
Molecular engineering of cyanine dyes to design a panchromatic response in co-sensitized dye-sensitized solar cells†
Giulio
Pepe
a,
Jacqueline M.
Cole
*ab,
Paul G.
Waddell‡
ac and
Scott
McKechnie
a
aCavendish Laboratory, University of Cambridge, J. J. Thomson Avenue, Cambridge, CB3 0HE, UK. E-mail: jmc61@cam.ac.uk
bArgonne National Laboratory, 9700 S. Cass Avenue, Argonne, IL 60439, USA
cAustralian Nuclear Science and Technology Organisation, Lucas Heights, New South Wales 2234, Australia
First published on 5th April 2016
Abstract
Cyanines are optically tunable dyes with high molar extinction coefficients, suitable for applications in co-sensitized dye-sensitized solar cells (DSCs); yet, barely thus applied. This might be due to the lack of a rational molecular design strategy that efficiently exploits cyanine properties. This study computationally re-designs these dyes, to broaden their optical absorption spectrum and create dye⋯TiO2 binding and co-sensitization functionality. This is achieved via a stepwise molecular engineering approach. Firstly, the structural and optical properties of four parent dyes are experimentally and computationally investigated: 3,3′-diethyloxacarbocyanine iodide, 3,3′-diethylthiacarbocyanine iodide, 3,3′-diethylthiadicarbocyanine iodide and 3,3′-diethylthiatricarbocyanine iodide. Secondly, the molecules are theoretically modified and their energetics are analyzed and compared to the parent dyes. A dye⋯TiO2 anchoring group (carboxylic or cyanoacrylic acid), absent from the parent dyes, is chemically substituted at different molecular positions to investigate changes in optical absorption. We find that cyanoacrylic acid substitution at the para-quinoidal position affects the absorption wavelength of all parent dyes, with an optimal bathochromic shift of ca. 40 nm. The theoretical lengthening of the polymethine chain is also shown to effect dye absorption. Two molecularly engineered dyes are proposed as promising co-sensitizers. Corresponding dye⋯TiO2 adsorption energy calculations corroborate their applicability, demonstrating the potential of cyanine dyes in DSC research.
1 Introduction
Dye-sensitized solar cells (DSCs) are becoming increasingly attractive for harvesting solar energy. The interest in this photovoltaic (PV) technology is driven, in part, by the low costs of production, stability and flexibility of design compared to other PV technologies.1 The capability of controlling their color and transparency gives them niche potential to be engineered in “smart windows”2–4 and they already outperform competitors in indoor applications.1 Yet, since their first application,5 DSCs have not seen a satisfactory rise in power conversion efficiency. However, recent developments have reported DSCs with 13% efficiency, achieved via a zinc-centred porphyrin dye,6 and a record-breaking cell with 14.3% efficiency,7 involving the co-sensitization of two metal-free organic dyes.The most commonly used sensitizers in DSCs are ruthenium-based complexes or dyes containing precious metals. Their strength chemically lies in metal-to-ligand charge transfer (MLCT), which broadens the optical absorption spectrum across most visible wavelengths.1,8,9 However, metal-free organic dyes have a number of advantages over metal-containing dyes, which makes them attractive for use in DSCs. By comparison, they are cheap, widely available and more environmentally sound. They have higher molar extinction coefficients (ε), which allows for greater light absorption at a given wavelength and they have readily tunable absorption wavelengths and high molecular design flexibility.1,10,11 Furthermore, they have found recent advances in device durability by chemically altering the molecular structure12 and have found applications in flexible DSCs.13
Despite this, a major disadvantage of organic dyes is their narrow absorption spectra. This substantially limits the light-harvesting capabilities of an associated DSC device, hampering the short-circuit current density and power conversion efficiency. An effective solution to this problem is to co-sensitize a TiO2 film with multiple dyes possessing different optical absorption spectra, thereby generating a panchromatic dye⋯TiO2 working electrode.14 This co-sensitization technique has found increasing popularity over the past few years, thanks to its promising enhanced light harvesting, and has generated the current world record in DSC efficiency.7 In order to successfully achieve panchromatic optical absorption, the combination of dyes employed in the co-sensitized solar cell must be carefully selected. Problems such as chemical compatibility and competition between dye molecules upon sensitization greatly hinder DSC performance. Molecular engineering of the sensitizers therefore opens an interesting prospect to rationally design a reliable combination of dyes for co-sensitization.
The molecular design of new dyes is restricted by two specific requirements that a sensitizer must meet in order to function properly in a DSC: (i) the lowest-unoccupied molecular-orbital (LUMO) energy of the dye must exceed the conduction-band energy of TiO2 to favor unidirectional charge injection into the TiO2. Likewise, the highest-occupied molecular-orbital (HOMO) energy of the dye must be lower than the electrolyte redox energy (Eredox) to successfully regenerate the dye after charge injection. A dye whose electronic structure does not meet these energetic requirements will fail to generate current in a DSC. (ii) The dye must possess an anchoring group that both stabilizes the dye on the TiO2 surface and allows for charge injection. The primal carboxylic acid, –COOH, and cyanoacrylic acid groups are, by far, the two most popular anchors.1
Other structure–property relationships have also been shown to benefit the function of DSCs. Firstly, dyes with a “push-pull” molecular framework are easier to design for use in DSCs. Here, an electron-donor moiety (D) is separated from an electron-acceptor group (A) via a π-linker, usually consisting of a π-conjugated bridge (D–π–A). The acceptor is typically situated in, or around, the anchoring group to ease charge injection. This D–π–A molecular dye architecture allows for fine tuning of its molar extinction coefficient, maximum optical absorption wavelength, and HOMO/LUMO energy levels, whilst enabling directed intra-molecular charge transfer (ICT). Furthermore, recent studies claim that separating the acceptor group from the anchoring group through adding an extra π-conjugation unit (to form the D–π–A–π-anchor motif) can lead to over 6.5 times higher power conversion efficiencies compared to the simple (D–π–A) structure.15 Secondly, high molar extinction coefficients, ε, permit the use of thinner films of TiO2 in the DSC working electrode, reducing unfavorable electron-recombination processes with the electrolyte.16 Thirdly, a combination of dyes that maximizes light harvesting in both the visible and near-IR region of the optical absorption spectrum is advantageous in order to achieve a panchromatic response, which increases the short-circuit current density in DSCs.17,18
Cyanines are a chemical family of well studied “push-pull” dyes which meet many of the structure–property requirements for DSC dyes. These chromophores have dominated the field of photography over the past century and have recently displayed potential as functional materials for a wide range of applications, such as lasers, optical data storage, photorefractive technology, antitumor agents, probes for biological systems and light harvesting devices.19 Their high molar extinction coefficients and simplicity of optical tuning makes them prime candidates for DSCs, particularly within the realm of co-sensitization. The generic cyanine consists of two nitrogen centers, located within hetero-cycles, one of which is positively charged and linked to the other nitrogen by a polymethine bridge that consists of an odd number of carbon atoms. The length of this π-conjugated chain constitutes the biggest contribution to the optical absorption spectrum of the dye. For example, the extension of the chromophore by one vinylene moiety (–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–) causes a bathochromic shift of approximately 100 nm in the absorption spectrum.19
CH–) causes a bathochromic shift of approximately 100 nm in the absorption spectrum.19
In the context of DSCs, cyanine dyes have not been extensively used as sensitizers or co-sensitizers. The earliest attempt to use cyanines in a DSC involves the co-sensitization of two cyanine dyes with an oxygen and sulfur center, respectively.20 This study realized the importance of using dyes with high molar extinction coefficients, which leads to thinner TiO2 films and reduced recombination. Subsequent studies by Sayama et al. analyzed DSCs co-sensitized with three cyanine dyes, focusing on the importance of co-sensitization for suppressing detrimental dye aggregation.21,22 An investigation of two carboxylated cyanine dyes reported the formation of a tightly-packed monolayer of cyanines, upon co-sensitization, reducing electron recombination with the electrolyte.23 Despite these experimental efforts to produce efficient co-sensitized DSCs with cyanine dyes, the reported power conversion efficiencies were between 2.5% and 3.5%, which is an unsatisfactory result in comparison with other organic dyes. Although these particular cyanine-based DSC trials did not meet the expectations conveyed by the aforementioned intrinsic optical properties that suggest their potential for DSC operation, a more systematic approach may be able to rectify this deficit.
Through a series of molecular adjustments, cyanines can be converted into efficient co-sensitizers for a DSC device. A key advantage of applying molecular engineering strategies to design co-sensitized DSCs lies in the capability to tailor a panchromatic response of the DSC by selecting a pair of (or more) dyes which exhibit complementary optical absorption spectra that, when combined, emulate the solar spectrum of light. For example, a panchromatic response can be achieved using a pair of dyes designed to exhibit optical absorption at opposite ends of the UV/vis spectral bandwidth to each other. Another beneficial feature of molecular engineering is the ability to shift optical absorption towards IR wavelengths. Near-IR is a rarely used portion of the solar spectrum for light harvesting and it makes possible translucent DSC devices with good optical penetration characteristics.24 Furthermore, co-sensitization of several chemical derivatives of the same parent dye tends to reduce the possibility of employing chemically incompatible dyes within the same DSC. To this end, four carbocyanines bearing complementary absorption spectra are herein investigated: 3,3′-diethyloxacarbocyanine iodide (1), 3,3′-diethylthiacarbocyanine iodide (2), 3,3′-diethylthiadicarbocyanine iodide (3) and 3,3′-diethylthiatricarbocyanine iodide (4).
This study involves the re-engineering of four cyanine-based laser dye molecules, 1–4, to generate DSC dyes via (i) adding a DSC-suitable anchoring group at an appropriate substitution point on each molecule to achieve a strong dye⋯TiO2 binding while affording ICT that promotes an advantageous shift in optical absorption energy; (ii) varying the length of the polymethine bridge in order to further tune the absorption energy. Experimental and computational methods are first employed to characterize the structure and optical properties of the parent dyes. In this respect, single-crystal X-ray diffraction on 1–4 is used to obtain their solid-state structure, while solution UV/vis absorption spectroscopy enables the determination of their absorption profiles. The initial data from these experiments serve as the pre-requisite knowledge-base for molecular engineering of cyanine-based DSC dyes. The computational efforts exploit density functional theory (DFT), time-dependent density functional theory (TD-DFT) and Laplace transformed density fitted local CC2 (LT-DF-LCC2) calculations. DFT predicts HOMO and LUMO energy levels of each molecule and the ICT properties. TD-DFT and LT-DF-LCC2 simulate the optical transitions of each dye. The best dye candidates, as judged by molecular structure and energy qualifiers, are taken forward for additional computational studies whereby dyes are adsorbed on a TiO2 surface as represented by a (TiO2)9 cluster. This modeling permits monitoring of plausible changes in the optoelectronic properties of the dye at the dye⋯TiO2 interface and enumeration of the associated adsorption energies. These molecular engineering steps are summarized in Fig. 1 and 2, the result of which is the material prediction of a pair of cyanine-based DSC co-sensitizers.
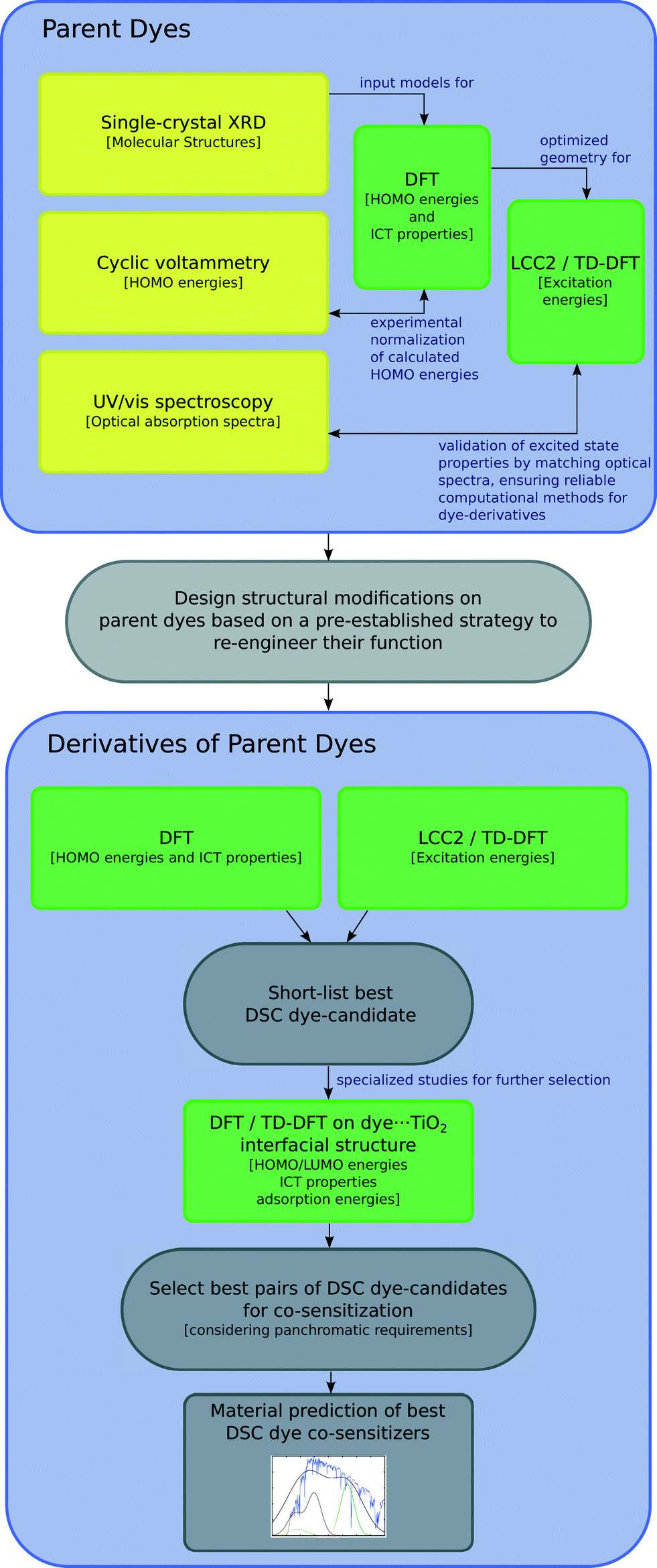 | ||
| Fig. 1 Schematics of the molecular engineering strategy. Experiments are in yellow, while computational methods are in green. | ||
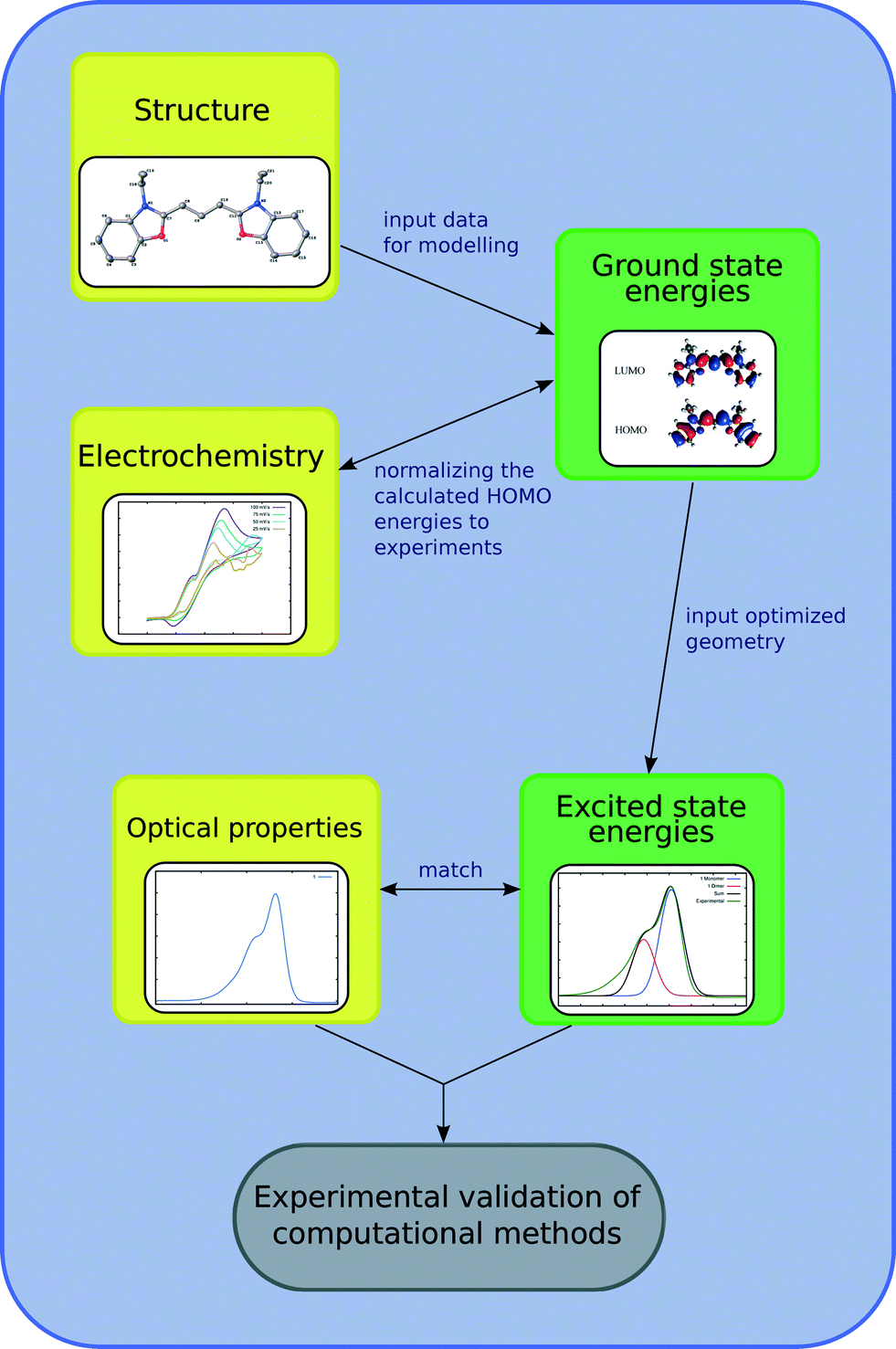 | ||
| Fig. 2 Schematic of the relationships between experimental and computational methods for the optoelectronic studies on the parent dyes, as illustrated for 1. Experiments are in yellow, while computational methods are in green boxes. | ||
2 Experimental and computational methods
2.1 Materials
Commercially available powders of dyes 1–4 were obtained from Sigma-Aldrich. These compounds are ionic, with I− serving as a neutralizing counter-ion for each dye. Skeletal formulae are shown in Fig. 3, showing 3,3′-diethyloxacarbocyanine iodide [C21H21N2O2][I] as 1, 3,3′-diethylthiacarbocyanine iodide [C21H21N2S2][I] as 2, 3,3′-diethylthiadicarbocyanine iodide [C23H23N2S2][I] as 3 and 3,3′-diethylthiatricarbocyanine iodide [C25H25N2S2][I] as 4.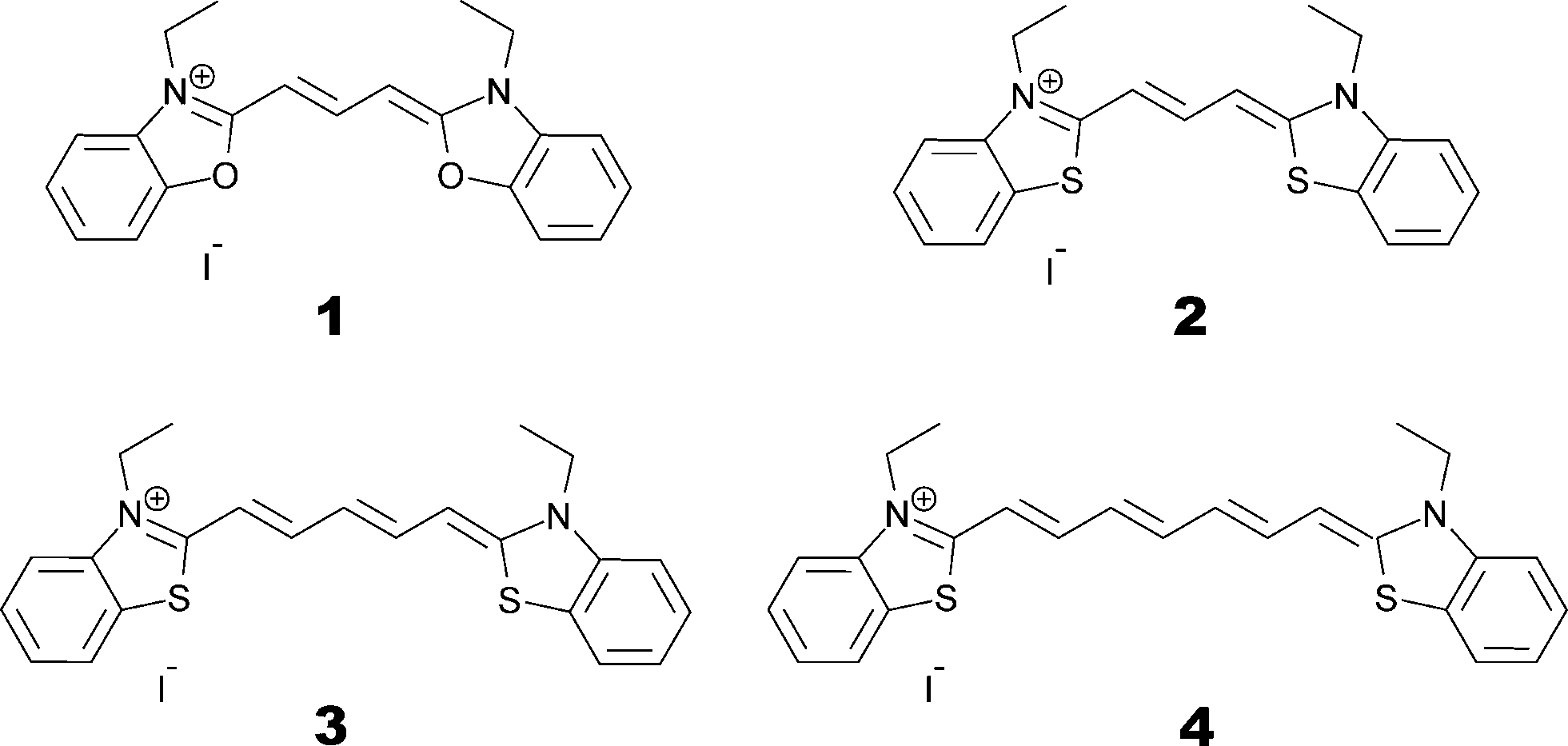 | ||
| Fig. 3 Skeletal chemical formulae of the parent dyes 1–4. | ||
2.2 Single crystal X-ray diffraction
Single crystals of dyes 1–4 were grown by slow evaporation of saturated methanol solutions at room temperature over the course of ten days. The resulting crystals were suitable for single-crystal X-ray diffraction measurements from which their crystal structures were realized. Diffraction data were collected at 120 K for all crystals, using a Rigaku Saturn 724+ CCD diffractometer, equipped with a molybdenum X-ray source (λMoKα = 0.71073 Å), SHINE Optics focusing and an Oxford Cryosystems CryostreamPlus open-flow N2 cooling device. Cell refinement, data collection and integration were undertaken via the Rigaku CrystalClear-SM Expert 2.0 software package,25 and an absorption correction was implemented using ABSCOR.26 Structures were solved by direct methods and refined by full-matrix least-squares methods on F2. All refinements were performed using SHELXS.27 Hydrogen atoms were positioned geometrically and refined as riding on their parent atoms. The crystal structure determination of 2 exhibited solvent accessible channels along the crystallographic [001] direction. The disordered solvent molecules therein could not be modeled rationally; hence, the contents were treated as a diffuse contribution to the overall scattering using SQUEEZE/PLATON.28 While the molecular geometry of 3 was clearly resolved from its best-fit crystal structure model, the refinement of this model against the data produced somewhat poor statistical qualifiers. Accordingly, use of this crystal structure of 3 was restricted to its provision of input molecular geometry for DFT calculations where this geometry was computationally optimized. A full summary of crystal data, crystallographic collection and refinement details are provided in ESI† S.1 and associated deposition material.2.3 UV/vis absorption spectroscopy
UV/vis absorption spectroscopy was performed on an Agilent Cary 300 UV-vis spectrophotometer. Absorption spectra were recorded in methanol (Fig. 4) at a concentration of 0.01 mM for all dyes. Peak molar extinction coefficients, ε, were calculated using the gradient of the regression line obtained from the measurement of peak absorbance over five different dye concentrations. Peak absorbance for each concentration was measured five times in order to spot potential systematic errors and obtain a standard uncertainty on the measurement. Dye aggregation was studied for dye 3, as it presented a number of shoulder peaks which could be attributed to dimer or trimer formation in solution; solution concentration and the solvent identity were modified in order to control the relative changes in peak intensity. The absorption spectrum of 3 was measured in methanol for 15 concentrations, halving it at every step from 5 × 10−4 M to 3 × 10−8 M and was further measured at 1.5 × 10−5 M in methanol, acetone, dimethyl sulfoxide, acetonitrile and dichloromethane (see ESI† S.2). A summary of the salient spectral information for all dyes is reported in Table 1.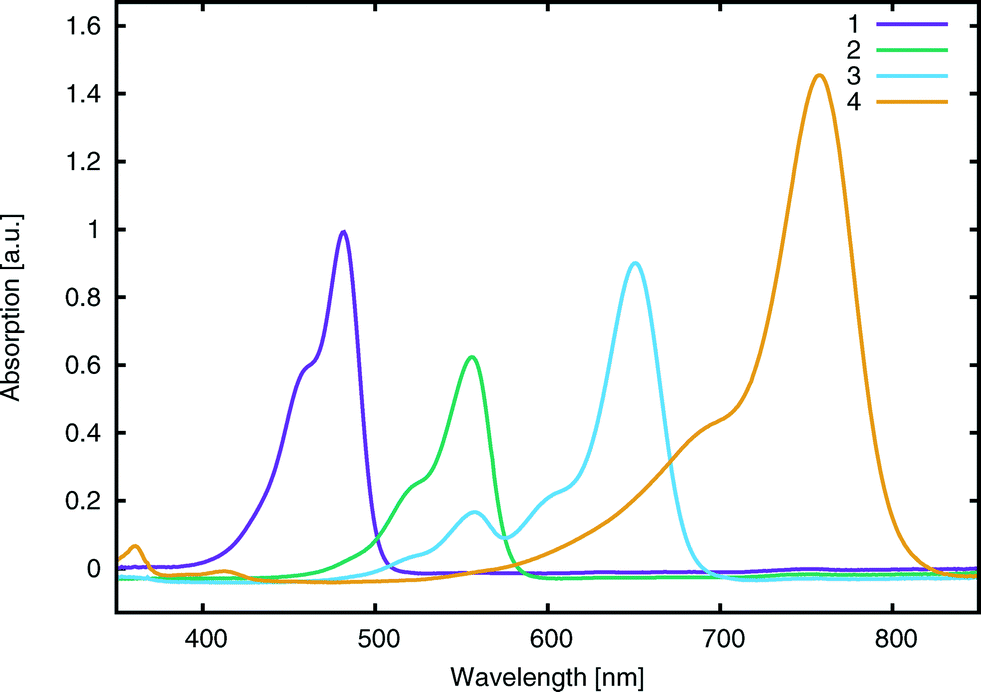 | ||
| Fig. 4 Experimental UV/vis absorption spectra for 1–4. Data were collected in methanol at 0.01 mM concentration. Spectra have been scaled by a common factor such that the absorption peaks are normalized with respect to 1. | ||
| Compound | E exptalV [eV] (λpeakmax [nm]) | E calcV [eV] | ε [×104 L mol−1 cm−2] | Λ [%] | HOMOexptal [eV] | HOMOcalc [eV] | E calcads [eV] |
|---|---|---|---|---|---|---|---|
| 1 | 2.57 ± 0.1 (482 ± 1) | 2.62 | 9.9 ± 0.6 | 69 | −5.30 ± 0.02 | −5.93 | −1.87 |
| 2 | 2.23 ± 0.1 (555 ± 1) | 2.41 | 6.4 ± 0.2 | 70 | −5.45 ± 0.02 | −5.83 | −1.86 |
| 3 | 1.90 ± 0.1 (652 ± 1) | 2.02 | 10.8 ± 0.2 | 71 | −5.43 ± 0.02 | −5.59 | −1.84 |
| 4 | 1.64 ± 0.1 (756 ± 1) | 1.72 | 11.0 ± 1.2 | 70 | −5.36 ± 0.02 | −5.41 | −1.54 |
2.4 Cyclic voltammetry
Cyclic voltammograms for 1–4 were recorded at scan rates of 25, 50, 75, and 100 mV s−1 in acetonitrile (0.5 mM) against an Ag/AgCl in KCl reference electrode, using an Autolab PGSTAT101 with a carbon working electrode and a platinum counter electrode. Prior to each measurement, analyte solutions were deoxygenated via nitrogen bubbling. The Ag/AgCl reference electrode was calibrated using a ferrocene/ferrocenium (Fc/Fc+) redox couple as an external standard. The HOMO energy was calculated from the voltammograms by taking the mean value between the oxidation and reduction peak. Energies were converted such that the vacuum served as reference, by subtracting 4.398 eV from the values where Ag/AgCl served as the reference (see ESI† S.3).2.5 Quantum chemical studies
Electronic structure calculations were performed on parent dyes 1–4 and their computationally engineered derivatives. These studies include DFT and TD-DFT calculations carried out using Gaussian 09,29 and ab initio equation-of-motion coupled-cluster methods (EOM-CC),30 using Molpro,31 which feature the EOM-CC singles and doubles approximation (EOM-CCSD)32 and the local coupled-cluster response method (LT-DF-LCC2).33,34The molecular structures of parent dyes 1–4, obtained from single crystal X-ray diffraction, were used as the input geometries for the calculations, which were geometrically optimized using the hybrid functional of Perdew, Burke, and Ernzerhof (PBE0)35–38 with the 6-31G(p) basis set.39 Solvent effects were treated via the polarizable continuum model (PCM)40,41 in methanol. The iodide counter-ion was excluded from calculations since its presence did not affect the geometry, nor the energies, of the molecule (see ESI† S.4.1 for full details). Vibrational frequencies were calculated for 1–4 and were all found to be positive, indicating that reliable ground-state structures were found.
Single-point DFT energy calculations were performed on the optimized geometries in order to determine HOMO energies and molecular-orbital spatial distributions. These calculations were performed at the PBE0/6-311+G(2d,p) level of theory42 in methanol using the PCM model. Orbitals were processed using cubgen29 and visualized in Avogadro43 at an isovalue of 0.02. Difference-density plots between the squares of the HOMO and LUMO plots were generated at an isovalue of 0.0007.
Excited state calculations were performed on the DFT-optimized geometry using the Laplace-transformed local CC2 coupled cluster approximation with density fitting and a double-zeta basis set (cc-pVDZ), with extended domains to account for the long-range behaviour of D–π–A dyes. This computational method was selected after extensive benchmarking of CC and TD-DFT methods, in order to establish an appropriate level of theory (see ESI† S.4.2 for full benchmarking details). The benchmarks included the local functional B3LYP and functionals with long-range correction: CAM-B3LYP and M06-HF, which overestimated excitation energies, compared to experiments and CC methods. In light of the results of the benchmarks, lowest-vertical excitation energies were calculated using LT-DF-LCC2, including solvent-shift effects.
3 Results and discussion
3.1 Establishing prerequisites to design optimal dyes
The efficient molecular design of dyes for applications in DSCs requires a rational strategy. The first step of this strategy involves an in-depth study of the structure and opto-electronic properties of the parent dyes, 1–4, from which DSC-functional chemical derivatives will be computationally engineered. This study on 1–4 consists of a synergistic combination of theoretical calculations and experiments. The chemical parameters to be investigated are the molecular structure, energy levels and intra-molecular charge transfer pathways of each dye. These chemical attributes can provide an understanding of the molecular origins of the chemical and optical dye characteristics. Identifying the key functional fragments of 1–4 involved in light absorption provides information about molecular modifications that might enhance the electron injection from the dye to the TiO2.44 The low-lying vertical excitation energies, EV, are also investigated in the first part of this molecular engineering study. The information gathered on the parent dyes is then used to design their molecular modifications. The benchmarking of calculations on 1–4 against their associated experimental data ensures that computed values for the parent dyes originated from reliable computational methods. This experimental validation provides confidence for further application of these methods to cyanines, via computation that re-engineers these dyes, in order to create DSC-functional chromophores. Fig. 2 illustrates the results for parent dye, 1, in terms of this synergy between computation and experiment; associated data for 2–4 are available in the ESI,† S.1–3.With the exception of 4, the dye molecules exhibit rotational symmetry with a C2 axis situated in the center of the molecule, perpendicular to the plane of the polymethine chain. The presence of ethyl groups prevents higher point group symmetries. The structure of 4 has mirror symmetry Cs, with the mirror plane lying normal to the polymethine chain. Given the nature of these symmetries, 1–4 have two donor units, which are situated on the nitrogen atoms. The central polymethine chain represents the π-conjugated fragment contributing to the optical absorption.19 Analysis of the bond-length alternation (BLA), defined as the difference between the average carbon–carbon single and double bond lengths,45,46 shows that the atomic separations in the polymethine chain differ by a maximum of 0.005 Å. C–N bond lengths vary between 1.347(4) and 1.359(6) Å, indicating a reduction in double bond character,47i.e. the lack of a definite resonance structure. The same observation holds for the DFT optimized structures. This concept is reinforced by a low bond-length alternation in the polymethine chain, suggesting π-conjugated electron density, which diffuses over the entire length of the chain, making its extent of delocalization close to the “cyanine limit”.48
These structure indicators were backed up by further analysis of bond geometries from the crystal structures using harmonic oscillator stabilization energies (HOSE) to determine the prevalent resonance structure.49,50 HOSE is a measure of the energy needed to transform the idealized bond geometry of a particular resonance structure to the geometry of a real molecule.51 It is defined as
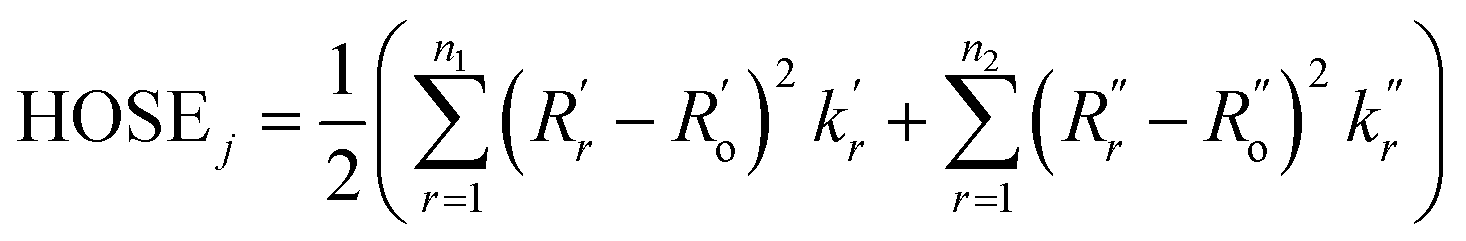 | (1) |
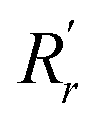 and
and 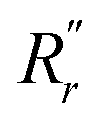 are, respectively, the measured single and double bonds;
are, respectively, the measured single and double bonds; 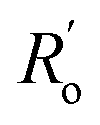 and
and 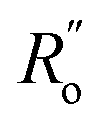 are single and double bonds from reference values. a and b are empirical constants, measured as: a = 44.39 × 104 Pa, b = −26.02 × 104 Pa, while
are single and double bonds from reference values. a and b are empirical constants, measured as: a = 44.39 × 104 Pa, b = −26.02 × 104 Pa, while 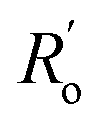 = 1.467 Å and
= 1.467 Å and 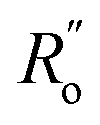 = 1.349 Å. The HOSE model can be used to compute the relative contributions of each canonical resonance structure to that of the real molecule. These contributions, Ci, are calculated according to:
= 1.349 Å. The HOSE model can be used to compute the relative contributions of each canonical resonance structure to that of the real molecule. These contributions, Ci, are calculated according to: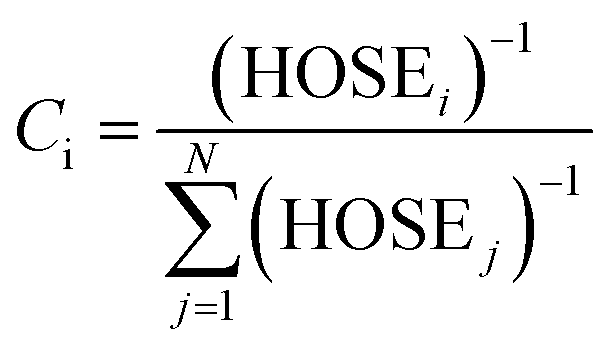 | (2) |
Density functional theory calculations on 1–4 provided complementary insight into the extent of the intra-molecular charge transfer and its possible bond pathways. In particular, visualizing the computed frontier orbitals enabled the regions of the molecule with greater electron density to be identified. HOMO and LUMO orbitals for 1–4 are depicted in Fig. 5. Given that these EV values pertain to HOMO → LUMO transitions, they represent the change in electron density from the ground state to the excited state. Corresponding difference orbital plots, calculated as LUMO2–HOMO2, highlight areas of electron density withdrawal and donation during the optical excitation process. The overall extent of intra-molecular charge transfer was enumerated via the orbital overlap measure (Λ),52 which quantifies the amount of orbital overlap between HOMO and LUMO orbitals across the dye molecule. As is evident from the orbital plots, there is a high degree of spatial overlap between the HOMO and LUMO orbitals, amounting to 69% < Λ < 71%. Values of Λ for 1–4 are reported in Table 1.
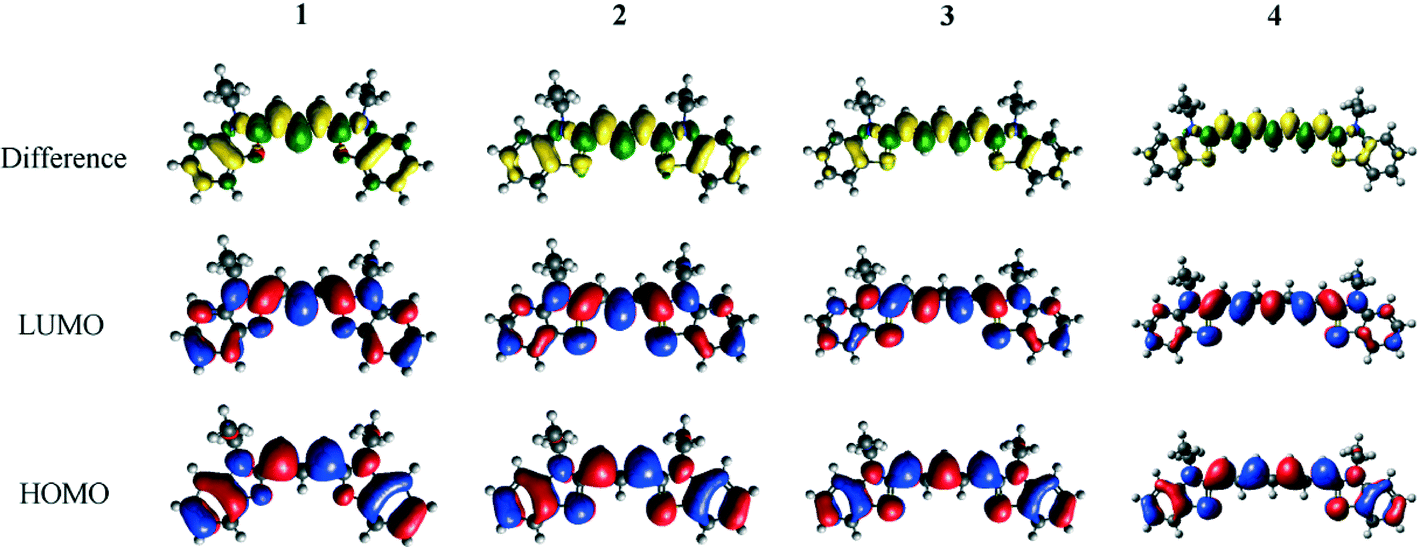 | ||
| Fig. 5 HOMO and LUMO Kohn–Sham orbitals of the parent dyes 1–4, generated from DFT calculations at the PBE0 level of theory with 6-311+G(2d,p) basis set and PCM. Corresponding difference density orbitals are also generated by subtracting the square of the LUMO orbitals from the square of the HOMO orbitals. Increasing electron density is represented in green, while decreasing electron density is represented in yellow. Orbitals have been drawn at an isovalue of 0.02 for HOMO and LUMO orbitals, and at an isovalue of 0.0007 for the difference orbitals. | ||
Molar extinction coefficients, ε, were also measured for all parent dyes (Table 1) which showed no sign of molecular aggregation in methanol, as judged by their correspondence with the Beer–Lambert law, in the context of relating changes in optical absorbance to variation in dye concentration. This is in spite of the fact that cyanines have been long known to form aggregates.19,53
Computational calculations, using LT-DF-LCC2 with solvent correction, complemented these experimental results. Calculated and experimental lowest-excitation energies, EV, differed by 0.12 eV (Table 1). The implicit inclusion of solvent in all computed models was deemed mandatory (see ESI† S.4.2).
3.2 Molecular engineering DSC dyes
The ICT pathways in 1–4 span the entire molecule, excluding the ethyl units since they lack π-conjugation to be able to transfer charge. This makes the ethyl groups an unsuitable place for adding an anchoring group. The difference density molecular orbital plots for 1–4 (Fig. 5) do not obviously suggest a favorable position for the anchoring group. On the one hand, an excess of electron density in the polymethine chain could suggest helpful chemical substitution along the chain; on the other hand, adding a carboxyl group at this point would impede successful dye⋯TiO2 anchoring because of steric hindrance. In contrast, the arene rings are sterically and electronically favorable fragments for an anchoring group. On each ring, there are two meta-, one para- and one ortho-possible substitution positions.
The chemical nature of the anchoring group is similarly important to that of its substitution position. Despite the plethora of research that details the effects of different anchoring groups on DSC performance,54 it has been hard to find the best anchoring group for any given DSC. This difficulty stems from the largely diverse effect of dye⋯TiO2 anchoring on its different molecular environments, where there is a large variety of dye molecules and electrolyte redox couples.54 To date, the primal carboxylic acid group, –COOH, has been the most used and reliable anchoring group. For this reason, it will be used in this study. The computational substitution of a chemically related, but larger, type of anchoring group, a cyanoacrylic acid,54 was also performed. Here, the points of substitution explored were pre-selected, choosing only the points of dye attachment which showed promise when anchor addition involved the –COOH group.
Given the nature of this molecular-design exploration of the anchoring group type and point of substitution in 1–4, a set of chemical derivatives was tailored for optical function using DFT, TD-DFT and LT-DF-LCC2 methods. These molecularly-engineered dyes were named as follows. Parent dyes (X = 1–4): X; carboxylic acid substitutions at the two meta-positions relative to the N donor: Xm and Xn; at the corresponding ortho-position: Xo; at the para-position: Xp; on the ethyl units: Xe; double anchoring at the para-position and on the ethyl moieties (as reported by Ehret et al.55): Xpe; for cyanoacrylic acid at the ortho-position: Xo* and cyanoacrylic acid at the para-position: Xp*. Fig. 6 provides a chemical schematic of these substituted dye molecules.
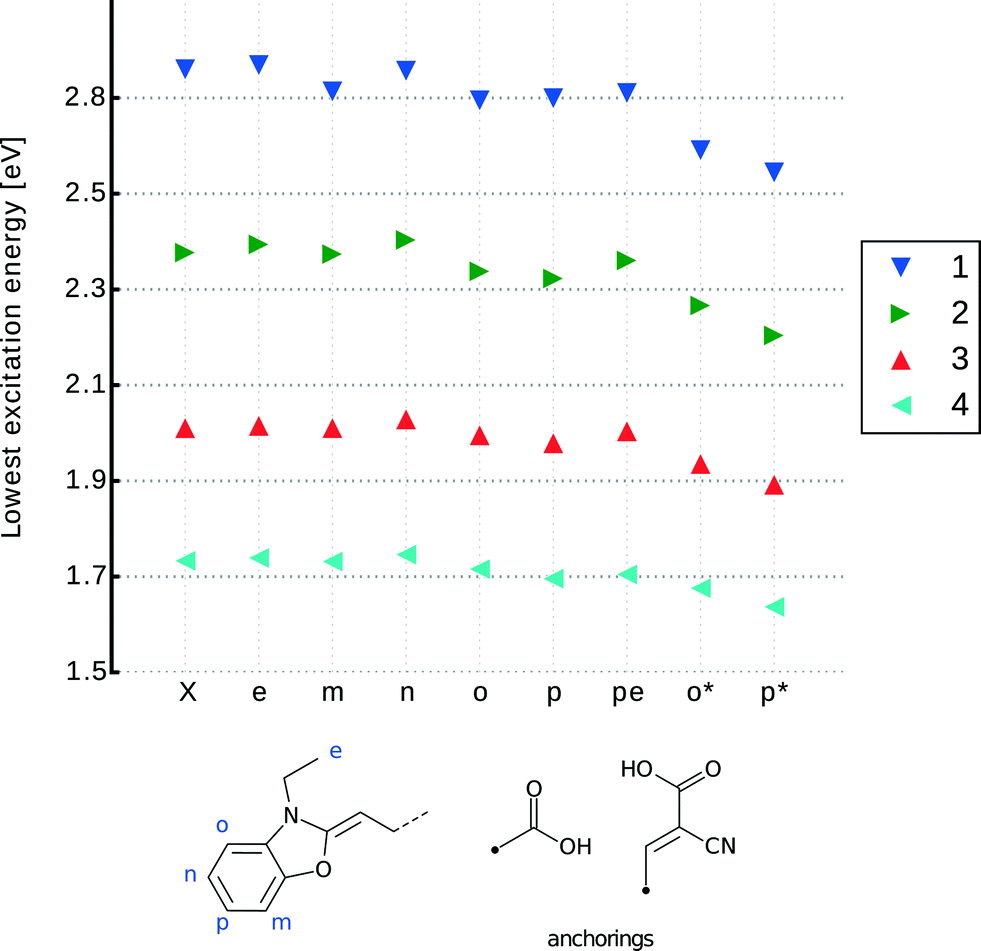 | ||
| Fig. 6 LT-DF-LCC2 calculated lowest-excitation energy for derivatives of 1–4 engineered with an anchoring group at different positions. X refers to the parent dye; e (ethylene), m (meta-1), n (meta-2), o (ortho), p (para) involve addition of the carboxylic acid anchoring group (COOH) at the indicated positions. pe involves the addition of COOH at both p and e positions. o* and p* are respectively the ortho- and para-substitutions of cyanoacrylic acid as the anchoring group. | ||
3.2.2.1 Adding an anchoring group. Visual inspection of the molecular geometry imposed by attaching either type of anchoring group at the ortho-position, Xo and Xo*, as defined in Fig. 6, shows that the process of anchoring the dye on the TiO2 would be prevented by substantial steric hindrance from the ethyl units. Nonetheless, single-point DFT energy calculations indicated that ortho- and para-positions are the most effective substitution points for the primal carboxylic acid attachment, given this substitution afforded increased electron density at this anchoring point for each of the parent dyes. An even stronger trend is observed where cyanoacrylic acid is employed as the anchoring moiety. Although unsuitable for a DSC device, attaching an anchoring group at the ortho-position shows the largest increase in electron density around this point of substitution. This is quantified by analyzing the index for spatial orbital overlap.52 For 1o*, Λ drops to 54% from a value of 69% for the parent dye, 1. By comparison, the corresponding para-substitution option for cyanoacrylic acid, 1p*, affords an orbital spatial overlap, Λ, of 62% and associated –COOH derivatives of 1 oscillate from 66% to 69%. Less variation is observed in parallel calculations featuring the other parent dye frameworks 2–4, with 4 showing the smallest variation. Derivatives 4o* and 4p* have, respectively, orbital overlap indices of 66% and 68% (see ESI† S.4.3). This trend is reflected in the difference orbital plots (see ESI† S.4.3) and replicated in the context of the lowest-excitation energies.
These lowest-excitation energies were analyzed for all derivatives using LT-DF-LCC2 calculations. By analyzing the shifts in peak maximum optical absorption wavelengths, Δλpeakmax, achieved upon anchoring group addition, a bathochromic shift was observed in all cases, except for those where the anchor was added to the ethyl group (Xe). This can be rationalized by noting that the substitutions are, by prior design, targeted on regions with high electron density. The addition of an electron withdrawing unit onto high electron density regions, such as the subject anchoring options, stabilizes each dye, lowering its excitation energy.56 Substituents on the ethyl groups do not lie in the ICT pathway and so cause small changes in excitation energy, which manifest in the hypsochromic direction; this can be explained by their proximity to the donor part of the molecule. For all dyes, the addition of a primal carboxylic acid at the meta-positions, Xm or Xn, or on the ethyl groups, Xe, did not cause a significant energy shift in lowest-excitation energy. Furthermore, the associated derivatives do not allow charge transfer from the donor moiety, so they will not be considered for co-sensitization. The difference in energy between ortho- and para-positions for the –COOH substitution is negligible. Considering the aforementioned negative steric effects of ortho-substitutions, only para-substitutions will be taken forward for co-sensitization.
With para-substitution in mind, it is possible to fine tune the choice of dye and anchoring group by considering the structure and energetic requirements of a DSC device in terms of achieving optimum dye⋯TiO2 adsorption characteristics for enabling electron injection, and realizing panchromatic optical absorption via co-sensitization. Visual inspection of the molecular geometry of 1p* shows that its anchoring groups lie perpendicular to the long π-conjugated axis of the molecule. This structure allows for double-anchoring onto the TiO2 surface. All other in silico generated derivatives of 1–4 display anchoring groups at a larger dihedral angle than 90°; as such, they can only be attached to the TiO2 by one of the two anchors.
The co-sensitization of 1p* with one of the singly-anchored dyes would remove any possibility of dye aggregation that would be detrimental to device functioning due to π⋯π stacking between dye molecules. Consulting the computed optical absorption spectra of each chemical derivative of 1–4, an optimal dye candidate for co-sensitizing with 1p* appears to be 4p. The λpeakmax of 4p is the farthest from the corresponding maximum absorption wavelength of 1p*, providing an absorption spectrum that extends across UV, visible and near-IR wavelengths when 1p* and 4p are combined via co-sensitization. Their computed absorption profiles present an excellent panchromatic match to that of the solar absorption spectrum, as simulated in Fig. 7. Furthermore, the presence of both types of anchoring groups explored for these dyes would perhaps be advantageous, as they can occupy different TiO2 adsorption sites, possibly preventing dye competition upon their sensitization onto TiO2.57 The orbital energy levels of 1p* and 4p, relative to those of the other material components of a DSC device, were also found to be suitable: as shown in Fig. 8, their HOMOs and LUMOs are lower and higher in energy, respectively, than that of the electrolyte or conduction-band edge of TiO2.
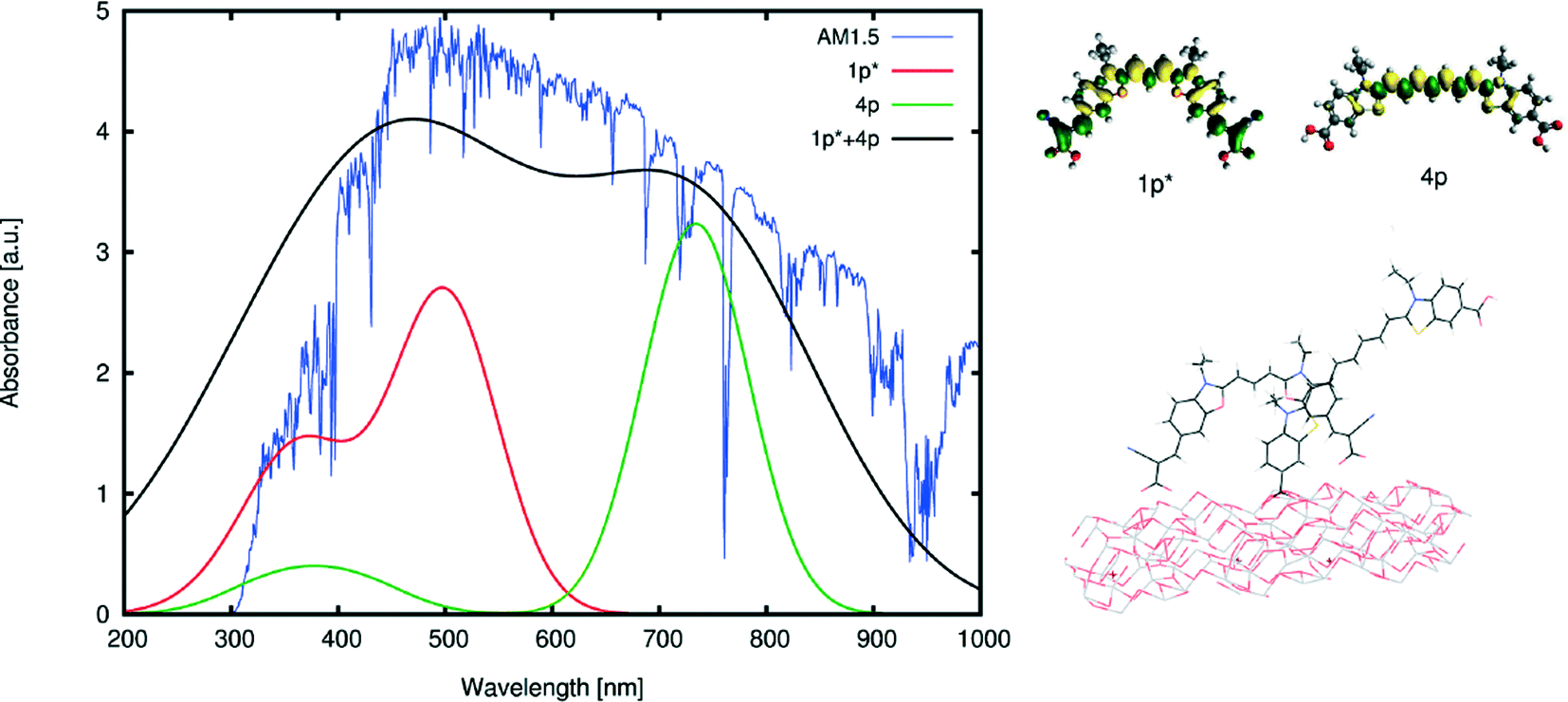 | ||
| Fig. 7 (Left) Calculated absorption spectra of the selected dyes for co-sensitization, 1p*, and 4p, along with their superposition and the solar emission spectrum at AM1.5. The absorption spectra are calculated by convoluting a Gaussian window function to the excitation energies, weighted by the oscillator frequencies. The excitation energies were calculated at the M06-HF level of theory (after a benchmark of different functionals) and shifted in order to match the lowest-excitation energy with the one calculated using LT-DF-LCC2. The superposed absorption spectrum of 1p* and 4p was calculated by convoluting a Gaussian window function with a larger full width at half maximum than the convoluted Gaussian function for single dyes, in order to visualize the broadening of the absorption spectrum for adsorbed dyes. (Right-Top) Difference molecular orbitals of the selected dyes. (Right-Bottom) Simulated adsorption modes for both dyes on a non-optimized slab of TiO2 particles (§ 3.3). | ||
 | ||
| Fig. 8 HOMO and LUMO energies for 1–4 and chemical derivatives where an anchoring group has been added. HOMO energies are all below the electrolyte reduction potential, while LUMO energies are all above the TiO2 conduction band. The identified co-sensitizers pairing, 1p* and 4p, are highlighted in red. Experimental values are represented in light blue, while calculated values are displayed in dark blue. Calculated LUMO energies are found by adding the lowest-vertical excitation energy (calculated using LT-DF-LCC2) to the HOMO energy. Experimental HOMO energies are found using cyclic voltammetry. Experimental LUMO energies are found by adding the lowest-vertical excitation energy (measured using UV/vis absorption spectroscopy) to the experimental HOMO energy. The TiO2 conduction band edge and electrolyte reduction potential are represented by two horizontal lines in red and green colors, respectively. | ||
3.2.2.2 Tuning the polymethine chain length. The length of the polymethine chain is well known to produce changes in peak optical absorption wavelengths, λpeakmax.58 Cyanine dyes are likely to be particularly susceptible to this structure–property relationship, owing to their unusually high levels of electron delocalization within their π-conjugated chains cf. the “cyanine limit”.48 Accordingly, the effect of progressively adding vinylene groups (up to 8 in total) to the π-conjugated chain on the λpeakmax of 1–4 was explored. Given that 1 contains oxygen and two vinylene groups while 2–4 contain sulfur and two, three or four vinylene units, respectively, computationally-generated dyes with polymethine chains containing n-vinylene variations were named as o3, o4, o5, o6, o7, o8 for π-conjugated extensions of 1, and s5, s6, s7, s8 for extensions of 2–4.
This study involved the same model and functional employed in the computation of the anchoring group substitutions. Visual inspection of the resulting molecular structures shows that ICT pathways were not modified by increasing the extent of π-conjugation. Their HOMO and LUMO orbitals have similar shapes for all derivatives and the only difference is found in the extended electron densities on the polymethine chain. A lengthened chain has a larger amount of electron density. This result did not seem to affect the amount of spatial overlap that is shared by the HOMO and LUMO orbitals. Orbital overlap indices for all derivatives were stable at around 70%. An associated HOSE analysis did not reveal a significant bias towards one of the two possible Kekulé resonant structures for any of the derivatives; this suggests that an energy-stabilized state exists irrespective of the polymethine chain length.
A clear relationship was observed, however, between the extent of molecular planarity of the sulfur-containing dyes (2–4 and s5–s8) and the polymethine chain length. Table 2 evidences a subtle, nonetheless consistent, monotonic decrease in the torsion angle between the arene rings in 2–4 and s5–s8 as a function of increasing polymethine chain length. This suggests an increase in the extent of molecular planarity for longer polymethine chains. The rate of change was best fitted by a cubic polynomial. Such a rational relationship was not replicated in the oxygen-based dyes, 1 and o3–o8. Nonetheless, the corresponding EV values show a consistent reduction with increasing π-conjugated chain length in O- and S-containing dyes. In all cases, HOMO and LUMO energies lie within the energy bounds associated with those of the other salient DSC device components (see Fig. 9). Thus, they are eminently adaptable to DSC applications. The trend towards lower EV values with increasing polymethine chain length means that a more panchromatic range of optical absorption energies is realized for dyes with longer π-conjugated units, while a corresponding energy shift in EV towards the NIR region of the absorption spectrum is also achieved.
| Vinylene units | E V (O) [eV] | E V (S) [eV] | Torsion angle (S) [°] |
|---|---|---|---|
| 2 | 2.62 | 2.41 | 4.42 |
| 3 | 2.31 | 2.02 | 3.47 |
| 4 | 1.94 | 1.72 | 2.77 |
| 5 | 1.67 | 1.48 | 2.40 |
| 6 | 1.44 | 1.28 | 2.22 |
| 7 | 1.27 | 1.13 | 2.13 |
| 8 | 1.11 | 1.00 | 2.10 |
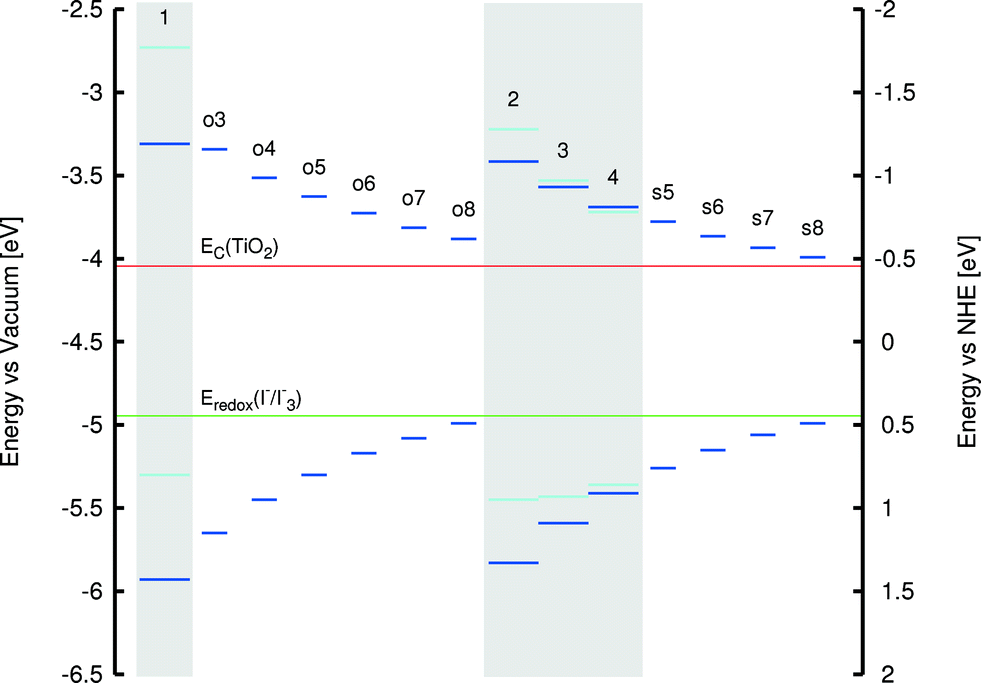 | ||
| Fig. 9 HOMO and LUMO energies for 1–4 and chemical derivatives, where the length of the polymethine chain has been modified. HOMO energies are all below the electrolyte reduction potential, while LUMO energies are all above the TiO2 conduction band. Experimental values are represented in light blue, while calculated values are displayed in dark blue. Calculated LUMO energies are found by adding the lowest-vertical excitation energy (calculated using LT-DF-LCC2) to the HOMO energy. Experimental HOMO energies are found using cyclic voltammetry. Experimental LUMO energies are found by adding the lowest-vertical excitation energy (measured using UV/vis absorption spectroscopy) to the experimental HOMO energy. The TiO2 conduction band edge and electrolyte reduction potential are represented by two horizontal lines in red and green colors, respectively. | ||
That said, a trade-off is observed between dyes with longer π-conjugated polymethine units and progressively NIR absorption wavelengths. As shown in Fig. 9, the LUMO and HOMO levels tend increasingly towards the TiO2 conduction band energy level and the electrolyte redox potential energy levels, respectively, as the polymethine chain lengthens. Detrimental reduction in driving forces for electron injection and dye regeneration may result. Therefore, since a sufficient panchromatic range was afforded by the 1p* and 4p co-sensitizers identified in the previous section, the polymethine chain length was not extended to avoid this compromising trade-off. Nonetheless, the generic findings of this trade-off effect are helpful to observe in the context of further molecular design studies of cyanine-based DSC dyes.
3.3 Modeling the dye⋯TiO2 interface
While computational modeling of isolated dyes in a gas or solution medium provides a rapid and accurate description of their opto-electronic properties, the dynamics of the dye at the dye⋯TiO2 interface may differ. Indeed, examples exist where the process of anchoring the dye onto the TiO2 changes the geometry and the energies of the molecular chromophore.54,59,60 Furthermore, the anchoring process may be energetically unfavorable for certain dye⋯TiO2 combinations, making dye adsorption difficult to achieve. More advanced computational methods that model the dye⋯TiO2 interface, via various binding modes, were therefore applied to a small selection of the more promising computationally designed dyes, 1p, 2p, 3p and 4p, as judged by the aforementioned gas and solution-phase dye studies (§ 3.1 and § 3.2).These advanced methods employed DFT and TD-DFT using a (TiO2)9 cluster predetermined by de Armas et al.61 (see ESI† S.4.4). Its binding to the dye was modeled by considering the three most common possible types of anchoring modes for a carboxylic acid: monodentate, bidentate chelating and bidentate bridging. Dyes bearing the primal carboxylic acid anchoring group were selected for the initial stage of this advanced modeling rather than the cyanoacrylic acid because its smaller molecular fragment size eases computational cost. Previous studies have shown that (TiO2)9 clusters are of a sufficient size that they present a good compromise between providing an accurate representation of the TiO2 surface and computational cost.62–64 The geometry of each dye⋯TiO2 interface was optimized using DFT at the B3LYP level of theory and 6-31G* basis set in ethanol (PCM). Single-point energy calculations and excitation energies were calculated using DFT and TD-DFT, respectively, at the B3LYP level of theory and 6-311+G* basis set in ethanol (PCM).
The exploration of the three possible anchoring modes on 1p revealed that the HOMO, EV and atomic orbital energies were invariant with anchoring mode. Differences between these modes were evident, however, in the associated adsorption energies which were calculated according to the formula:
| Eads = Edye + ETiO2 − Edye⋯TiO2 | (3) |
The dye⋯TiO2 models that employ the bidentate chelating mode of 1p, 2p, 3p and 4p onto TiO2 were then computed and compared with those of their respective 1p, 2p, 3p and 4p dyes in solution. HOMO energies were 0.1–0.2 eV higher for the dyes in solution, while the vertical excitation energies were equivalent. The HOMO → LUMO transition, involved in EV, shows some increased electron density on the anchoring group, similar to the behavior in solution. A study of non-frontier energetic transitions shows that there are changes between the dye at the dye⋯TiO2 interface and when in solution. These were assigned to higher electronic transitions such as HOMO → LUMO+1, HOMO → LUMO+2 and HOMO → LUMO+3. Visual inspection of the orbital plots shows that these higher-order transitions involve both orbitals of the dye and the TiO2, as depicted in Fig. 10. This plot also shows that the electron density progressively shifts from the dye to the semiconductor as a function of increasing optical absorption energy, as denoted by the changing molecular orbital population trends from HOMO to LUMO+3. This suggests that the electron injection required to initiate the electrical current in a DSC is eminently viable. Whilst all dyes show similar trends in the atomic orbitals, the adsorption energies varied between dyes by up to 0.3 eV (Table 1). Based on these considerations, the most suitable para-substituted COOH– bearing dye for anchoring proved to be 1p, which proceeds via bidentate chelating bearing an adsorption energy of −1.87 eV.
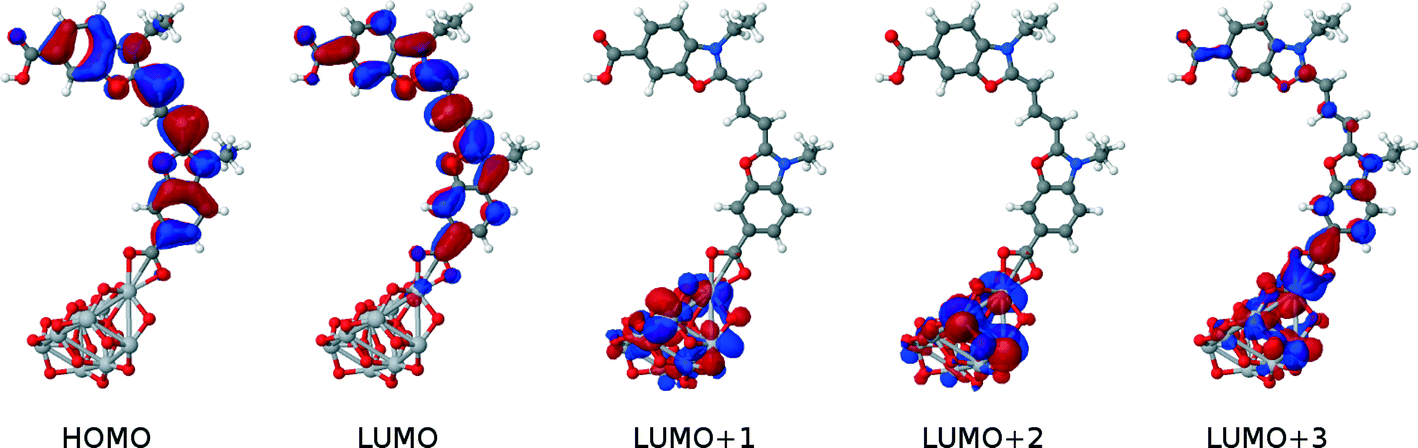 | ||
| Fig. 10 Molecular orbitals of 1p attached to a slab of (TiO2)9, involved in electronic transitions upon photoexcitation. Excited states show a deposition of charge on the TiO2 cluster, which hints at a successful electron injection. Plots for 1p*, 2p, 3p, 4p are essentially equivalent to 1p. | ||
The dye⋯TiO2 model for the dye derivatives earlier selected for co-sensitization, 4p and 1p*, was then computed employing the bidentate chelating mode. Lowest-vertical excitation energies and HOMO energies were equivalent to the isolated dye in solution medium and the molecular orbitals involved in the transition were HOMO → LUMO, as for 1p–3p. Orbital plots showed a similar trend as the one depicted in Fig. 10, indicating the viability of electron injection for these dyes. Adsorption energies for 4p and 1p* were −1.54 eV and −1.71 eV, respectively, indicating a favorable and stable adsorption of the dyes onto the TiO2. This computational study of the dye⋯TiO2 interface for 4p and 1p* therefore corroborated the suitability of these dyes for application in a co-sensitized DSC.
4 Conclusions
Four cyanine dyes 1–4 have been studied experimentally and computationally, and subsequently re-engineered using calculations that are designed to functionalize their structure and enhance their optical response for DSC applications. The study adopted a synergistic combination of experimental and computational methods to first reveal the structure and opto-electronic properties of the parent dyes. A molecular engineering strategy was then rationally designed and performed on these parent cyanine dyes in order to make them suitable for use in DSCs and broaden their absorption spectrum for co-sensitization. Two different types of anchoring groups, the primal carboxylic acid and cyanoacrylic acid, were strategically tested at different substitution positions on 1–4. The effect of lengthening the central polymethine chain was also explored and the dye⋯TiO2 interface was analyzed via further in silico calculations.The central polymethine chain was extended up to 8 vinylene units. All modified dyes showed successful features for applications in DSCs and absorption spectra reaching wavelengths beyond the NIR region. The best reported chemical substitution points for both anchoring types were found to be at the para-position. Dye⋯TiO2 interface studies on four derivatives with –COOH anchoring groups at the para-position showed successful adsorption and charge injection onto a modeled (TiO2)9 cluster. Two of the molecularly-engineered dyes, 1p* and 4p, were selected for co-sensitization on the basis of their suitability in a real DSC, mitigation of unfavorable aggregation and complementarity of the absorption spectrum; their simulated absorption spectra spanned UV, visible and NIR regions.
We have shown that molecular engineering provides a cheap and useful rational tool in screening refunctionalized dye candidates and fine tuning their properties, through molecular modifications, to achieve the best performing dye co-sensitized solar cell for a given class of dyes. This is one of few examples of “bottom up” rational dye design,66 which complements the “top down” approach associated with large-scale data mining for materials discovery of new classes of dyes.67 Such strategies allow one to move away from previous efforts where new dyes have been found via trial-and-error, which is very costly and time consuming. This systematic design concept is even more important when considering the recent interest in co-sensitization techniques. Co-sensitization introduces additional molecular design factors that leaves the possibility of finding a successful combination of dyes through blind search and experiments to pure chance. Complications such as chemical compatibility, dye competition and complementarity of the absorption spectrum arise in these types of DSCs. This study shows that these problems can be efficiently and cheaply tackled through systematic design and computational screening, which will become essential for future development of efficient co-sensitized DSCs.
Acknowledgements
G. P. thanks the EPSRC for a DTA Studentship (Reference: EP/K503009/1). J. M. C. is grateful to the 1851 Royal Commission for the 2014 Design Fellowship, and Argonne National Laboratory where work done was supported by DOE Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. The Bragg Institute at ANSTO is gratefully acknowledged for funding (for P. G. W.). S. M. is grateful to King's College, University of Cambridge, UK, and the EPSRC (Grant No. EP/P505445/1) for Ph.D. funding. The authors thank the EPSRC UK National Service for Computational Chemistry Software (NSCCS) and acknowledge contributions from its staff in supporting this work.Notes and references
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef CAS PubMed.
- Z. Xie, et al. , Chem. Commun., 2014, 50, 608 RSC.
- C. Bechinger, S. Ferrere, A. Zaban, J. Sprague and B. A. Gregg, Nature, 1996, 383, 608 CrossRef CAS.
- K.-S. Ahn, S. J. Yoo, M.-S. Kang, J.-W. Lee and Y.-E. Sung, J. Power Sources, 2007, 168, 533 CrossRef CAS.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737 CrossRef.
- A. Yella, et al. , Science, 2011, 334, 629 CrossRef CAS PubMed.
- K. Kakiage, et al. , Chem. Commun., 2015, 51, 15894 RSC.
- M. Kimura, H. Nomoto, N. Masaki and S. Mori, Angew. Chem., 2012, 124, 4447 CrossRef.
- G. Boschloo and A. Hagfeldt, Acc. Chem. Res., 2009, 42, 1819 CrossRef CAS PubMed.
- G. D. Sharma, S. P. Singh, R. Kurchania and R. J. Ball, RSC Adv., 2013, 3, 6036 RSC.
- T. Bessho, S. M. Zakeeruddin, C.-Y. Yeh, E. W.-G. Diau and M. Grätzel, Angew. Chem., 2010, 122, 6796 CrossRef.
- S. Maniam, et al. , Org. Lett., 2015, 17, 4022 CrossRef CAS PubMed.
- D. Pugliese, et al. , Org. Electron., 2014, 12, 3715 CrossRef.
- G. D. Sharma, M. S. Roy and S. P. Singh, J. Mater. Chem., 2012, 22, 18788 RSC.
- B.-G. Kim, K. Chung and J. Kim, Chem. - Eur. J., 2013, 19, 5220 CrossRef CAS PubMed.
- B. E. Hardin, H. J. Snaith and M. D. McGehee, Nat. Photonics, 2012, 6, 162 CrossRef CAS.
- W. Wu, F. Meng, J. Li, X. Teng and J. Hua, Synth. Met., 2009, 159, 1028 CrossRef CAS.
- A. Burke, L. Schmidt-Mende, S. Ito and M. Graetzel, Chem. Commun., 2007, 234 RSC.
- A. Mishra, R. K. Behera, P. K. Behera, B. K. Mishra and G. B. Behera, Chem. Rev., 2000, 100, 1973 CrossRef CAS PubMed.
- A. Ehret, L. Stuhl and M. T. Spitler, J. Phys. Chem. B, 2001, 105, 9960–9965 CrossRef CAS.
- K. Sayama, et al. , Sol. Energy Mater. Sol. Cells, 2003, 80, 47–71 CrossRef CAS.
- M. Guo, P. Diao, Y. Ren, F. Meng, H. Tian and S. Cai, Sol. Energy Mater. Sol. Cells, 2005, 88, 23–35 CrossRef CAS.
- W. Wu, F. Meng, J. Li, X. Teng and J. Hua, Synth. Met., 2009, 159, 1028–1033 CrossRef CAS.
- N. Robertson, Angew. Chem., Int. Ed., 2006, 45, 2338 CrossRef CAS PubMed.
- CrystalClear-SM Expert, 2.0 Software Package, 2009 Search PubMed.
- A. Messerschmidt, M. Schneider and R. Huber, J. Appl. Crystallogr., 1990, 23, 436 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2007, 64, 112 CrossRef PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian∼09 Revision D.01, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- J. F. Stanton and R. J. Bartlett, J. Chem. Phys., 1993, 98, 7029 CrossRef CAS.
- H.-J. Werner, et al., MOLPRO, version 2012.1, a package of ab initio programs, 2012 Search PubMed.
- J. F. Stanton and J. Gauss, J. Chem. Phys., 1994, 100, 4695 CrossRef CAS.
- D. Kats and M. Schutz, J. Chem. Phys., 2009, 131, 124117 CrossRef PubMed.
- D. Kats, T. Korona and M. Schütz, J. Chem. Phys., 2006, 125, 104106 CrossRef PubMed.
- B. Hammer, L. Hansen and J. Norskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982 CrossRef CAS.
- C. Adamo and C. Barone, Chem. Phys. Lett., 1998, 298, 113 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213 CrossRef CAS.
- D. M. Chipman, J. Chem. Phys., 2000, 112, 5558 CrossRef CAS.
- S. Miertus, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS.
- M. D. Hanwell, et al. , J. Cheminf., 2012, 4, 17 CAS.
- F. A. Y. N. Schröder, J. M. Cole, P. G. Waddell and S. McKechnie, Adv. Energy Mater., 2015, 5, 1401728 Search PubMed.
- J. L. Brédas, J. Chem. Phys., 1985, 82, 3808–3811 CrossRef.
- C. B. Gorman and S. R. Marder, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 297 CrossRef.
- F. H. Allen, et al. , J. Chem. Soc., Perkin Trans. 2, 1987, 2, S1 RSC.
- F. Meyers, S. R. Marder, B. M. Pierce and J. L. Bredas, J. Am. Chem. Soc., 1994, 116, 10703 CrossRef CAS.
- T. M. Krygowski, R. Anulewicz and J. Kruszewski, Acta Crystallogr., Sect. B: Struct. Sci., 1983, 39, 732 CrossRef.
- X. Liu, Z. Xu and J. M. Cole, J. Phys. Chem. C, 2013, 117, 16584 CAS.
- J. Karolak-Wojciechowska, Acta Crystallogr., Sect. B: Struct. Sci., 1987, 43, 574 CrossRef.
- M. J. G. Peach, P. Benfield, T. Helgaker and D. J. Tozer, J. Chem. Phys., 2008, 128, 044118 CrossRef PubMed.
- W. West and S. Pearce, J. Phys. Chem., 1965, 69, 1894 CrossRef CAS.
- L. Zhang and J. M. Cole, ACS Appl. Mater. Interfaces, 2015, 7, 3427 CAS.
- A. Ehret, L. Stuhl and M. T. Spitler, J. Phys. Chem. B, 2001, 105, 9960 CrossRef CAS.
- K. Higashiguchi, et al. , Eur. J. Org. Chem., 2005, 25, 91 CrossRef.
- P. Hirva and M. Haukka, Langmuir, 2010, 26, 17075 CrossRef CAS PubMed.
- T. A. Merz, P. G. Waddell and J. M. Cole, J. Phys. Chem. C, 2013, 117, 8429 CAS.
- J. McCree-Grey, J. M. Cole and P. J. Evans, ACS Appl. Mater. Interfaces, 2015, 7, 16404 CAS.
- J. M. Cole, K. S. Low and Y. Gong, ACS Appl. Mater. Interfaces, 2015, 7, 27646 CAS.
- R. S. de Armas, M. A. S. Miguel, J. Oviedo and J. F. Sanz, Phys. Chem. Chem. Phys., 2012, 14, 225 RSC.
- R. S. de Armas, et al. , J. Chem. Theory Comput., 2010, 6, 2856 CrossRef PubMed.
- R. S. de Armas, M. A. San-Miguel, J. Oviedo, A. Marquez and J. F. Sanz, Phys. Chem. Chem. Phys., 2011, 13, 156 Search PubMed.
- R. S. de Armas, J. Oviedo, M. A. S. Miguel and J. F. Sanz, J. Phys. Chem. C, 2011, 115, 11293 Search PubMed.
- L. Zhang, J. M. Cole and C. Dai, ACS Appl. Mater. Interfaces, 2014, 6, 7535 CAS.
- S. L. Bayliss, J. M. Cole, P. G. Waddell, S. McKechnie and X. Liu, J. Phys. Chem. C, 2014, 118, 14082 CAS.
- J. M. Cole, et al. , Phys. Chem. Chem. Phys., 2014, 16, 26684 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6me00014b |
| ‡ Current address: School of Chemistry, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. |
| This journal is © The Royal Society of Chemistry 2016 |