
Enabling method to design versatile biomaterial systems from colloidal building blocks†
S.
Saxena
*ab and
L. A.
Lyon
*c
aSchool of Materials Science and Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA. E-mail: ssaxena30@gatech.edu; Tel: +1 301 760 0805
bPetit Institute for Biosciences and Bioengineering, Georgia Institute of Technology, Atlanta, GA 30332, USA
cSchmid College of Science and Technology, Chapman University, Orange, CA 92866, USA. E-mail: lyon@chapman.edu; Tel: 714 997 6930
First published on 8th June 2016
Abstract
Development of materials with fine spatial control over topographical, mechanical, or chemical features has been investigated for a variety of applications. Here we present a method to fabricate an array of polyelectrolyte constructs including two-dimensionally and three-dimensionally patterned assemblies using both compressible and incompressible colloidal building blocks. This method eliminates prior constraints associated with specific chemistries, and can be used to develop modular, multi-component, patterned assemblies. In particular, development of constructs were investigated using microgels, which are colloidally stable hydrogel microparticles, polystyrene (PS) beads, and PS-microgel core–shell building blocks in conjunction with the polycation poly(ethyleneimine) (PEI). The topography, mechanical properties, and microstructure of these materials were characterized via bright field microscopy, laser scanning confocal microscopy, atomic force microscopy (AFM), and AFM nanoindentation. Cellular studies demonstrate that such patterned film constructs can be used as model systems to investigate and direct cellular adhesion and spreading. Finally, this fabrication method is expanded to develop bulk polyelectrolyte gels that can be used to develop cell-laden gels.
Design, System, ApplicationMaterials with fine spatial control over topographical, mechanical, or chemical features are highly sought after in a variety of applications. Here we present a facile centrifugation method that can be exploited to develop an array of two-dimensional and three-dimensional polyelectrolyte constructs, both with and without patterning. Though simple and easily accessible, this method is a powerful tool that can be leveraged to develop assemblies with a high degree of control over feature characteristics such as shape, size, topography, surface chemistry, and mechanical properties. The basic building blocks of this system, hydrogel nanoparticles, or microgels, can be finely tuned based on their chemistries, synthetic conditions, and post-synthesis modifications to achieve specific chemical, structural, mechanical, and optical features. A variety of other particles, and even live cells, can be used as additional building blocks with unique material properties in this centrifugation method to design modular, multi-component, hierarchical constructs. In particular, this investigation demonstrates the use of this method to design assemblies that can be used to evaluate cellular responses for tissue engineering and regenerative medicine applications. |
Introduction
Development of two-dimensional films with fine spatial control over mechanical properties and chemical functionalities has become increasingly important for a variety of applications including membranes, sensors, tissue engineering scaffolds, and biomedical devices. Previously, development of films with detailed patterns or hierarchical ordering have been achieved via either a “bottom-up” approach, where patterns are self-assembled,1,2 or a “top-down” approach where a continuous material is deposited followed by removal of targeted areas. Well-established, versatile “top-down” nanolithography approaches include electron-beam direct-write lithography,3 nanoimprint lithography,4 and photolithography;5 these techniques have been demonstrated to produce nanoscale patterns for a variety of materials including inorganic and organic materials.In the field of biomaterials in particular, hydrogels represent a class of materials under investigation for their potential use in biomedical applications including drug delivery,6 tissue engineering scaffolds,7 and biosensing.8 A variety of two-dimensionally and three-dimensionally patterned hydrogel systems have been explored for use in tissue engineering applications9–13 and biosensing.14–16 Though many approaches have targeted both patterning of the surface17,18 and of the bulk,19–21 they are often limited by the use of highly specialized chemistries and equipment, which can limit their wide-spread usage. For example, Azagarsamy et al. demonstrated the ability to employ photodriven click reactions to develop patterned cellular microenvironments.22 Additionally, Mosiewics et al. demonstrated the ability to use photoinduced uncaging of thiols to modulate hydrogel stiffness.23 Thus, the development of a facile method to pattern hydrogel materials with spatial control over chemical functionality and/or mechanical properties without the need for complex chemistries and equipment could be greatly beneficial for the advancement of patterned biomaterials in biomedical research.
Hydrogel nanoparticles, also known as microgels, are discrete hydrogel particles that can be used as modular building blocks to fabricate tunable two-dimensional films and three-dimensional scaffolds for a range of biomaterial applications.24,25 Tunable hydrogel nanoparticle properties have been broadly investigated resulting in the development of microgels that respond to stimuli such as temperature, ionic strength, pH, molecular bonding, light, magnetic fields, and enzyme activities.26,27 In particular, hydrogel nanoparticles composed of the thermoresponsive polymer poly(N-isopropylacrylamide) (pNIPAm) have received a great deal of attention in the past and are well-characterized. In aqueous media, pNIPAm exhibits a lower critical solution temperature around 31 °C. When incorporated into hydrogel nanoparticles, this responsive behavior translates into a thermally-triggered microgel phase transition from a swollen gel state to a collapsed globule. Investigators have demonstrated the ability to synthesize hydrogel nanoparticles at various sizes with low polydispersity using precipitation polymerization; this control over polydispersity is of interest because fabrication of a monodisperse population of soft particles can often be difficult to achieve for other natural and synthetic polymers. Moreover, researchers have previously demonstrated the ability to synthesize hydrogel nanoparticles with complex architectures including core–shell microgels and hollow microgels, for added functionality.28–31
Hydrogel nanoparticle assemblies have previously been investigated for a variety of applications including drug delivery,32,33 non-fouling coatings,34 and sensing.35,36 The layer-by-layer (LbL) technique is the established method for fabricating polyelectrolyte microgel films.37 In the past, our group has specifically built films using a repetitive process of centrifugal deposition of anionic microgels onto a positively charged substrate followed by passive adsorption of a polycation.38 Though this LbL method has been well-established, it can also be limiting in the development of some films, particularly those with more complex architectures. Development of patterned microgel films, for example, presents a problem to the current fabrication strategy. Few investigators have successfully developed patterned microgel assemblies. Most notably, Peng et al. demonstrated the ability to pattern microgel monolayers via cool microcontact printing.39 Additionally, Lord et al. demonstrated the ability to cast a solution of carbon disulfide and microgels prepared from RAFT derived polystyrene in order to produce films with a porous honeycomb structure.40 However, these investigations are limited in the film compositions that can be achieved.
In the present contribution, an enabling film fabrication technique is investigated wherein polyanionic and polycationic components are mixed, forming a polyelectrolyte complex, and immediately deposited via centrifugation. Using five building blocks, including compressible microgels, incompressible latex beads, core–shell PS-microgel spheres (raspberry-like particles), and live cells, we demonstrate the wide-scale applicability of this method to develop modular material platforms (Scheme 1). The film fabrication technique is compared to the traditional LbL approach. The simplicity and versatility of this technique is also demonstrated through the development of several constructs not previously feasible using the traditional LbL technique, including lateral and perpendicularly patterned polyelectrolyte microgel constructs. It is demonstrated that these constructs can be used as biological interfaces to control cellular behavior. Finally, it is demonstrated that this method can also be used to assemble bulk poly-electrolyte gels, which can function as another scaffold to direct cellular behavior. Together, these studies demonstrate that this method can be a facile route to fabricate a variety of architectures that can be used as model experimental systems to investigate fundamental film properties, elicit basic cellular responses, and develop biomaterials.
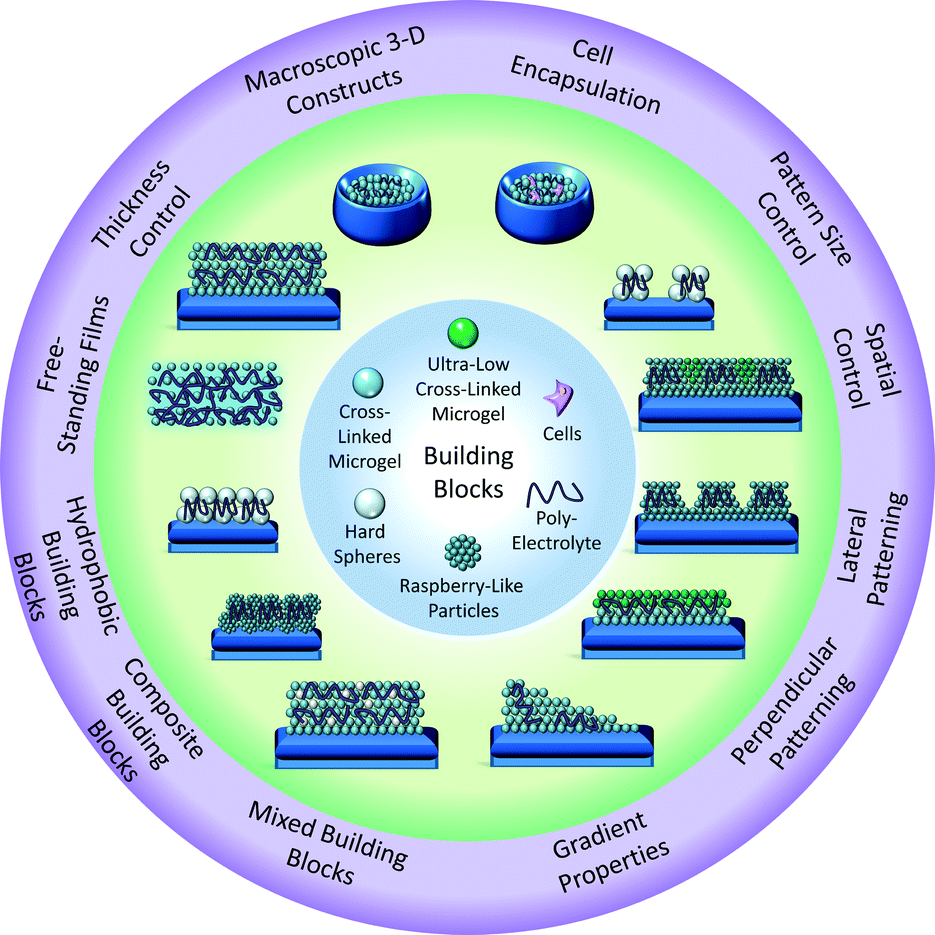 | ||
| Scheme 1 Depiction of the range of building blocks and resultant structures prepared using the current approach. | ||
Experimental
MaterialsAll reagents were purchased from Sigma-Aldrich (St. Louis, MO) and used as received, unless otherwise noted. Reagents N,N′-methylenebis(acrylamide) (BIS), acrylic acid (AAc), sodium dodecyl sulfate (SDS), ammonium persulfate (APS), 1-ethyl-2-[2-dimethylaminopropyl]carbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), 3-aminopropyltrimethoxysilane (APTMS), ethanol (EtOH), and polyethylenimine (PEI) were all used as received. The monomer N-isopropylacrylamide (NIPAm) were recrystallized from hexanes (VWR International, West Chester, PA) and dried in vacuo prior to use. Carboxyl modified polystyrene (PS) spheres were purchased from Polysciences, Inc. (Warrington, PA). Methacryloxyethyl thiocarbamoyl rhodamine B (Rho-B) and 4-acrylamidofluorescein (AFA) were used as fluorophores. Deionized water used in all reactions, purifications, and buffer preparations was purified to a resistance of 18 MΩ (Barnstead E-Pure system), and filtered through a 0.2 μm filter to remove particulate matter. A 10 mM phosphate buffer solution (PBS) was prepared with 150 mM ionic strength of NaCl and a pH of 7.4. 10 mM formate buffers (pH 3.3) were prepared with 25 mM, 100 mM, or 150 mM NaCl. 10 mM MES buffers (pH 5.5) were prepared with 25 or 150 mM NaCl.
Microgel synthesis
Microgels were synthesized as previously described using precipitation polymerization.26 For all microgels, the total monomer concentration was 100 mM with molar compositions of 66 mol% pNIPAm, 4 mol% BIS, and 30 mol% AAc. Monomer, cross-linker, and surfactant (SDS) were dissolved in 30 mL of water. For fluorescent microgels, less than 1 mg of the fluorophore Rho-B or AFA was additionally added. The solution was filtered through a 0.2 μm Acrodisc syringe filter to remove any undissolved solids. For ultra-low cross-linked microgels, no cross-linker was added to the solution.41 The solution was placed in a 3-neck round bottom flask equipped with a condenser, heated to 70 °C while mixing with a magnetic stir bar (stir speed 500 RPM) while being purged with N2 for approximately 1 h. Finally, 1 mL of the initiator APS (1 mM) was added via syringe. The solution was held at ∼70 °C for at least 4 h, and then cooled to room temperature. The microgel solution was filtered through glass wool, purified via sedimentation, and lyophilized for storage. Microgel compositions used in these studies are listed in Table 1. Microgel hydrodynamic radius (RH) values were determined using a DynaPro Dynamic Light Scattering (DLS) instrument (Wyatt, Technology, Santa Barbara, CA).| Sample | pNIPAm (mol%) | BIS (mol%) | AAc (mol%) | Other |
|---|---|---|---|---|
| 1 | 66 | 4 | 30 | N/A |
| 2 | 66 | 4 | 30 | ∼0.1 mol% Rho-B |
| 3 | 70 | 0 | 30 | ∼0.1 mol% of AFA |
Glass coverslip functionalization
All glass coverslips were cleaned sequentially by sonication in an Alconox solution (30 min), DI water (30 min), acetone, 95% (v/v) ethanol, and isopropanol (15 min each). All glass coverslips were functionalized in a solution of 1% (v/v) APTMS in absolute EtOH solution on a shaker table for 2 h. Coverslips were rinsed with deionized water and placed in the bottom of a well-plate.Preparation of microgel polyelectrolyte films using the single-step method
Microgel solutions were suspended on a shaker overnight before each experiment was performed. To prepare films composed of polymeric, colloidal building blocks, our lab has established a method of centrifugal deposition. In this method, a solution with the colloidal building blocks, along with a substrate (typically glass) are transferred into a well-plate.42 The well-plate is then placed on a plate rotor and centrifuged. A 0.086 g mL−1 stock solution of branched PEI was prepared in PBS. PEI stock solution was made 2–12 h prior to fabrication to ensure that the PEI solution was well-mixed. Microgels solutions were also prepared in PBS for a stock solution concentration of 10 mg mL−1. Within each well, a solution was prepared containing PBS, PEI, and microgels and was immediately mixed. Various amounts of microgel dispersions and PEI were used to prepare the constructs, but all components had a consistent volumetric ratio of PBS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PEI:Microgel 2:1:1 (i.e. 200 μL PBS + 100 μL PEI + 100 μL microgels). Samples were centrifuged at 2250 × g for 20 min. Liquid was removed using a transfer pipette. Samples were removed and dipped in DI water to rinse excess film transferred from the surrounding area of the well-plate. Samples were then allowed to air dry.
:PEI:Microgel 2:1:1 (i.e. 200 μL PBS + 100 μL PEI + 100 μL microgels). Samples were centrifuged at 2250 × g for 20 min. Liquid was removed using a transfer pipette. Samples were removed and dipped in DI water to rinse excess film transferred from the surrounding area of the well-plate. Samples were then allowed to air dry.
Preparation of microgel polyelectrolyte layer-by-layer (LbL) films
Microgel solutions were suspended on a shaker overnight before each experiment was performed. Films were equilibrated in PBS for 30 min. Film growth occurred through the deposition of 1 mL of a 0.1 mg mL−1 solution of microgels suspended in PBS. Films were washed, placed in a solution of PEI (8.6 mg mL−1) in PBS for 30 min on a shaker, and subsequently washed again. This process was repeated until four layers of microgels and three layers of polycation were added.Characterization of microgel film topography and thickness
Film topography was characterized for a four layer (4L) LbL film and for single-step films prepared with 1 mg, 2 mg, or 4 mg of microgels via an MFP-3D AFM (Asylum Research, Santa Barbara, CA). Images in air were collected in AC mode using silicon SPM probes with Al reflex coating, and a 42 N m−1 nominal force constant (Nanoworld, NCHR). Dry film thickness was characterized using a NOVA 200 focused ion beam/scanning electron microscope system (FEI, Hillsboro, OR). Samples were adhered to vertical metal stubs using copper tape and were coated with gold/palladium using a Hummer V Sputterer (Anatech, USA, Union City, CA) for 2–3 min.Characterization of microgel film mechanical properties
AFM nanoindentation was performed to determine the Young's modulus of films. AFM has been used previously to probe the mechanical properties of a substrate and/or film at the micro-scale, using a technique where the AFM tip is brought into contact with the surface to act as a nano-indenter.40 AFM nanoindentation was performed using an MFP-3D AFM (Asylum, Santa Barbara, CA). These studies were performed in liquid using silicon nitride probes with Cr/Au reflex coating, and a 0.09 N m−1 nominal spring constant (Asylum, BL-TR400PB) mounted in the iDrive cantilever holder. For soft samples (E < 10 kPa), a colloidal surface probe was used that had a 3.5 μm diameter polystyrene bead attached to a 200 μm length triangular cantilever with a 0.08 N m−1 force constant (NanoAndMore USA, Watsonville, CA). All force curves were collected in contact mode. The samples were allowed to equilibrate in 10 mM, pH 7.4 PBS with 100 mM NaCl ionic strength for a minimum of 30 min prior to analysis. For samples where silicon nitride probes were used, exact spring constants were determined using the GetReal Probe program provided by Asylum. For samples where colloidal surface probe microscopy was required, exact spring constants were determined on a clean glass coverslip by a combination of force curves and thermal spectrum calibration using methods in the MFP-3D software. Force maps were collected as 32 × 32 arrays of force curves with trigger point = 0.3 V, except for samples with an E < 10 kPa, which were collected as a 10 × 10 array due to decreased velocity required for measurements. Elastic moduli were determined via the MFP-3D analysis tools using the Hertz model, assuming a Poisson ratio of 0.5, and assigning the appropriate tip geometry as a cone or a sphere for the silicon nitride probes and polystyrene probes, respectively. Reported values were calculated by averaging 108 data points collected from three samples with 36 points taken from 2–3 spots on each individual sample and calculating a standard deviation for all points. Statistical analysis was performed with Prism software program (GraphPad, San Diego CA). Data were analyzed using the Kruskal–Wallis test with Dunn post-test due to the significant differences between sample variations where *** = p < 0.001; all sample points were pooled together for statistical analysis.Microgel film swelling
Polyelectrolyte microgel films were prepared in a 24-well-plate using circular, 12 mm diameter glass coverslips functionalized with APTMS. 150 μL of microgels (10 mg mL−1), 150 μL PEI (0.086 g mL−1), and 300 μL PBS were mixed and deposited via centrifugation (20 min, 2250 × g). Films were then air-dried overnight. A razorblade was used to score the edge of the glass coverslip to detach the film prepared on the top of the coverslip from remnant film on the bottom of the coverslip. Films were then placed into a 6-well-plate with either formate buffer (10 mM formate, pH 3), MES buffer (10 mM MES, pH 5), or PBS (10 mM phosphate, pH 7.4) with either 150 mM NaCl (HIS) or 25 mM NaCl (LIS), and left on a shaker for 1.5 hours. After that time, films were incubated in buffer for several months. Polyelectrolyte microgel films that were found to detach from glass coverslips were imaged using a camera. The increase in diameter was determined via Image J (NIH) analysis of three films. To characterize how film thickness and film area influence film swelling, films were prepared on either 12 mm or 22 mm diameter circular coverslips that had been functionalized with APTMS. Microgel samples containing 1.5, 3, or 6 mg of microgel (using a 10 mg mL−1 stock solution in PBS) were mixed with an equal amount of the polycation and deposited via centrifugation (20 min, 2250 × g). Film swelling was then characterized by placing all samples in formate buffer with 100 mM NaCl concentration. Films were placed on a shaker for 24 hours; after that time, films were removed and left on an immobile surface.Preparation and characterization of laterally patterned microgel polyelectrolyte films
Using square, 9 mm × 9 mm glass coverslips, 1 mL of 0.1 mg mL−1 microgels (either microgel 1 or microgel 2, see Table 1) was added to each well and deposited via centrifugation at 2250 × g for 10 min. Samples were washed with deionized water to remove excess microgel solution. Microgel monolayers were cross-linked to the APTMS functionalized surface via carbodiimide coupling. Films were cross-linked overnight in 1 mL of MES buffer containing 2 mM EDC and 5 mM NHS. Films were then washed with deionized water, and 1 mL of PBS was added to each well, followed by the addition of 0.5 mL of PEI (8.6 mg mL−1) to each well that was mixed with the PBS. A Veco-CU TEM grid (Ted Pella, Redding, CA) was then immersed in the solution and applied to the surface of the coverslip. Finally, 0.5 mL of a 1 mg mL−1 microgel solution was added and mixed with the existing solution. Microgels and PEI were deposited via centrifugation at 2250 × g for 20 min. Samples were carefully removed and dipped in deionized water to remove excess film that had detached during the transfer process. Patterned microgel films were imaged using an Olympus IX-70 inverted microscope (Olympus, Center Valley, PA) equipped with a PixelFly black and white CCD camera (Andor, South Windsor, UK).Preparation and characterization of perpendicularly patterned polyelectrolyte microgel film
Microgel films exhibiting patterns normal to the substrate were created using microgels 1, 2, and 3 as listed in Table 1. Using square, 9 × 9 mm glass coverslips, each layer was added using the single-step film fabrication process detailed previously in this section. For films containing microgels 1 and 2, the construct was patterned as follows: 2:1:2:1:2. For films containing microgels 2 and 3, the construct was patterned as follows: 2:3:2. For all films, 0.5 mL of a 0.1 mg mL−1 solution of microgels was mixed with 0.5 mL of PEI (8.6 mg mL−1). Samples were imaged via a 700-405 Zeiss Laser Scanning Confocal Microscope (Zeiss, Thornwood, NY).
Preparation and characterization of polyelectrolyte raspberry-like particles films
Raspberry-like particles (RLPs) were prepared by immersing aminobenzophenone (ABP)-functionalized PS cores (4.5 μm diameter) into a concentrated microgel dispersion by scaling up a previously described method.41 1.25 mL PBS was added to the well, followed by 0.5 mL of PEI (8.6 mg mL−1), and the solution was mixed. Finally, 0.250 mL of RLPs suspended in PBS was added and gently mixed with a pipette. This solution was then deposited via centrifugation at 2250 × g for 30 min. Samples were gently rinsed with deionized water and left on a benchtop to dry overnight. Films were imaged using an Olympus IX-70 inverted microscope (Olympus, Center Valley, PA) equipped with a PixelFly black and white CCD camera (Andor, South Windsor, UK).Preparation and characterization of polyelectrolyte carboxyl-functionalized PS films
1.25 mL PBS was added to the well, followed by 0.5 mL of PEI (8.6 mg mL−1), and the solution was then mixed. Finally, 0.250 mL of 4.5 μm diameter, carboxyl-functionalized PS (0.25% w/v) was suspended in PBS and gently mixed with a pipette. This solution was then deposited via centrifugation at 2250 × g for 30 min. Films were gently rinsed with deionized water and left to dry overnight. Films were imaged using an Olympus IX-70 inverted microscope (Olympus, Center Valley, PA) equipped with a PixelFly black and white CCD camera (Andor, South Windsor, UK).Preparation and characterization of laterally patterned polystyrene films
A monolayer of microgels was deposited by centrifuging 1 mL of 0.1 mg mL−1 microgels for 10 min at 2250 × g. PEI was added first, followed by addition of PBS, a TEM grid was then applied, and finally a solution of carboxyl-functionalized PS beads (0.25% w/v) were added before centrifugation for 30 min at 2250 × g. Samples were removed from the well, gently rinsed with deionized water to remove grids, and air-dried overnight. For initial characterization, film composition was identical to PS monolayers. Samples prepared for cell studies were fabricated using 0.5 mL PEI (8.6 mg mL−1), 0.2 mL PS (4.5 μm diameter), and 0.3 mL PBS. Films were imaged using an Olympus IX-70 inverted microscope (Olympus, Center Valley, PA) equipped with a PixelFly black and white CCD camera (Andor, South Windsor, UK).Preparation and characterization of bulk gels
To prepare bulk polyelectrolyte gels, 0.5 mL of 10 mg mL−1 of microgels suspended in PBS were mixed with 0.5 mL of PEI (0.086 g mL−1) suspended in PBS and 1 mL of PBS in a microcentrifuge tube. This solution was mixed and centrifuged at 2250 × g for 20 min. Excess liquid was removed using a transfer pipette. For cross-linked samples, 0.2/0.5 mM, 2/5 mM, or 20/50 mM EDC/NHS (2 h) was used. For bulk gels containing a 1:1 ratio of 4% BIS non-fluorescent microgels and AFA containing ULC microgels, 0.25 mL of non-fluorescent microgels and 0.25 mL of ULC microgels were used in each sample. Bulk gel mechanical properties were investigated using an Anton Paar rheometer with a cone-plate geometry. Samples were subjected to a pre-shearing protocol to eliminate material memory before each experiment. Oscillatory experiments were performed: a strain sweep at a constant frequency was initially applied to determine the dependence of both the elastic and viscous moduli (G′ and G′′, respectively). The linear regime of the gel was then identified. Next, a frequency sweep at a steady strain in the linear regime (γ = 0.8) was undertaken to observe the viscoelastic properties of the gel in a wide range of time-scales.
Analysis of fibroblast cell attachment and spreading on microgel films
NIH-3T3 fibroblasts were purchased from ATCC and were maintained in DMEM media supplemented with 10% bovine calf serum (BCS) and 1% penicillin/streptomycin. Cells were cultured to 70–80% confluency prior to experiments. Cells were trypsinized and then membranes were stained with either CellTracker Green CMFDA or Deep Red Dye (Life Technologies). NIH-3T3 cells stably transfected with an m-Emerald Actin construct were utilized for analysis of actin distribution and alignment on microgel films. Cells were plated at a density of 5000 cells per cm2, and following 24 h in culture, samples were fixed and mounted. Samples were imaged using a 700-405 Zeiss Laser Scanning Confocal Microscope (Zeiss, Thornwood, NY). Cell attachment, circularity, aspect ratio and area were determined through Image J (NIH freeware). All statistical analyses were performed with Prism software program (GraphPad, San Diego CA). Data were analyzed using a Kruskal–Wallis test with Dunn post hoc test; cells were not expected to exhibit a Gaussian distribution in response to the film patterns.Analysis of ATII cell attachment and spreading on microgel films
RLE-6TN ATII cells (ATII) were cultured in DMEM/F12 media supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S). To analyze the effect of patterned films on ATII cell attachment and spreading, RLE-6TN cells were trypsinzied, membranes were stained with CellTracker Green CMFDA Dye (Life Technologies), and cells were then plated on films at a density of 25000 cells per cm2. Cells were cultured on films for 24 hours in DMEM/F12 media supplemented with 10% FBS and 1% P/S. Cells were washed with PBS and then fixed with 4% formaldehyde for 20 minutes at room temperature. At this point, any cells that that were not stained with CellTracker Green CMFDA (Life Technologies) were permeabilized with 0.1% Triton-X 100 for 5 minutes, and then actin was stained with Texas-red phalloidin (Life Technologies). Samples were subsequently mounted and imaged with a 700-405 Zeiss Laser Scanning Confocal Microscope (Carl Zeiss).
Preparation and characterization of bulk gels with embedded NIH-323 fibroblasts and ATII cells
Bulk gels were prepared by adding 0.3 mL of a 10 mg mL−1 solution of microgels, 0.3 mL of a PEI solution (0.086 g mL−1), and 0.6 mL of PBS to a 2 mL microcentrifuge tube. The solution was then mixed and immediately centrifuged at 2250 × g for 20 min. Excess solution was removed from the tube. NIH-3T3 fibroblasts and ATII cells were labeled with the membrane dye CellTracker Green CMFDA and then washed with PBS to remove excess dye. 50 μL of stained ATII cells at approximately 7 × 106 cells per mL or 25 μL of stained NIH-3T3 fibroblasts at approximately 1.9 × 106 cells per mL were added to each microgel-containing tube. Samples for each cell type were prepared using two protocols; samples were centrifuged for either 500 RPM for 1 min or 1000 RPM for 30 s. Samples were fixed overnight in a solution of 5% glutaraldehyde in PBS. Samples were imaged via a 700-405 Zeiss Laser Scanning Confocal Microscope (Zeiss, Thornwood, NY).Results and discussion
Films were prepared by combining well-mixed solutions of an anionic polymer building block and the polyelectrolyte, branched PEI, in a well-plate before centrifugation onto a functionalized glass coverslip using the “single-step” fabrication technique (Scheme 2). Microgel film fabrication was investigated using two microgel building blocks: pNIPAm microgels containing 30 mol% acrylic acid (AAc) that were either cross-linked with 4 mol% BIS or self-cross-linked (no exogenous cross-linker added), termed ultra-low-cross-linked (ULC) microgels. Though both BIS and ULC microgels are considered soft materials, they exhibit significantly different mechanical properties. The Lyon group has previously demonstrated that ULC microgels have a Young's modulus <10 kPa and are an order of magnitude softer than typical BIS-containing microgel constructs, which have a Young's modulus closer to ∼100 kPa.34,42 To complete these studies, three microgel populations were synthesized and characterized (Table 2). Films were prepared with 0.5 mg, 1 mg, 2 mg, or 4 mg of microgels, while maintaining a constant ratio of PEI by weight. | ||
| Scheme 2 Depiction of the single-step fabrication technique. | ||
| Sample | Chemical composition | R H (nm) | ||||
|---|---|---|---|---|---|---|
| pNIPAm (mol%) | AAc (mol%) | BIS (mol%) | Rho-B (mol%) | AFA (mol%) | ||
| μgels | 66 | 30 | 4 | 0 | 0 | 509 ± 10 |
| Rho-B μgels | 66 | 30 | 4 | ∼0.1 | 0 | 288 ± 53 |
| AFA μgels | 70 | 30 | 0 | 0 | ∼0.1 | 620 ± 10 |
In order to investigate film growth, SEM imaging of film cross-sections was performed using dried films of 4% BIS microgels (Fig. 1A). LbL films prepared with 0.4 mg of microgel exhibit a thickness of 630 nm ± 130 nm. In contrast, single-step films prepared using 0.1, 0.5, 1, 2, or 4 mg of microgel exhibit film thicknesses of 1.4 μm ± 390 nm, 4.1 μm ± 640 nm, 9.3 μm ± 2.0 μm, 19.5 μm ± 3.1 μm, and 55.6 μm ± 12.2 μm, respectively. These experiments demonstrate that the single-step method enables the fabrication of thick microgel films not typically feasible using an LbL approach. These studies also reveal that single-step films containing only 0.1 mg of microgels, one-fourth the amount used for a typical 4L LbL film, are almost twice as thick as a typical 4L LbL film. Thus, the single-step film fabrication technique uses material more efficiently than the LbL method. Quantitative analysis of SEM images reveals that film formation occurs in a linear fashion. Additionally, film growth of ULC microgels differs from 4% BIS microgels when >2 mg of microgels are used (Fig. 1B).
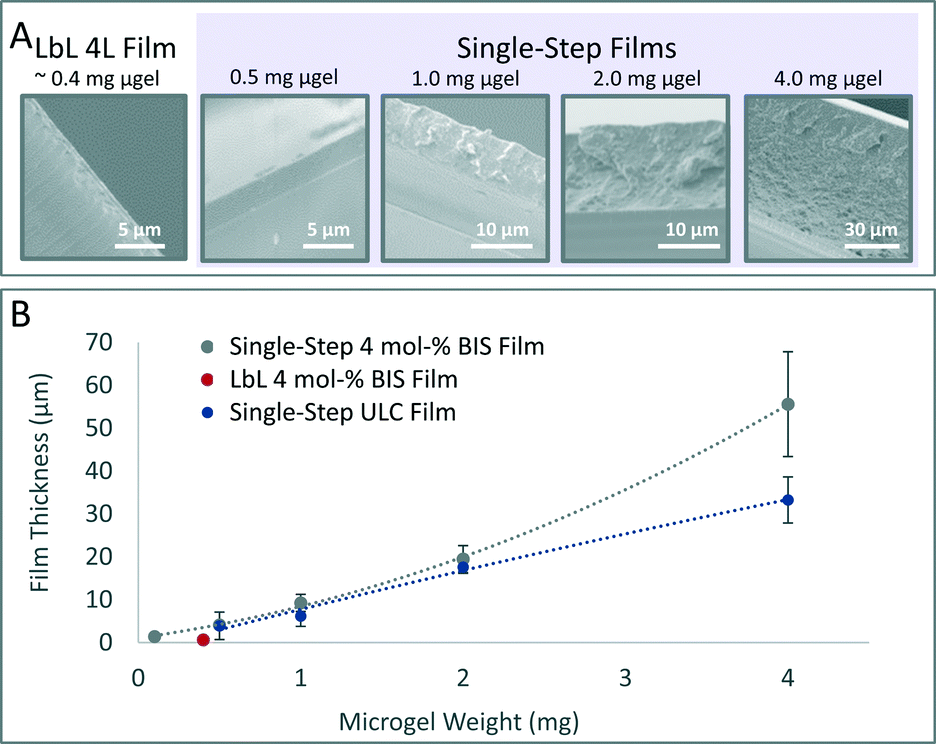 | ||
| Fig. 1 Characterization of microgel film growth. (A) Cross-sections of microgel films prepared with varied weights of 4% BIS microgels and PEI were imaged via SEM. Dried films were adhered to vertical stubs using copper tape. (B) Film thickness was determined using Image J. Error bars represent plus or minus one standard deviation about the average value of 9 measurements taken in distinct spots on each film. | ||
To explore the effect of the single-step fabrication technique on film topography, microgel films were imaged via AFM (Fig. 2A). In these studies, films were prepared using the microgels containing 4 mol% BIS. AFM images reveal that microgels are highly packed and exhibit uniform spacing at the surface of the film. The degree of microgel packing appears consistent regardless of the amount of microgels used. However, films prepared with increasing amounts of microgels exhibit greater uniformity as can be seen by the decreased variability in film topography via AFM. In contrast, AFM images of films prepared using the established LbL method reveal a higher degree of heterogeneity. Films appear to contain a microgel layer with partial coverage at the surface; as is common for LbL microgel films,36 individual microgels do not appear integrated into a single layer, appearing less packed overall compared to single-step films.
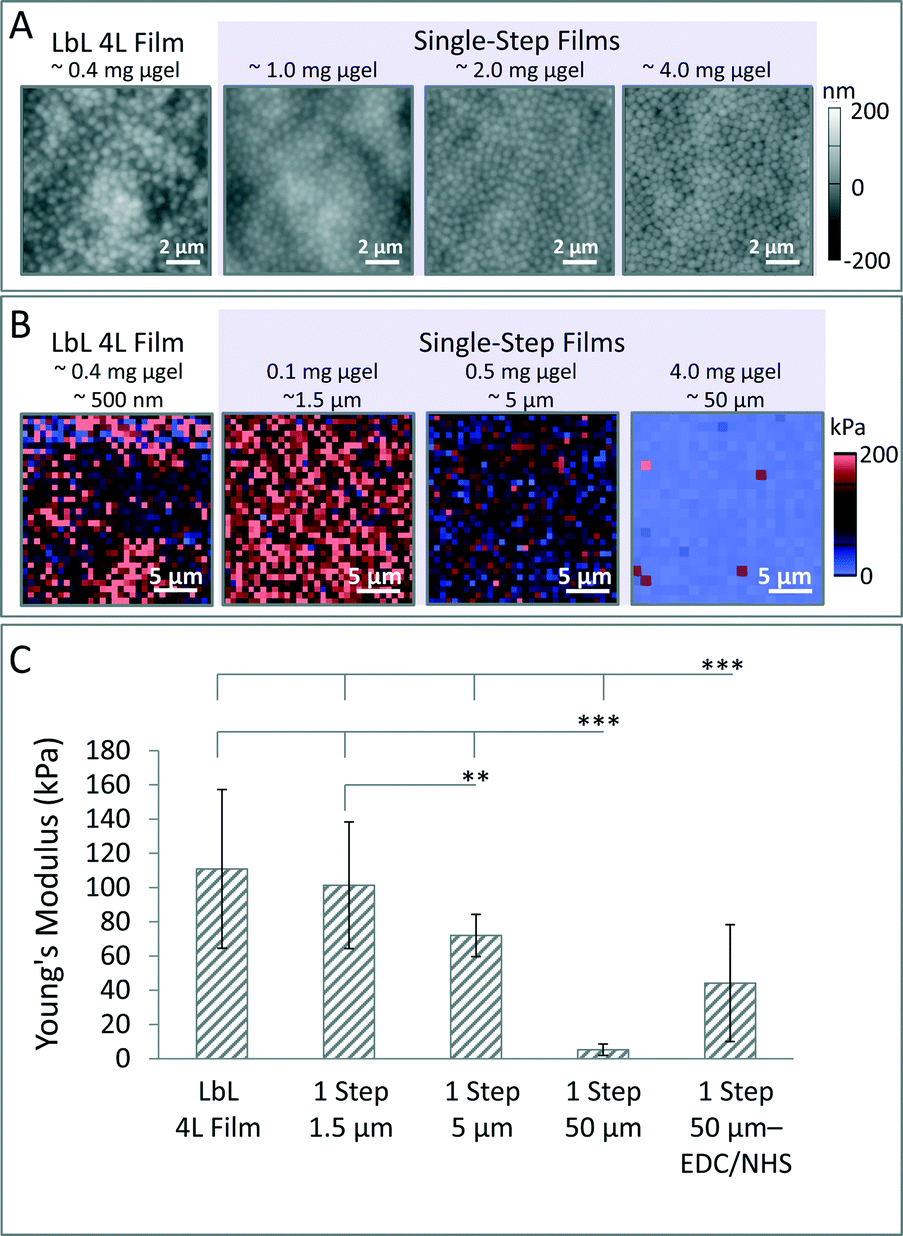 | ||
| Fig. 2 Characterization of microgel topography and mechanical properties via AFM. (A) AFM height retraces of dry films fabricated with 66 mol% pNIPAm, 30 mol% AAc, and 4 mol% BIS. (B) Representative AFM nanoindentation force maps are displayed. (C) Young's modulus was quantified. Error bars represent plus or minus the standard deviation of the average values from three independent samples. All comparisons of samples to either the single-step (1 step) 50 μm or single-step 50 μm EDC/NHS samples are statistically significant (p < 0.001). Comparison between the single-step 1.5 μm and single-step 5 μm films is also statistically significant (p < 0.01). Statistical analysis was performed using the Kruskal–Wallis test with Dunn post-test due to the significant differences between sample variations where *** = p < 0.001 and ** = p < 0.01; all sample points were pooled together for statistical analysis. | ||
Next, AFM nanoindentation studies were performed on films prepared using 4 mol% BIS microgels to characterize the micro-mechanical properties (Fig. 2B and C). Films prepared using the single-step method were fabricated to be roughly 1.5 μm, 5 μm, or 50 μm in dry thickness; when solvated, these films exhibit a Young's moduli of 101 kPa ± 37 kPa, 72 kPa ± 12 kPa, and 5 kPa ± 3 kPa, respectively. Films prepared with a dry thickness of approximately 50 μm and subsequently subjected to EDC/NHS cross-linking exhibit an increased Young's modulus of 44 kPa μm ± 34 kPa. In comparison, a typical 4L LbL film has a Young's modulus of 111 kPa ± 46 kPa. Carbodiimide coupling is also demonstrated to be a facile method to tune the mechanical properties of these films (Fig. 2C and Scheme S1†). These studies also reveal that for single-step films, stiffness is inversely related to film thickness. The thickest microgel films (∼55 μm in thickness) exhibit a Young's modulus of ∼5 kPa, which is significantly lower than 4 layer LbL films previously prepared and characterized in the Lyon group.34 As such, the single-step fabrication technique enables the development of constructs that have mechanical properties in the range of cell sensitivity (1–50 kPa).43 The large influence that film thickness has on mechanical properties can likely be attributed to film swelling characteristics, which we describe below.
Previously, we demonstrated the ability to obtain free-standing films by exposing microgel films to acidic buffers (pH 2).44 In these studies, we prepared our films via the LbL method using pNIPAm-co-AAc microgels and PEI as the polycation. In order to assess the ability to form free-standing films, controlled swelling studies were performed where films were placed in either 10 mM formate buffer (pH 3.3), 10 mM MES buffer (pH 5.5) or 10 mM PBS (pH 7.4) containing either 25 mM or 150 mM NaCl. Swelling studies reveal that films prepared on circular 12 mm diameter glass coverslips detach from the glass substrate when placed in acidic buffer conditions; specifically, films detach in 10 mM formate buffer (pH 3.3) and/or 10 mM MES buffer (pH 5.5) both with 150 mM NaCl, forming stable free-standing films (Fig. S1†). This delamination process is influenced by a strong force caused by the swelling of the film that is further promoted by changes in protonation of the acid groups on the microgels; below pH 4.6, the solution equilibrium shifts towards the protonated form of the AAc comonomer. These results are in line with the previous study exploring development of free-standing films conducted in our group; films assembled at pH 7.4 exhibit in-plane swelling in acidic conditions (pH 2), resulting in delamination from the substrate due to a rapid increase in film area.44 Here we also see a rapid increase in film area that is likely driving the delamination process. The key difference in these studies concerns the substrate preparation. The former free-standing film study required treatment of the glass with lightly boiled piranha solution in order to promote film detachment. In this case, the boiled piranha treatment likely decreases interactions between the substrate and the film, aiding the delamination process. These new results reveal that such strong substrate pre-treatments may not be necessary to form free-standing polyelectrolyte microgel films, depending on the buffer conditions employed to drive delamination; it also suggests that the buffer range over which delamination occurs is larger than was previously thought.
Further swelling studies were performed to investigate the influence of film thickness and film area on the detachment process. In one of these studies, films were prepared on circular 22 mm diameter coverslips from 1.5 mg, 3 mg, or 6 mg microgel; films were then solvated in buffer conditions (10 mM formate, pH 3.3, 100 mm NaCl) on a shaker for 24 h (Fig. S2†). All films prepared at the smallest thickness detach from the glass, tearing into pieces in the process. All films prepared with 3 mg of microgels were found to detach, but they do not exhibit appreciable tearing. Finally, approximately 33% of films prepared using 6 mg of microgels detach completely. In this case, the films exhibit rolling, because the film has swollen to a size that is physically confined by walls of the 6-well-plate, while remaining intact. A swelling study was also performed using 12 mm diameter coverslips (Fig. S3†), and in these studies, films prepared with 3 mg or 6 mg of microgels did not completely detach during the 24 h period of solvation in 10 mM formate (pH 3.3) buffer with 100 mM NaCl. Lack of film detachment may have occurred for several reasons. The thickness of the microgel films may inhibit the rate of swelling to an appreciable degree; an insufficient lateral swelling force could be responsible for the lack of film detachment. Stability studies reveal that if a film remains intact during the detachment process, the film will exhibit minimal degradation when left standing in buffer for more than 3 months; during that time, films in neutral buffer slowly begin to detach from the glass (Fig. 3). Overall, these studies demonstrate that preparation of microgel films using the single-step method followed by solvation in an acidic buffer is a simple route to obtain free-standing microgel films.
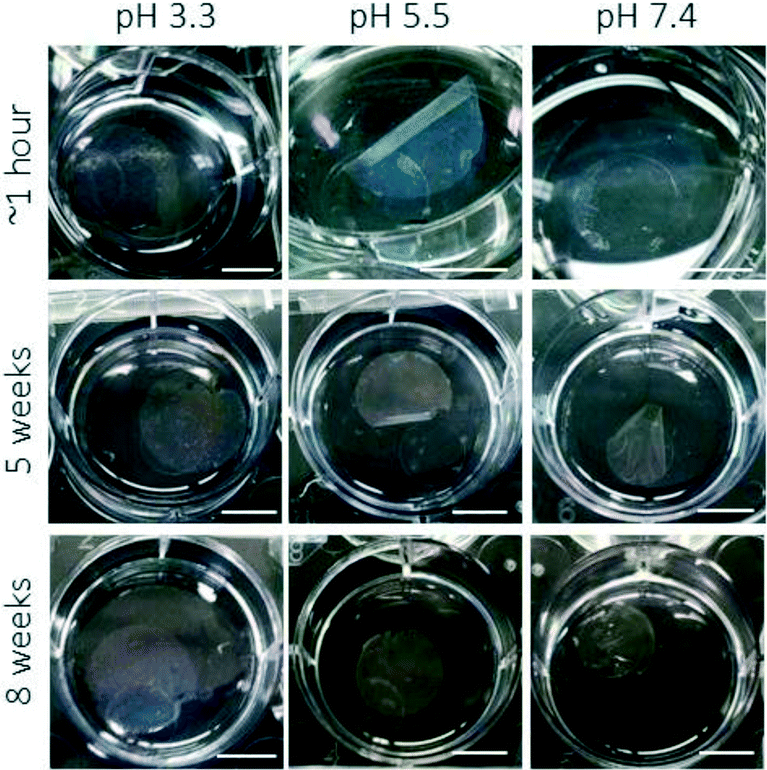 | ||
| Fig. 3 Characterization of film degradation over two months. Films that remain intact after detachment process remain stable in solution for several weeks. Scale bars represent 10 mm. | ||
The single-step technique can be extended to include an array of constructs achieving unique topographical and bulk properties through combination of dissimilar modular building blocks and clever experimental designs (Fig. S4†). Microgel film wedges were fabricated simply based upon placement of coverslip and polyelectrolyte mixture in an outer well of a well-plate during centrifugation; this results in fabrication of a film that is thickest at one end with a gradual decrease in material. Such wedge constructs could be of value to investigate cellular responses to materials with varied pitches or gradient mechanical properties. The use of incompressible, carboxyl-functionalized polystyrene (PS) beads and raspberry-like particles were also explored as alternative building blocks to fabricate films. Films of raspberry-like particles exhibit hierarchical patterning, adding another dimension to these two-dimensional materials. Finally, fabrication of films composed of microgels with polystyrene beads distributed throughout a continuous gel phase was demonstrated using the same approach.
Next, the development of both laterally and perpendicularly patterned constructs was explored using modified versions of the single-step technique. In the “grid method,” laterally patterned constructs were fabricated by depositing a monolayer of microgels onto a glass substrate, applying a TEM grid on top of the monolayer, depositing a solution of microparticles mixed with the polycation via centrifugation, and finally removing the TEM grid via gentle rinsing or through the use of tweezers (Scheme 3A). Using the grid method, fabrication of patterned microgel films with multiple micron-scale square sizes was demonstrated (Fig. 4A). Additionally, the grid method can also be expanded to include incompressible anionic building blocks including polystyrene spheres. Fabrication of patterned PS films with multiple pattern features including squares and channels was demonstrated (Fig. 4B). Films prepared using both soft and hard building blocks exhibit well-defined pattern edges. To fabricate perpendicularly patterned microgel constructs (Scheme 3B), microgels mixed with a polycation are deposited onto a glass coverslip to build the first layer. This step is then repeated with alternating microgel building blocks to build a pattern perpendicular to the substrate. Because film thickness is controlled by the amount of microgels deposited, this technique can be used to create patterns with varied layer thicknesses, if desired.
 | ||
| Scheme 3 Depiction of method to fabricate (A) laterally and (B) perpendicularly patterned films. | ||
 | ||
| Fig. 4 Characterization of laterally. (A) Laterally patterned microgel films characterized via brightfield microscopy using TEM grids with (i) 85 μm × 85 μm and (ii and iii) 283 μm × 283 μm meshes. (B) Laterally patterned PS films characterized via laser scanning confocal microscopy using TEM grids with (i) channels, (ii) slits, (iii) 30 μm × 30 μm, (iv) 85 μm × 85 μm, (v) 283 μm × 283 μm meshes. | ||
To develop perpendicularly patterned films, two populations of microgels with distinct compositions were deposited in alternating layers through repetition of the single-step method (Scheme 3B). The development of perpendicularly patterned microgel assemblies using two similar microgels was explored to demonstrate the ability to create distinct microgel layers (Fig. 5A). The two populations of microgels used were composed of pNIPAm (66 mol%), AAc (30 mol%), and BIS (4 mol%); one population was unlabeled while the other was labeled red, using Rho-B. These films exhibit a perpendicular pattern containing five layers, three fluorescent red layers and two non-fluorescent layers, establishing the efficacy of this patterning method. The ability to pattern microgels with varied mechanical properties and/or density was also explored due to the functionality of such constructs for tissue engineering applications. In these studies, development of patterned constructs was explored using Rho-B labeled microgels and ultra-low-cross-linked (ULC) pNIPAm microgels containing 30 mol% AAc and labeled with AFA. These two microgel populations have different densities and stiffness due to the differing degrees of network cross-linking. For this study, a mixture of microgels and polycation was deposited with layers in the following order: (1) Rho-B labeled microgels, (2) ULC microgels, (3) Rho-B labeled microgels. However, laser scanning confocal microscopy imaging revealed that only two microgel layers exist instead of three layers (Fig. 5B). The absence of three layers suggests that the denser 4 mol% BIS microgels penetrate the weaker ULC microgel layer, resulting in a single layer of each particle. Thus, use of building blocks with highly dissimilar mechanical properties and densities is possible, but may be limited by the number of layers formed.
 | ||
| Fig. 5 Perpendicularly patterned microgel films characterized via laser scanning confocal microscopy. (A) Films were fabricated using alternating layers of pNIPAm microgels containing 30 mol% AAc and 4 mol% BIS. Five distinct layers are visible, alternating red-blank-red-blank-red above the substrate. (B) Films were fabricated using alternating layers of the ULC microgels and the 4 mol% BIS microgels. Two distinct layers are visible. | ||
These patterned microgel constructs resemble biomaterials explored for tissue engineering applications. When designing tissue engineering tools to direct cellular behavior, major interactions to consider are cell–cell interactions and cell–substrate interactions. Over the past several decades, interest in understanding basic cell–substrate interactions has increased significantly.45 A growing field of literature has focused on development of constructs that mimic the varied micro/nano-topography and surface chemistry of the extra-cellular matrix, the cell’s natural environment.46 Numerous scientists have characterized cellular responses to micro-and nano sized lines, wells, holes, pillars, and more complex geometries.47–49 These studies indicate that surface topography can significantly influence cellular orientation, morphology, adhesion, spreading, migration, proliferation, etc., independent of substrate chemistry. Integration of this knowledge has become a key aspect of biomaterials design for tissue engineering applications.
In particular, the innovative methods described in this publication could provide an inexpensive and fast method to produce patterned material constructs that can be used to investigate and direct cellular behavior. Depending on material properties, such patterned constructs could either promote or inhibit cellular adhesion and spreading. In the field, hydrogels have already been extensively explored for development of patterned constructs for tissue engineering applications.50–53 Microgel-based materials have also been explored for development of cell scaffolds. Tsai et al. previously fabricated biocompatible substrates by patterning pNIPAM microgels onto polystyrene substrates using dip coating. This method resulted in a substrate with stripes of densely packed microgels separated by areas of sparsely populated microgels, all in monolayer form. NIH-3T3 fibroblasts seeded onto substrates with varied line spacing (50–100 μm features) preferentially adhered within the spacing and proliferated to form a confluent cell layer three days after seeding. In the Lyon group, we also explored methods to modulate fibroblast adhesion and spreading on microgel constructs.34 Our studies focused on understanding how fibroblasts adhere and spread on microgel multilayers, in comparison to monolayers, like those investigated by Tsai et al. Building upon these studies, we explored the laterally patterned microgel films as an intriguing construct to interrogate the influence of microenvironments, containing both microgel multilayers and monolayers, on fibroblast adhesion and spreading. Laterally patterned microgel films were prepared using the grid method to assess fibroblast adhesion and spreading. NIH-3T3 fibroblasts were seeded onto the laterally patterned films and incubated for 24 h (Fig. 6A). When fibroblasts were seeded onto films with smaller pattern sizes (85 μm × 85 μm microgel islands), cells preferentially attach to the microgel monolayer. In addition, cells exhibit spreading in between the microgel islands. In contrast, when fibroblasts were plated on patterned microgel films with large islands (283 μm × 283 μm), fewer cells attach overall. Cells that are present preferentially attach to the monolayer; however, in this case, cells do not spread, exhibiting a round morphology. Quantitative image analysis corroborates the finding that pattern size influences fibroblast behavior (Fig. S5†). Patterned microgel films were also found to affect the attachment and spreading of more sensitive epithelial cells (Fig. S6†). Overall, these studies demonstrate that patterned microgel films present a unique experimental tool to assess cellular responses to a microenvironment for further development of platforms that either promote or inhibit cellular adhesion. Patterned microgel films with smaller feature sizes can be used to obtain greater cellular adhesion and spreading; these films may also be used to direct cellular spreading. In contrast, the patterned microgel films containing larger features may exhibit a higher degree of film swelling due to the larger film area, which could contribute to the low cell attachment numbers and lack of cell spreading.
 | ||
| Fig. 6 Fibroblast behavior is influenced by film patterns. Cells were plated at a density of 5000 cells per cm2 on laterally patterned microgel films with either (Ai) 85 μm × 85 μm squares or (Aii) 283 μm × 283 μm squares. NIH-3T3 cells stably transfected with an m-Emerald Actin construct were utilized for visualization of spreading on microgel films. (B) Cells were plated at a density of 5000 cells per cm2 on laterally patterned PS films. NIH-3T3 cells stably transfected with m-Emerald Actin (green) were stained with CellTracker Deep Red Dye (Life Technologies) to visualize the cell membrane (red). Following 24 h in culture, samples were fixed and mounted. Image saturation was enhanced to allow visualization of the cells. | ||
The adhesion and spreading of fibroblasts on laterally patterned polystyrene films was also explored. Using the grid method, laterally patterned polystyrene films were prepared using carboxyl functionalized PS beads with a diameter of either 3 μm, 4.5 μm, or 6 μm and TEM grids with either square or slit patterns of varied sizes. NIH-3T3 fibroblasts were plated onto the films and incubated for 24 h to assess adhesion and spreading (Fig. 6B). When fibroblasts were seeded on films with polystyrene channels, fibroblasts adhere preferentially to the polystyrene beads and elongate along the polystyrene channels. For the polystyrene square patterned films, fibroblasts adhere preferentially to the polystyrene squares with larger pattern features. However, when pattern features approach the lengthscale of cells (30 μm × 30 μm PS square sizes), fibroblasts are more likely to bridge multiple PS squares (Fig. 6Bii). Quantitative analysis of cell area, cell circularity, and cell aspect ratio corroborate the finding that fibroblast spreading is influenced by pattern features, pattern sizes, and building block sizes (Fig. S7 and S8†). These responses suggest that these constructs could be a suitable tissue engineering platform to direct cellular adhesion and spreading that can be further targeted/modulated to investigate wound healing responses and develop therapeutic tools.
Finally, the development of bulk polyelectrolyte constructs was explored (Scheme 4). Bulk gels were formed by depositing a mixture of microgels and polycation via centrifugation into a microcentrifuge tube and subsequently removing the supernatant. This procedure can be further modified to enable cell encapsulation by centrifuging a solution of cells over the bulk gel. In this case, a solution of cells is added on top of the pre-formed bulk gel before centrifuging the sample for a short time (1 min or less) to distribute cells throughout the gels. Bulk gels were fabricated from typical pNIPAm-co-AAc microgels (Fig. S9†) containing 4 mol% BIS. EDC/NHS cross-link was utilized to chemically cross-link the polycation to the microgels. Composite bulk gels were also fabricated from two separate microgel components, including typical microgels containing 4 mol% BIS cross-linker as well as the ULC microgels. Oscillatory rheology was performed to investigate the mechanical properties of these gels (Fig. S10–S15†). Rheological characterization did not yield significant differences in mechanical properties of the different gels. Studies indicate that all samples have a higher elastic modulus (G′) than viscous modulus (G′′). Overall, mechanical properties are similar and reminiscent to an elastic system on the linear regime where experiments were performed. Polyelectrolyte microgel bulk gels that contain differing degrees of intra-microgel cross-links (through varied cross-linker content) or inter-microgel cross-links through cross-linking of the polycation to the microgels, likely exhibit varied nano- or micro-mechanical properties due to the discrete nature of these colloidal building blocks. When designing material interfaces to interrogate or direct cellular responses, these differences can be leveraged. However, it is difficult to capture such differences in mechanical properties via traditional rheological characterization of bulk-scale properties.
 | ||
| Scheme 4 Development of bulk polyelectrolyte gels. Bulk gels are created by (1) centrifuging a mixture of polyelectrolyte and microgels within a microcentrifuge tube. Gels with encapsulated cells are then prepared by (2) centrifuging a solution of suspended cells into a fabricated gel. | ||
Due to the facile method used to fabricate these bulk gels, further studies were conducted exploring the use of these constructs as a tissue engineering scaffold in which cells could be encapsulated. In the field of tissue engineering, encapsulation of living cells in soft polymers is an opportunistic route that has been explored for regeneration and rehabilitation of functional tissues; this technical approach enables cells to be physically separated from surrounding tissues, providing protection from unwanted attack by the host immune system, while maintaining transport of critical gas, nutrients, wastes, and therapeutic molecules.54 Currently scientists have developed microsystems able to encapsulate cells in beads,55 sheets,56 fibers,57 and more advanced structures,58,59 with fine control over size and cell density. Microgels have already been used in this area, particularly for development of injectable hydrogel scaffolds.24,60 Though several routes exist to encapsulate cells, barriers limit their functional applicability, including high cost, long fabrication or manufacturing times, material limitations, and need for highly skilled workers. Herein, we propose that cells could be encapsulated into bulk microgel-based polyelectrolyte gels, through addition of a simple sedimentation processes. To test this theory, microgel-based bulk gels were fabricated using the single-step approach, and embedded with either fibroblasts or ATII cells using a rapid centrifugation step at a low centrifugal force (Fig. 7). Cell Tracker green CMFDA dye was used to label cells to mimic typical long-term cell tracking experiments used to design and evaluate embedded cell scaffolds. 3D laser scanning confocal microscopy studies indicate centrifugation is an effective method to distribute cells through the gel. This proof-of-principle study establishes a fast, facile route to prepare cell-laden gels.
 | ||
| Fig. 7 Fibroblasts and ATII cells were encapsulated in bulk gels. Laser scanning confocal microscopy was used to image z-stacks of gels containing (A) fibroblasts or (B) ATII cells that were labeled with the membrane dye CellTracker Green CMFDA. Both top-view projections (Ai and Bi) and orthogonal cross-sections are shown (Aii, Aiii, Bii, Biii). Imaging reveals that the distribution of cells throughout the gel is highly dependent on the cell type due to differences in size and density. | ||
This approach allows for the development of a truly versatile platform to design and investigate tissue engineering scaffolds. At the most rudimentary level, one could envision a cell-laden, spreadable gel that could be applied to a topographical wound, such as a burn, to augment wound healing. However, these microgel-based materials show promise to compete with other highly controlled systems, as well. Due to the modular nature of these bulk gels, composed of microgel building blocks, this approach provides an intriguing avenue for the design of tissue engineering scaffolds with a high degree of synthetic control of the material properties. As the Lyon group has demonstrated numerous times before, microgels themselves can be altered to have certain chemical, mechanical, and physical properties.61 Through careful selection of microgel composition, the porosity and network structure can be altered, influencing the diffusion of small molecules and macromolecules, and the nano-scale rigidity and stiffness can be controlled. Through post-synthesis bio-conjugation, microgels can be functionalized to promote or inhibit cellular interactions. Furthermore, combination of dissimilar microgels can be exploited to form uniquely tailored scaffolds to tackle highly complex problems, ranging from investigation of fundamental cell–substrate interactions to development of functional, therapeutic materials.
Conclusions
This investigation details the versatility of a facile method to rapidly fabricate an array of polyelectrolyte constructs with varying levels of hierarchical structure. Overall, this method is fast, inexpensive and enabling, eliminating many of the constraints of previous techniques. Specifically, the efficacy of this method to fabricate both two-dimensionally and three-dimensionally patterned constructs from compressible and incompressible colloidal building blocks was established. Microgel films were fabricated using both the established LbL technique and the new “single-step” fabrication technique. Comparison between microgel films indicates that single-step films exhibit less heterogeneity in topographical features and mechanical properties than 4L LbL films. Investigation of single-step film swelling and degradation reveals that single-step films exhibit high degrees of swelling; depending on film thickness, single-step microgel films solvated in acidic conditions are able to form stable free-standing microgel films. The versatility of this method was also demonstrated through development of films using alternative building blocks. A “grid method” was established to fabricate laterally patterned microgel multilayer films and laterally patterned PS films. Laterally patterned films exhibit a variety of micron-scale features. The single-step method was used in a repetitive manner to successfully fabricate perpendicularly patterned microgel films using microgels of varied chemical and mechanical properties.Laterally patterned microgel films and patterned PS films were used to investigate cellular responses to patterned materials with varied mechanical properties. For patterned microgel films, cellular adhesion and spreading are influenced by feature size. Laterally patterned PS films influence cell adhesion and spreading as well. Cellular adhesion studies demonstrate the utility of these constructs as competitive experimental tools to investigate fundamental responses of cells to varied chemical, mechanical, or topographical features. Bulk gels were fabricated and characterized, revealing overarching similarities in mechanical properties. A proof-of-principle study was also conducted demonstrating the ability to produce cell-laden gels. Polyelectrolyte materials fabricated using this method can be used for a variety of applications including hemostatic materials, environmental sensing coatings, self-healing coatings, non-adherent coatings, drug delivery scaffolds, tissue engineering scaffolds, cell encapsulation scaffolds, underwater adhesives, medical adhesives, and further experimental tools.
Acknowledgements
We would like to thank the Georgia Institute of Technology, School of Chemistry and Biochemistry for providing funding for this research. In addition, we would like to acknowledge A. Brown for her kind generosity in assisting with routine experimental procedures and manuscript preparation. Finally, we would like to acknowledge M. A. P Fernandez and A. Fernandez-Nieves for their generous support with the rheological characterization of the bulk gels.References
- S. Krishnamoorthy, C. Hinderling and H. Heinzelmann, Mater. Today, 2006, 9, 40–47 CrossRef CAS.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed.
- S. J. Randolph, J. D. Fowlkes and P. D. Rack, Crit. Rev. Solid State Mater. Sci., 2006, 31, 55–89 CrossRef CAS.
- J. D. Hoff, L. J. Cheng, E. Meyhofer, L. J. Guo and A. J. Hunt, Nano Lett., 2004, 4, 853–857 CrossRef CAS.
- M. D. Levenson, N. S. Viswanathan and R. A. Simpson, IEEE Trans. Electron Devices, 1982, 29, 1828–1836 CrossRef.
- Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2001, 53, 321–339 CrossRef CAS PubMed.
- D. Rana, T. S. S. Kumar and M. Ramalingam, J. Biomater. Tissue Eng., 2014, 4, 507–535 CrossRef CAS.
- K. Gawel, D. Barriet, M. Sletmoen and B. T. Stokke, Sensors, 2010, 10, 4381–4409 CrossRef CAS PubMed.
- H. Tsutsui, E. Yu, S. Marquina, B. Valamehr, I. Wong, H. Wu and C. M. Ho, Ann. Biomed. Eng., 2010, 38, 3777–3788 CrossRef PubMed.
- Y. Lee, S. Park, S. W. Han, T. G. Lim and W. G. Koh, Biosens. Bioelectron., 2012, 35, 243–250 CrossRef CAS PubMed.
- A. M. Greiner, P. Hoffmann, K. Bruellhoff, S. Jungbauer, J. P. Spatz, M. Moeller, R. Kemkemer and J. Groll, Macromol. Biosci., 2014, 14, 1547–1555 CrossRef CAS PubMed.
- T. W. Hsiao, P. A. Tresco and V. Hlady, Biomaterials, 2015, 39, 124–130 CrossRef CAS PubMed.
- R. J. McMurtrey, J. Neural Eng., 2014, 11, 066009 CrossRef PubMed.
- W. G. Koh, in Biological Microarrays: Methods and Protocols, Methods Mol. Biol., ed. A. Khademhosseini, K. Y. Suh and M. Zourob, 2011, vol. 671, pp. 133–145 Search PubMed.
- J. E. Meiring, S. Lee, E. A. Costner, M. J. Schmid, T. B. Michaelson, C. G. Willson and S. M. Grayson, Opt. Eng., 2009, 48, 37201.1–37201.14 CrossRef.
- V. A. Pedrosa, J. Yan, A. L. Simonian and A. Revzin, Electroanalysis, 2011, 23, 1142–1149 CrossRef CAS.
- Z. B. Hu, Y. Y. Chen, C. J. Wang, Y. D. Zheng and Y. Li, Nature, 1998, 393, 149–152 CrossRef CAS.
- M. S. Hahn, L. J. Taite, J. J. Moon, M. C. Rowland, K. A. Ruffino and J. L. West, Biomaterials, 2006, 27, 2519–2524 CrossRef CAS PubMed.
- R. G. Wylie, S. Ahsan, Y. Aizawa, K. L. Maxwell, C. M. Morshead and M. S. Shoichet, Nat. Mater., 2011, 10, 799–806 CrossRef CAS PubMed.
- D. P. J. Cotton, A. Popel, I. M. Graz and S. P. Lacour, J. Appl. Phys., 2011, 109 Search PubMed.
- K. A. Mosiewics, L. Kolb, A. J. van der Vlies, M. M. Martino, P. S. Lienemann, J. A. Hubbell, M. Ehrbar and M. P. Lutolf, Nat. Mater., 2013, 12, 1072–1078 CrossRef PubMed.
- M. A. Azagarsamy, I. A. Morazas, S. Spaans and K. S. Anseth, ACS Macro Lett., 2016, 5, 19–23 CrossRef CAS.
- K. A. Mosiewics, L. Kolb, A. J. van der Vlies and M. P. Lutolf, Biomater. Sci., 2014, 2, 1640–1651 RSC.
- Y. Guan and Y. Zhang, Soft Matter, 2011, 7, 6375–6384 RSC.
- L. A. Lyon, Z. Y. Meng, N. Singh, C. D. Sorrell and A. S. John, Chem. Soc. Rev., 2009, 38, 865–874 RSC.
- G. R. Hendrickson, M. H. Smith, A. B. South and L. A. Lyon, Adv. Funct. Mater., 2010, 20, 1697–1712 CrossRef CAS.
- T. Hoare and R. Pelton, Macromolecules, 2004, 37, 2544–2550 CrossRef CAS.
- X. B. Hu, Z. Tong and L. A. Lyon, Macromol. Rapid Commun., 2011, 32, 1461–1466 CrossRef CAS PubMed.
- I. Berndt and W. Richtering, Macromolecules, 2003, 36, 8780–8785 CrossRef CAS.
- B. P. Tripathi, N. C. Dubey and M. Stamm, ACS Appl. Mater. Interfaces, 2014, 6, 17702–17712 CAS.
- S. M. Lee and Y. C. Bae, Macromolecules, 2014, 47, 8394–8403 CrossRef CAS.
- M. J. Serpe, K. A. Yarmey, C. M. Nolan and L. A. Lyon, Biomacromolecules, 2005, 6, 408–413 CrossRef CAS PubMed.
- C. M. Nolan, M. J. Serpe and L. A. Lyon, Macromol. Symp., 2005, 227, 285–294 CrossRef CAS.
- S. Saxena, M. W. Spears, H. Yoshida, J. C. Gaulding, A. J. Garcia and L. A. Lyon, Soft Matter, 2014, 10, 1356–1364 RSC.
- L. Hu and M. J. Serpe, Polymer, 2012, 4, 134–149 CAS.
- C. D. Sorrell, M. C. D. Carter and M. J. Serpe, Adv. Funct. Mater., 2011, 21, 425–433 CrossRef CAS.
- G. Decher, Science, 1997, 277, 1232–1237 CrossRef CAS.
- M. J. Serpe, C. D. Jones and L. A. Lyon, Langmuir, 2003, 19, 8759–8764 CrossRef CAS.
- J. Peng, D. Zhao, X. Tang, F. Tong, L. Guan, Y. Wang, M. Zhang and T. Cao, Langmuir, 2013, 29, 11809–11814 CrossRef CAS PubMed.
- H. T. Lord, J. F. Quinn, S. D. Angus, M. R. Whittaker, M. H. Stenzel and T. P. Davis, J. Mater. Chem., 2003, 13, 2819–2824 RSC.
- H. Bachman, A. C. Brown, K. C. Clarke, K. S. Dhada, A. Douglas, C. E. Hansen, E. Herman, J. S. Hyatt, P. Kodlekere, Z. Meng, S. Saxena, M. W. Spears, N. Welsch and L. A. Lyon, Soft Matter, 2015, 11, 2018–2028 RSC.
- A. B. South, R. E. Whitmire, A. J. Garcia and L. A. Lyon, ACS Appl. Mater. Interfaces, 2009, 1, 2747–2754 CAS.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS PubMed.
- L. Zhang, M. W. Spears and L. A. Lyon, Langmuir, 2014, 30, 7628–7634 CrossRef CAS PubMed.
- P. E. Hockberger, B. Lom, A. Soekarno, C. H. Thomas and K. E. Healy, Nanofabr. Biosyst., 1996, 276–299 CAS.
- N. Sniadecki, R. Desai, S. Ruiz and C. Chen, Ann. Biomed. Eng., 2006, 34, 59–74 CrossRef PubMed.
- E. Martínez, E. Engel, J. A. Planell and J. Samitier, Ann. Anat., 2009, 191, 126–135 CrossRef PubMed.
- R. G. Flemming, C. J. Murphy, G. A. Abrams, S. L. Goodman and P. F. Nealey, Biomaterials, 1999, 20, 573–588 CrossRef CAS PubMed.
- A. Curtis and C. Wilkinson, Biomaterials, 1997, 18, 1573–1583 CrossRef CAS PubMed.
- W. Jing, Y. Chunxi, W. Yizao, L. Honglin, H. Fang, D. Kerong and H. Yuan, Soft Mater., 2013, 11, 173–180 CrossRef.
- L. Y. Jiang and Y. Luo, Soft Matter, 2013, 9, 1113–1121 RSC.
- M. J. Poellmann, P. A. Harrell, W. P. King and A. J. Wagoner Johnson, Acta Biotechnol., 2010, 6, 3514–3523 CAS.
- S. J. Bryant, J. L. Cuy, K. D. Hauch and B. D. Ratner, Biomaterials, 2007, 28, 2978–2986 CrossRef CAS PubMed.
- A. Kang, J. Park, J. Ju, G. S. Jeong and S. H. Lee, Biomaterials, 2014, 35, 2651–2663 CrossRef CAS PubMed.
- K. J. Park, K. G. Lee, S. Seok, B. G. Choi, M. K. Lee, T. J. Park, J. Y. Park, D. H. Kim and S. J. Lee, Lab Chip, 2014, 14, 1873–1879 RSC.
- A. Kobayashi, K. Yamakoshi, Y. Yajima, R. Utoh, M. Yamada and M. Seki, J. Biosci. Bioeng., 2013, 116, 761–767 CrossRef CAS PubMed.
- J. Olmos Buitrago, R. A. Perez, A. El-Fiqi, R. K. Singh, J. H. Kim and H. W. Kim, Acta Biotechnol., 2015, 28, 183–192 CAS.
- P. Bajaj, D. Marchwiany, C. Duarte and R. Bashir, Adv. Healthcare Mater., 2013, 2, 450–458 CrossRef CAS PubMed.
- M. Akbari, A. Tamayol, V. Laforte, N. Annabi, A. H. Najafabadi, A. Khademhosseini and D. Juncker, Adv. Funct. Mater., 2014, 24, 4060–4067 CrossRef CAS PubMed.
- D. R. Griffin, W. M. Weaver, P. O. Scumpia, D. Di Carlo and T. Segura, Nat. Mater., 2015, 14, 737–744 CrossRef CAS PubMed.
- S. Saxena, C. E. Hansen and L. A. Lyon, Acc. Chem. Res., 2014, 47, 2426–2434 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6me00026f |
| This journal is © The Royal Society of Chemistry 2016 |