
Using carbon nanodots as inexpensive and environmentally friendly sensitizers in mesoscopic solar cells†‡
J. T.
Margraf
ab,
F.
Lodermeyer
a,
V.
Strauss
a,
P.
Haines
a,
J.
Walter
cd,
W.
Peukert
cd,
R. D.
Costa
a,
T.
Clark
*bd and
D. M.
Guldi
*ad
aDepartment of Chemistry and Pharmacy & Interdisciplinary Center for Molecular Materials (ICMM), Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), Egerlandstrasse 3, 91058 Erlangen, Germany. E-mail: dirk.guldi@fau.de; Fax: +49-9131-852-8307
bComputer-Chemie-Centrum & Interdisciplinary Center for Molecular Materials (ICMM), Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), Nägelsbachstr. 25, 91058 Erlangen, Germany
cInstitute of Particle Technology (LFG), Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), Cauerstr. 4, 91058 Erlangen, Germany
dCenter of Functional Particle Systems, Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), Haberstr. 9a, 91058 Erlangen, Germany
First published on 9th March 2016
Abstract
We discuss the use of carbon nanodots (CNDs) as sensitizers in mesoscopic solar cells. The CNDs are synthesized using a one-step, bottom-up microwave approach with citric acid, urea, and formic acid as precursors in aqueous media. Their light-harvesting capabilities can be tuned by adjusting the synthetic parameters. Comprehensive spectroscopic and theoretical studies allow us to rationalize the nature of their absorption features. Promising power conversion efficiencies (η) of 0.24% can be achieved from these cheap and eco-friendly sensitizers by optimizing the solar-cell assembly process. Interestingly, we found that extending the light absorption towards longer wavelengths does not necessarily improve the performance of the solar cells, since the longer-wavelength absorption features hardly contribute to the cells' photo-action spectra, so that the overall power conversion efficiency is actually worse. The origin of the lower performance is corroborated in transient absorption spectroscopy and photovoltage decay measurements. Our work points, on one hand, to the limits of as-synthesized CNDs as photosensitizers and, on the other hand, to possible improvements.
Conceptual insightsDye-sensitized solar cells (DSSC) and related concepts bear great potential as the next-generation green energy sources. As the technology has matured, it has also overcome important obstacles towards main-stream applicability, such as low power conversion efficiency and device lifetime. However, this progress has led to increased complexity (and cost) of the light-harvesting molecules. Light absorbing inorganic quantum dots can typically be synthesized in a single step at a fraction of the cost of high performance molecular dyes, but usually contain rare and toxic elements like lead, cadmium or indium. In this work, we use carbon nanodots (CND) as novel light harvesters for DSSCs. CNDs combine the simple synthesis of an inorganic material with the environmental benefits of organic molecules. We found that the assembly of CNDs on TiO2 electrodes can be controlled via the pH of the solution, leading to twofold improved solar cell performance, compared to previous reports. Interestingly, further extending the absorption of CNDs into the visible did not improve the device performance. A comprehensive investigation of the current generation mechanism indicate that this is because the extended absorption is related to trap states, which do not contribute to the photogenerated current. Our work shows that the effects of modifying the spectroscopic properties of CNDs on device performance can be counterintuitive, and that investigation of devices is essential to obtain high performance CND solar cells. |
Since their introduction in 1991, dye-sensitized solar cells (DSSC) have matured into a technology on the brink of widespread commercialization.1 Recent advances such as novel dyes and (solid-state) electrolytes have shown that long-standing issues such as low efficiency and insufficient stability can be overcome.2 The emergence of organo-lead-halide perovskite absorbers in mesoscopic solar cells has further accelerated this trend, although it remains to be seen whether this promising new technology can meet the goal of providing cheap, long-term stable, environmentally benign, and efficient solar cells.3
The downside of these advances is an increase in the complexity of the device and/or the presence of rare and toxic elements. For example, the synthesis of the YD2-o-C8 dye with reported efficiencies of 13% takes nine separate steps and requires noble-metal catalysis.4 It is doubtful whether this dye can be synthesized in the amounts necessary for the large-scale production of solar panels at a reasonable price. In addition, the most efficient reported cells rely on co-sensitization with different dyes, which complicates the fabrication process.4
Colloidal semiconductor quantum dots (QDs) are an attractive alternative in this respect.5,6 Synthesis of QDs via the hot-injection method is a one-step process; the reaction is typically completed within minutes or hours and the QDs produced feature high extinction coefficients (ca. 105 cm−1 M−1 for CdSe) and tunable band gaps. Recent years have seen a steep rise in the reported efficiencies of QD-based solar cells, both in the DSSC and depleted heterojunction architectures.7–11 Unfortunately, the systems reported all employ QDs based on cadmium, lead and/or indium.
This could potentially be circumvented by using carbon nanomaterials such as graphene quantum dots (GQDs) and carbon nanodots (CNDs), which are free from these environmental concerns. More typically, carbon materials are used as metal-free electrodes or to facilitate charge transport in solar cells.12–15 We have seen that CNDs are prone to undergo photoinduced charge-transfer in nanohybrid materials, indicating that they are also suitable as photoactive components.16–18 Indeed, several studies have shown that the principle of using such materials as sensitizers in mesoscopic solar cells is sound, although the reported power conversion efficiencies cannot yet compete with inorganic QD solar cells.19–24
We here classify GQDs as structurally well-defined, carbon-rich organic molecules that generally require multi-step synthetic procedures in contrast to CNDs, which are usually synthesized in a single step but do not exhibit a well-defined molecular structure. This differentiation is, however, not universally accepted in the literature, where the terms graphene quantum dot, carbon nanodot and carbon dot are often used interchangeably.
The highest reported efficiencies of CND solar cells range from 0.06 to 0.13%.18–20 This relatively modest performance can be attributed to low coverage of the photoelectrodes, non-optimized electrolytes, and an incomplete understanding of the electronic structure of the CND.
In this work, we focus on the performance of CND solar cells using a newly developed synthesis. The latter enables, on one hand, broader absorptions throughout the visible part of the spectrum and, on the other hand, optimized device-assembly procedures. As such, the performance of the resulting devices with η of 0.24% is significantly better than in previously reported CND-based devices, albeit still more than an order of magnitude below the performance of typical dye or inorganic QD-based ones. To understand the origin of the low performance, the role of trap states in these CNDs is explored using spectroscopic techniques and molecular modeling. From this we reach important conclusions regarding the relationship between the CND's electronic structure and the charge injection, recombination and regeneration processes under device operation conditions.
CNDs were synthesized in a domestic microwave using urea (U), citric acid (CA), and formic acid (FA) as precursors in aqueous solutions (see Experimental details). This is a slight modification of our previously reported procedure, which leads to larger, more polydisperse CNDs (see below).16 The absorption spectra of the products feature two prominent peaks at approximately 350 and 430 nm. By varying the molar ratio of the precursors, the position and relative intensities of the peaks are tunable. Furthermore, a very broad absorption feature spanning beyond 700 nm can be obtained, depending on the synthetic procedure. In Fig. 1, the absorption spectra of two representative CND batches are shown. CND1 (CA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :U:FA 1:1:2) feature relatively sharp absorption peaks and only weak absorption beyond 550 nm, while CND2 (CA:U:FA 1:1:20) show significantly broader and red-shifted features. Furthermore, the absorption beyond 450 nm is amplified in CND2.
:U:FA 1:1:2) feature relatively sharp absorption peaks and only weak absorption beyond 550 nm, while CND2 (CA:U:FA 1:1:20) show significantly broader and red-shifted features. Furthermore, the absorption beyond 450 nm is amplified in CND2.
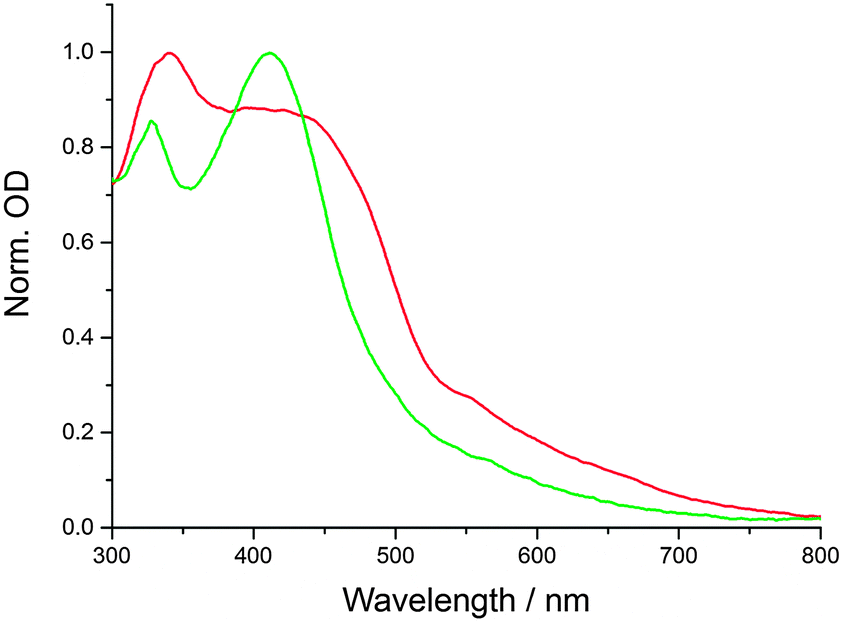 | ||
| Fig. 1 Normalized absorption spectra of CND1 (green) and CND2 (red) in H2O at room temperature. | ||
Both samples feature excitation-wavelength-dependent emission spectra, a well-known property of poly-disperse CNDs – see the ESI,‡ Fig. S4 and S5. For both samples, the emission intensities follow the absorption spectra, i.e.CND1 display the most intense emission upon excitation at around 420 nm, and CND2 upon excitation at 360 nm.
The particle size of CNDs was determined via analytical ultracentrifugation. As previously reported, this technique allows the accurate determination of particle sizes and size distributions of CNDs and other carbon allotropes.16,35 The 2-dimensional distributions of the sedimentation (s) and diffusion coefficients (D) for both CNDs are provided in the ESI,‡ Fig. S1–S3 (ESI‡). A much more pronounced polydispersity is found for the CND1 and CND2 compared to the predominantly UV-absorbing pCND studied previously.16 Moreover, a slight bimodality is present for CND1 and CND2. The mean s and D values are 0.389 × 10−13 s and 5.272 × 10−6 cm2 s−1 for CND1 and 0.470 × 10−13 s and 4.325 × 10−6 cm2 s−1 for CND2. The corresponding diameters derived from the Stokes–Einstein equation reached from 0.5 to 2.0 nm for CND1 and 0.7 to 2.5 nm for CND2. These results are in line with the expectation that red-shifted and broader absorption and emission features are due to larger, more polydisperse CNDs.
CND sensitized photoelectrodes were constructed by dipping mesoporous TiO2 electrodes into CND solutions (1 mg mL−1) for several hours. To check for ideal conditions, we used aqueous nitric acid solutions with pH between 1 and 7 (pure water) as solvent. Lower pH was found to lead to significantly improved solar cell performance (see below). Ideal sensitization times were 6–8 h – Fig. S6 (ESI‡). The solar cells were completed with a standard I−/I3− electrolyte and Pt counter-electrodes (see Experimental details in the ESI‡).
With the optimized conditions described above, CND1 sensitized solar cells display power conversion efficiencies of 0.24% – Fig. 2. Using acidic CND solutions for sensitizing the electrodes strongly improves the performance of the solar cells – Fig. S2 (ESI‡). Incident-Photon-to-Current Efficiency (IPCE) spectra as displayed in Fig. 3 show that this improved performance is due to increased photocurrent generation by the CNDs. This, in turn, is due to improved coverage of the electrodes by CNDs at low pH, as shown by the absorption spectra of the photoelectrodes in Fig. 3.
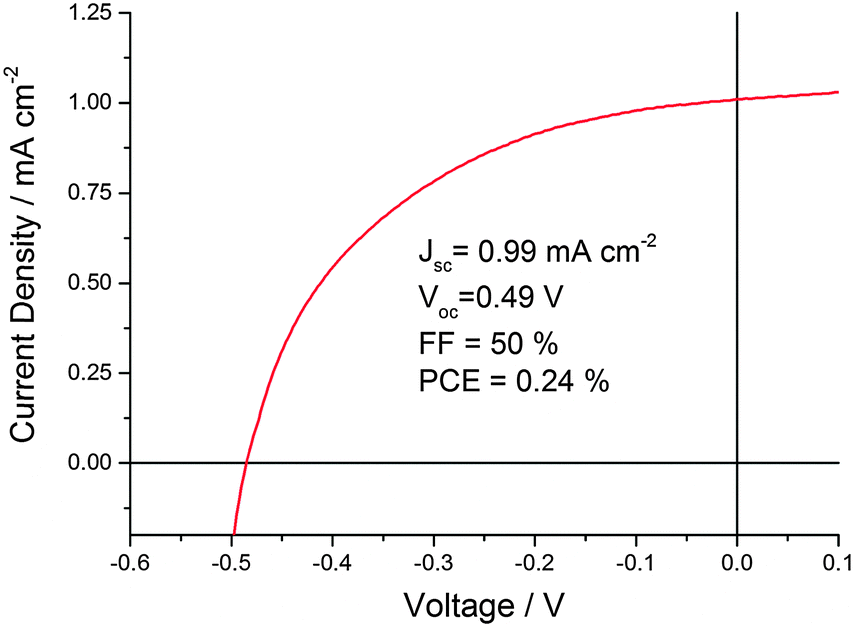 | ||
| Fig. 2 Representative J–V-curve of CND1 solar cells sensitized at pH 1 for 6 h. | ||
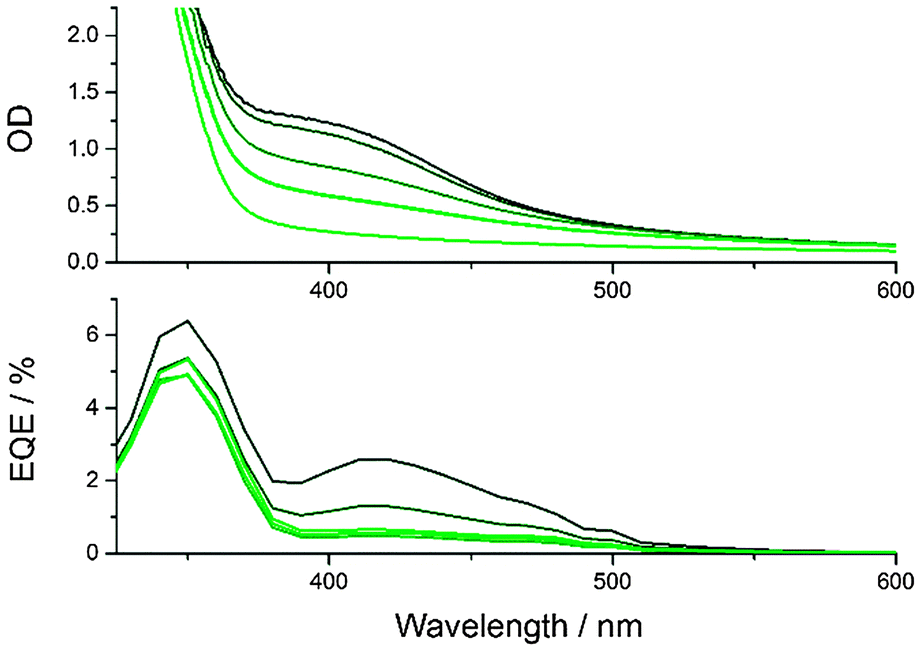 | ||
| Fig. 3 Absorption (above) and IPCE spectra (below) of TiO2 photoelectrodes sensitized using water with pH 7 (light green) to 1 (black) as solvent. The lightest green line in the top graph is the absorption spectrum of a bare TiO2 electrode. | ||
The increased CND coverage at low pH-values can be explained by several factors. Firstly, the solubility of CNDs decreases as they are protonated, shifting the equilibrium towards the adsorbed species. Additionally, protonation of the TiO2 surface increases its hydrophilicity, allowing improved penetration of the mesoporous network during the soaking process.
To test the impact of the pH on TiO2, the morphology of the electrodes was characterized. Scanning electron microscopy images of sensitized TiO2 electrodes show that their morphology is unchanged even after immersion into a pH 1 solution for 6 h – Fig. S10 (ESI‡). Profilometry shows that the film thickness is also unaffected.
Because CNDs are negatively charged in neutral aqueous solutions, the absolute value of their Zeta potential also decreases with decreasing pH, approaching zero at pH 1, but does not become positive – Fig. S11 (ESI‡). This shows that there is no electrostatic repulsion between CNDs and the positively charged TiO2-surface at low pH. Even at long soaking times (40 h) the CND coverage is around twice as high at pH 1 than at pH 7 – Fig. S8 (ESI‡).
As mentioned above, the synthetic procedure can be adjusted to obtain CNDs with enhanced absorption beyond 550 nm (CND2), which might be expected to be better suited for solar energy conversion applications. Interestingly, the performance of CND2 in solar cells is poorer than that of CND1 – please compare Fig. 4 and Fig. S9 (ESI‡). The lower performance cannot be attributed to inferior light harvesting, since the absorption spectra of CND1- and CND2-loaded TiO2 electrodes reveal equally strong absorptions at the CND peak – Fig. S12 (ESI‡). This implies similar CND coverage of the electrodes in both cases. In addition, CND2-loaded TiO2 electrodes absorb more light in the longer-wavelength region. The corresponding external quantum efficiency (EQE) spectra suggest that the absorbance beyond 550 nm hardly contributes to the current generated by the solar cell.
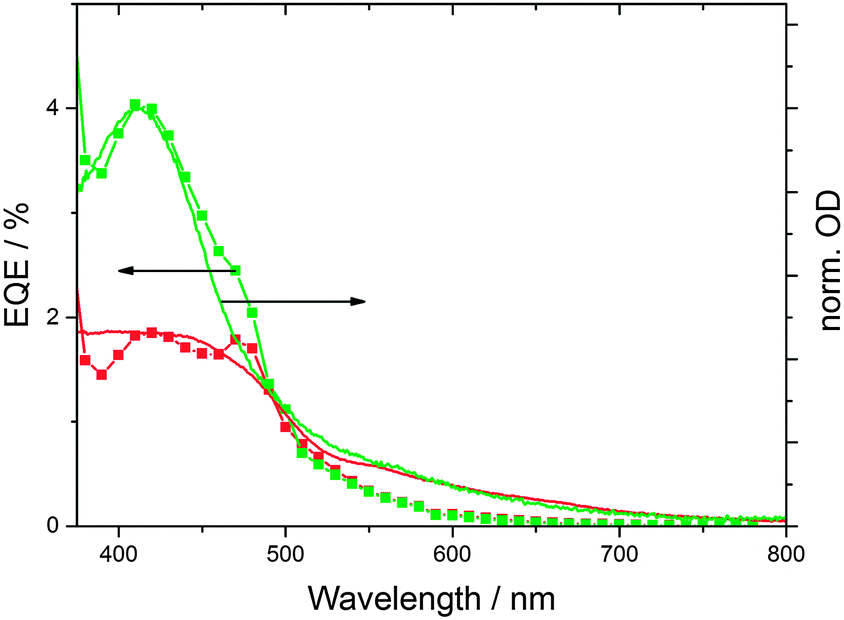 | ||
| Fig. 4 IPCE and absorption spectra of solar cells sensitized with CND1 (green) and CND2 (red) (at pH 1 for 16 h). The absorption spectra were scaled to match the external quantum efficiency (EQE) curves. | ||
In summary, optimized assembly conditions have led to devices that feature improved performance when compared to the recent literature on CND solar cells. Still, more established technologies like dye- or QD-sensitized solar cells reach values, which are more than an order of magnitude higher. Pushing the absorbance of CNDs into the visible does not improve the performance. The following section helps to shed light onto this phenomenon and to indentify possible directions for future improvements. Firstly, we discuss the role of trap states in the excited-state dynamics of CNDs and, secondly, we rationalize the current-generation mechanism in terms of charge injection, recombination and regeneration under device operating conditions.
The trailing absorption beyond 550 nm is qualitatively different from the main absorption peaks at 430 nm, in that it consists of a broad distribution of excited states with, however, low oscillator strength. It has been proposed that such trap states in the bandgap play an important role for the photoluminescence of CNDs,16,25,26 and that the trapping effect consists of energy transfer between π-conjugated subdomains within single particles or with the recombination of photogenerated charges at defect centers.27,28
We found that the fluorescence intensity of CND1 is significantly quenched under acidic conditions (pH 1). Similarly, the absorption in the visible region is reduced – Fig. 5. We therefore performed pH-dependent spectroscopic investigations of CND1 in order to understand the nature of these states better.
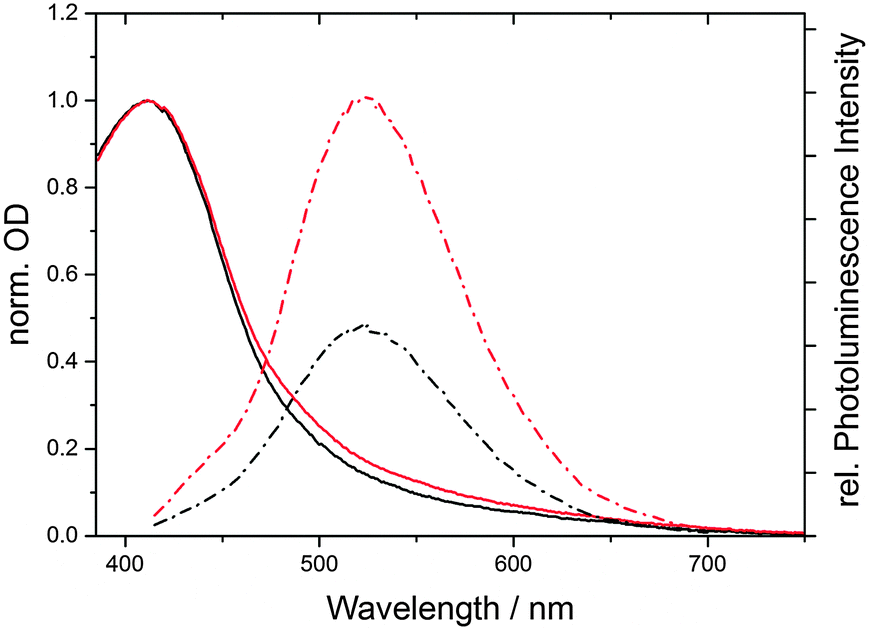 | ||
| Fig. 5 Normalized absorption (solid) and emission (dashed) spectra of equally concentrated CND1 in neutral aqueous solution (red) and at pH 1 (black). | ||
The excited-state decay of CND1 was probed by time-correlated single-photon counting (TCSPC) in acidic aqueous solutions at pH values between 1 and 7 – Fig. S13 (ESI‡). Two major decay components of 7.5 ns (44%) and 3.5 ns (56%) were derived from the fluorescence-time profiles in the pH range between 3 and 7. A noticeable impact of the acidity on the excited-state deactivation was found on approaching pH 1. Here, the lifetimes were best fit bi-exponentially with time-constants 5.5 ns (29%) and 1.2 ns (71%).
Femtosecond transient absorption measurements were used to help understand the excited-state dynamics of CND1. Neutral aqueous solutions of CND1 were subjected to 387 nm laser pulses and transient absorption spectra were recorded with time delays between 0 and 7500 ps. The transient absorption spectra in Fig. 6 are dominated by two features, that is, positive and negative transients at ∼470 nm and ∼560 nm, respectively. A negative feature in the high energy region <450 nm is only seen as an onset due to the 430 to 800 nm limits of the white light. It is attributed to ground-state bleaching. The broad positive signal in the long-wavelength region at >700 nm originates from excited-state absorptions. These decay rapidly with two lifetimes of <1 ps and ∼8 ps and give rise to a 560 nm negative feature, which is due to fluorescence in analogy with previous reports.29,30 Accordingly, we assign the 8 ps lifetime to the population of trap states, from which the fluorescence takes place. A small contribution to the decay of the fluorescence feature, with a lifetime of ∼50 ps, was assigned to exciton recombination. The main components of this transient feature lifetimes of >3 ns and reflect trap-mediated radiative decays.
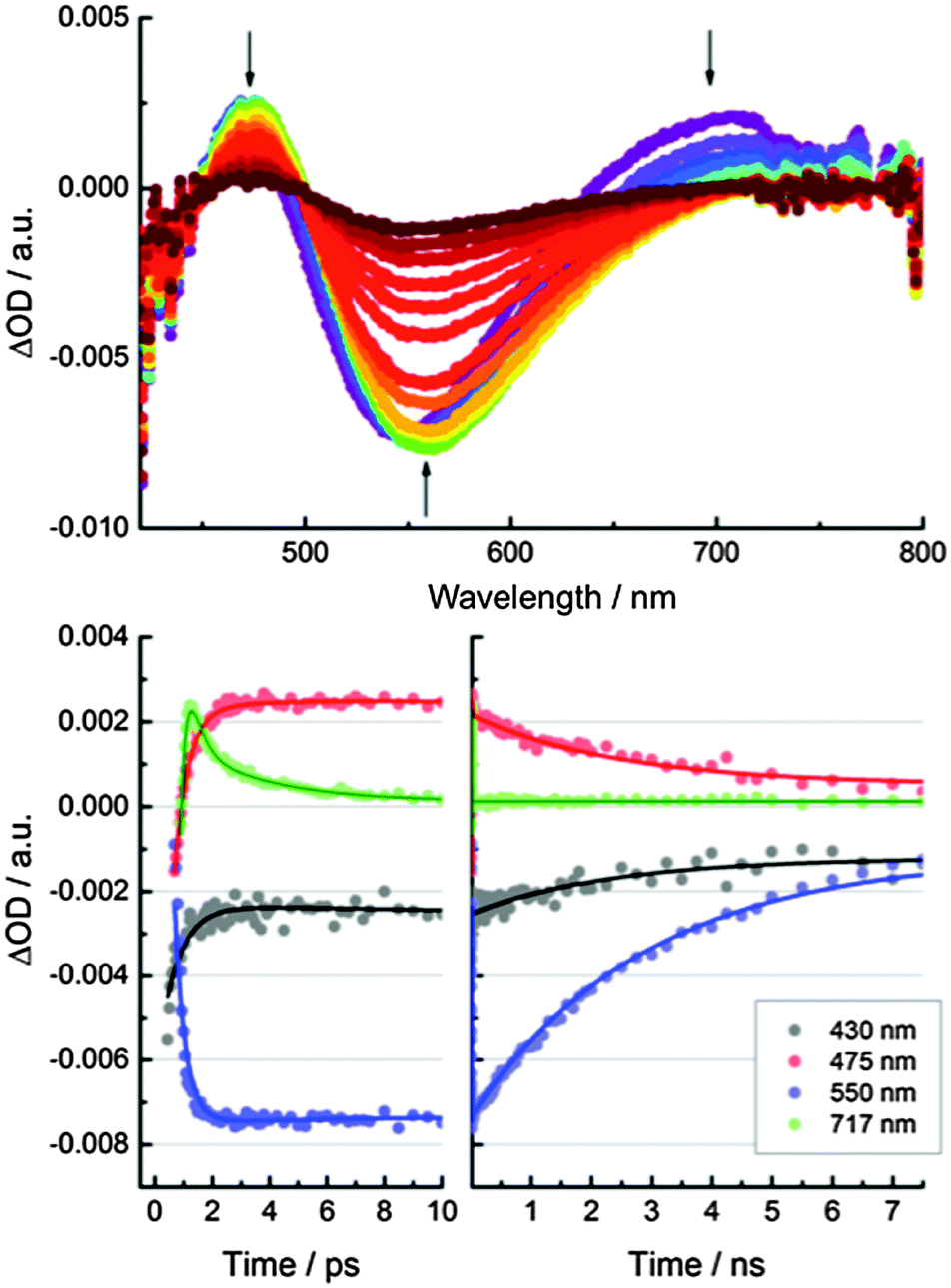 | ||
| Fig. 6 Above: Differential absorption spectra obtained upon femtosecond pump probe experiments (387 nm) of CND1 with several time delays between 0 and 7500 ps. Below: Time absorption profiles of the spectra shown in the upper part at 430 nm (black), 475 nm (red), 550 nm (blue), and 717 nm (green) monitoring the excited state decay. | ||
Significant differences in the kinetics were observed at pH 1, while the spectral features remain unchanged in comparison to experiments performed at pH 7. Most notably, the trap-related features at 475 and 550 nm deactivate faster – Fig. S14 (ESI‡). We derive three lifetimes of 4.4, 17, and 1700 ps from the excited-state decays at 475 and 550 nm. As for the excited-state deactivation at pH 7, the short lifetime (4.4 ps) represents the population of the trap states and the long lifetime (1700 ps) corresponds to trap-state mediated radiative processes. The excitonic fluorescence follows the intermediate lifetime (17 ps). The considerably shortened lifetimes at pH 1 indicate a lower population of long-lived, radiatively decaying trap states.
Steady-state and time-resolved measurements therefore point to a decrease in the trap density at low pH values. We turned to molecular modeling to elucidate the molecular origin of this effect. We recently reported a theoretical study of amorphous carbon nanodots.31 The disordered structure of these systems makes them good models of CND surfaces. For this study we modified an amorphous CND model with carboxylate groups and studied its protonated and deprotonated forms. This model contains nine carboxylate groups in different chemical environments, reflecting the complex surface chemistry of real CNDs. To obtain good sampling of the structures' conformational freedom, we performed direct Born–Oppenheimer quantum mechanical molecular-dynamics (MD) simulations with EMPIRE14, using the semiempirical AM1 Hamiltonian (see Computational details in the ESI‡).32–34
Fig. 7 shows a histogram of the HOMO–LUMO gap of the protonated and deprotonated structures taken from a 3.6 ps MD run. Note that the energy gap for the deprotonated CND is smaller and the variation of the energy levels is larger. This can be understood by comparing the electronic density of states of the two structures. The deprotonated species contains additional states within the band gap that are absent in the protonated case – Fig. 7. The molecular orbitals corresponding to these states are localized on the carboxylate groups and adjacent atoms. Note that not all trap states are necessarily localized on carboxylate groups, as we have shown for CNDs that consist entirely of carbon and hydrogen.31
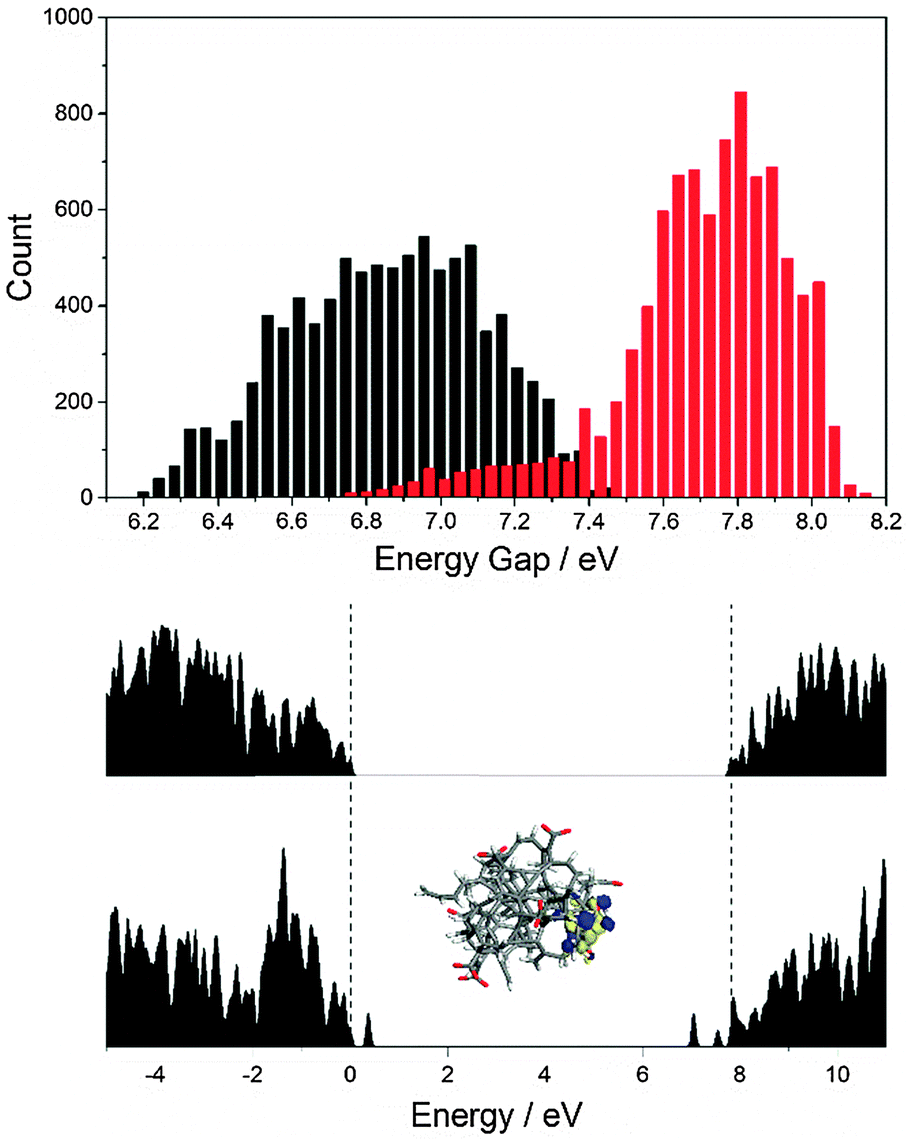 | ||
| Fig. 7 Above: Histogram of electronic gaps extracted from a 3.6 ps MD trajectory of a protonated (red) and deprotonated (black) CND. Below: Representative density-of-states plot for the protonated and deprotonated species. | ||
To explore how the electronic structures of CND1 and CND2 affect the photocurrent generation in the solar cells, we performed transient absorption spectroscopy with CND-sensitized TiO2 electrodes. 480 nm photoexcitation measurements were carried out in the presence of I−/I3− electrolyte and in pure acetonitrile as reference (see Fig. 8 and Fig. S15–S17, ESI‡). All of the differential spectra display distinct ground-state bleaching with a minimum at around 470 nm as well as a broad featureless transient from 600 to 1200 nm. In line with previous reports, we ascribe the latter to trapped charge carriers and free electrons in the conduction band of TiO2.36 The trap-related CND transients, as observed in solution – Fig. 6 – appear in the same region and are likely to contribute to the differential absorption. Considering the lack of spectroscopic signature for the CND radical cation, the current generation process was followed via the evolution of the CND ground state bleaching at 465 nm.17,18 The respective kinetics are best fit tri-exponentially with lifetimes on the order of 1, 10, and 1000 ps for all measurements – Table 1. The short and intermediate lifetimes, that is, 1 and 10 ps, are attributed to non-radiative recombination, population of trap states, and charge carrier injection. In acetonitrile, the longest lifetime, is assigned to the recombination of injected charges with the CND radical cation. In the presence of the electrolyte, we assign it to the charge regeneration process.
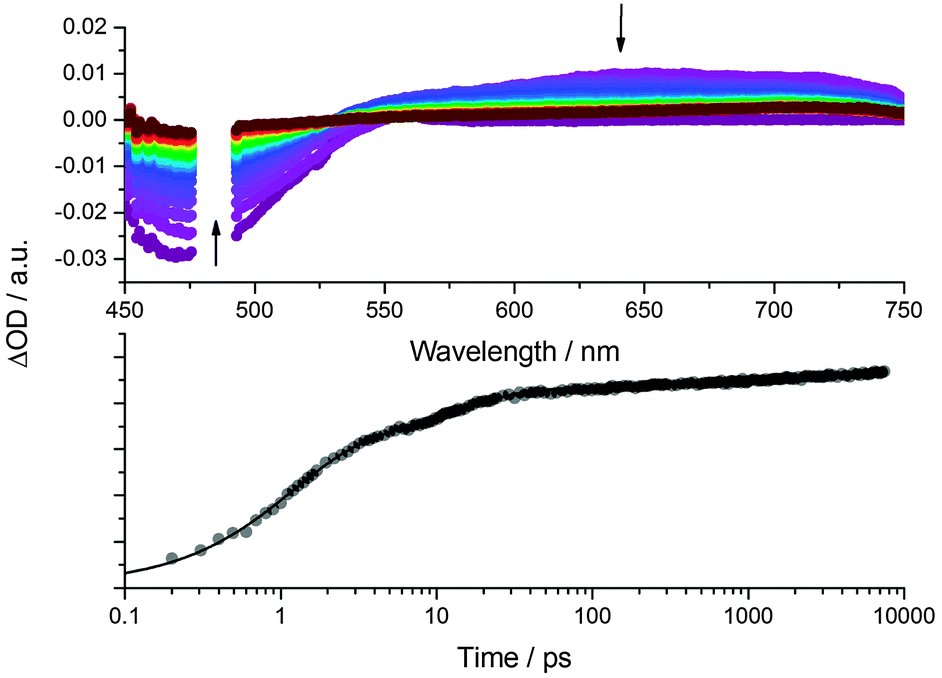 | ||
| Fig. 8 Above: Differential absorption spectra obtained upon femtosecond pump probe experiments (λex = 480 nm) with a CND1 sensitized TiO2 electrode in the presence of I−/I3− electrolyte with several time delays between 0 (violet) and 7500 ps (red). Below: Time absorption profile of the spectra shown in the upper part at 465 nm, monitoring the decay of the CND ground state bleaching. | ||
| Acetonitrile (ps) | I−/I3− electrolyte (ps) | |
|---|---|---|
| CND1 | τ 1 = 1.3 ± 0.04 | τ 1 = 1.1 ± 0.02 |
| τ 2 = 11 ± 0.4 | τ 2 = 13 ± 0.4 | |
| τ 3 = 550 ± 30 | τ 3 = 1200 ± 100 | |
| CND2 | τ 1 = 1.3 ± 0.04 | τ 1 = 1.1 ± 0.03 |
| τ 2 = 13 ± 0.5 | τ 2 = 9 ± 0.3 | |
| τ 3 = 600 ± 30 | τ 3 = 700 ± 60 | |
In none of the experiments do we find any impact of the electrolyte – regardless of using CND1 or CND2 – on the short and intermediate lifetimes. In contrast, the long lifetimes are around 600 ps for CND1 and CND2 in the absence of the I−/I3− electrolyte. From this finding we postulate that the TiO2–CND charge recombination is not affected by the higher trap density in CND2. As to the presence of the I−/I3− electrolyte, the lifetime extends to 1200 and 700 ps for CND1 and CND2, respectively. Implicit is that the presence of the electrolyte slows down the charge carrier recombination dynamics – a trend that is significantly stronger for CND1. As such, an improved charge regeneration seems to correlate with an improved performance for CND1 solar cells.
The impact of the trap states under device operation was investigated by comparing the kinetics of the ground state bleaching in CND2 electrodes at 465 and 550 nm – Fig. S18 (ESI‡). Please note that the bleaching at 550 nm reflects the broad absorption feature seen in steady-state absorption and, in turn, is assigned to trap states. The decay at 550 nm, which is rather fast with lifetimes of 0.2 and 8 ps, indicates a fast relaxation to the ground state rather than a contribution to the photocurrent.
As a complement, we investigated the open-circuit voltage decay of CND1 and CND2 solar cells upon illumination with a pulsed LED – Fig. S19 (ESI‡). In agreement with our transient absorption measurements, the photovoltage decays are significantly faster for CND2 than for CND1, pointing to a higher electron recombination rate. This indicates that a rather poor charge carrier management evolves as the main cause for the relatively low device performance.
Conclusions
We have shown that CNDs made from citric acid, urea, and formic acid in a one-step microwave synthesis can be used as sensitizers in mesoscopic solar cells. Using acidic solutions during the sensitization improves the CND-coverage of the photoelectrode, and, consequently, the performance of the solar cells. Increasing the light-harvesting range of the CNDs towards the red end of the spectrum failed to improve the solar-cell performance because these additional low-energy excited states do not inject electrons into TiO2 efficiently.The nature of the low-energy, low-oscillator-strength trap states was investigated via pH-dependent spectroscopic measurements and molecular modeling. Both indicate that protonation reduces the number of trap states, specifically of long-lived, light-emitting ones. Quantum mechanical MD simulations show that carboxylate groups conjugated to adjacent sp2-carbon domains form states in the band gap, which are removed upon protonation.
Time-resolved investigations of the photocurrent generation indicate that the balance between the charge recombination and regeneration processes is not ideal in these devices. We also demonstrated that CND trap states deactivate in the device on an ultrafast timescale, and, in turn, fall short in terms of contributing to the overall photocurrent generation. These traps do not promote faster charge recombination between TiO2 and CNDs. For future work on CND solar cells, it is imperative to improve their excitonic, that is, non-trap related, absorptions. Additionally, chemical surface modification seems a promising way to improve the charge-injection and regeneration kinetics under device operating conditions.
Acknowledgements
This work was supported by the Deutsche Forschungsgemein-schaft as part of the Excellence Cluster “Engineering of Advanced Materials” and the Collaborative Research Centre SFB 953 “Synthetic Carbon Allotropes”. The Bavarian government is also kindly acknowledged for funding granted as part of the “Solar Technologies go Hybrid” initiative. J. W. and W. P. would like to acknowledge financial support through the DFG project PE 427/28-1 and NSF grant ACI-1339649 to Borries Demeler from University of Texas, USA, as well as computing time (HER21) at the Jülich Supercomputing Centre (JSC), granted by the John von Neumann Institute for Computing (NIC).Notes and references
- B. O'Regan and M. Graetzel, Nature, 1991, 353, 737–740 CrossRef.
- M. A. Green, K. Emery, H. Yoshihiro, W. Warta and E. D. Dunlop, Prog. Photovoltaics, 2015, 23, 1–9 Search PubMed.
- M. Grätzel, Nat. Mater., 2014, 13, 838–842 CrossRef PubMed.
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Graetzel, Science, 2011, 334, 629–634 CrossRef CAS PubMed.
- I. J. Kramer and E. H. Sargent, ACS Nano, 2011, 5, 8506–8514 CrossRef CAS PubMed.
- P. V. P. Kamat, J. Phys. Chem. Lett., 2013, 4, 908–918 CrossRef CAS PubMed.
- Z. Pan, I. Mora-Seró, Q. Shen, H. Zhang, Y. Li, K. Zhao, J. Wang, X. Zhong and J. Bisquert, J. Am. Chem. Soc., 2014, 136, 9203–9210 CrossRef CAS PubMed.
- J. Luo, H. Wei, F. Li, Q. Huang, D. Li, Y. Luo and Q. Meng, Chem. Commun., 2014, 50, 3464–3466 RSC.
- S. Jiao, Q. Shen, I. Mora-Seró, J. Wang, Z. Pan, K. Zhao, Y. Kuga, X. Zhong and J. Bisquert, ACS Nano, 2015, 9, 908–915 CrossRef CAS PubMed.
- X. Lan, S. Masala and E. H. Sargent, Nat. Mater., 2014, 13, 233–240 CrossRef CAS PubMed.
- Z. Ning, O. Voznyy, J. Pan, S. Hoogland, V. Adinolfi, J. Xu, M. Li, A. R. Kirmani, J. Sun, J. Minor, K. W. Kemp, H. Dong, L. Rollny, A. Labelle, G. Carey, B. Sutherland, I. Hill, A. Amassian, H. Liu, J. Tang, O. M. Bakr and E. H. Sargent, Nat. Mater., 2014, 13, 4–10 CrossRef PubMed.
- F. Lodermeyer, R. D. Costa, R. Casillas, F. T. U. Kohler, P. Wasserscheid, M. Prato and D. M. Guldi, Energy Environ. Sci., 2015, 8, 241–246 CAS.
- R. D. Costa, S. Feihl, A. Kahnt, S. Gambhir, D. L. Officer, G. G. Wallace, M. I. Lucio, M. A. Herrero, E. Vázquez, Z. Syrgiannis, M. Prato and D. M. Guldi, Adv. Mater., 2013, 25, 6513–6518 CrossRef CAS PubMed.
- R. Casillas, F. Lodermeyer, R. D. Costa, M. Prato and D. M. Guldi, Adv. Energy Mater., 2014, 4, 1301577 Search PubMed.
- S. Kirner, M. Sekita and D. M. Guldi, Adv. Mater., 2014, 26, 1482–1493 CrossRef CAS PubMed.
- V. Strauss, J. T. Margraf, C. Dolle, B. Butz, T. J. Nacken, J. Walter, W. Bauer, W. Peukert, E. Spiecker, T. Clark and D. M. Guldi, J. Am. Chem. Soc., 2014, 136, 17308–17316 CrossRef CAS PubMed.
- V. Strauss, J. T. Margraf, T. Clark and D. M. Guldi, Chem. Sci., 2015, 6, 6878–6885 RSC.
- V. Strauss, J. T. Margraf, K. Dirian, Z. Syrgiannis, M. Prato, C. Wessendorf, A. Hirsch, T. Clark and D. M. Guldi, Angew. Chem., Int. Ed., 2015, 54, 8292–8297 CrossRef CAS PubMed.
- P. Mirtchev, E. J. Henderson, N. Soheilnia, C. M. Yip and G. A. Ozin, J. Mater. Chem., 2012, 22, 1265–1269 RSC.
- J. Briscoe, A. Marinovic, M. Sevilla, S. Dunn and M. Titirici, Angew. Chem., Int. Ed., 2015, 54, 4463–4468 CrossRef CAS PubMed.
- M. Sun, S. Qu, W. Ji, P. Jing, D. Li, L. Qin, J. Cao, H. Zhang, J. Zhao and D. Shen, Phys. Chem. Chem. Phys., 2015, 17, 7966–7971 RSC.
- K. J. Williams, C. Nelson, X. Yan, L.-S. Li and X. Zhu, ACS Nano, 2013, 7, 1388–1394 CrossRef CAS PubMed.
- M. Dutta, S. Sarkar, T. Ghosh and D. Basak, J. Phys. Chem. C, 2012, 116, 20127–20131 CAS.
- X. Yan, X. Cui, B. Li and L. S. Li, Nano Lett., 2010, 10, 1869–1873 CrossRef CAS PubMed.
- V. Nguyen, J. Si, L. Yan and X. Hou, Carbon, 2015, 95, 659–663 CrossRef CAS.
- X. Wang, L. Cao, F. Lu, M. J. Meziani, H. Li, G. Qi, B. Zhou, B. A. Harruff, F. Kermarrec and Y. P. Sun, Chem. Commun., 2009, 3774–3776 RSC.
- M. Fu, F. Ehrat, Y. Wang, K. Z. Milowska, C. Reckmeier, A. L. Rogach, J. K. Stolarczyk, A. S. Urban and J. Feldmann, Nano Lett., 2015, 15, 6030–6035 CrossRef CAS PubMed.
- S. Ghosh, A. M. Chizhik, N. Karedla, M. O. Dekaliuk, I. Gregor, H. Schuhmann, M. Seibt, K. Bodensiek, I. A. T. Schaap, O. Schulz, A. P. Demchenko, J. Enderlein and A. I. Chizhik, Nano Lett., 2014, 14, 5656–5661 CrossRef CAS PubMed.
- S. Zhu, Y. Song, X. Zhao, J. Shao, J. Zhang and B. Yang, Nano Res., 2015, 8, 355–381 CrossRef CAS.
- L. Wang, S.-J. Zhu, H.-Y. Wang, Y.-F. Wang, Y.-W. Hao, J.-H. Zhang, Q.-D. Chen, Y.-L. Zhang, W. Han, B. Yang and H.-B. Sun, Adv. Opt. Mater., 2013, 1, 264–271 CrossRef.
- J. T. Margraf, V. Strauss, D. M. Guldi and T. Clark, J. Phys. Chem. B, 2015, 119, 7258–7265 CrossRef CAS PubMed.
- M. Hennemann and T. Clark, J. Mol. Model., 2014, 20, 2331–2341 CrossRef PubMed; J. T. Margraf, M. Hennemann, B. Meyer and T. Clark, J. Mol. Model., 2015, 21, 144 CrossRef PubMed.
- M. J. S. Dewar, E. G. Zoebisch, E. F. Healy and J. J. P. Stewart, J. Am. Chem. Soc., 1985, 107, 3902–3909 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- J. Walter, K. Löhr, E. Karabudak, W. Reis, J. Mikhael, W. Peukert, W. Wohlleben and H. Cölfen, ACS Nano, 2014, 8, 8871–8886 CrossRef CAS PubMed.
- A. Kahnt, C. Oelsner, F. Werner, D. M. Guldi, S. P. Albu, R. Kirchgeorg, K. Lee and P. Schmuki, Appl. Phys. Lett., 2013, 102, 233109 CrossRef.
Footnotes |
| † Dedicated to Jonathan L. Sessler on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nh00010j |
| This journal is © The Royal Society of Chemistry 2016 |