
Alloy oxidation as a route to chemically active nanocomposites of gold atoms in a reducible oxide matrix†
P.
Sutter
*a,
S. A.
Tenney
b,
F.
Ivars-Barcelo
b,
L.
Wu
c,
Y.
Zhu
c and
E.
Sutter
d
aDepartment of Electrical and Computer Engineering, University of Nebraska – Lincoln, Lincoln, Nebraska 68588, USA. E-mail: psutter@unl.edu
bCenter for Functional Nanomaterials, Brookhaven National Laboratory, Upton, New York 11973, USA
cCondensed Matter Physics and Materials Science Department, Brookhaven National Laboratory, Upton, New York 11973, USA
dDepartment of Mechanical and Materials Engineering, University of Nebraska – Lincoln, Lincoln, Nebraska 68588, USA
First published on 12th February 2016
Abstract
While nanoparticles are being pursued actively for a number of applications, dispersed atomic species have been explored far less in functional materials architectures, primarily because composites comprising dispersed atoms are challenging to synthesize and difficult to stabilize against sintering or coarsening. Here we show that room temperature oxidation of Au–Sn alloys produces nanostructures whose surface is terminated by a reducible amorphous oxide that contains atomically dispersed Au. Analysis of the oxidation process shows that the dispersal of Au in the oxide can be explained by predominant oxygen anion diffusion and kinetically limited metal mass transport, which restrict phase separation due to a preferential oxidation of Sn. Nanostructures prepared by oxidation of nanoscale Au–Sn alloys with intermediate Au content (30–50%) show high activity in a CO-oxidation probe reaction due to a cooperative mechanism involving Au atoms as sites for CO adsorption and reaction to CO2 embedded in a reducible oxide that serves as a renewable oxygen reservoir. Our results demonstrate a reliable approach toward nanocomposites involving oxide-embedded, atomically dispersed noble metal species.
Conceptual insightsWhereas nanoparticles of semiconductors, metals, and alloys have been explored extensively for diverse applications, materials at the extreme limit that derive their functionality from individual atomic species are relatively rare. This is at least in part due to the difficulty of synthesizing materials comprising dispersed atoms, which are sufficiently stable against coarsening to maintain their function over extended periods. The oxidation of noble/non-noble metal binary alloys can produce nanocomposites in which noble metal species are atomically dispersed in an oxide matrix. This concept is demonstrated here with the example of Au–Sn alloys. Room temperature oxidation of Au–Sn nanostructures, governed by a predominant oxygen anion migration and limited mass transport of metal cations, produces amorphous Sn-oxides with embedded, atomically dispersed Au. The functional properties of such composites are demonstrated via oxidation probe reactions. Samples with optimal concentration of Au atoms in the surface of the reducible oxide show room temperature CO to CO2 conversion efficiencies close to 100% that are stable over extended time periods, and are thus promising for applications in CO oxidation for air pollution prevention. Our results highlight a novel avenue toward functional materials architectures comprising atomically dispersed species, which show promise for catalytic applications and may find broader potential uses in areas including electronics, photonics, sensing, energy conversion and storage. |
Bimetallic nanoparticles promise tunable properties by selection of the constituent metals and by adjusting other variables, such as composition, structure, size, or shape. Recent progress in nanoparticle synthesis has provided access to a large family of particle configurations, including alloys,1–4 core–shell structures,4–9 and Janus particles,10 among others. Given the very different chemical, electronic, and optical properties of metals and metal oxides, the oxidation of bimetallic systems promises further extended tunability of multifunctional nanomaterials for applications in plasmonics,11 imaging,12 energy storage,13 surface chemistry,14 and electrocatalysis.15–17 Binary alloys that consist of a noble and a non-noble metal component are of particular interest in chemistry because their oxidation can give rise to a variety of chemically active configurations, such as oxide supported noble metal nanoparticles,18–20 core–shell structures,21–23etc.
Gold is a noble metal whose chemical properties are particularly intriguing. Bulk Au is the only transition metal that is not reactive, except in isolated cases.24 The inability of bulk Au surfaces to activate O2 is the main cause for their low activity in oxidation reactions. If, however, a supply of atomic oxygen is established, Au can become exceptionally active. Examples are bulk Au surfaces with chemisorbed oxygen,25 surface or bulk Au-oxides,26 composites of Au nanoparticles on reducible metal oxide supports,27,28 and Au particles on non-reducible oxides.29 Several studies, e.g., for Au nanoparticles on TiO2(110)30 and dispersed Au on carbon nanotubes,31 concluded that the reactivity is highest for a specific Au cluster size and that larger or smaller Au particles, including atomically dispersed Au, are less active. In that case, the synthesis of materials with optimal properties requires tight size control, and later sintering needs to be avoided since the associated size increase would cause a loss of chemical activity.
Here, we show that the room temperature oxidation of alloys of Au and a non-noble metal (Sn) produces chemically active nanocomposites that consist of a reducible, amorphous Sn oxide with embedded Au atoms. This structure is obtained because the oxidation proceeds by the predominant diffusion of oxygen anions through the growing oxide whereas metal (Sn, Au) mass transport is kinetically limited.21 The behavior in a CO-oxidation probe reaction demonstrates a high chemical activity of these nanocomposites over a limited composition range of the initial Sn–Au alloy. Further characterization shows that this reactivity involves the amorphous Sn oxide as a reducible oxygen reservoir, whereas the embedded Au atoms locally destabilize the oxide matrix and act as low-coordinated sites for CO adsorption and reaction to CO2 with oxygen from the surrounding oxide.
Fig. 1 summarizes the structure and morphology of AuxSn1−x alloys produced by sequential vacuum deposition of Au and Sn. Transmission electron microscopy (TEM) of a representative AuxSn1−x alloy (x = 0.5) on amorphous C shows the typical morphology we observe for a wide alloy composition range on different supports (e.g., Ge, see ESI,† Fig. S1) following room temperature oxidation in air. Low magnification images (Fig. 1a) show a high surface area morphology of elongated, meandering nanostructures. From high-resolution TEM (Fig. 1b and c) we conclude that the darker meandering ridges are mostly crystalline, whereas the brighter interspersed areas are amorphous, consistent with a spontaneous alloying of the sequentially deposited metals (Au followed by Sn) at room temperature32–37 and a partial dewetting of the Au–Sn alloy into nanoscale ridges, between which the amorphous C support is exposed. Following air exposure an additional component is found near the surface of the Au–Sn ridges, which does not show any crystalline order. Further characterization, discussed below, identifies these areas as an amorphous oxide shell containing both Au and Sn. Electron diffraction (Fig. 1d) confirms a majority AuSn (i.e., Au0.5Sn0.5) phase in the crystalline alloy cores, along with a detectable Au-rich minority phase (Au5Sn) whose presence indicates a slight increase in the Au content of the alloy core during oxidation.32–37
 | ||
| Fig. 1 (a and b) TEM images of the structure of a Au–Sn sample prepared by sputtering of 6 nm Au and 6 nm Sn onto an amorphous carbon support. The resulting AuSn bimetallic alloy nanostructures were oxidized at room temperature in air. Scale bars: (a) 50 nm; (b) 5 nm. (c) High-resolution TEM image of a crystalline AuSn grain terminated by an amorphous surface oxide. Scale bar: 5 nm. (d) Electron diffraction pattern (DP) of the AuSn sample. Comparison of the experimental DP (left) with simulated patterns (right) confirm a majority AuSn phase. Additional diffraction intensity is detectable from a Au5Sn minority phase. Simulated DPs calculated using the software package JEMS for nanoparticles with the hexagonal AuSn38 and Au5Sn39 structures. Scale bar: 2 nm−1. (e) High-angle annular dark-field (HAADF) STEM image of two sections of meandering oxidized AuSn ridges, clearly showing the metallic core and darker oxide shell of the nanostructures. Scale bar: 5 nm. (f) HAADF-STEM image and simultaneously recorded chemical (Sn, O, Au) maps, acquired in the rectangular area outlined in (e), containing two adjacent AuSn structures separated by their oxide shells. Scale bar: 2 nm. | ||
To further characterize these core–shell structures, we performed high-angle annular dark-field scanning-TEM (HAADF-STEM, i.e., Z-contrast imaging, Fig. 1e; ESI,† Fig. S2) and simultaneous chemical mapping using electron energy-loss spectroscopy (EELS) and energy-dispersive X-ray spectroscopy (EDS) (see Methods and ESI,† Fig. S3). Atomic-resolution Z-contrast imaging shows distinct contrast between the crystalline bimetallic cores (bright; high average atomic number, Z) and the amorphous oxide shells (darker) terminating their surface (Fig. 1e; Fig. S2, ESI†). Interspersed dark zones represent the bare carbon support (low Z). The distribution of Sn, O, and Au is captured in chemical maps, in which the intensity represents elemental abundance (Fig. 1f). Sn is distributed throughout but has highest concentration in the cores, whereas the O content is largest in the oxide shell. The concentration of Au is also highest in the bimetallic cores, but Au is clearly detectable throughout the oxide shells as well.
Sn, similar to In40,41 and Pb,42 oxidizes at room temperature by preferential oxygen anion migration through the oxide.43 The diffusion of metal cations is kinetically limited. For alloys between Sn and a noble metal (e.g., Au), the equilibrium configuration – a core–shell structure, in which the less noble species is preferentially oxidized at the surface and the noble metal becomes concentrated in the core – may not be realized.21 Instead, the system can remain trapped in a metastable state in which the oxide shell contains both metals, thus generating a composite oxide with potentially interesting functional properties, e.g., chemical reactivity. Such a scenario for the oxidation of nanostructured AuxSn1−x alloys is corroborated by X-ray photoelectron spectroscopy (XPS) and low-energy ion scattering spectroscopy (LEIS), measured on oxidized high surface area AuSn on Ge supports. Sn 3d XPS spectra (Fig. S4a, ESI†) were used to determine the oxidation rate of AuSn in comparison with a Sn nanoparticle reference (Fig. S4b, ESI†). Sn 3d5/2 spectra of both samples can be deconvoluted into two peaks, assigned to metallic Sn0 (∼485–485.5 eV) and Sn oxide (∼487 eV), respectively.44 For AuSn, the Sn0 component is shifted by 0.3 eV to higher binding energy, in agreement with previous results for δ-AuSn alloys.45
Room temperature oxidation of Sn nanoparticles forms a Sn(II)-oxide shell.43 Hence, we assign the Sn 3d5/2 peak at ∼487 eV to Sn2+ in both the Sn reference and AuSn, which is supported by a quantitative analysis of Sn 3d5/2 and O 1s XPS spectra (see ESI†) that yields an Sn:O ratio close to 1 in the oxide. A major difference between AuSn and Sn lies in the evolution of oxidized Sn (Fig. 2a). Using the atomic densities, the oxide thickness has been calculated (Fig. 2b; see also ESI†). After extended air exposure Sn nanoparticles are covered by ∼5 nm oxide shells.43 For AuSn, little additional oxidation is observed beyond a limiting oxide thickness of 1.5 to 2 nm. Hence, AuSn alloys are substantially more oxidation resistant than pure Sn.
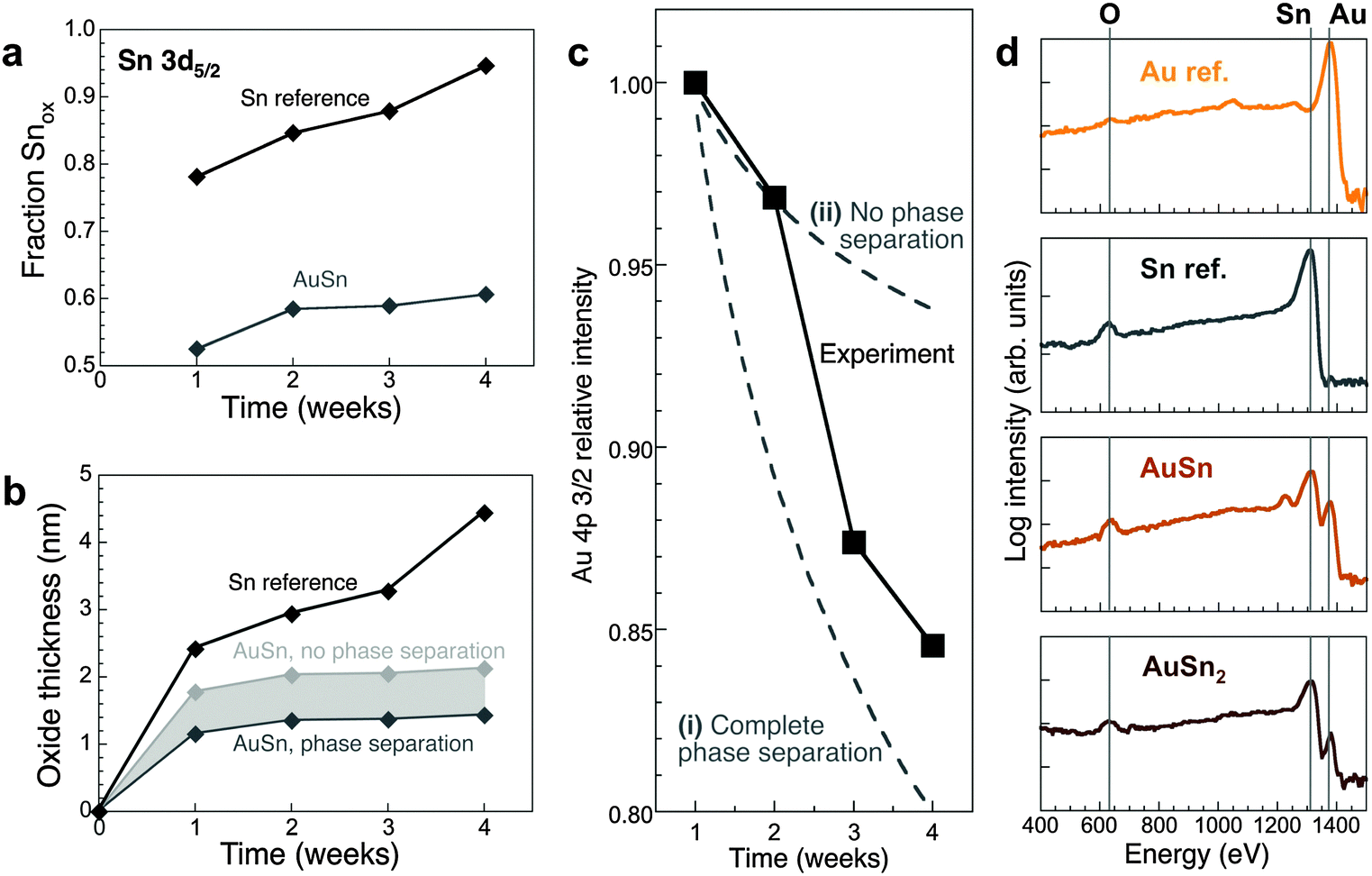 | ||
Fig. 2 (a) Area fraction of the Sn-oxide component of Sn 3d5/2 XPS spectra of oxidized AuSn and a Sn reference (see ESI,† Fig. S4) as a function of oxidation time. (d) Evolution of the oxide thickness. For the AuSn alloy, two extreme cases are shown (see ESI†). (b) Time evolution of the measured Au 4p 3/2 XPS intensity of AuSn (black symbols), compared with two different scenarios for the oxidation process: (i) preferential oxidation of Sn to form a SnO shell and concentration of Au in the metallic core; and (ii) formation of a (Au–Sn) oxide with unchanged Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Sn ratio. (c) LEIS spectra (incident He+ ions at 1.5 keV primary energy) of AuSn2 and AuSn, oxidized at room temperature in air, as well as Sn and Au reference samples. :Sn ratio. (c) LEIS spectra (incident He+ ions at 1.5 keV primary energy) of AuSn2 and AuSn, oxidized at room temperature in air, as well as Sn and Au reference samples. | ||
Au 4f XPS spectra (Fig. S4c, ESI†) can be fitted by 4f7/2 and 4f5/2 components centered at 84.8 eV and 88.5 eV, respectively, shifted ∼0.6–0.8 eV to higher binding energy compared to pure Au due to coordination to Sn in the alloy.45 The Au 4f7/2 and 4f5/2 binding energies remain constant and no additional Au 4f components appear during oxidation. The time dependent intensity of the Au 4p 3/2 core level was used to track the concentration of Au in the growing oxide (Fig. 2c; ESI†). Phase separation (i.e., Sn oxidation and Au concentration in the alloy core) is initially suppressed. At later stages, the overall Au content of the oxide decreases, consistent with the detection of Au-rich phases in the core by electron diffraction. However, a pure Sn-oxide shell is never achieved but instead an oxide containing both Au and Sn is formed. LEIS used to probe the atomic species present in the outermost layer of the oxide confirms this conclusion. The surface of oxidized AuSn and AuSn2 nano-alloys contains O, Sn, and Au (Fig. 2d), and the Au:Sn peak intensity ratios correlate with the Au concentration in the Au–Sn alloy. Specifically, comparison of different samples (e.g., AuSn, AuSn2; Fig. 2d) shows a larger Au:Sn ratio (i.e., higher Au atom fraction) in the outermost surface layer for samples with higher Au content of the initial alloy (IAu:ISn = 0.18 and 0.07 for AuSn, AuSn2, respectively).
The state of Au in the oxide, which defines key functional properties such as chemical reactivity, was probed by aberration-corrected high-resolution TEM (AC-HRTEM). AC-HRTEM shows neither Au nanoparticles nor clusters in the oxide, but instead detects an ensemble of strong scatterers with uniform size dispersed throughout the oxide shell (Fig. 3a and b). The contrast due to incoherent electron scattering by these heavy scattering centers embedded in the amorphous oxide matrix is very similar to that of individual Au atoms in free-standing atomic chains46 and on graphene supports.47 The conclusion that these species are dispersed Au atoms is confirmed by line scans through a large number of them in our AC-HRTEM images, which show nearly identical full width at half maximum size (0.203 ± 0.022 nm, Fig. 3c and d) as single Au atoms on graphene (0.201 nm).47 Thus room temperature oxidation of Au–Sn alloy nanostructures produces core–shell morphologies with Au atomically dispersed in an amorphous oxide shell. Previously, Zhang et al. used sub-Å resolution HAADF-STEM to identify atomically dispersed Pt in a crystalline FeOx matrix.48 For dispersed Au atoms in an amorphous oxide, considered here, combined chemical mapping and AC-HRTEM can unambiguously identify single Au atoms by detecting the heavy atoms via incoherent scattering in AC-HRTEM in which the amorphous matrix contributes approximately uniform background intensity.
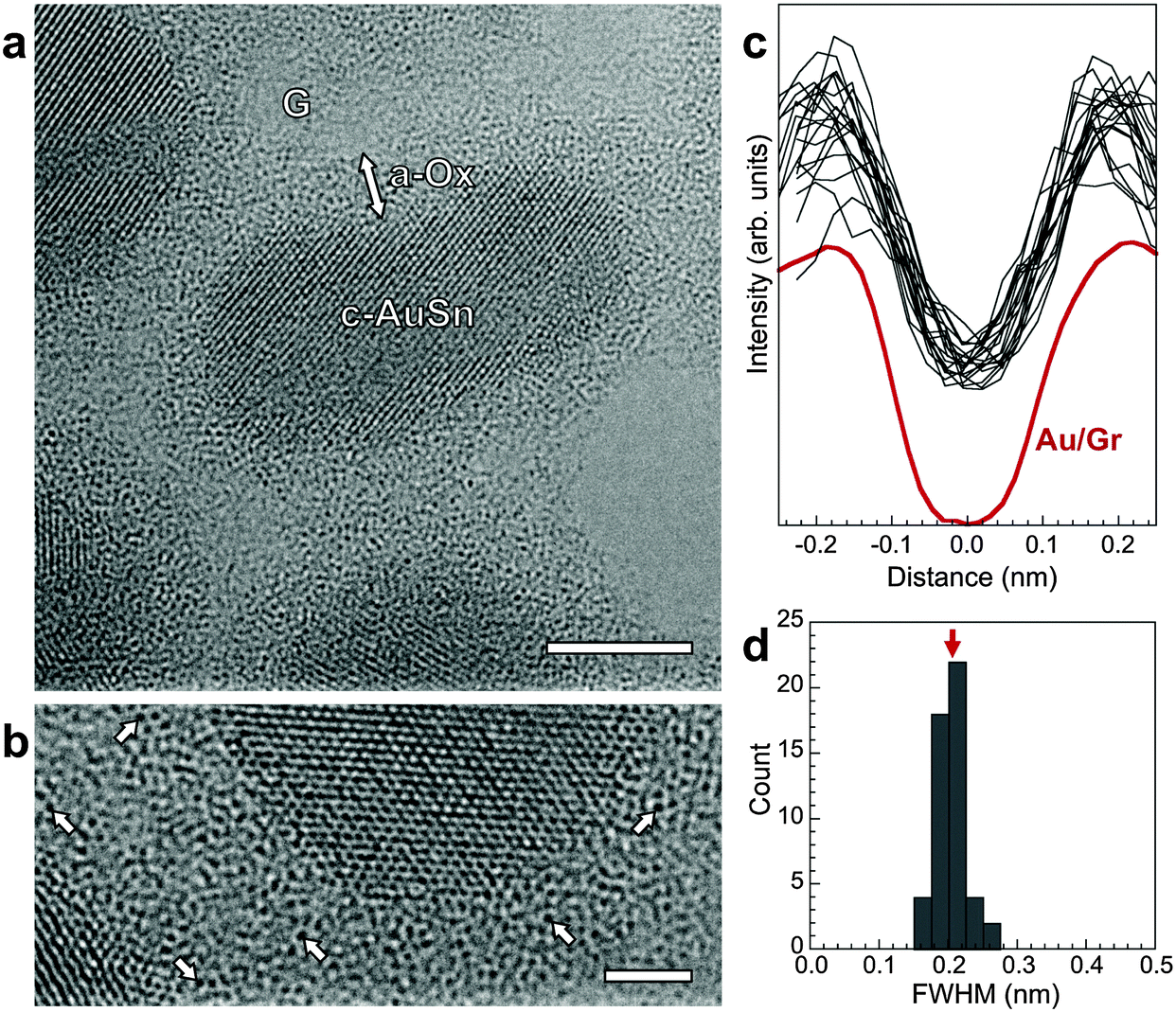 | ||
| Fig. 3 (a) AC-HRTEM image of oxidized AuSn nanostructures, showing the crystalline metal alloy cores (C-AuSn) surrounded by an amorphous oxide shell (a-Ox). The support is a 3-layer graphene grid (G). Scale bar: 5 nm. (b) Detailed view of a section of the amorphous oxide shell, showing discrete point-like scattering centers, some of which are marked by arrows. Scale bar: 2 nm. (c) Line scans through 20 of the dark scatterers (black lines), in comparison with the AC-HRTEM line profile of a single Au atom on graphene (Au/G, ref. 47). (d) Histogram of the full width at half maximum (FWHM) of 50 scatterers dispersed in the amorphous oxide shell, giving a mean value of (0.203 ± 0.022) nm. The AC-HRTEM FWHM of a single Au atom on graphene is 0.201 nm (red arrow). | ||
To explore the properties of these (Au–Sn) oxide nanocomposites, we examined the chemical reactivity of different oxidized Au1−xSnx alloys (Au2Sn, AuSn, AuSn2, and AuSn4, with different Au:Sn concentrations in the oxide surface) along with a Sn reference, using CO + 1/2O2 → CO2 as a probe reaction. CO2 production was probed at room temperature by dosing different (CO + O2) gas mixtures (Fig. 4). In situ near-ambient pressure synchrotron XPS (NAP-XPS) was obtained simultaneously at pressures up to several Torr.
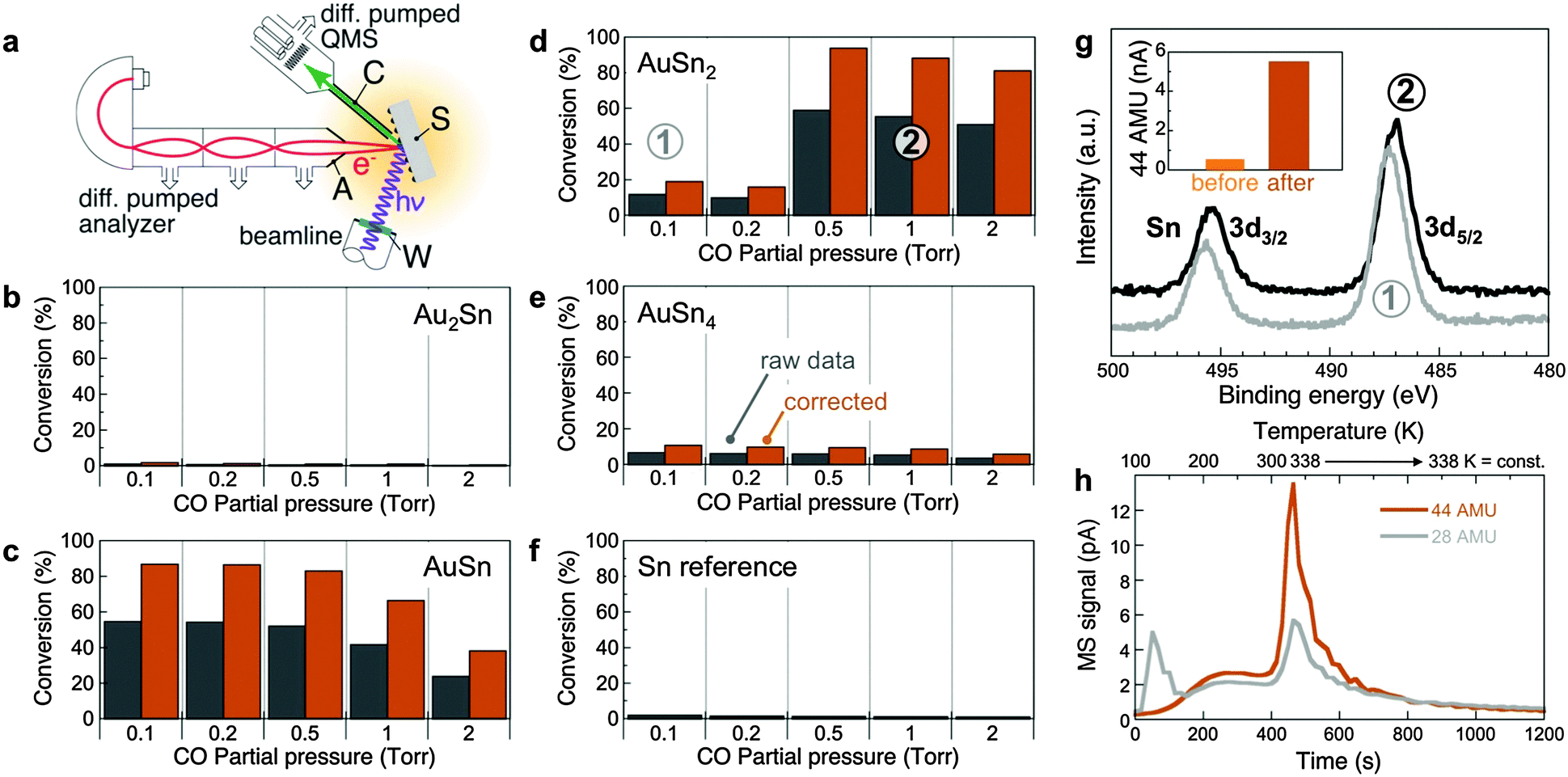 | ||
| Fig. 4 (a) Schematic setup used to probe CO oxidation at near-ambient pressures with simultaneous chemical analysis of the oxidized Au–Sn samples by synchrotron NAP-XPS. S: Sample; C: Gas sampling capillary; A: Analyzer entrance aperture; W: X-ray window. Shading indicates the zone at near-ambient gas pressure. (b–f) Chemical reactivity (CO to CO2 conversion efficiency) of samples with different compositions: Au2Sn (b), AuSn (c), AuSn2 (d), AuSn4 (e), and Sn reference (f). Measurements performed by mass spectrometry using a sampling capillary with entrance aperture placed close to the sample surface. O2 partial pressure was fixed at 0.1 Torr; CO pressure was varied up to a maximum of 2 Torr. Gray: raw data; colored: corrected, taking into account the actual active sample area (see ESI† for details). (g) Surface-sensitive in-situ synchrotron AP-XPS (photon energy: 729 eV) from AuSn2 for different gas compositions: (1) 0.1 Torr O2; 0.1 Torr CO. (2) 0.1 Torr O2; 1 Torr CO. Inset: CO2 production before and after activation under reducing conditions, i.e., high CO partial pressure. (h) Temperature-programmed desorption of CO (28 AMU) and CO2 (44 AMU), following CO adsorption on AuSn2 at T = 100 K. The temperature was ramped from 100 K to 338 K at constant rate and then held fixed at 338 K. | ||
Oxidized Sn produces no detectable CO2, but already a Sn-rich AuSn4 alloy shows a small amount of CO2 product. On the Au-rich side, Au2Sn does not show any measurable conversion to CO2. The samples at intermediate composition (AuSn, AuSn2) are highly active, reaching between 80% and close to 100% conversion efficiency at room temperature. Clear trends among the active samples point to the underlying reaction mechanism. Whereas oxidized AuSn is active over a broad range of CO:O2 ratios, it deactivates under CO-rich conditions (Fig. 4c). Fully oxidized AuSn2, on the other hand, shows low conversion efficiency at low CO:O2 ratios and only becomes activated after exposure to reducing (CO-rich) conditions, but subsequently shows the highest conversion efficiency of all compositions probed here (Fig. 4d) with stable CO2 production over extended time periods (several hours).
The reactivity measurements suggest that the Au-containing oxide matrix plays an active role in CO oxidation as a reservoir with variable oxygen content, which functions optimally when in a partially reduced state. For AuSn, the initial oxide already contains sufficient oxygen vacancies. Oxidized AuSn2, on the other hand, is activated by exposure to reducing conditions. Surface-sensitive synchrotron NAP-XPS probing only the oxide shell (kinetic energy ∼240 eV; Sn 3d escape depth ∼0.6 nm) of AuSn2-based samples shows a shift of the Sn 3d core level to lower binding energy (487.3 to 487.0 eV), i.e., toward lower oxidation state, with increasing CO partial pressure (Fig. 4g). Under the conditions of our reactivity experiments the oxide is only partially reduced, and there is no Sn 3d intensity due to metallic Sn0 (485.0–485.5). Further support for the active role of the mixed oxide is provided by temperature-programmed desorption/reaction (TPD/R), in which CO is adsorbed at 100 K and the temperature is subsequently increased while following CO (28 AMU) and CO2 (44 AMU) desorption by mass spectrometry (Fig. 4h). The measurements show a substantial evolution of CO2 near room temperature, i.e., confirm that CO can be oxidized in the absence of an external oxygen source by consuming oxygen supplied by the oxide matrix. Thus, oxidized nanostructured Au–Sn alloys derive their chemical reactivity from atomically dispersed Au embedded in a reducible oxide that acts as an oxygen reservoir. The Au atoms play a dual role, namely: (i) to provide active, low-coordinated sites for CO adsorption and reaction to CO2 with oxygen from the surrounding oxide surface (see also ESI,† Fig. S8);49–51 and (ii) to locally destabilize the oxide matrix (i.e., enhance its reducibility), as indicated by substantially lower oxidation rates of Au–Sn alloys compared to Sn (Fig. 2a and b).
Our results highlight the ability of simple room-temperature oxidation processes to generate complex functional materials architectures. For the chosen system, nanostructured alloys comprising a noble metal (Au) and a non-noble species (Sn), oxidation by predominant oxygen anion diffusion and a limited mobility of the metal species lead to the formation of core–shell structures in which a residual crystalline bimetallic core is surrounded by a non-crystalline oxide. In the surface region of this oxide shell, dispersed Au atoms and a surrounding oxygen reservoir work in concert to provide chemical reactivity in low-temperature oxidation reactions. The catalytic activity is determined by the concentration of surface Au-atoms in the oxide, which can be selected via the Au content of the initial bimetallic Au–Sn alloy; the highest activity is realized at intermediate Au contents, specifically AuSn and AuSn2. The active state has been obtained here by slow oxidation at room temperature in air, but for practical applications the process could likely be accelerated by exposure to higher oxygen pressures. While our study used vacuum evaporation for forming bimetallic alloy nanostructures without controlling size or morphology, room temperature oxidation could be combined with solvothermal or sol–gel synthesis of size- and shape-controlled bimetallic clusters to achieve specific high-surface area configurations of (Au–Sn) oxide nanocomposites. Aside from the demonstrated high CO to CO2 conversion efficiencies of these composite catalysts, which are important for applications in air pollution prevention, oxide-embedded dispersed Au atoms may represent a pathway toward Au-based catalysts with enhanced stability against deactivation and may have potential for other oxidation reactions, such as preferential CO oxidation (PROX) or partial oxidation reactions.
Methods
Bimetallic Au1−xSnx alloy nanostructures were prepared in a high-vacuum chamber by sequential evaporation of controlled amounts of Au and Sn at room temperature onto different substrates: amorphous C films supported on Cu transmission electron microscopy (TEM) grids for TEM, and oxidized Ge(111) wafers for surface characterization and reactivity measurements. Reference samples of pure Sn nanoparticles were prepared following the same procedure. The as-synthesized samples were exposed at room temperature to ambient air, and their evolution during oxidation in air was followed with time for up to 1 month. Structure and morphology of the samples were analyzed by TEM and scanning TEM (STEM), using a JEOL 2100F field emission microscope. STEM imaging used a Gatan high-angle annular dark field detector (Gatan 806 HAADF) for incoherent HAADF (Z-contrast) imaging with smallest probe size of 0.2 nm. Sample composition was probed locally in STEM mode using energy dispersive X-ray spectrometry (EDS, Oxford Energy TEM 250). Samples on bulk Ge were analyzed by scanning electron microscopy (SEM) in a JEOL 7600F field emission microscope to compare the morphology with that observed by TEM/STEM for samples on amorphous C supports. Aberration-corrected high-resolution TEM (AC-HRTEM) was performed to identify the state of Au in the amorphous oxide shells of the Au–Sn nanostructures. These experiments used a FEI Titan 80–300 microscope equipped with a CEOS Cs-corrector, at 300 keV electron energy.High-resolution STEM imaging and chemical mapping were performed in a double aberration-corrected JEOL-ARM200F microscope with a cold field emission gun, operated at 200 kV. The microscope is equipped with JEOL and Gatan HAADF detectors for incoherent HAADF (Z-contrast) imaging, Gatan GIF Quantum Energy Filter with dualEELS for EELS and Oxford Instruments large area silicon drift detector for EDS. HAADF images, EELS spectrum images with the Sn-M4,5, O–K and Au–M4,5 edges, as well as EDS spectrum images with the Sn–L, Lβ and Au–Mα and Mβ peaks were simultaneously acquired in scanning mode to ensure all the signals were recorded from the same area. Au maps were obtained both from the signal intensity of Au–M4,5 edges in EELS spectrum images and the signal intensity of Au–Mαβ peaks in EDS spectrum images. Both types of images were acquired simultaneously and showed consistent results. Sn and O maps were obtained from EELS spectrum images. Due to the proximity of the Sn–M4,5 and O–K edges in EELS spectra (see Fig. S3, ESI†), it is difficult to obtain Sn and O maps by directly integrating the signal intensity for Sn and O in EELS. We therefore used the multiple linear least-squares fitting (MLLS) method to obtain the Sn and O mapping, as described in Fig. S3 (ESI†). Sn maps also extracted from Sn-Lαβ peaks in EDS spectrum images, and results were consistent with those in EELS.
Laboratory X-ray photoelectron spectra (XPS) were acquired at room temperature using a Specs Phoibos 100 MCD hemispherical analyzer, with excitation by Al Kα (hν = 1486.6 eV) radiation at 300 W (15 keV and 20 mA). The spectra were acquired at 25 eV pass energy and 0.05 eV energy step. The base pressure in the analysis chamber was in the range of 2–6 × 10−9 Torr. The C 1s binding energy (284.8 eV) was used as internal reference. Laboratory XPS was complemented by in situ synchrotron near-ambient pressure XPS (NAP-XPS) at pressures up to several Torr, performed at the National Synchrotron Light Source using a differentially pumped hemispherical analyzer (Specs Phoibos 150 NAP) equipped with a CEM 9 channeltron detector and a 300 μm entrance aperture to the first differential pumping stage. The sample was positioned >0.5 mm from the aperture to ensure that the local pressure at the surface was not significantly affected by differential pumping through the aperture. The analyzer was positioned at an angle of 70° from the incident X-ray beam and 20° from the surface normal of the sample. The photon energy was kept fixed during the course of each experiment. Peak shape analysis used fitting with a combination of Gaussian and Lorentzian functions after Shirley background subtraction in the analysis region. For quantitative analysis, spectrometer transmission function, cross-sections of different core levels and inelastic mean free path values were used as given in the CasaXPS software package.
Reactivity measurements were carried out in the NAP-XPS system, by backfilling the chamber with variable gas mixtures and detecting products using a sampling capillary (0.75 mm inner diameter) brought within 1 mm from the sample surface; the gases were analyzed using a mass spectrometer (Pfeiffer Prisma Plus) with single stage differential pumping. A constant partial pressure of O2 (0.1 Torr) was maintained while the CO partial pressure was varied in discrete steps from ∼0 (i.e., the CO background pressure in the UHV system) to 2 Torr. The partial pressures for masses 28 AMU (CO), 32 AMU (O2), and 44 AMU (CO2) were followed by mass spectrometry. The reported data represent steady-state values of the CO to CO2 conversion efficiency calculated from measurements at masses 28 AMU and 44 AMU, recorded after 10 minutes following each change in the CO flow rate (stabilization of the gas pressures to <5%).
Acknowledgements
This research used resources of the Center for Functional Nanomaterials, which is a U.S. DOE Office of Science Facility at Brookhaven National Laboratory under Contract No. DE-SC0012704. The authors thank X. Tong for technical assistance.References
- R. E. Schaak, A. K. Sra, B. M. Leonard, R. E. Cable, J. C. Bauer, Y. F. Han, J. Means, W. Teizer, Y. Vasquez and E. S. Funck, Metallurgy in a beaker: nanoparticle toolkit for the rapid low-temperature solution synthesis of functional multimetallic solid-state materials, J. Am. Chem. Soc., 2005, 127(10), 3506–3515 CrossRef CAS PubMed.
- X. Huang, H. Zhang, C. Guo, Z. Zhou and N. Zheng, Simplifying the Creation of Hollow Metallic Nanostructures: One-Pot Synthesis of Hollow Palladium/Platinum Single-Crystalline Nanocubes, Angew. Chem., Int. Ed., 2009, 48(26), 4808–4812 CrossRef CAS PubMed.
- J. Wu, A. Gross and H. Yang, Shape and Composition-Controlled Platinum Alloy Nanocrystals Using Carbon Monoxide as Reducing Agent, Nano Lett., 2011, 11(2), 798–802 CrossRef CAS PubMed.
- Y. Wu, S. Cai, D. Wang, W. He and Y. Li, Syntheses of Water-Soluble Octahedral, Truncated Octahedral, and Cubic Pt–Ni Nanocrystals and Their Structure–Activity Study in Model Hydrogenation Reactions, J. Am. Chem. Soc., 2012, 134(21), 8975–8981 CrossRef CAS PubMed.
- S. E. Habas, H. Lee, V. Radmilovic, G. A. Somorjai and P. Yang, Shaping binary metal nanocrystals through epitaxial seeded growth, Nat. Mater., 2007, 6(9), 692–697 CrossRef CAS PubMed.
- F.-R. Fan, D.-Y. Liu, Y.-F. Wu, S. Duan, Z.-X. Xie, Z.-Y. Jiang and Z.-Q. Tian, Epitaxial Growth of Heterogeneous Metal Nanocrystals: From Gold Nano-octahedra to Palladium and Silver Nanocubes, J. Am. Chem. Soc., 2008, 130(22), 6949–6951 CrossRef CAS PubMed.
- M. Jiang, B. Lim, J. Tao, P. H. C. Camargo, C. Ma, Y. Zhu and Y. Xia, Epitaxial overgrowth of platinum on palladium nanocrystals, Nanoscale, 2010, 2(11), 2406–2411 RSC.
- L. Zhang, W. Niu, W. Gao, L. Qi, J. Lai, J. Zhao and G. Xu, Synthesis of Convex Hexoctahedral Palladium@Gold Core–Shell Nanocrystals with 431 High-Index Facets with Remarkable Electrochemiluminescence Activities, ACS Nano, 2014, 8(6), 5953–5958 CrossRef CAS PubMed.
- D. Kim, Y. W. Lee, S. B. Lee and S. W. Han, Convex Polyhedral Au@Pd Core–Shell Nanocrystals with High-Index Facets, Angew. Chem., Int. Ed., 2012, 51(1), 159–163 CrossRef CAS PubMed.
- A. Walther, A. H. Muller and E. Janus, Particles: Synthesis, Self-Assembly, Physical Properties, and Applications, Chem. Rev., 2013, 113(7), 5194–5261 CrossRef CAS PubMed.
- X. H. Xia, Y. Wang, A. Ruditskiy and Y. N. Xia, 25th Anniversary Article: Galvanic Replacement: A Simple and Versatile Route to Hollow Nanostructures with Tunable and Well-Controlled Properties, Adv. Mater., 2013, 25(44), 6313–6333 CrossRef CAS PubMed.
- Y. N. Xia, W. Y. Li, C. M. Cobley, J. Y. Chen, X. H. Xia, Q. Zhang, M. X. Yang, E. C. Cho and P. K. Brown, Gold Nanocages: From Synthesis to Theranostic Applications, Acc. Chem. Res., 2011, 44(10), 914–924 CrossRef CAS PubMed.
- Y.-C. Chen, J.-M. Chen, Y.-H. Huang, Y.-R. Lee and H. C. Shih, Size effect of tin oxide nanoparticles on high capacity lithium battery anode materials, Surf. Coat. Technol., 2007, 202(4–7), 1313–1318 CrossRef CAS.
- F. Tao, M. E. Grass, Y. Zhang, D. R. Butcher, J. R. Renzas, Z. Liu, J. Y. Chung, B. S. Mun, M. Salmeron and G. A. Somorjai, Reaction-Driven Restructuring of Rh–Pd and Pt–Pd Core–Shell Nanoparticles, Science, 2008, 322(5903), 932–934 CrossRef CAS PubMed.
- M. H. Shao, T. Huang, P. Liu, J. Zhang, K. Sasaki, M. B. Vukmirovic and R. R. Adzic, Palladium monolayer and palladium alloy electrocatalysts for oxygen reduction, Langmuir, 2006, 22(25), 10409–10415 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Trends in electrocatalysis on extended and nanoscale Pt-bimetallic alloy surfaces, Nat. Mater., 2007, 6(3), 241–247 CrossRef CAS PubMed.
- H. Zhang, M. S. Jin and Y. N. Xia, Enhancing the catalytic and electrocatalytic properties of Pt-based catalysts by forming bimetallic nanocrystals with Pd, Chem. Soc. Rev., 2012, 41(24), 8035–8049 RSC.
- R. A. Koeppel, A. Baiker, C. Schild and A. Wokaun, Carbon-Dioxide Hydrogenation over Au/ZrO2 Catalysts from Amorphous Precursors – Catalytic Reaction-Mechanism, J. Chem. Soc., Faraday Trans., 1991, 87(17), 2821–2828 RSC.
- S. H. Zhou, Z. Ma, H. F. Yin, Z. L. Wu, B. Eichhorn, S. H. Overbury and S. Dai, Low-Temperature Solution-Phase Synthesis of NiAu Alloy Nanoparticles via Butyllithium Reduction: Influences of Synthesis Details and Application As the Precursor to Active Au-NiO/SiO2 Catalysts through Proper Pretreatment, J. Phys. Chem. C, 2009, 113(14), 5758–5765 CAS.
- S. H. Zhou, H. F. Yin, V. Schwartz, Z. L. Wu, D. Mullins, B. Eichhorn, S. H. Overbury and S. Dai, In Situ Phase Separation of NiAu Alloy Nanoparticles for Preparing Highly Active Au/NiO CO Oxidation Catalysts, ChemPhysChem, 2008, 9(17), 2475–2479 CrossRef CAS PubMed.
- E. A. Sutter, X. Tong, K. Jungjohann and P. W. Sutter, Oxidation of nanoscale Au–In alloy particles as a possible route toward stable Au-based catalysts, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(26), 10519–10524 CrossRef CAS PubMed.
- J. C. Bauer, D. Mullins, M. J. Li, Z. L. Wu, E. A. Payzant, S. H. Overbury and S. Dai, Synthesis of silica supported AuCu nanoparticle catalysts and the effects of pretreatment conditions for the CO oxidation reaction, Phys. Chem. Chem. Phys., 2011, 13(7), 2571–2581 RSC.
- K. Yu, Z. Wu, Q. Zhao and B. Li; Y. Xie, High-Temperature-Stable Au@SnO2 Core/Shell Supported Catalyst for CO Oxidation, J. Phys. Chem. C, 2008, 112, 2244–2247 CAS.
- E. R. Klobukowski, R. J. Angelici and L. K. Woo, Bulk Gold-Catalyzed Oxidations of Amines and Benzyl Alcohol Using Amine N-Oxides as Oxidants, Catal. Lett., 2012, 142(2), 161–167 CrossRef CAS.
- B. K. Min, A. R. Alemozafar, D. Pinnaduwage, X. Deng and C. M. Friend, Efficient CO oxidation at low temperature on Au(111), J. Phys. Chem. B, 2006, 110(40), 19833–19838 CrossRef CAS PubMed.
- B. K. Min and C. M. Friend, Heterogeneous gold-based catalysis for green chemistry: low-temperature CO oxidation and propene oxidation, Chem. Rev., 2007, 107(6), 2709–2724 CrossRef CAS PubMed.
- M. Haruta, Size- and support-dependency in the catalysis of gold, Catal. Today, 1997, 36(1), 153–166 CrossRef CAS.
- T. V. W. Janssens, B. S. Clausen, B. Hvolbaek, H. Falsig, C. H. Christensen, T. Bligaard and J. K. Norskov, Insights into the reactivity of supported Au nanoparticles: combining theory and experiments, Top. Catal., 2007, 44(1–2), 15–26 CrossRef CAS.
- D. Gajan, K. Guillois, P. Delichere, J.-M. Basset, J.-P. Candy, V. Caps, C. Coperet, A. Lesage and L. Emsley, Gold Nanoparticles Supported on Passivated Silica: Access to an Efficient Aerobic Epoxidation Catalyst and the Intrinsic Oxidation Activity of Gold, J. Am. Chem. Soc., 2009, 131(41), 14667–14669 CrossRef CAS PubMed.
- M. Valden, X. Lai and D. W. Goodman, Onset of Catalytic Activity of Gold Clusters on Titania with the Appearance of Nonmetallic Properties, Science, 1998, 281(5383), 1647–1650 CrossRef CAS PubMed.
- A. Corma, P. Concepció, M. Boronat, M. J. Sabater, J. Navas, M. J. Yacaman, E. Larios, A. Posadas, M. A. López-Quintela, D. Buceta, E. Mendoza, G. Guilera and A. Mayoral, Exceptional oxidation activity with size-controlled supported gold clusters of low atomicity, Nat. Chem., 2013, 5(9), 775–781 CrossRef CAS PubMed.
- L. Buene, Characterization of evaporated gold–tin films, Thin Solid Films, 1977, 43(3), 285–294 CrossRef CAS.
- L. Buene, Interdiffusion and phase formation at room temperature in evaporated gold–tin films, Thin Solid Films, 1977, 47(2), 159–166 CrossRef CAS.
- L. Buene, T. Finstad, K. Rimstad, O. Lönsjö and T. Olsen, Alloying behavior of Au–In and Au–Sn films on semiconductors, Thin Solid Films, 1976, 34(1), 149–152 CrossRef CAS.
- C. Ghosh, Interdiffusion study in binary gold–tin system, Intermetallics, 2010, 18(11), 2178–2182 CrossRef CAS.
- B. Hugsted, L. Buene, T. Finstad, O. Lönsjö and T. Olsen, Interdiffusion and phase formation in Au/Sn thin film couples with special emphasis on substrate temperature during condensation, Thin Solid Films, 1982, 98(2), 81–94 CrossRef CAS.
- S. Nakahara, R. J. McCoy, L. Buene and J. M. Vandenberg, Room temperature interdiffusion studies of Au/Sn thin film couples, Thin Solid Films, 1981, 84(2), 185–196 CrossRef CAS.
- J. S. Charlton, M. Cordey-Hayes and I. R. Harris, A study of the 119Sn mössbauer isomer shifts in some platinum–tin and gold–tin alloys, J. Less-Common Met., 1970, 20(2), 105–112 CrossRef CAS.
- K. Osada, S. Yamaguchi and M. Hirabayashi, An Ordered Structure of Au5Sn, Trans. Jpn Inst. Met., 1974, 15(4), 256–260 CrossRef CAS.
- E. Sutter and P. Sutter, Enhanced oxidation of nanoscale In particles at the interface with a Si nanowire, Appl. Phys. Lett., 2012, 100(23), 231602 CrossRef.
- E. Sutter and P. Sutter, Size-Dependent Room Temperature Oxidation of In Nanoparticles, J. Phys. Chem. C, 2012, 116(38), 20574–20578 CAS.
- R. Nakamura, D. Tokozakura, H. Nakajima, J. G. Lee and H. Mori, Hollow oxide formation by oxidation of Al and Cu nanoparticles, J. Appl. Physiol., 2007, 101(7), 074303 CrossRef.
- E. Sutter, F. Ivars-Barcelo and P. Sutter, Size-Dependent Room Temperature Oxidation of Tin Particles, Part. Part. Syst. Charact., 2014, 31, 879–885 CrossRef CAS.
- NIST X-ray Photoelectron Spectroscopy Database, Version 4.1, National Institute of Standards and Technology: Gaithersburg, 2012.
- J. A. Taylor, S. M. Merchant and D. L. Perry, Study of the oxidation of gold–tin preforms using X-ray photoelectron spectroscopy, J. Appl. Physiol., 1995, 78(9), 5356–5361 CrossRef CAS.
- P. Z. Coura, S. B. Legoas, A. S. Moreira, F. Sato, V. Rodrigues, S. O. Dantas, D. Ugarte and D. S. Galvão, On the Structural and Stability Features of Linear Atomic Suspended Chains Formed from Gold Nanowires Stretching, Nano Lett., 2004, 4(7), 1187–1191 CrossRef CAS.
- B. Westenfelder, J. C. Meyer, J. Biskupek, S. Kurasch, F. Scholz, C. E. Krill and U. Kaiser, Transformations of Carbon Adsorbates on Graphene Substrates under Extreme Heat, Nano Lett., 2011, 11(12), 5123–5127 CrossRef CAS PubMed.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Single-atom catalysis of CO oxidation using Pt1/FeOx, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- T. W. Janssens, B. Clausen, B. Hvolbæk, H. Falsig, C. Christensen, T. Bligaard and J. Nørskov, Insights into the reactivity of supported Au nanoparticles: combining theory and experiments, Top. Catal., 2007, 44(1–2), 15–26 CrossRef CAS.
- S. H. Overbury, V. Schwartz, D. R. Mullins, W. Yan and S. Dai, Evaluation of the Au size effect: CO oxidation catalyzed by Au/TiO2, J. Catal., 2006, 241(1), 56–65 CrossRef CAS.
- D. Widmann and R. J. Behm, Active Oxygen on a Au/TiO2 Catalyst: Formation, Stability, and CO Oxidation Activity, Angew. Chem., Int. Ed., 2011, 50(43), 10241–10245 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nh00123d |
| This journal is © The Royal Society of Chemistry 2016 |