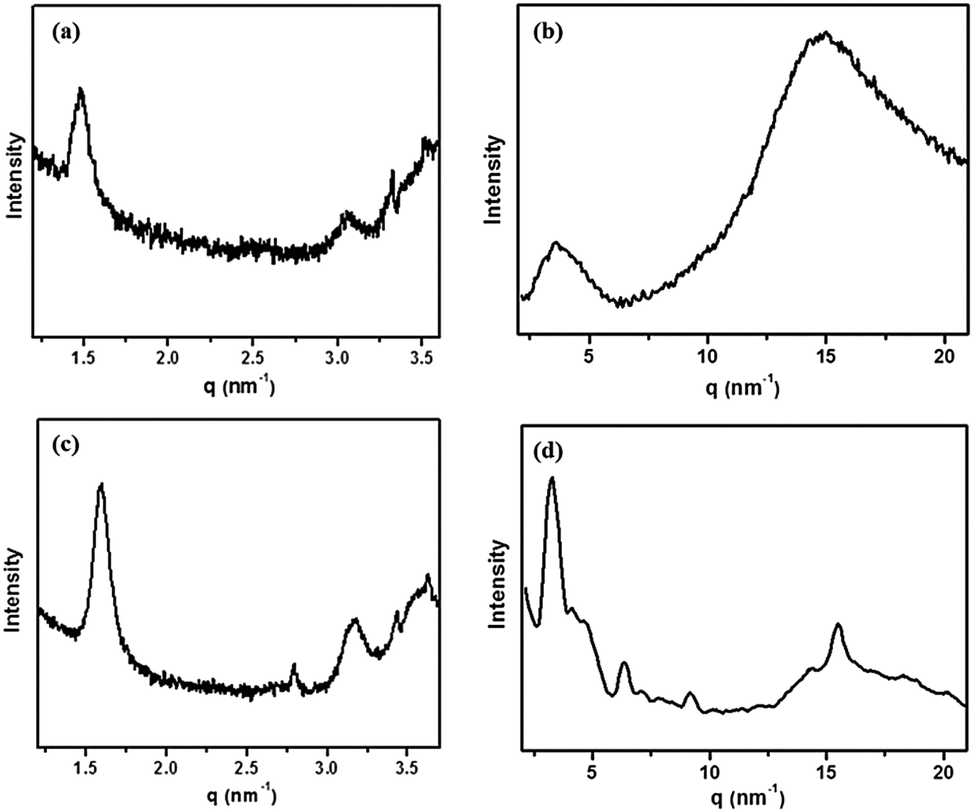
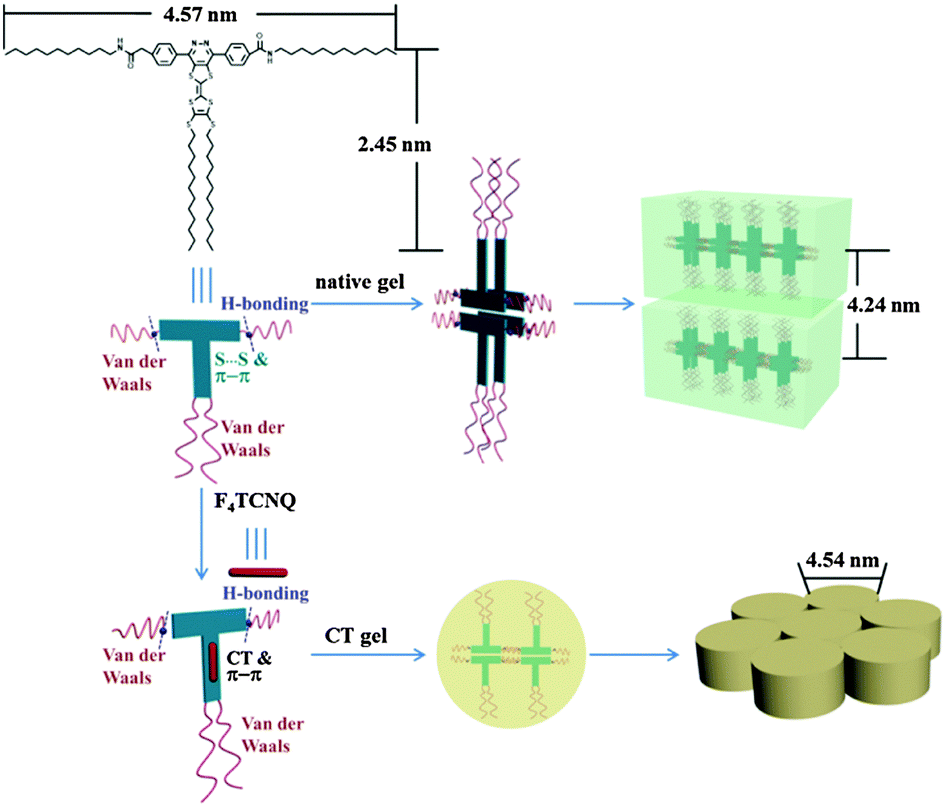
To gain further insight into the structures of the self-assemblies, small-angle X-ray diffraction (SAXRD) and wide-angle X-ray diffraction (WAXRD) measurements were performed. Taking 1b in DMF as an example, as shown in Fig. 6a, in the SAXRD spectrum of the native xerogel, the scattering pattern of the xerogel was characterized by reflection peaks at 4.19 and 2.09 nm with a scattering vector ratio of 1:2. The result illustrated that a lamellar organization with 4.19 nm of interlayer distance was formed in the gel phase. Considering that the molecular length of 1b was about 2.45 nm along the pyrazine–TTF side (by the CPK model), a bilayer arrangement in the gel matrix with the flexible chains interdigitated or inclined was expected. The d-spacing of 0.39 nm in the wide-angle region was a typical π–π stacking distance (Fig. 6b). The reasonable packing of the gel of 1b in DMF is schematically represented in Fig. 7.
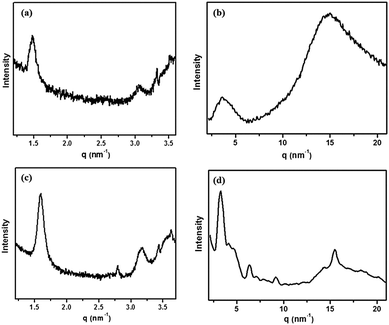 |
| | Fig. 6 XRD patterns of xerogels of 1b (a and b) and 1b/F4TCNQ complex (c and d) from DMF. | |
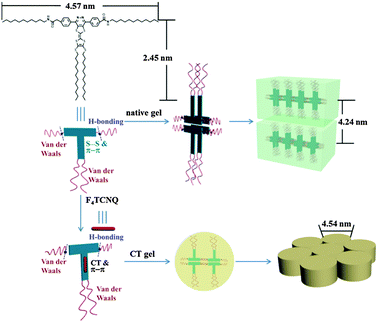 |
| | Fig. 7 Cartoon representation of the self-assembly of 1b and 1b/F4TCNQ (mole ratio = 1:1) in DMF. | |
To reveal the change of molecular packing mode in CT complex gels, XRD analyses were conducted and the results are shown in Fig. 6c. Taking these XRD spectral data into account, the packing mode of 1b with F4TCNQ in the CT complex gel was discussed. The CT complex gel showed a significant change and showed a series of scattering peaks with d-spacings of 1.60, 2.80, and 3.17 nm in the small-angle region, with a scattering vector ratio of 1:√3:2. It strongly reveals the hexagonal columnar structure of the CT complex gel with a column diameter of 4.54 nm. The distance corresponding to the first scattering peak was shorter than the length of two molecules, showing that the flexible chains of molecules interdigitated in the columns. The number of molecules in a disk was calculated to be approximately 4. Given the packing structural differences between the gels, we could presume that the CT interaction had a significant effect on their packing modes. In the wide-angle region, the strong diffraction peak corresponds to a d-spacing of 0.38 nm that could be a typical π–π stacking distance (Fig. 6d), indicating that the π–π stacking also existed in between TTF and F4TCNQ except for CT interactions in the gel matrix. The detailed aggregation mode of the CT complex gel is shown in Fig. 7.
Driving-force analysis
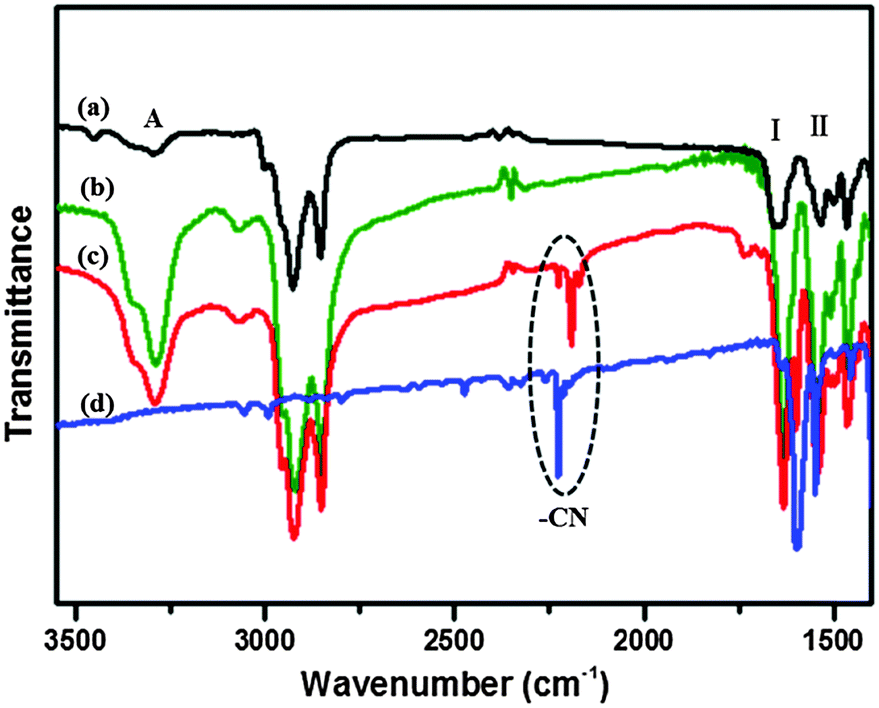
In order to ascertain how the T-shaped organic conjugate molecules aggregated into the gels, the participation of amide moieties in hydrogen-bonding interactions during gelation was examined by recording FTIR spectra of the xerogel of 1b from DMF and the non-self-assembled state of 1b in CHCl3. The FTIR spectrum of 1b in CHCl3 showed transmission bands at 3448, 3295, 1650 and 1535 cm−1, which correspond to the non-hydrogen-bonded ν N–H (amide A), ν C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (amide I), and δ N–H (amide II) modes, respectively (Fig. 8a and b). However, the transmission bands for DMF xerogel of 1b, which appeared at 3288 cm−1 (amide A) and 1631 cm−1 (amide I), red-shifted by 7 and 19 cm−1, respectively, and the band at 1540 cm−1 (amide II) blue-shifted by 5 cm−1. The shifts of transmission bands indicate the involvement of hydrogen bonding between the carbonyl group and the amide N–H group in the gel state, possibly via intermolecular interactions.50
O (amide I), and δ N–H (amide II) modes, respectively (Fig. 8a and b). However, the transmission bands for DMF xerogel of 1b, which appeared at 3288 cm−1 (amide A) and 1631 cm−1 (amide I), red-shifted by 7 and 19 cm−1, respectively, and the band at 1540 cm−1 (amide II) blue-shifted by 5 cm−1. The shifts of transmission bands indicate the involvement of hydrogen bonding between the carbonyl group and the amide N–H group in the gel state, possibly via intermolecular interactions.50
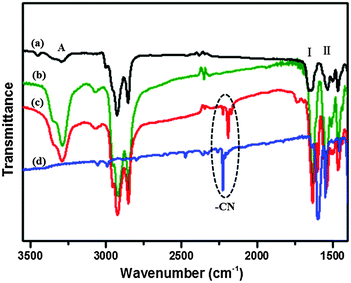 |
| | Fig. 8 FT-IR spectra of 1b in CHCl3 solution (a), native xerogel of 1b from DMF (b), the CT complex gel of 1b with F4TCNQ (mole ratio = 1:1) from DMF (c) and the F4TCNQ powder (d). | |
By contrast, we focused our attention on the CN stretching band in the CT complex gels in the FT-IR spectra (Fig. 8c and d). The CN stretching band in the 1b–F4TCNQ CT complex gel from DMF appeared at 2192 cm−1, exhibited a low frequency, and shifted by 34 cm−1 in comparison with that of the powder of F4TCNQ, indicating that CT interaction had a key role in the formation of the CT complex gels. However, we did not observe any shifts of the amide A, amide I and amide II bands in the CT complex gel with respect to the native 1b gel, which suggests that the intermolecular hydrogen-bonding interactions between the gelator molecules remain unperturbed even after the integration of F4TCNQ within the gel matrix. The native and CT complex xerogels from toluene had also been testified and similar conclusions could be drawn by studying their FT-IR spectra shown in Fig. S4 (ESI†).
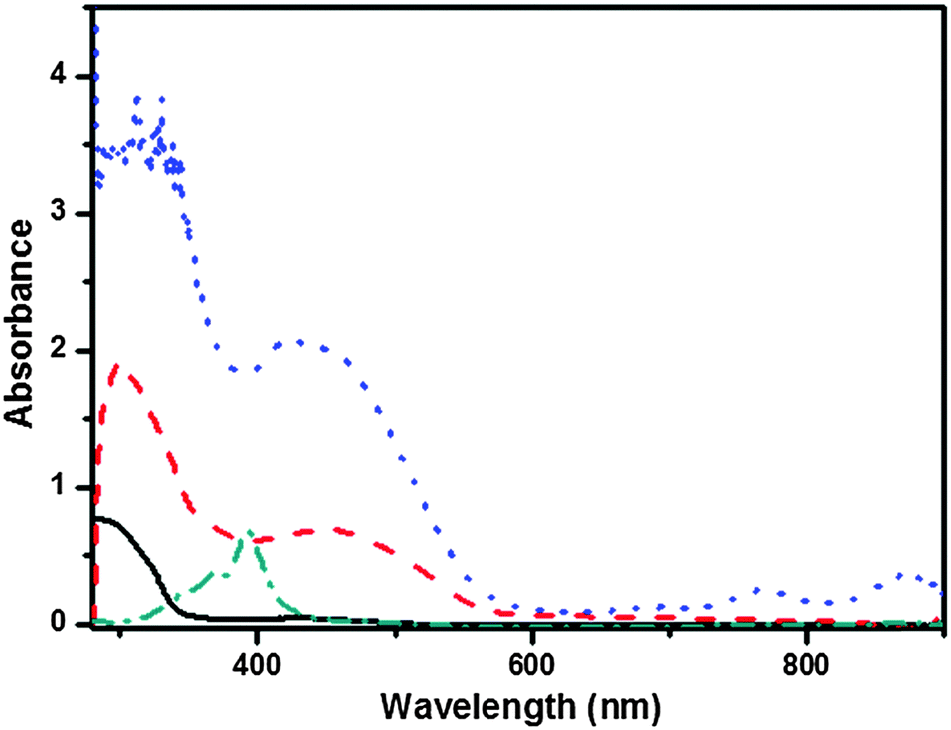
Furthermore, we were also eager to know the other types of non-covalent interactions that facilitate the self-assembly of these T-shaped gelators. The 1H NMR spectra of compound 1b clearly show concentration dependent features in CDCl3. By increasing the concentration from 7 mg mL−1 to 30 mg mL−1, the signals at 6.28 ppm ascribed to the amide groups gradually shifted downfield to 6.56 ppm, consistent with the formation of intermolecular hydrogen-bonding interactions. Besides, the resonance signal corresponding to the aromatic protons displayed an upfield shift with the increase in the concentration of gelator in CDCl3, from 7.95 ppm to 7.88 ppm, which strongly revealed the π–π stacking between the aromatic moieties (Fig. S5, ESI†), while no change was observed in the aliphatic protons. UV-Vis spectra also provided information about the π–π stacking. Firstly, the T-shaped 1b molecule showed maximum absorption at 291 nm in a dilute DMF solution (1 × 10−5 M), which could be assigned to the π–π* transitions of the molecular backbone. When the concentration was increased to 5 × 10−4 M, the absorption bands underwent a significant red shift to 297 nm, indicating the existence of certain supramolecular interactions at a higher concentration (Fig. S6, ESI†). Next, the UV-Vis absorption spectrum of the DMF gel of 1b was measured and compared with that of its dilute CHCl3 solution (2 × 10−5 M) in which the molecules may be considered to be in a monomer state. As shown in Fig. 9, the peak assignable to molecular backbone absorption gave a much red-shifted band (∼19 nm) in gel phase than that in the CHCl3 solution, indicative of the formation of J-aggregation within the gel state. The red shift results from UV-Vis spectra strongly indicated that the π–π interaction played a key role in the formation of the gels.
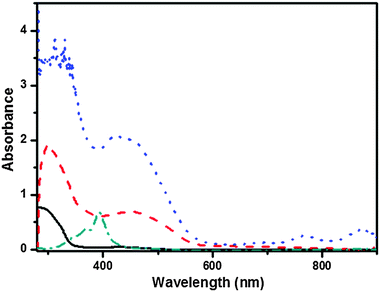 |
| | Fig. 9 Absorption spectra of 1b in CHCl3 (2 × 10−5 M, solid line), native gel of 1b in DMF (dashed line), CT complex gel of 1b with F4TCNQ in DMF (dotted line) and F4TCNQ in CHCl3 (2 × 10−5 M) solution (dash dotted line). | |
By comparison, the UV-Vis spectrum of the CT complex gel of 1b with F4TCNQ showed two sharp absorption bands at λmax 768 and 875 nm and a shoulder-like band at 691 nm (Fig. 9). The two former absorption bands corresponded to the progressive formation of the F4TCNQ radical anion and the latter band corresponded to the TTF radical cation,47 and proved that the CT complex gel was really formed and the CT interaction of TTF˙+ with F4TCNQ˙− contributed to the self-assembly process of CT complexes.
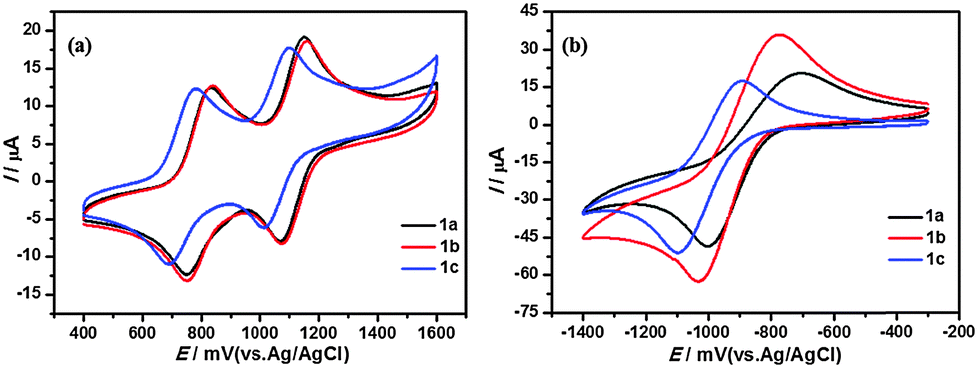
Electrochemical properties
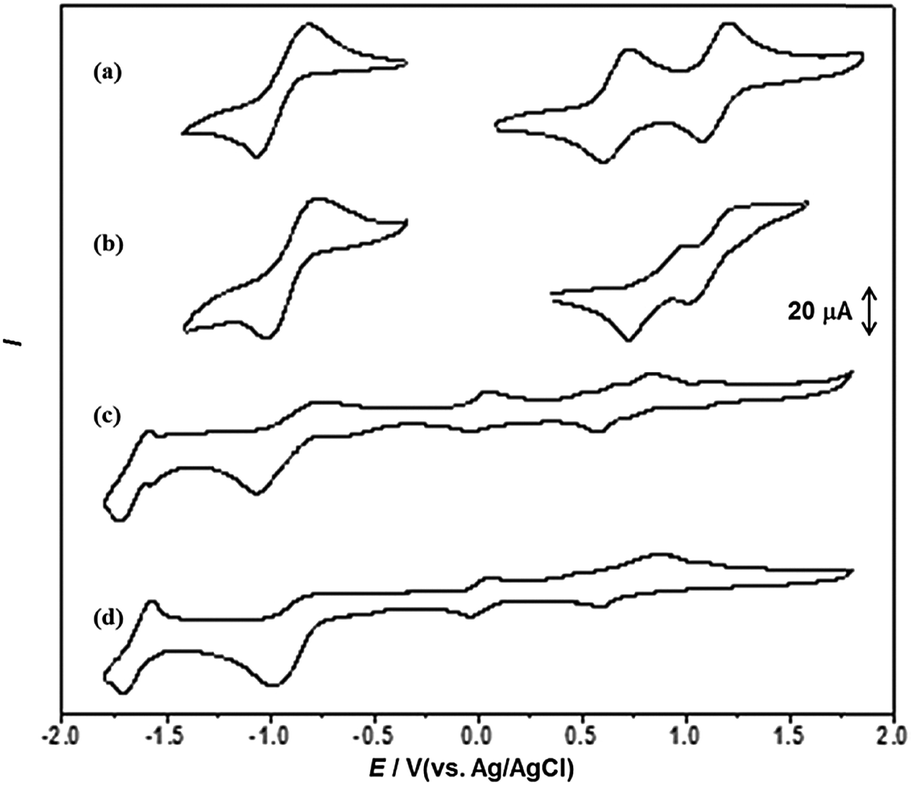
The electrochemical properties of T-shaped organogelators 1a–c were studied by cyclic voltammetry (CV) in benzonitrile solution (1 mM, vs. Ag/AgCl). As shown in Fig. 10, all gelators exhibited two reversible single-electron oxidation waves around 0.80 and 1.11 V. The first oxidation wave should be attributed to the bis(alkylthio) group substituted 1,3-dithiole unit, while the second oxidation wave arises from the pyridazine-annulated 1,3-dithiole unit. Moreover, the compounds showed a quasi-reversible reduction process around −1.03 V, which can be assigned to one-electron reduction of the pyridazine unit.44 The length of the alkyl chains had some effect on the redox processes. As seen in Fig. 10, the redox potentials of TTF and pyridazine units were lowered with increasing alkyl chain length. But in general, the intramolecular charge transfer from the TTF unit to the pyridazine moiety decreases the electron density of the TTF ring; as a consequence, these systems are expected to be more stable to oxygen exposure.
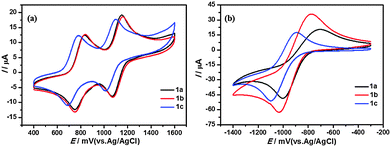 |
| | Fig. 10 Cyclic voltammograms of compounds 1a–c (1 mM) in benzonitrile containing 0.1 M Bu4NPF6. Scan rate was 100 mV s−1, with Pt as the counter electrode, glass carbon as the working electrode and Ag/AgCl electrode (saturated KCl) as the reference electrode. | |
Next, we examined the electrochemical behaviour of the gels of 1b and its CT complex, both obtained from DMF. The native gel of 1b exhibited two quasi-reversible oxidation waves at 1.00 and 1.24 V, respectively, which are much higher than those of the solution of 1b under the same conditions (0.84 and 1.16 V for the first and second oxidation potentials, respectively). And the reduction potential of the pyridazine moiety anodically shifted by 47 mV compared with solution. These findings were probably due to the stabilization effect arising from the strong π–π and S⋯S interactions (Fig. 11a and b). It is worth noting that the first and second oxidation potentials (0.83 and 1.13 V) in the CT complex xerogel assigned to the TTF unit were relatively low compared with the solution of 1b, which were interpreted to be the result of the CT interaction between TTF•+ (electron acceptor) and F4TCNQ•− (electron donor) that increased the electron density of the pyridazine–TTF moiety (Fig. 11c and d and Fig. S7, ESI†). Nevertheless, the reduction potentials of F4TCNQ at approximately E = −1.72, −1.07, −0.04 and 0.57 V were lower than those of F4TCNQ itself in the solution (−1.70, −0.99, −0.03 and 0.59 V). Therefore, the gelation of the CT complex was driven by both hydrogen-bonding and CT interactions.
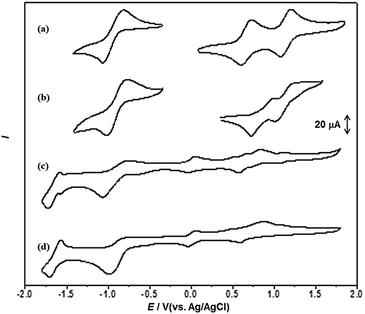 |
| | Fig. 11 The CV curves of (a) 1b (1 mM), (b) native xerogel of 1b from DMF, (c) CT complex xerogel of 1b with F4TCNQ and (d) F4TCNQ (1 mM) in benzonitrile. Scan rate was 100 mV s−1, with Pt as the counter electrode, glass carbon as the working electrode and Ag/AgCl electrode (saturated KCl) as the reference electrode, and Bu4NPF6 (0.1 M) as the supporting electrolyte. | |
Anion responsive studies
With reference to previous studies, amide NH units are well known anion binding sites, owing to the polarization by π conjugation with the neighboring carbonyl moiety.51 If the hydrogen bonding donor NH sites in the amide units interacted with anions, the gelators may not form stable assemblies, resulting in the transformation of the gels into a solution. Therefore the response of the DMF gel of 1b towards various anions (such as tetrabutylammonium salt) was investigated. As shown in Fig. 12, after 2 equiv. of solid TBAF was carefully added on the top of the 1b DMF gel, as time passed, the gel gradually collapsed and transformed into a suspension without changing the color of the system. The result indicated that the collapse of the gel phase arises from the anion-binding interactions rather than the deprotonation. The UV-Vis spectra of gelator 1b in CHCl3 (5 × 10−5 M) solution in the presence of an increasing amount of fluoride ions also demonstrated the interaction mechanism (Fig. S8, ESI†). However, the gel could be preserved after adding the same amount of other anions (Cl−, Br−, I−, H2PO4−, HSO4−, AcO−) under the same conditions. These results supported the assumption that the F− not the TBA cation was responsible for the transformation of the organogel. Therefore, we believed that the DMF gel of 1b could be used as a selectively naked-eye sensor system for fluoride anions. On the other hand, the toluene gel of 1b showed an opposite phenomenon. As shown in Fig. S9 (ESI†), the gel was transformed into a clear yellow solution after the addition of 2 equiv. of F− or Cl− and into a suspension upon the addition of 2 equiv. of Br− or I−. We thought that these results were due to the weak hydrogen bonding interaction between the gelator molecules in toluene.52 Unfortunately, the above transition processes of DMF or toluene gels did not undergo regelation even on addition of protic solvents, such as methanol or water.
 |
| | Fig. 12 Photographs of the 1b organogel (DMF, 1.5 mg mL−1) upon the addition of 2.0 equiv. of each anion. From left to right is native gel, addition of F−, Cl−, Br−, I−, H2PO4−, HSO4−, AcO−. | |
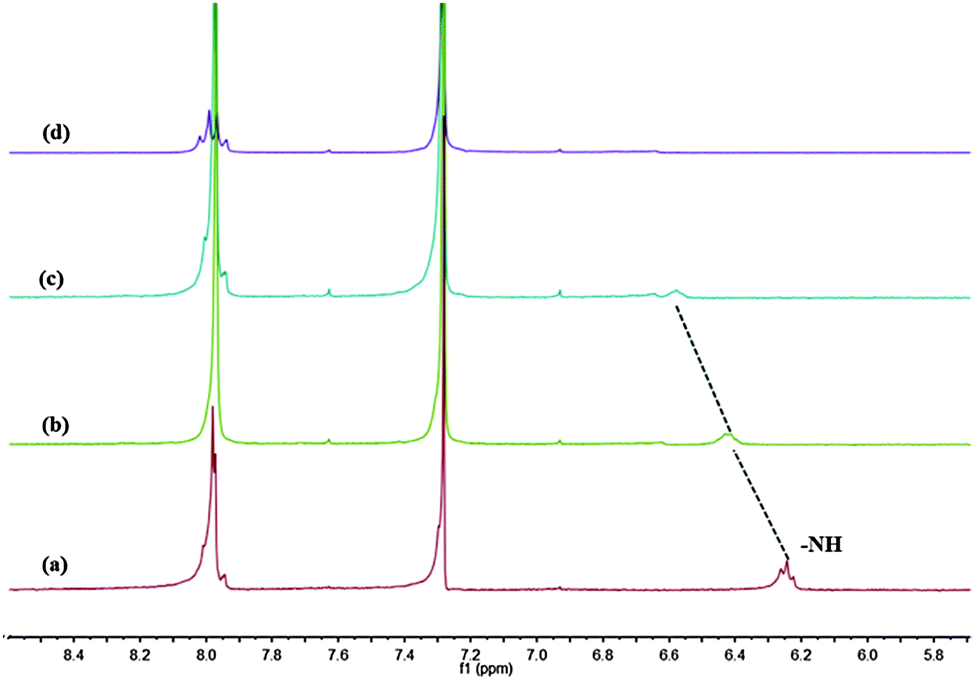
Additional evidence for the anion-responsive mechanism was obtained from 1H NMR experiments in CDCl3 (Fig. 13). Before the addition of F−, the proton signal assigned to the amide –NH at 6.24 ppm shifted downfield to 6.58 ppm and broadened with increasing fluoride anion concentration from 0 to 1.5 equiv. When 2 equiv. of fluoride was added, the NH proton signal finally disappeared, but no new signal emerged at 16–17 ppm, which suggested that the NH groups did not undergo deprotonation reaction.
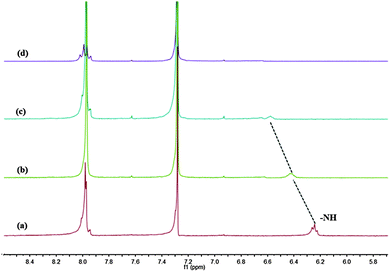 |
| | Fig. 13 Partial 1H NMR spectra of 1b (2 mM) in CDCl3 upon the addition of F−: (a) free; (b) 0.5 equiv.; (c) 1.0 equiv.; and (d) 2.0 equiv. | |
Conclusion
In summary, we have reported a novel series of T-shaped low-molecular-mass organic π-conjugates, monopyridazine–TTFs, and demonstrated the supramolecular self-assembly process in common organic solvents. Spectroscopic and electrochemical studies revealed that the pyridazine unit greatly facilitates electronic communication between the TTF moiety and the acceptor unit, thus leading to intramolecular charge transfer and enhancing the stability in the air. The gelation test results suggested that gelator 1b had the lowest CGC in DMF. Furthermore, the DMF gel of 1b could be transformed into suspension only by the addition of F− through the disruption of intermolecular hydrogen bonds. But the toluene gel exhibited relatively complicated responses to anion stimuli, which could be destroyed by halogen anions. Importantly, the gelators could form stable CT complex gels with the stronger oxidant F4TCNQ rather than TCNQ, indicating that introduction of a pyrazine ring to the TTF skeleton was effective in further reducing the electron-donating property. The CT complex gels were further confirmed by various spectroscopy, microscopy, XRD and electrochemistry techniques. The present results of the T-shaped molecular skeleton provide a new branch of TTF chemistry, and will expand the excellent intelligent soft materials with potential applications in the optoelectronics areas.
Acknowledgements
The authors acknowledge financial support from the National Natural Science Foundation of China (grant no. 21262039).
Notes and references
-
(a) H. Engelkamp, S. Middelbeek and R. J. M. Nolte, Science, 1999, 284, 785 CrossRef CAS PubMed;
(b) D. Díaz, J. J. Cid, P. Vázquez and T. Torres, Chem. – Eur. J., 2008, 14, 9261 CrossRef PubMed.
-
(a) J.-L. Pozzo, G. M. Clavier and J.-P. Desvergne, J. Mater. Chem., 1998, 8, 2575 RSC;
(b) J.-P. Hong, M.-C. Um, S.-R. Nam, J.-I. Hong and S. Lee, Chem. Commun., 2009, 310 RSC.
- J. Reichwagen, H. Hopf, A. Del Guerzo, C. Belin, H. Bouas-Laurent and J.-P. Desvergne, Org. Lett., 2005, 7, 971 CrossRef CAS PubMed.
-
(a) K. Sugiyasu, N. Fujita and S. Shinkai, Angew. Chem., Int. Ed., 2004, 43, 1229 CrossRef CAS PubMed;
(b) F. Würthner, B. Hanke, M. Lysetska, G. Lambright and G. S. Harms, Org. Lett., 2005, 7, 967 CrossRef PubMed.
- X. Dou, W. Pisula, J. Wu, G. J. Bodwell and K. Müllen, Chem. – Eur. J., 2008, 14, 240 CrossRef CAS PubMed.
- D.-C. Lee, K. K. McGrath and K. Jang, Chem. Commun., 2008, 3636 RSC.
-
(a) J. Sinha, S. J. Lee, H. Kong, T. W. Swift and H. E. Katz, Macromolecules, 2013, 46, 708 CrossRef CAS;
(b) L. Tan, Y. Guo, Y. Yang, G. Zhang, D. Zhang, G. Yu, W. Xu and Y. Liu, Chem. Sci., 2012, 3, 2530 RSC.
-
(a) M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910 CrossRef CAS PubMed;
(b) N. Aratani, D. Kim and A. Osuka, Acc. Chem. Res., 2009, 42, 1922 CrossRef CAS PubMed;
(c) P. D. Frischmann, K. Mahata and F. Wìrthner, Chem. Soc. Rev., 2013, 42, 1847 RSC.
- F. S. Kim, G. Ren and S. A. Jenekhe, Chem. Mater., 2011, 23, 682 CrossRef CAS.
-
(a) A. P. Kulkarni, C. J. Tonzola, A. Babel and S. A. Jenekhe, Chem. Mater., 2004, 16, 4556 CrossRef CAS;
(b) S. C. Lo and P. L. Burn, Chem. Rev., 2007, 107, 1097 CrossRef CAS PubMed.
-
(a) C. D. Dimitrakopoulos and P. R. L. Malenfant, Adv. Mater., 2002, 14, 99 CrossRef CAS;
(b) A. R. Murphy and J. M. J. Fréchet, Chem. Rev., 2007, 107, 1066 CrossRef CAS PubMed;
(c) H. Klauk, Chem. Soc. Rev., 2010, 39, 2643 RSC.
-
(a) S. Günes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324 CrossRef PubMed;
(b) Y. J. Cheng, S. H. Yang and C. S. Hsu, Chem. Rev., 2009, 109, 5868 CrossRef CAS PubMed.
-
(a) S. Matile, A. V. Jentzsch, J. Montenegro and A. Fin, Chem. Soc. Rev., 2011, 40, 2453 RSC;
(b) B. Zhu, H. X. Chen, W. Lin, Y. Ye, J. Wu and S. J. Li, J. Am. Chem. Soc., 2014, 136, 15126 CrossRef CAS PubMed;
(c) S. Li, J. Huang, F. Zhou, T. R. Cook, X. Yan, Y. Ye, B. Zhu, B. Zheng and P. J. Stang, J. Am. Chem. Soc., 2014, 136, 5908 CrossRef CAS PubMed.
-
(a) B. Jeong, S. W. Kim and Y. H. Bae, Adv. Drug Delivery Rev., 2002, 54, 37 CrossRef CAS;
(b) A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002 CrossRef CAS PubMed.
-
(a) Y. Gao, Y. Kuang, Z.-F. Guo, Z.-H. Guo, I. J. Krauss and B. Xu, J. Am. Chem. Soc., 2009, 131, 13576 CrossRef CAS PubMed;
(b) J. Boekhoven, M. Koot, T. A. Wezendonk, R. Eelkema and J. H. van Esch, J. Am. Chem. Soc., 2012, 134, 12908 CrossRef CAS PubMed.
- E. Carretti, M. Bonini, L. Dei, B. H. Berrie, L. V. Angelova, P. Baglioni and R. G. Weiss, Acc. Chem. Res., 2010, 43, 751 CrossRef CAS PubMed.
-
(a) Q. Wei and S. L. James, Chem. Commun., 2005, 1555 RSC;
(b) X. Wu, S. Ji, Y. Li, B. Li, X. Zhu, K. Hanabusa and Y. Yang, J. Am. Chem. Soc., 2009, 131, 5986 CrossRef CAS PubMed;
(c) B.-T. Li, L.-M. Tang, L. Qiang and K. Chen, Soft Matter, 2011, 7, 963 RSC.
- D. Díaz, D. Khbecka and R. J. Koopmansc, Chem. Soc. Rev., 2011, 40, 427 RSC.
- E. Krieg, H. Weissman, E. Shirman, E. Shimoni and B. Rybtchinski, Nat. Nanotechnol., 2011, 6, 141 CrossRef CAS PubMed.
-
(a) S. Kiyonaka, K. Sada, I. Yoshimura, S. Shinkai and N. Katoand I. Hamachi, Nat. Mater., 2004, 3, 58 CrossRef CAS PubMed;
(b) E. F. Banwell, E. S. Abelardo, D. J. Adams, M. A. Birchall, A. Corrigan, A. M. Donald, M. Kirkland, L. C. Serpell, M. F. Butler and D. N. Woolfson, Nat. Mater., 2009, 8, 596 CrossRef CAS PubMed;
(c) V. Jayawarna, M. Ali, T. A. Jowitt, A. E. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611 CrossRef CAS.
- J. A. Foster, M. O. M. Piepenbrock, G. O. Lloyd, N. Clarke, J. A. K. Howard and J. W. Steed, Nat. Chem., 2010, 2, 1037 CrossRef CAS PubMed.
-
(a) J. H. van Esch and B. L. Feringa, Angew. Chem., Int. Ed., 2000, 39, 2263 CrossRef CAS;
(b) N. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821 RSC;
(c) M. George and R. Weiss, Acc. Chem. Res., 2006, 39, 489 CrossRef CAS PubMed;
(d) R. Weiss, Langmuir, 2009, 25, 8369 CrossRef CAS PubMed;
(e) P. Dastidar, Chem. Soc. Rev., 2008, 37, 2699 RSC;
(f) M. Piepenbrock, G. Lloyd, N. Clarke and J. Steed, Chem. Rev., 2010, 110, 1960 CrossRef CAS PubMed;
(g) G. Yu, X. Yan, C. Han and F. Huang, Chem. Soc. Rev., 2013, 42, 6697 RSC.
-
(a) C. Maity, W. E. Hendriksen, J. H. van Esch and R. Eelkema, Angew. Chem., Int. Ed., 2015, 54, 998 CrossRef CAS PubMed;
(b) Z. Qiu, H. Yu, J. Li, Y. Wang and Y. Zhang, Chem. Commun., 2009, 3342 RSC;
(c) J. W. Chung, S. J. Yoon, S. J. Lim, B. K. An and S. Y. Park, Angew. Chem., Int. Ed., 2009, 48, 7030 CrossRef CAS PubMed.
-
(a) K. Isozaki, H. Takay and T. Naota, Angew. Chem., Int. Ed., 2007, 46, 2855 CrossRef CAS PubMed;
(b) J. Wu, T. Yi, T. Shu, M. Yu, Z. Zhou, M. Xu, Y. Zhou, H. Zhang, J. Han, F. Li and C. Huang, Angew. Chem., Int. Ed., 2008, 47, 1063 CrossRef CAS PubMed;
(c) D. Bardelang, F. Camerel, J. Margeson, D. Leek, M. Schmutz, M. Zaman, K. Yu, D. Soldatov, R. Ziessel, C. Ratcliffe and J. Ripmeester, J. Am. Chem. Soc., 2008, 130, 3313 CrossRef CAS PubMed.
-
(a) H. Komatsu, S. Matsumoto, S. Tamaru, K. Kaneko, M. Ikeda and I. Hamachi, J. Am. Chem. Soc., 2009, 131, 5580 CrossRef CAS PubMed;
(b) Z. Ge, J. Hu, F. Huang and S. Liu, Angew. Chem., Int. Ed., 2009, 48, 1798 CrossRef CAS PubMed;
(c) S. Dong, Y. Luo, X. Yan, B. Zheng, X. Ding, Y. Yu, Z. Ma, Q. Zhao and F. Huang, Angew. Chem., Int. Ed., 2011, 50, 1905 CrossRef CAS PubMed.
-
(a) Y. Gao, F. Zhao, Q. Wang, Y. Zhang and B. Xu, Chem. Soc. Rev., 2010, 39, 3425 RSC;
(b) J. W. Sadownik, J. Leckie and R. V. Ulijn, Chem. Commun., 2011, 47, 728 RSC.
-
(a) M. Shirakawa, N. Fujita and S. Shinkai, J. Am. Chem. Soc., 2005, 127, 4164 CrossRef CAS PubMed;
(b) X. Huang, S. Raghavan, P. Terech and R. Weiss, J. Am. Chem. Soc., 2006, 128, 15341 CrossRef CAS PubMed;
(c) W. Weng, J. B. Beck, A. Jamieson and S. Rowan, J. Am. Chem. Soc., 2006, 128, 11663 CrossRef CAS PubMed;
(d) Z. Qi and C. Schalley, Acc. Chem. Res., 2014, 47, 2222 CrossRef CAS PubMed.
-
(a) J. Webb, M. Crossley, P. Turner and P. Thordarson, J. Am. Chem. Soc., 2007, 129, 7155 CrossRef CAS PubMed;
(b) H. Maeda, Y. Haketa and T. Nakanishi, J. Am. Chem. Soc., 2007, 129, 13661 CrossRef CAS PubMed;
(c) Z. Zhang, N. Krishna, M. Lettinga, J. Vermant and E. Grelet, Langmuir, 2009, 25, 2437 CrossRef CAS PubMed.
-
(a) Z. Sun, Z. Li, Y. He, R. Shen, L. Deng, M. Yang, Y. Liang and Y. Zhang, J. Am. Chem. Soc., 2013, 135, 13379 CrossRef CAS PubMed;
(b) M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2011, 2, 511 CrossRef PubMed;
(c) J. Liu, P. He, J. Yan, X. Fang, J. Peng, K. Liu and Y. Fang, Adv. Mater., 2008, 20, 2508 CrossRef CAS.
-
(a) M. Mas-Torrent and C. Rovira, J. Mater. Chem., 2006, 16, 433 RSC;
(b) X. Gao, W. Wu, Y. Liu, W. Qiu, X. Sun, G. Yu and D. Zhu, Chem. Commun., 2006, 2750 RSC;
(c) S. Arumugam, D. Cortizo-Lacalle, S. Rossbauer, S. Hunter, A. L. Kanibolotsky, A. R. Inigo, P. A. Lane, T. D. Anthopoulos and P. J. Skabara, ACS Appl. Mater. Interfaces DOI:10.1021/am5080562;
(d) M. Mas-Torrent, S. Masirek, P. Hadley, N. Crivillers, N. Crivillers, P. Reuter, J. Veciana, C. Rovira and C. Rovira, Org. Electron., 2008, 9, 143 CrossRef CAS.
-
(a) H. C. Li, J. O. Jeppesen, E. Levillain and J. Becher, Chem. Commun., 2003, 846 RSC;
(b) X. W. Xiao, W. Xu, D. Q. Zhang, H. Xu, H. Y. Lu and D. B. Zhu, J. Mater. Chem., 2005, 15, 2557 RSC;
(c) E. Gomar-Nadal, J. Veciana, C. Rovira and D. B. Amabilino, Adv. Mater., 2005, 17, 2095 CrossRef CAS;
(d) P. T. Chiang, P. N. Cheng, C. F. Lin, Y. H. Liu, C. C. Lai, S. M. Peng and S. H. Chiu, Chem. – Eur. J., 2006, 12, 865 CrossRef CAS PubMed.
-
(a) M. R. Bryce, Chem. Soc. Rev., 1991, 20, 355 RSC;
(b) J. Puigmartí-Luis, A. Minoia, S. Lei, V. Geskin, B. Li, R. Lazzaroni, S. D. Feyter and D. B. Amabilino, Chem. Sci., 2011, 2, 1945 RSC;
(c) Y. Qiao, Y. Lin, S. Liu, S. Zhang, H. Chen, Y. Wang, Y. Yan, X. Guo and J. Huang, Chem. Commun., 2013, 49, 704 RSC.
-
(a) H. Lu, W. Xu, D. Zhang and D. Zhu, Chem. Commun., 2005, 4777 RSC;
(b) K. R. Larsen, C. Johnsen, O. Hammerich and J. O. Jeppesen, Org. Lett., 2013, 15, 1452 CrossRef CAS PubMed.
-
(a) M. Katsuhara, I. Aoyagi, H. Nakajima, T. Mori, T. Kambayashi, M. Ofuji, Y. Takanishi, K. Ishikawa, H. Takezoe and H. Hosono, Synth. Met., 2005, 149, 219 CrossRef CAS;
(b) R. Hou, K.-L. Zhong, Z. Huang, L. Yi Jin and B. Yin, Tetrahedron, 2011, 67, 1238 CrossRef CAS.
- J. Puigmartí-Luis, A. Minoia, Á. Pérez del Pino, G. Ujaque, C. Rovira, A. Lledós, R. Lazzaroni and D. B. Amabilino, Chem. – Eur. J., 2006, 12, 9161 CrossRef PubMed.
- G. Ono, A. Izuoka, T. Sugawara and Y. Sugawara, J. Mater. Chem., 1998, 8, 1703 RSC.
-
(a) M. Jørgensen, K. Bechgaard, T. Bjørnholm, O. Sommer-Larsen, L. G. Hansen and K. Schaumburg, J. Org. Chem., 1994, 59, 5877 CrossRef;
(b) D. R. Talham, Chem. Rev., 2004, 104, 5479 CrossRef CAS PubMed;
(c) J. Sly, P. Kasák, E. Gomar-Nadal, C. Rovira, L. GGrriz, P. Thordarson, D. B. Amabilino, A. E. Rowan and R. J. M. Nolte, Chem. Commun., 2005, 1255 RSC.
-
(a) T. Kitahara, M. Shirakawa, S.-i. Kawano, U. Beginn, N. Fujita and S. Shinkai, J. Am. Chem. Soc., 2005, 127, 14980 CrossRef CAS PubMed;
(b) T. Kitamura, S. Nakaso, N. Mizoshita, Y. Tochigi, T. Shimomura, M. Moriyama, K. Ito and T. Kato, J. Am. Chem. Soc., 2005, 127, 14769 CrossRef CAS PubMed;
(c) J. Puigmartí-Luis, V. Laukhin, Á. Pérez del Pino, J. Vidal-Gancedo, C. Rovira, E. Laukhina and D. B. Amabilino, Angew. Chem., Int. Ed., 2007, 46, 238 CrossRef PubMed.
-
(a) J. Puigmartí-Luis, Á. P. del Pino, E. Laukhina, J. Esquena, V. Laukhin, C. Rovira, J. Vidal-Gancedo, A. G. Kanaras, R. J. Nichols, M. Brust and D. B. Amabilino, Angew. Chem., Int. Ed., 2008, 47, 1861 CrossRef PubMed;
(b) T. Kitahara, M. Shirakawa, S.-I. Kawano, U. Beginn, N. Fujita and S. Shinkai, J. Am. Chem. Soc., 2005, 127, 14980 CrossRef CAS PubMed.
-
(a) C. Wang, D. Zhang and D. Zhu, J. Am. Chem. Soc., 2005, 127, 16372 CrossRef CAS PubMed;
(b) C. Wang, Q. Chen, F. Sun, D. Zhang, G. Zhang, Y. Huang, R. Zhao and D. Zhu, J. Am. Chem. Soc., 2010, 132, 3092 CrossRef CAS PubMed.
- S. A. Odom, M. M. Caruso, A. D. Finke, A. M. Prokup, J. A. Ritchey, J. H. Leonard, S. R. White, N. R. Sottos and J. S. Moore, Adv. Funct. Mater., 2010, 20, 1721 CrossRef CAS.
- Y. Tatewaki, T. Hatanaka, R. Tsunashima, T. Nakamura, M. Kimura and H. Shirai, Chem. – Asian J., 2009, 4, 1474 CrossRef CAS PubMed.
-
(a) Y. Liu, N. Zheng, H. Li and B. Yin, Soft Matter, 2013, 9, 5261 RSC;
(b) Y. Liu, N. Zheng, T. Chen, L. Jin and B. Yin, Org. Biomol. Chem., 2014, 12, 6927 RSC.
- N. Zheng, H. Li, G. Sun, K. Zhong and B. Yin, Org. Biomol. Chem., 2013, 11, 5100 CAS.
- D. K. Maiti and A. Banerjee, Chem. – Asian J., 2013, 8, 113 CrossRef CAS PubMed.
- P. Jana, S. Maity, S. K. Maity, P. K. Ghorai and D. Haldar, Soft Matter, 2012, 8, 5621 RSC.
- S. Bivaud, J.-Y. Balandier, M. Chas, M. Allain, S. Goeb and M. Sallé, J. Am. Chem. Soc., 2012, 134, 11968 CrossRef CAS PubMed.
- J. Seo, J. W. Chung, J. E. Kwon and S. Y. Park, Chem. Sci., 2014, 5, 4845 RSC.
- B. Adhikari, J. Nanda and A. Banerjee, Chem. – Eur. J., 2011, 17, 11488 CrossRef CAS PubMed.
- S. Mukherjee, T. Kar and P. Kumar Das, Chem. – Asian J., 2014, 9, 2798 CrossRef CAS PubMed.
- H. Maeda, Chem. – Eur. J., 2008, 14, 11274 CrossRef CAS PubMed.
- J. E. A. Webb, M. J. Crossley, P. Turner and P. Thordarson, J. Am. Chem. Soc., 2007, 129, 7155 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures with structural characterization, UV-Vis spectra after addition of F4TCNQ and TCNQ, tuning the gel formation in toluene, TGA curves, FT-IR spectra of native and CT xerogels from toluene, variable-concentration 1H-NMR and UV-Vis spectra and anion stimuli response of the toluene gel. See DOI: 10.1039/c5nj02322j |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.