
Charge generation in organic photovoltaics: a review of theory and computation
Kenley M.
Pelzer
a and
Seth B.
Darling
*ab
aCenter for Nanoscale Materials, Argonne National Laboratory, 9700 Cass Ave., Argonne, IL 60439, USA. E-mail: darling@anl.gov
bInstitute for Molecular Engineering, University of Chicago, 5640 S. Ellis Ave., Chicago, IL 60637, USA
First published on 29th February 2016
Abstract
Due to their amenability to highly scalable fabrication and steadily improving efficiencies, organic photovoltaics (OPVs) offer great potential as an alternative to carbon-based fuel sources. With recently reported power conversion efficiencies of 11–12%, OPVs are on the brink of economic viability. However, to push these technologies forward into widespread use, further optimizations of efficiency are needed. The process of exciton dissociation and charge separation at donor/acceptor interfaces is a major factor influencing the power conversion efficiency of these devices, with loss of useful energy occurring if the charges fail to separate. However, this process of exciton dissociation and separation at OPV heterojunctions is not fully understood, and experimental efforts to optimize these processes via trial and error are costly. Thus, theoretical modeling plays a key role in understanding and improving the rate of charge generation in OPVs. Here we review key theoretical approaches to modeling the process of exciton dissociation and charge separation and their contributions to the challenge of optimizing OPV technology.
 Kenley M. Pelzer | Kenley Pelzer completed her Ph.D. in Chemistry at the University of Chicago in 2014. She is currently an Aneesur Rahman Named Fellow at the Center for Nanoscale Materials at Argonne National Laboratory, where her research focuses on theoretical modeling of charge transport in organic photovoltaics. Other research interests include exciton transport in photosynthetic systems, combustion chemistry, and energy transfer in nanoscale devices. |
 Seth B. Darling | Seth B. Darling is a Scientist at Argonne National Laboratory and a Fellow at the Institute for Molecular Engineering at the University of Chicago. His group's research is motivated by humankind's grand challenges and centers around molecular engineering with a particular emphasis on solar energy and water treatment. Dr. Darling has published over 100 papers and a popular book on climate change, holds several patents, and lectures widely on topics related to energy, climate, and water. |
Introduction
In the quest for carbon-neutral, renewable energy sources, photovoltaics have been the focus of a vast amount of research. Because of their composition from abundant materials and the availability of large-scale, low-energy fabrication methods,1 organic photovoltaics (OPVs) have attracted much attention. This scalable manufacturing combined with steadily improving efficiencies provide hope that they may serve as an economically viable renewable energy technology.1–15 The efficiency of OPVs has been steadily increasing, with reported values of power conversion efficiency recently reaching 11–12%.16–18A crucial step in the photovoltaic process is the conversion of excitons (bound electron/hole pairs) into free charges. The process of charge generation begins when a photon is absorbed by an electron-donating molecule and an electron is excited to form an exciton. If generation of free charges is to occur, first the exciton must diffuse to a donor/acceptor heterojunction. If the exciton reaches the heterojunction, it is possible (but not certain) that an electron will transfer to the acceptor molecule creating a “charge transfer state.” After creation of a charge transfer state, it is possible, but again uncertain, that the separation of this electron/hole pair will create the free charges that are collected by the electrodes. These free charges are described as the “charge separation state,” and the charges are then free to travel via donor-rich or acceptor-rich domains to be collected at the electrodes. It is generally believed that these free charges deform the position of the surrounding nuclei to form “polarons”, which can be thought of as pseudoparticles comprised of a charge and phonons. We refer readers to ref. 19–23 for more detailed discussion of exciton diffusion in OPVs and ref. 24–28 for more discussion of charge transport.
We pause here to present the architecture of a typical bulk heterojunction OPV to provide a clearer context for the charge generation process that we discuss (noting that the development of new architectures is a focus of constant research). Such cells are generally comprised of five layers. In the traditional stacking, the first is a transparent anode through which light enters. The second is referred to as an “interfacial layer” or “hole-transporting layer (HTL)”, comprised of some hole-transporting material that prevents unwanted negative charge carriers from reaching the anode. This is followed by the active layer comprised of a combination of electron-donating and electron-accepting materials, in which the steps of light absorption, charge transfer, charge separation, and charge transport occur. This is followed by a second interfacial layer (“electron-transporting layer (ETL)”) and a cathode. Most OPV research focuses on optimization of the active layer to maximize the number of charge carriers reaching the electrode. Determining the active layer morphology of an OPV is an endeavor in itself, but generally speaking the active layer is comprised of donor-rich regions through which positive polarons travel and acceptor-rich regions through which negative polarons travel.29,30 The structure of an example bulk heterojunction OPV, comprised of commonly used materials, is schematically depicted in Fig. 1.
 | ||
| Fig. 1 Schematic of the layers in a traditional (non-inverted) organic photovoltaic device with typical materials provided in parentheses. | ||
The efficiency of charge transfer and charge separation plays a key role in determining the overall efficiency of the solar cell.31–38 However, our understanding of these processes is quite limited; there is no straightforward way to predict the efficiency of transfer and separation for potential OPV technologies (or even to fully describe these processes in existing solar cells). Experimentally assessing rates of charge transfer and separation is far from trivial, thus making theoretical modeling of transfer and separation a crucial tool in designing efficient photovoltaic materials. In this review, we describe a range of theoretical approaches that both qualitatively and quantitatively elucidate this process in the donor/acceptor molecules of OPVs.
Some loss of energy occurs even before charge transfer can occur: after excitation, the exciton must diffuse from the site of excitation to the heterojunction before geminate electron/hole recombination occurs. A diffusion length of ∼10 nm and an exciton lifetime between 100 ps and 1 ns is typical for OPVs, although some progress has been made in developing materials with a larger exciton diffusion lengths.39–45 After charge separation, major losses can occur due to non-geminate (or bimolecular) recombination, when oppositely charged carriers that were generated from different excitons encounter one another at heterojunctions and recombine. Here we will concern ourselves with the possible energy loss—or possible success in charge generation—when an exciton has reached the heterojunction and must achieve charge separation before geminate recombination can occur. Energy loss via geminate recombination may occur if the exciton recombines before transferring an electron to an acceptor molecule, or loss may occur after a charge transfer state has been achieved. In this state, if the strong Coulombic interaction between the electron and hole significantly exceeds kBT, the excitation may lead to electron–hole pairs that are bound to one another at the heterojunction and do not achieve charge separation.39 At room temperature, exciton binding energies (estimated at 0.1–0.5 eV in OPVs,39 with values as low as 0.05 eV and as high as ∼1–2 eV calculated for other organic systems46–50) far exceed kBT ∼ 0.026 eV.39,51 Thus, the competition between charge separation and geminate recombination for charge transfer states is a major focus of the literature that we review here. We note that there is some variation in nomenclature for these Coulombically bound pairs, with OPV literature using labels including charge transfer (CT) states,39,52–61 geminate pairs,51,53,56,62–64 exciplexes,65–67 or bound polarons.68 In this review we will use the term “CT state,” to be distinguished from the charge separation (CS) state where the charges are fully separated and free from one another's Coulombic attraction. We will also use “charge transfer” to refer specifically to transfer of a charge over a heterojunction (although in other OPV literature this term is sometimes used more broadly to refer to any movement of an electron or hole). We note that this use of “charge transfer” is synonymous with “exciton dissociation” (assuming that the dissociation places the electron and hole on the acceptor/donor molecules), and these terms are usually used interchangeably.
Before proceeding we must acknowledge some complexities and disagreements in the study of charge generation. First, although it is usually assumed that donor molecules are excited by photons, excitation of acceptor molecules is possible and perhaps significant for some systems.39,69 Some work also suggests that there may be significant generation of free carriers within large fullerene domains or in single-phase organic systems.53,54 However, for simplicity, this review focuses on the generally assumed case of exciton creation in donor domains and dissociation at donor/acceptor heterojunctions. Second, we note that there is controversy about the significance of the CT state: while much work on charge photogeneration in OPVs describes the CT state as a necessary precursor to the CS state, other work argues that efficient photovoltaics bypass a state in which the electron and hole are bound at the interface (with these CT states serving mostly as a loss mechanism rather than as a precursor to separated charges).34,70–72 For modeling of charge photogeneration this is essentially an issue of semantics, with theoretical models built upon the physical properties of the electron and hole and not the words that describe them. However, we note that there is disagreement (due likely to the system-dependent nature of this issue) about whether the CT state should be thought of as a significant source of separated charges.
This review is organized as follows. First, we will present a very brief overview of the energetics involved in charge transfer and separation. Next, we will discuss four of the key classes of models that have been applied to these processes in OPVs: Onsager theory, Marcus theory, models that are based on frontier orbital energies and electron density distributions, and Monte Carlo modeling. We note that these models are not mutually exclusive, and their principles are often combined in OPV modeling. We close with a discussion of density functional theory (DFT), which is used to derive parameters for nearly every model of charge generation in OPVs.
Energetics of charge transfer and separation
To predict the likelihood of charge separation for a particular OPV system, one must consider the Coulombic interaction that binds the charge transfer state, which is calculated as:39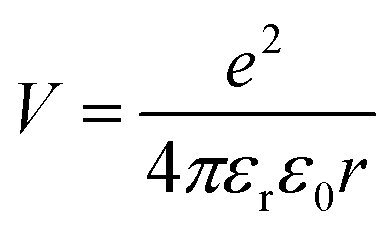 | (1) |
In addition to the Coulombic interaction, electron–electron and electron–lattice interactions contribute to the processes of charge transfer and charge separation.39,97 Electron–lattice interactions may promote charge transfer through the formation of polarons:70,98 when the nuclei around a CT state relax around the electron and hole to form lower energy states (with this stabilizing relaxation lowering the energy of the electron and hole), the energy of the lowest energy CT state (CT0) is relaxed relative to the energy of the exciton, making charge transfer more likely.70 Some authors argue that polaron effects can be disregarded due to the timescales involved, pointing out that while polaron formation occurs on greater than picosecond timescales,70 timescales for charge transfer are generally believed to be ultrafast (∼100 fs or less)34,51,55,71,86,99–101 and temperature-independent73 (where temperature-dependence would be expected in a process strongly linked to vibrational properties).
Assuming that substantial relaxation and polaron formation is significant for some OPV systems, the situation remains unclear: some authors argue that a large reorganization energy leading to a lower energy and more localized CT state inhibits charge separation, which may ultimately lead to fewer free charge carriers. Smith and Chin51 argue that relaxation may lower the energy of the ground charge transfer state without affecting the energies of higher energy, delocalized eigenstates. Because excitation to such eigenstates allows charge separation, this polaron formation may cause charges to remain bound at the donor/acceptor interface. The authors note that this phenomenon is sensitive to both temperature and the delocalization of the ground charge transfer state, with greater delocalization lowering reorganization energy and a higher temperature allowing thermal fluctuations that are large enough to overcome reorganization energies. Thus, understanding the timescales of the charge transfer and separation process, as well as thermal effects of the environment, is an important goal for theoretical work.
Onsager theory
One model that has been influential in the study of charge separation is Onsager theory,102 which has frequently been applied to charge separation in organic photovoltaics103–110 as well as to the problem of geminate recombination.111 Details of the theory, its advantages and limitations, and variations of the model are well reviewed by ref. 39 and 112–114 and we will provide only a brief summary here. Onsager theory works with a picture of a “hot” electron that, via thermal energy, travels some distance away from its geminate hole. The distance between the electron and hole is referred to as the thermalization length a. Onsager proposed a Coulomb capture radius rc at which the Coulombic interaction between the electron and hole equals the thermal energy kBT: | (2) |
 | (3) |
 | ||
| Fig. 2 Schematic of exciton diffusion and charge separation processes in a polymer-fullerene bulk heterojunction active layer showing Onsager parameters. | ||
We see qualitatively that a higher temperature promotes separation via smaller rc. We also note that this theory captures the simple intuition given in eqn (1): That an electron and hole that are located further apart (with the distance given by r in eqn (1), a in eqn (3)) are more likely to achieve complete charge separation. Typical values of these parameters for organic materials have been estimated to be ∼4 nm for r and 0.5–1 nm for a,39,115 with fullerenes (the most common electron acceptor in OPVs) having a diameter of ∼0.7 nm. Essentially all models of charge separation in some way incorporate this electron/hole distance, although models vary significantly in their hypotheses of how and why electron/hole distances vary between systems. We also see the importance of a low dielectric constant increasing the capture radius of the electron/hole pair, demonstrating why there has been recent theoretical54,116 and experimental117,118 research interest in engineering OPVs with greater dielectric constants. Intuitively, an increase in the macroscopic electric field that enters into E should promote charge separation. However, we note that in the case of OPVs this field is not necessarily influential; although some work argues that the electric field plays a significant role in charge separation,53,119 this is a controversial topic, with some research indicating that the fields in OPVs are not strong enough to have a significant effect.39
Braun115 modified Onsager theory to incorporate the finite lifetime of charge transfer states in solids; while the original Onsager theory assumed recombination when the distance between the hole and electron reached zero, Braun's theory incorporates the fact that the hole and electron may remain at a heterojunction for some time with the possibility of separation. The probability of escape is then given by
| P(E) = kd(E)τ(E) | (4) |
In its application to OPVs, Onsager theory and its modifications have been successful in making predictions consistent with experimental data. Mihailetchi et al. apply Onsager–Braun theory to charge separation in OC1C10-PPV:PCBM and calculate a field and temperature dependence of photocurrent consistent with experimental results103 as well as a correct dependence of photocurrent on effective applied voltage.108 Jeong et al. apply the Onsager–Braun model to exciton dissociation in (CuPc)/C60 and achieve a good match to experimental data with the voltage dependence of photocurrent.107
Marcus theory
Marcus theory121 and its variations are widely used in the study of charge transfer and separation in organic photovoltaics.22,39,52,55,122–127 The reader is referred to ref. 128 and 129 for a general discussion of Marcus theory and in particular the “inverted region” of Marcus theory (the inverted region, which is often argued to be important for OPVs, refers to a state in which a more exergonic reaction counter-intuitively corresponds to lower electron transfer rates, as we will discuss later). Ref. 39 and 130 provide discussions of Marcus theory in the context of charge transfer in organic systems. Here we briefly present basic Marcus theory and discuss its relevance to the study of charge transfer and separation in OPVs.The basic rate for electron transfer, as given by Marcus theory, is:130
 | (5) |
 | (6) |
Marcus theory as presented above treats a discrete acceptor state. To treat a band of acceptor states, the rate can be integrated over available states39 as described by Schmickler.122 A good review of techniques for calculating λ and |Hab| is provided by Zhao et al.131
Above we noted that Onsager theory works with the concept of a “hot” electron where the thermalization of the electron plays a major role. A similar concept has been explored in Marcus theory in Nan et al.’s study of “hot” excitons.52 The label of a “hot” charge-transfer exciton refers to an excited state generated by above-gap excitations. There has been much speculation about the role of hot excitons in free carrier generation.71,132–135 While some experimental studies have suggested that hot excitons promote charge separation,66,100,101,136,137 others have found the hot CT state to be insignificant.138–141 After using Marcus theory to calculate the rate of electron hopping in systems with hot CT states, they find that the crystallinity of the donor phase determines the relationship between the energy of the CT state and the electron/hole distance, while acceptor crystallinity influences charge separation efficiency. This is consistent with other work showing the importance of crystallinity in charge separation,51,86,142,143 including work that used a qualitative Marcus theory argument to examine the effects of crystalline domain size on electron transfer.143
One of the most striking and extensively researched features of Marcus theory is the fact that the activation energy has a non-monotonic relationship with ΔG. With λ being positive, at −ΔG < λ, as −ΔG becomes larger (the reaction being more exergonic), the activation energy ΔG‡ becomes smaller and electron transfer increases until the activation energy reaches a minimum of 0 at λ = ΔG. At this point electron transfer is maximized. As the reaction becomes more exergonic and −ΔG becomes larger than λ, the term (λ + ΔG)2 once again becomes positive, and a more exergonic reaction counter-intuitively leads to a higher activation energy and less electron transfer. This region where a more negative ΔG leads to a higher ΔG‡ is referred to as the inverted region, and we see that the span of this region is determined by the reorganization energy λ. The quantities ΔG and λ are defined by the potential energy curves for the reactant and product states as depicted in Fig. 3.
 | ||
| Fig. 3 Here we see potential energy curves (along the reaction coordinate) for the reactant and product states. (a) Depicts the normal region, where −ΔG < λ. (b) Depicts the special case where –ΔG = λ and there is no activation energy to serve as a barrier to transfer. (c) Shows the inverted regime where −ΔG > λ. | ||
The reorganization energy is the change in energy as the environment surrounding the charge rearranges in response to the transfer event. It can be separated into the “inner” contribution comprised of changes in the nuclear geometry, and an “outer” contribution from changes in polarization of the surrounding medium.39 While this seems at first glance to be purely a property of the surrounding molecules, in the case of ultrafast charge transfer, the very fast rate at which the transfer occurs becomes a consideration: if transfer occurs on timescales that are much faster than the timescale at which relaxation of the environment can occur, the transfer event effectively occurs with low reorganization energy, and is then in the far-inverted regime. As mentioned above, Wu et al.73 argue that the ultrafast nature of the charge transfer allows one to disregard polaron effects, where “polaron effects” refer to the same rearrangement of nuclei that partly determines the reorganization energy.
Many authors have argued that charge transfer in OPVs occurs in the inverted regime.39,55,74,144–146 The ultrafast nature of charge transfer in Marcus theory and its placement in the inverted regime have lead to some protest about whether Marcus theory should be used at all for charge transfer in OPVs. In a detailed review of charge transfer research, Barbara et al. point out that ultrafast reactions are particularly sensitive to non-equilibrium dynamical effects of the nuclei in the surrounding environment, leading to a breakdown of many models of charge transfer kinetics.128,147–156 With Marcus theory rates being derived from measurements of the environment at equilibrium (where the reorganization energy and exergonicity are both derived from energies of the pre-transfer and post-transfer equilibrium states), the ultrafast nature of charge transfer in most OPVs is cause for concern.
In an effort to provide an improved treatment of transfer rates, there has been work applying Fermi's Golden Rule (FGR)72,131,157–160 to obtain charge transfer rates for OPVs and other organic donor/acceptor systems, where Marcus theory is the high-temperature, short-time limit of the FGR.74,79,145,146 The authors argue that a full treatment of Fermi's Golden Rule allows a proper treatment of the inverted regime, where activation barriers may be so high that much charge transfer occurs via nuclear tunneling, which traditional Marcus theory does not include. The tutorial review of Zhao et al. provides a thorough discussion of Fermi's Golden Rule and its relationship to the theories of Marcus and others, as well as deriving several other rate theories that are beyond the scope of this paper.131 An extension of Marcus theory, the Marcus–Levich–Jortner (M–L–J) equation,161 explicitly involves the effect of vibrational modes and allows tunneling. The M–L–J method has been applied to OPV charge transfer by ref. 127, 162 and 163. These more precise methods are promising in allowing calculations of charge transfer rates that avoid the questionable approximations of Marcus theory.
Recently, the argument that charge transfer belongs in the inverted regime has been questioned by Savoie et al.72 These authors propose that the most useful OPVs bypass a charge transfer state, with excitons coupled directly to charge separation states (a controversy mentioned above). The authors argue that for this mechanism of charge separation, the inverted regime will not be observed.
In spite of these concerns and controversies, Marcus theory is widely used and has had some success in matching experimental predictions in studies of charge separation in OPVs. Liu et al.127 and Leng et al.125 apply Marcus theory to charge separation and recombination for P3HT/PCBM and PCPDTBT/fullerene systems, respectively, and find reasonable agreement with experimentally measured rates. Another study by Yi et al. found that Marcus theory could predict the experimentally observed poorer performance when perylenetetracarboxydiimide (PDI) electron acceptors were used rather than fullerene acceptors in α-sexithienyl solar cells.124
Evaluation of orbital energies and energy distributions
In this section we discuss methods that derive their predictions from orbital energies and/or inspection of electronic densities. To separate this discussion from the other models is rather artificial, since all models obviously deal (at least indirectly) with energies and electron distributions. However, in an effort to organize the vast amount of literature devoted to charge transfer in OPVs, we have devoted this section to methods that derive predictions directly from these quantities without use of models such as Marcus or Onsager theory rates.Frontier orbital energies (the energies of the highest occupied molecular orbital, HOMO, and the lowest unoccupied molecular orbital, LUMO) can be used to approximate many OPV properties including ionization energies, electron affinities,164 open circuit voltage,165 electronic band gap, and the driving force for charge separation.66,126,164,166 These approximations are frequently looked to for design principles: for example, the commonly used suggestion that ELUMO,D − ELUMO,A gives a driving force for dissociation suggests that a lower-lying acceptor LUMO is advantageous for charge separation, but work by Scharber et al. demonstrated empirically that open-circuit voltage VOC is a function of |EHOMO,D| − |ELUMO,A|, showing that VOC suffers from lower-lying acceptor LUMOs.165 These methods are highly convenient and computationally tractable, with these quantities usually measured as the Kohn–Sham eigenvalues of a DFT calculation. However, in all cases the relationship between the orbital energy and the OPV property that is being predicted involves some approximation, and these approximations are controversial.
The most common approximation involving frontier orbitals is to assume that EHOMO is equivalent to the ionization potential (IP), an argument based upon Koopman's theorem.167 Although Koopman's theorem dealt only with the IP, the argument is commonly extended to the idea that −ELUMO is equivalent to the electron affinity (EA). After several other approximations regarding the relationship of frontier orbitals to physical properties, one can derive the approximation noted above that EHOMO,D − ELUMO,A represents the driving force for charge transfer. If this driving force exceeds the exciton binding energy, charge transfer will occur.98
Although this approximation provides an opportunity to predict an important physical property in a computationally tractable way, there are several problems with this method of predicting charge transfer. The appropriateness of using HOMO and LUMO energies to predict charge transfer behavior in OPVs is discussed in detail by Savoie et al., who conclude that the errors in the various assumptions are so severe as to render even qualitative interpretations inaccurate.98 These authors point out that it is extremely difficult to accurately calculate ELUMO with any existing electronic structure methods. They also point out that equating the EA and IP with orbital energies leads to inaccurate enthalpies and free energies due to the neglect of zero point energies and entropies of formation, which are rarely included due to computational cost. Kanai et al. point out that calculating the LUMO energies of the donor and acceptor via calculations of the isolated molecules fails to incorporate the fact that as electron transfer to the acceptor occurs, the electrostatic potential changes and alters the energy levels near the heterojunction,168 and Armstrong et al. note the significance of interface dipoles in altering frontier orbital energies.91 Even if EHOMO and −ELUMO provided accurate measurements of ionization potentials and electron affinities, there are other questionable assumptions in the derivation of the conclusion that EHOMO,D −ELUMO,A is equal (or nearly equal) to the driving force for charge transfer. We refer the reader to Savoie et al.98 for a more thorough discussion of this derivation and its problems.
In spite of the problems in using HOMO and LUMO energies to approximate IPs and EAs, the HOMO and LUMO are widely applied in determining parameters for other models, usually in calculating energy differences between states. Some work goes beyond this to consider LUMO+1 energies as important in describing some excitations.52,169 Many authors argue that the situation is not so dismal in utilizing frontier orbital energies, with Phillips et al.170 reporting good agreement between frontier orbital energies and experimentally determined IPs and EAs when using range-separated functionals (these functionals, which have attracted much attention for their accuracy in modeling OPVs, are discussed below). Other authors suggest that using the LUMO to approximate the energy of an excited electron is acceptable for more compact molecules, although the approximation deteriorates in less compact charge transfer states.171 Risko et al. use DFT to calculate the HOMO and LUMO energies of donor and acceptor copolymers and find respectable agreement with experimentally measured adiabatic EAs and IPs.164
Rather than measuring the electron affinity as −ELUMO, it is also simple and computationally tractable to simply measure electron affinity as the difference in the total energy between the neutral and anionic states (the more rigorous definition of the EA). Fortunately, studies have found DFT calculations (regardless of functional) to be very effective in capturing the high EA of fullerenes, a crucial factor in the efficiency of polymer/fullerene OPVs.168,172
Some work has moved beyond the focus on orbital and total energies and used a more qualitative inspection of orbital densities to gain insight into charge transfer. In a DFT calculation incorporating both the donor and acceptor molecules, Kanai et al. inspected the electron distribution of the single-particle states of an excited electron in a DFT calculation.168 They describe a “bridging state” that is derived from the LUMO orbitals of the donor and acceptor, demonstrating how insight can be attained by comparing calculations of the isolated donor and acceptor molecules with calculations of the donor/acceptor compound. Inspection of charge densities also played a key role in the work of Bakulin et al.,34 who studied the “hot” excitonic states discussed above and concluded via atomistic modeling that these states had more delocalized charge densities, thus increasing the electron/hole separation as predicted by other work. Few et al. also inspect charge densities in their study of higher energy CT states and hole delocalization.173
In closing this section we note a few studies that have performed quantum dynamics to model charge transfer and separation, defining their parameters through DFT. These models are more complex than many of the studies cited here in the fact that they treat vibrational modes explicitly. Tamura et al. present a model of charge transfer and separation using non-perturbative quantum dynamics, with their Hamiltonian explicitly including the effects of intramolecular phonon modes in addition to the energies and couplings between the states.86 Smith et al. also perform quantum dynamics using a Hamiltonian with explicit vibrational modes and use their model to predict the effect of vibrational relaxation on the delocalization of the electronic states involved in charge separation.51 Xie et al. apply the multi-configuration time-dependent Hartree (MCTDH) quantum dynamics method174,175 to model four electronic states and 246 vibrational degrees of freedom to study electron transfer between anthracene and C60. MCTDH was also applied to thiophene/fullerene interfaces by Tamura et al.176 While the original MCTDH model could only treat tens of vibrational modes, the multilayer (ML) MCTDH method and improvements in its implementation have led to the ability to treat thousands of degrees of freedom,177–183 making it a potentially useful method for dealing with the large molecules involved in OPV charge transfer. Xie et al. demonstrate that decay in the donor population is strongly affected by the inclusion of vibrational modes in the model; however, they note that because of the ultrafast nature of charge transfer, low-frequency vibrations can be ignored.58 Given the uncertainty about properties such as polaron formation that are a function of vibrational modes, treatments that include vibrational modes explicitly are a promising direction for future work.
Monte Carlo modeling
A method that is ubiquitous in OPV research is Monte Carlo modeling, which was in fact combined with Onsager theory in references.64,109 Although there is no universally agreed upon definition of “Monte Carlo,” it generally refers to work that simulates physical processes using the properties of random number distributions. It has been applied to charge transport184 and charge separation185 in disordered polymer systems, geminate111,186,187 and non-geminate188 recombination in OPVs, and charge separation in OPVs.56,64,90,109,119,189,190There are many ways in which the likelihood of a particular charge movement can be treated stochastically in Monte Carlo, and a full review of them is beyond the scope of this paper. However, we note that one of the most common methods used in Monte Carlo simulations of OPVs is the Miller–Abrahams equation191 for the probability of electron/hole movement. This model, frequently used for Monte Carlo simulations of organic semiconductors,56,119,185,186,189,192 can be used to provide probabilities v of movement of an electron or hole:
 | (7) |
Monte Carlo is perhaps best thought of not as a model in itself, but as a numerical technique used to implement other models and achieve quantitative predictions. The accuracy of Monte Carlo theory is essentially a function of the accuracy of the models chosen for its parameters; for example, the accuracy of assessing the energies necessary to construct ΔEij. With DFT used heavily to define Monte Carlo parameters such as energy changes, we will discuss the use of DFT in OPV charge separation research in detail below.
Density functional theory
In almost every model described above, DFT calculations are used as a tool to provide the necessary parameters (with higher-level electronic structure methods usually inaccessible due to the large size of the donor and acceptor molecules in OPVs). For a concise explanation of the theory behind DFT calculations, we refer to the reader to Sumpter et al.’s review of computational methods for OPV research.193 Although the most frequent use of DFT is to provide the energies of states or of frontier orbitals, DFT is also used for properties such as couplings,70,79,194,195 normal modes,58,79 polarizabilites,5,54 dipole moments,5,54,89 electron/hole Coulombic interactions,54 polaron delocalization,196 and descriptions of the electron density distribution.34,168 Constrained DFT (CDFT), in which a charge can be constrained to a given set of atoms, is useful for building the diabatic energy curves used in Marcus theory.130,197,198 Time-dependent DFT199 (TD-DFT) also offers the ability to treat excited states, allowing calculation of charge transfer state energies.200–202 A method that goes beyond the TD-DFT approach to calculating excited states is ensemble density functional theory, which models an ensemble of fractionally occupied states. The state-averaged spin-restricted ensemble referenced (SA-REKS) method and the state-interaction SA-REKS (SI-SA-REKS) method treat the ground and a single excited state, while the recently developed 4SI-3SA-REKS method treats HOMO→LUMO+1 transitions, an extension that may be very useful for charge transfer studies.57,203–210In spite of its wide use, there are many difficulties in applying DFT to OPV charge transfer and separation. One issue is the challenge of performing calculations that are meaningful for the solid state, in which the geometries and electronic properties of each molecule are surely influenced by the surrounding environment. Unfortunately, due to computational cost, most of the surroundings cannot be treated with first principles. Because of the disordered nature of OPVs, there are many possible environments for a given molecule, making this challenge even more daunting. Possible inaccuracies in geometries are concerning given the well-known sensitivity of donor/acceptor systems to small changes in geometry. Previous work has shown that OPV performance is affected by the relative orientation of donor and acceptor molecules,123,124,146,211 and charge transport in organic semiconductors is known to be sensitive to small changes in intermolecular orientation.25,26,212,213 This sensitivity to small changes in orientations is consistent with reports that changes in phase, such as aggregation into a mixed phase, cause changes in excitation energies for both polymers and fullerenes in OPV materials.142,214–217 EPR studies have shown that the electronic structure of C60 anions is sensitive to geometric distortions, consistent with the sensitivities to geometry that we observe in organic semiconducting devices.218
One cost-effective approach that can been used to model the solid state environment in OPVs is to use an implicit solvent model such as PCM (“Polarizable Continuum Model”),219 CPCM (“Conductor-like Polarizable Continuum Model”),220,221 or COSMO (“Conductor-like Screening Model”).222 All of these methods treat the environment as a continuum rather than treating surrounding atoms explicitly, using a polarizable (CPM) or conductor-like (CPCM and COSMO) continuum. CPCM has been used in the modeling of charge transfer in OPVs,74,79 and CPM has been used in measuring frontier orbital energies in organic photovoltaics.170,223 We note that CPCM is also useful in its ability to represent the dielectric properties of the medium,55 which, as discussed above, directly affects the probability of charge separation. However, Lee et al.79 (who use CPCM in their study of charge transfer in photovoltaics) note that a continuum dielectric approach is not adequate to explore the relationship between crystalline structure and charge delocalization, and suggest that a QM/MM approach might be helpful for this problem. Another possibility is to treat a larger region of space using periodic boundary conditions. Although this method is by definition limited in how much disorder it can include, geometry optimizations using periodic boundary conditions for P3HT/C60 solar cells were successful in reproducing the orientation between C60 and the thiophene ring found in NMR studies.168
A variety of coarse-grained methods have been proposed as alternatives to purely ab initio approaches such as DFT, which we briefly summarize here. Savoie et al. explore delocalized states in fullerene aggregates via a fragment method in which the unoccupied states of large PC61BM aggregates are treated with a basis consisting of the three lowest unoccupied orbitals of a single PC61BM molecule.72 A similar method is applied to the treatment of an excess electron in PCBM by Cheung et al., who use a multiscale approach in which molecular dynamics (MD) is used to treat morphology.224 Use of MD for geometry optimization, with application of DFT to calculate charge densities at the heterojunction, was also utilized by Bakulin et al.,34 while Liu et al. use MD to treat geometries prior to using TDDFT to derive parameters for M–L–J theory (discussed above) for charge separation at the interface.162 Huang et al. combined three methods in their calculations of interfacial states at semiconductor heterojunctions, utilizing molecular mechanics and DFT to treat morphology and the Intermediate Neglect of Differential Overlap (INDO) method,225 a semi-empirical method, to treat charge distributions, excitation energies, and transition dipoles.226 Another approach to delocalization was presented by Raos et al., in which only the HOMO and LUMO orbitals of donor/acceptor molecules are considered.227Ref. 72, 224 and 227 and their role in treating charge separation are treated in more detail by Few et al.96Ref. 5, 95 and 228 have proposed various QM/MM approaches to modeling electrostatic effects of the environment on charge transfer and separation. With first-principles treatment of the environment still far out of computational reach, and implicit solvent models limited in their ability to provide the relevant information about surrounding environment, coarse-grained and multiscale methods are an important direction for future studies of charge generation.
Even beyond the challenges of dealing with the solid state, it is well known that there are serious issues with applying DFT to the study of charge transfer states,171,229–243 which is believed to be due to self-interaction errors and failure to treat derivative discontinuities, causing inaccuracies at long ranges.233–235,238,244 In addition to difficulties in calculating energies of charge transfer states, there are problems with the delocalization of charges: as discussed above, Coulombic electron/hole interactions are directly impacted by the delocalization of the charges, and DFT calculations tend to over-delocalize charge densities.
A major development in the use of DFT to study charge transfer is the development of range-separated functionals. Self-interaction error and over-delocalization can be helped significantly by using a range-separated functional that separates the exchange functional into a DFT exchange term and Hartree–Fock (HF) exchange term.244,245 This separation has been applied to the study of donor/acceptor conjugated polymers and showed consistency with experimental excitation energies, and these functionals are now frequently used in DFT calculations of OPVs.52,57,70,73,74,79,86,125,126,166,246,247 The term usually referred to as the “range-separation parameter” determines the mixture of DFT/HF exchange. It is well-known that the optimal value of the range-separation is system-dependent and “tuning” the range-separation parameter for a particular system improves accuracy;233,245,248–256 thus, the vast majority of studies employing range-separated functionals tune the range-separation parameter. The tuning process generally involves finding the value of the range-separation parameter at which the Kohn–Sham eigenvalues best satisfy Koopman's theorem167 and/or Janak's theorem,257 as suggested by ref. 245, 255, 258 and 259. Although the tuning process may add significantly to the computational cost of the project, the gain in accuracy likely makes the additional cost worthwhile.
We note that although range-separated functionals offer increased accuracy, there are some uses of DFT for charge generation modeling that are less sensitive to functional effects. In the quantum dynamics work cited above, Xie et al. tested the performance of the PBE260,261 functional relative to the B3LYP262–264 and LC-ωPBE261,265–268 functionals in the DFT calculations that built their Hamiltonian and found that their results were not particularly sensitive to the choice of functional. Given the increased cost of using range-separated functionals, this insensitivity may be an advantage in treating large systems.
In addition to choosing range-separated functionals, many studies of charge transfer and separation in organic materials add dispersion effects to account for van der Waals interactions.54,73,79,166 Such dispersion effects are incorporated into some functionals, such as ωB97X-D,244,269 which incorporates the dispersion model of Grimme et al.,270 or B3LYP-D3(BJ) that incorporates the dispersion model of Grimme et al.271 With other functionals these dispersion models can be added to the calculation with common software packages.
Other than the range-separated functionals, B3LYP is used frequently (sometimes in combination with range-separated functionals, with different functionals applied to different steps in the calculations).98,166 In geometry optimizations of an OPV heterojunction B3LYP was shown to perform comparably with the mW1PW91272 functional, which, although not a range-separated functional, is known to exhibit improved long-range behavior.127 In surveying theoretical work on charge generation in OPVs and similar donor/acceptor systems, we saw use of the following functionals (sometimes tested against one another for accuracy). The range-separated functional with dispersion effects ωB97X-D244,269 was used by ref. 73, 74 and 79 with ref. 247 using the non-dispersion variation ωB97X. Other range-separated functionals are LC-BLYP263,264,273,274 used by ref. 57 and 86, CAM-B3LYP262–264,275 used by ref. 57, 125 and 126, LC-ωPBE261,265–268 used by ref. 125, and BNL276 used by ref. 13 and 223. Non-range-separated functionals were B3LYP262–264 used by ref. 57, 173, 196 and 277, SVWN262,278 used by ref. 89, M06HF279–281 used by ref. 125, and BLYP263,264,273 and BH&HLYP282 used by ref. 57.
Before closing our discussion of DFT, we note a few ways in which the accuracy of DFT applications for charge generation can be improved (unfortunately most with added computational cost).
First, the distinction between diabatic and adiabatic excitations must be carefully considered. Simulation of a diabatic excitation (frequently referred to as a “vertical excitation”) maintains a fixed nuclear geometry, while simulation of an adiabatic excitation allows optimization of the geometry of the excited state. Diabatic energies are sufficient if ionization occurs on a timescale much faster than nuclear motion, but if ionization occurs on a timescale comparable to nuclear motions, adiabatic calculations are appropriate.98 Confusion between diabatic and adiabatic excitations is not only harmful to theoretical calculations per se, but can also cause error in the comparison of theoretical and experimental results if diabatic (adiabatic) theoretical excitations are compared with data on adiabatic (diabatic) excitations.
Second, in one-electron treatments dealing with ionization potentials and electron affinities, high-level electronic structure calculations such as CCSD(T) with large basis sets are helpful in computing accurate energies (as opposed to performing the much more affordable option of DFT with a smaller basis set).98 This was demonstrated in a comparison of many theoretical methods in the calculation of electron affinities in benzenes and linear acenes,283 and is likely to be important in the treatment of other organic systems as well. When methods such as CCSD(T) are not computationally accessible and DFT is used, Savoie et al. note that the range-separated functionals discussed above offer some hope of greater accuracy.98
Third, we note that although many studies treat bare fullerenes for maximum generalizability (and simplicity), adding functional groups may significantly disrupt the orbital energies of a fullerene molecule and should be considered if possible. Although this energetic disruption was found to be minimal when Kanai et al. compared DFT calculations of C60 and PCBM,168 efforts to predict quantitatively the properties of an OPV structure should ideally include fullerene functional groups.
Fourth, we note that the generally neglected entropic effects may be important. Most models of charge separation focus on enthalpies; however, it is important to consider entropic contributions to the free energy change with charge separation. As the charges separate, there are more possible locations for the electron and hole, and entropy increases.39 Some studies have considered free energy as a driving force,60 and a focus on free energies may provide an important contribution to future work.
Conclusions
With the high cost of experimentally optimizing OPVs, theory plays a crucial role in predicting the properties of possible OPV materials. In this review we have presented the fundamentals of past and current theoretical modeling of charge transfer and separation.Looking to the future, the accuracy of all methods described above will benefit greatly from progress in the field of density functional theory. Although range-separated functionals have contributed significantly to DFT's accuracy in treating charge-transfer states, there is still much room for improvement in resolving the issues that we discuss above in applying DFT to charge separation. As computational resources evolve, it may increasingly be possible to incorporate multireference electronic structure methods into charge generation research, and new methods such as the variational subspace valence bond (VSVB) method284 may offer a cost-effective alternative to DFT. There is also a need for continued experimental validation of the assumptions inherent in models such as Onsager theory and Marcus theory that are commonly used to treat rates of charge transfer. As OPV technology evolves and new donor/acceptor molecules are employed, the approximations involved in these theories may become less (or more) appropriate. Studies of transfer and separation will also benefit from the development of more multiscale methods that incorporate effects that are beyond the reach of ab initio methods. Using multiscale methods to provide a more explicit treatment of the surrounding environment and the extent of its disorder or crystallinity may provide important insights into the charge generation process. As theoretical approaches improve, methods of modeling charge transfer and separation may offer both qualitative trends as well as truly quantitative predictions regarding charge generation that guide the design of the next generation of organic photovoltaics.
Acknowledgements
This work was performed at the Center for Nanoscale Materials, a U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences User Facility under Contract no. DE-AC02-06CH11357. K. M. Pelzer was funded by the Aneesur Rahman Fellowship of Argonne National Laboratory.References
- S. B. Darling and F. You, RSC Adv., 2013, 3, 17633 RSC.
- N. S. Lewis, Science, 2007, 315, 798–801 CrossRef CAS PubMed.
- A. J. Heeger, Adv. Mater., 2014, 26, 10–28 CrossRef CAS PubMed.
- C. W. Lee, O. Y. Kim and J. Y. Lee, J. Ind. Eng. Chem., 2014, 20, 1198–1208 CrossRef CAS.
- H. D. D. Gier, F. Jahani, R. Broer, J. C. Hummelen and R. W. A. Havenith, J. Phys. Chem. A, 2015 DOI:10.1021/acs.jpca.5b09279.
- L. Li, W. Niu, X. L. Zhao, X. N. Yang and S. W. Chen, Sci. Adv. Mater., 2015, 7, 2021–2036 CrossRef CAS.
- L. Y. Lu, M. A. Kelly, W. You and L. P. Yu, Nat. Photonics, 2015, 9, 491–500 CrossRef CAS.
- H. Youn, H. J. Park and L. J. Guo, Small, 2015, 11, 2228–2246 CrossRef CAS PubMed.
- I. Etxebarria, J. Ajuria and R. Pacios, Org. Electron., 2015, 19, 34–60 CrossRef CAS.
- I. Etxebarria, J. Ajuria and R. Pacios, J. Photonics Energy, 2015, 5, 057214 CrossRef.
- K. A. Mazzio and C. K. Luscombe, Chem. Soc. Rev., 2015, 44, 78–90 RSC.
- D. Joshi, R. Shivanna and K. S. Narayan, J. Mod. Opt., 2014, 61, 1703–1713 CrossRef CAS.
- J. Yu, Y. Zheng and J. Huang, Polymers, 2014, 6, 2473–2509 CrossRef.
- W. R. Cao and J. G. Xue, Energy Environ. Sci., 2014, 7, 2123–2144 CAS.
- M. C. Scharber and N. S. Sariciftci, Prog. Polym. Sci., 2013, 38, 1929–1940 CrossRef CAS PubMed.
- A. R. B. M. Yusoff, D. Kim, H. P. Kim, F. K. Shneider, W. J. D. Silva and J. Jang, Energy Environ. Sci., 2015, 8, 303–316 CAS.
- M. A. Green, K. Emery, Y. Hishikawa, W. Warta and E. D. Dunlop, Progr. Photovolt.: Res. Appl., 2015, 23, 1–9 CrossRef.
- C.-C. Chen, W.-H. Chang, K. Yoshimura, K. Ohya, J. You, J. Gao, Z. Hong and Y. Yang, Adv. Mater., 2014, 26, 5670–5677 CrossRef CAS PubMed.
- T. Stübinger and W. Brütting, J. Appl. Phys., 2001, 90, 3632–3641 CrossRef.
- J. J. M. Halls, K. Pichler, R. H. Friend, S. C. Moratti and A. B. Holmes, Appl. Phys. Lett., 1996, 68, 3120–3122 CrossRef CAS.
- D. E. Markov, J. C. Hummelen, P. W. M. Blom and A. B. Sieval, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 045216 CrossRef.
- A. Zhugayevych, O. Postupna, R. C. B. II, G. C. Welch, G. C. Bazan and S. Tretiak, J. Phys. Chem. C, 2013, 117, 4920–4930 CAS.
- R. R. Lunt, J. B. Benziger and S. R. Forrest, Adv. Mater., 2010, 22, 1233–1236 CrossRef CAS PubMed.
- T. Kirchartz and J. Nelson, in Multiscale Modelling of Organic and Hybrid Photovoltaics, ed. D. Beljonne and J. Cornil, Springer-Verlag, Berlin Heidelberg, 2014, pp. 279–324 Search PubMed.
- V. Coropceanu, J. Cornil, D. A. D. S. Filho, Y. Olivier, R. Silbey and J.-L. Brédas, Chem. Rev., 2007, 107, 926–952 CrossRef CAS PubMed.
- K. M. Pelzer, M. K. Y. Chan, S. K. Gray and S. B. Darling, J. Phys. Chem. C, 2014, 118, 21785–21797 CAS.
- Y. N. Li, P. Sonar, L. Murphy and W. Hong, Energy Environ. Sci., 2013, 6, 1684–1710 CAS.
- V. Burtman, Z. Zelichonok and A. V. Pakoulev, Int. J. Mol. Sci., 2011, 12, 173–225 CrossRef CAS PubMed.
- W. Chen, M. P. Nikiforov and S. B. Darling, Energy Environ. Sci., 2012, 5, 8045 CAS.
- W. Chen, T. Xu, F. He, W. Wang, C. Wang, J. Strzalka, Y. Liu, J. Wen, D. J. Miller, J. Chen, K. Hong, L. Yu and S. B. Darling, Nano Lett., 2011, 11, 3707–3713 CrossRef CAS PubMed.
- C. Deibel, T. Strobel and V. Dyakonov, Adv. Mater., 2010, 22, 4097–4111 CrossRef CAS PubMed.
- R. D. Pensack and J. B. Asbury, J. Am. Chem. Soc., 2009, 131, 15986–15987 CrossRef CAS PubMed.
- J. Guo, H. Ohkita, H. Benten and S. Ito, J. Am. Chem. Soc., 2010, 132, 6154 CrossRef CAS PubMed.
- A. A. Bakulin, A. Rao, V. G. Pavelyev, P. H. M. V. Loosdrecht, M. S. Pshenichnikov, D. Niedzialek, J. Cornil, D. Beljonne and R. H. Friend, Science, 2012, 335, 1340–1344 CrossRef CAS PubMed.
- Y. Yuan, T. J. Reece, P. Sharma, S. Poddar, S. Ducharme, A. Gruverman, Y. Yang and J. Huang, Nat. Mater., 2011, 10, 296–302 CrossRef CAS PubMed.
- P. K. Nayak, K. L. Narasimhan and D. Cahen, J. Phys. Chem. Lett., 2013, 4, 1707–1717 CrossRef CAS PubMed.
- H. Tamura and I. Burghardt, J. Phys. Chem. C, 2013, 117, 15020–15025 CAS.
- D. Caruso and A. Troisi, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13498–13502 CrossRef CAS PubMed.
- T. M. Clarke and J. R. Durrant, Chem. Rev., 2010, 110, 6736 CrossRef CAS PubMed.
- Y. Terao, H. Sasabe and C. Adachi, Appl. Phys. Lett., 2007, 90, 103515 CrossRef.
- S. R. Forrest, MRS Bull., 2005, 30, 28–32 CrossRef CAS.
- P. Peumans and S. R. Forrest, Appl. Phys. Lett., 2001, 79, 126–128 CrossRef CAS.
- H. Tamura and Y. Matsuo, Chem. Phys. Lett., 2014, 598, 81–85 CrossRef CAS.
- S. M. Menke, W. A. Luhman and R. J. Holmes, Nat. Mater., 2012, 12, 152–157 CrossRef PubMed.
- Z. Li, X. Zhang and G. Lu, J. Phys.: Condens. Matter, 2014, 26, 185006 CrossRef CAS PubMed.
- P. K. Nayak, Synth. Met., 2013, 174, 42–45 CrossRef CAS.
- A. Dkhissi, Synth. Met., 2011, 161, 1441–1443 CrossRef CAS.
- K. Hummer and C. Ambrosch-Draxl, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 081202 CrossRef.
- J.-W. V. D. Horst, P. A. Bobbert, M. A. J. Michels and H. Bässler, J. Chem. Phys., 2001, 114, 6950–6957 CrossRef.
- M. Knupfer, Appl. Phys. A: Mater. Sci. Process., 2003, 77, 623–626 CrossRef CAS.
- S. L. Smith and A. W. Chin, Phys. Chem. Chem. Phys., 2014, 16, 20305 RSC.
- G. Nan, X. Zhang and G. Lu, J. Phys. Chem. C, 2015, 119, 15028–15035 CAS.
- G. F. Burkhard, E. T. Hoke, Z. M. Beiley and M. D. McGehee, J. Phys. Chem. C, 2012, 116, 26674–26678 CAS.
- S. Kraner, R. Scholz, C. Koerner and K. Leo, J. Phys. Chem. C, 2015, 119, 22820–22825 CAS.
- H. Kuang, M. J. Janik and E. D. Gomez, J. Polym. Sci., Part B: Polym. Phys., 2015, 53, 1224–1230 CrossRef CAS.
- T. M. Burke and M. D. McGehee, Adv. Mater., 2014, 26, 1923–1928 CrossRef CAS PubMed.
- M. Filatov, M. Huix-Rottlant and I. Burghardt, J. Chem. Phys., 2015, 142, 184104 CrossRef PubMed.
- Y. Xie, J. Zheng and Z. Lan, J. Chem. Phys., 2015, 142, 084706 CrossRef PubMed.
- M. A. Loi, S. Toffanin, M. Muccini, M. Forster, U. Scherf and M. Scharber, Adv. Funct. Mater., 2007, 17, 2111–2116 CrossRef CAS.
- D. Veldman, S. C. J. Meskers and R. A. J. Janssen, Adv. Funct. Mater., 2009, 19, 1939–1948 CrossRef CAS.
- K. Tvingstedt, K. Vandewal, A. Gadisa, F. Zhang, J. Manca and O. Inganäs, J. Am. Chem. Soc., 2009, 131, 11819–11824 CrossRef CAS PubMed.
- C. R. McNeill, S. Westenhoff, C. Groves and R. H. Friend, J. Phys. Chem. C, 2007, 111, 19153 CAS.
- A. C. Morteani, P. Sreearunothai, L. M. Herz, R. H. Friend and C. Silva, Phys. Rev. Lett., 2004, 92, 247402 CrossRef PubMed.
- P. Peumans and S. R. Forrest, Chem. Phys. Lett., 2004, 398, 27–31 CrossRef CAS.
- T. Offermans, P. A. V. Hal and S. C. J. Meskers, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 045213 CrossRef.
- H. Ohkita, S. Cook, Y. Astuti, W. Duffy, S. Tierney, W. M. Zhang, M. Heeney, L. McCulloch, J. Nelson, D. D. C. Bradley and J. R. Durrant, J. Am. Chem. Soc., 2008, 130, 3030–3042 CrossRef CAS PubMed.
- C. Yin, T. Kietzke, D. Neher and H.-H. Hörhold, Appl. Phys. Lett., 2007, 90, 092117 CrossRef.
- J. J. Benson-Smith, L. Goris, K. Vandewal, J. Haenen, J. V. Manca, D. Vanderzande, D. D. C. Bradley and J. Nelson, Adv. Funct. Mater., 2007, 17, 451–457 CrossRef CAS.
- A. A. Bakulin, S. D. Dimitrov, A. Rao, P. C. Y. Chow, C. B. Nielsen, B. C. Schroeder, I. McCulloch, H. J. Bakker, J. R. Durrant and R. H. Friend, J. Phys. Chem. Lett., 2013, 4, 209–215 CrossRef CAS PubMed.
- B. M. Savoie, N. E. Jackson, L. X. Chen, T. J. Marks and M. A. Ratner, Acc. Chem. Res., 2014, 47, 3385–3394 CrossRef CAS PubMed.
- A. E. Jailaubekov, A. P. Willard, J. R. Tritsch, W.-L. Chan, N. Sai, R. Gearba, L. G. Kaake, K. J. Williams, K. Leung, P. J. Rossky and X.-Y. Zhu, Nat. Mater., 2012, 12, 66–73 CrossRef PubMed.
- B. M. Savoie, A. Rao, A. A. Bakulin, S. Gelinas, B. Movaghar, R. H. Friend, T. J. Marks and M. A. Ratner, J. Am. Chem. Soc., 2014, 136, 2876–2884 CrossRef CAS PubMed.
- Q. Wu, J. Phys. Chem. B, 2015, 119, 7321–7327 CrossRef CAS PubMed.
- D. E. Wilcox, M. H. Lee, M. E. Sykes, A. Niedringhaus, E. Geva, B. D. Dunietz, M. Shtein and J. P. Ogilvie, J. Phys. Chem. Lett., 2015, 6, 569–575 CrossRef CAS PubMed.
- H. Gommans, D. Cheyns, T. Aernouts, C. Girotto, J. Poortmans and P. Heremans, Adv. Funct. Mater., 2007, 17, 2653–2658 CrossRef CAS.
- A. F. Hebard, R. C. Hadden, R. M. Fleming and A. R. Kortan, Appl. Phys. Lett., 1991, 59, 2109–2111 CrossRef CAS.
- B. Pevzner, A. F. Hebard and M. S. Dresselhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 439–449 CrossRef.
- G. J. Dutton and S. W. Robey, J. Phys. Chem. C, 2012, 116, 19173–19181 CAS.
- M. H. Lee, B. D. Dunietz and E. Geva, J. Phys. Chem. Lett., 2014, 5, 3810–3816 CrossRef CAS PubMed.
- R. O. Loutfy and J. H. Sharp, J. Chem. Phys., 1979, 71, 1211–1217 CrossRef CAS.
- A. Sussman, J. Appl. Phys., 1967, 38, 2738–2748 CrossRef CAS.
- F.-R. Fan and L. R. Faulkner, J. Chem. Phys., 1978, 69, 3334–3340 CrossRef CAS.
- R. Pandey, A. A. Gunawan, K. E. Mkhoyan and R. J. Holmes, Adv. Funct. Mater., 2012, 22, 617–624 CrossRef CAS.
- U. Zhokhavets, R. Goldhan, G. Gobsch, M. Al-Ibrahim, H.-K. Roth and S. Sensfuss, Thin Solid Films, 2003, 444, 215–220 CrossRef CAS.
- K. Harigaya and S. Abe, Optical Response of C60 and C70 Fullerenes: Exciton and Lattice Fluctuation Effects, 2013, arXiv: chem-ph/9407002 Search PubMed.
- H. Tamura and I. Burghardt, J. Am. Chem. Soc., 2013, 135, 16364–16367 CrossRef CAS PubMed.
- http://www.pveducation.org/pvcdrom/materials/general-properties-of-silicon, (accessed 12–23-15, 2015).
- B. S. Rolczynski, J. M. Szarko, H. J. Son, Y. Liang, L. Yu and L. X. Chen, J. Am. Chem. Soc., 2012, 134, 4142–4152 CrossRef CAS PubMed.
- C. F. N. Marchiori and M. Koehler, Synth. Met., 2010, 160, 643–650 CrossRef CAS.
- M. Koehler, M. C. Santos and M. G. E. D. Luz, J. Appl. Phys., 2006, 99, 053702 CrossRef.
- N. R. Armstrong, W. Wang, D. M. Alloway, D. Placencia, E. Ratcliff and M. Brumbach, Macromol. Rapid Commun., 2009, 30, 717–731 CrossRef CAS PubMed.
- T. Xu, L. Lu, T. Zheng, J. M. Szarko, A. Schneider, L. X. Chen and L. Yu, Adv. Funct. Mater., 2014, 24, 3432–3437 CrossRef CAS.
- L. Lu and L. Yu, Adv. Mater., 2014, 26, 4413–4430 CrossRef CAS PubMed.
- S. Verlaak, D. Beljonne, D. Cheyns, C. Rolin, M. Linares, F. Castet, J. Cornil and P. Heremans, Adv. Funct. Mater., 2009, 19, 3809–3814 CrossRef CAS.
- S. R. Yost and T. V. Voorhis, J. Phys. Chem. C, 2013, 117, 5617–5625 CAS.
- S. Few, J. M. Frost and J. Nelson, Phys. Chem. Chem. Phys., 2015, 17, 2311–2325 RSC.
- J.-L. Brédas, J. Cornil and A. J. Heeger, Adv. Mater., 1996, 8, 447–452 CrossRef.
- B. M. Savoie, N. E. Jackson, T. J. Marks and M. A. Ratner, Phys. Chem. Chem. Phys., 2013, 15, 4538–4547 RSC.
- L. G. Kaake, D. Moses and A. J. Heeger, J. Phys. Chem. Lett., 2013, 4, 2264–2268 CrossRef CAS.
- G. Grancini, M. Maiuri, D. Fazzi, A. Petrozza, H.-J. Egelhaaf, D. Brida, G. Cerullo and G. Lanzani, Nat. Mater., 2013, 12, 29–33 CrossRef CAS PubMed.
- S. Gélinas, A. Rao, A. Kumar, S. L. Smith, A. W. Chin, J. Clark, T. S. V. D. Poll, G. C. Bazan and R. H. Friend, Science, 2014, 343, 512–516 CrossRef PubMed.
- L. Onsager, Phys. Rev., 1938, 54, 554 CrossRef CAS.
- V. D. Mihailetchi, L. J. A. Koster, J. C. Hummelen and P. W. M. Blom, Phys. Rev. Lett., 2004, 93, 216601 CrossRef CAS PubMed.
- L. J. A. Koster, E. C. P. Smits, V. D. Mihailetchi and P. W. M. Blom, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 085205 CrossRef.
- G. A. Buxton and N. Clarke, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 085207 CrossRef.
- C. Deibel, A. Wagenpfahl and V. Dyakonov, Phys. Status Solidi A, 2008, 2, 175 CAS.
- W.-I. Jeong, Y. E. Lee, H.-S. Shim, T.-M. Kim, S.-Y. Kim and J.-J. Kim, Adv. Funct. Mater., 2012, 22, 3089–3094 CrossRef CAS.
- V. D. Mihailetchi, L. J. A. Koster and P. W. M. Blom, Appl. Phys. Lett., 2004, 85, 970–972 CrossRef CAS.
- M. Wojcik, P. Michalak and M. Tachiya, Appl. Phys. Lett., 2010, 96, 162102 CrossRef.
- V. D. Mihailetchi, L. J. A. Koster, P. W. M. Blom, C. Melzer, B. D. Boer, J. K. J. V. Duren and R. A. J. Janssen, Adv. Funct. Mater., 2005, 15, 795–801 CrossRef CAS.
- C. Groves, R. A. Marsh and N. C. Greenham, J. Chem. Phys., 2008, 129, 114903 CrossRef CAS PubMed.
- K. Feron, W. J. Belcher, C. J. Fell and P. C. Dastoor, Int. J. Mol. Sci., 2012, 13, 17019–17047 CrossRef CAS PubMed.
- C. Deibel and V. Dyakonov, Rep. Prog. Phys., 2010, 73, 096401 CrossRef.
- A. Baumann, Charge Transport and Recombination Dynamics in Organic Bulk Heterojunction Solar Cells, Dissertation, University of Würzburg, Würzburg, 2011 Search PubMed.
- C. L. Braun, J. Chem. Phys., 1984, 80, 4157–4161 CrossRef CAS.
- S. Kraner, C. Koerner, K. Leo, E. Bittrich, K.-J. Eichhorn, Y. Karpov, A. Kiriy, M. Stamm, K. Hinrichs and M. Al-Hussein, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 195202 CrossRef.
- S. Torabi, F. Jahani, I. V. Severen, C. Kanimozhi, S. Patil, R. W. A. Havenith, R. C. Chiechi, L. Lutsen, D. J. M. Vanderzande, T. J. Cleij, J. C. Hummelen and L. J. A. Koster, Adv. Funct. Mater., 2015, 25, 150–157 CrossRef CAS.
- N. Cho, C. W. Schlenker, K. M. Knesting, P. Koelsch, H.-L. Yip, D. S. Ginger and A. K.-Y. Jen, Adv. Energy Mater., 2014, 4, 1301857 Search PubMed.
- T. Offermans, S. C. J. Meskers and R. A. J. Janssen, Chem. Phys., 2005, 2005, 125–133 CrossRef.
- M. Tachiya, J. Chem. Phys., 1988, 89, 6929–6935 CrossRef CAS.
- R. A. Marcus, Rev. Mod. Phys., 1993, 65, 599–610 CrossRef CAS.
- W. Schmickler, Interfacial Electrochemistry, Oxford University Press, Oxford, 1996 Search PubMed.
- Y. Yi, V. Coropceanu and J. Brédas, J. Am. Chem. Soc., 2009, 131, 15777 CrossRef CAS PubMed.
- Y. Yi, V. Coropceanu and J.-L. Brédas, J. Mater. Chem., 2011, 21, 1479–1486 RSC.
- C. Leng, H. Qin, Y. Si and Y. Zhao, J. Phys. Chem. C, 2014, 118, 1843–1855 CAS.
- Y. Li, T. Pullerits, M. Zhao and M. Sun, J. Phys. Chem. C, 2011, 115, 21865–21873 CAS.
- T. Liu and A. Troisi, J. Phys. Chem. C, 2011, 115, 2406–2415 CAS.
- P. F. Barbara, T. J. Meyer and M. A. Ratner, J. Phys. Chem., 1996, 100, 13148–13168 CrossRef CAS.
- G. Grampp, Angew. Chem., Int. Ed. Engl., 1993, 32, 691–693 CrossRef.
- B. Kaduk, T. Kowalczyk and T. V. Voorhis, Chem. Rev., 2011, 112, 321 CrossRef PubMed.
- Y. Zhao and W.-Z. Liang, Chem. Soc. Rev., 2012, 41, 1075–1087 RSC.
- C. Silva, Nat. Mater., 2013, 12, 5–6 CAS.
- X.-Y. Zhu, Q. Yang and M. Muntwiler, Acc. Chem. Res., 2009, 42, 1779–1787 CrossRef CAS PubMed.
- A. Armin, Y. Zhang, P. L. Burn, P. Meredith and A. Pivrikas, Nat. Mater., 2013, 12, 593–593 CrossRef CAS PubMed.
- G. Grancini, M. Binda, L. Criante, S. Perissinotto, M. Maiuri, D. Faqzzi, A. Petrozza, H.-J. Egelhaaf, D. Brida, G. Cerullo and G. Lanzani, Nat. Mater., 2013, 12, 594–595 CrossRef CAS PubMed.
- S. D. DImitrov, A. A. Bakulin, C. B. Nielsen, B. C. Schroeder, J. P. Du, H. Bronstein, I. McCulloch, R. H. Friend and J. R. Durrant, J. Am. Chem. Soc., 2012, 134, 18189–18192 CrossRef CAS PubMed.
- K. Chen, A. J. Barker, M. E. Reish, K. C. Gordon and J. M. Hodgkiss, J. Am. Chem. Soc., 2013, 135, 18502–18512 CrossRef CAS PubMed.
- K. Vandewal, S. Albrecht, E. T. Hoke, K. R. Graham, J. Widmer, J. D. Douglas, M. Schubert, W. R. Mateker, J. T. Bloking, G. F. Burkhard, A. Sellinger, J. M. J. Fréchet, A. Amassian, M. K. Riede, M. D. McGehee, D. Neher and A. Salleo, Nat. Mater., 2014, 13, 63–68 CrossRef CAS PubMed.
- A. Armin, M. Velusamy, P. Wolfer, Y. L. Zhang, P. L. Burn, P. Meredith and A. Pivrikas, ACS Photonics, 2014, 1, 173–181 CrossRef CAS.
- T. G. J. V. D. Hofstad, D. D. Nuzzo, M. V. D. Berg, R. A. J. Janssen and S. C. J. Meskers, Adv. Energy Mater., 2012, 2, 1095–1099 CrossRef.
- J. Lee, K. Vandewal, S. R. Yost, M. E. Bahlke, L. Goris, M. A. Baldo, J. V. Manca and T. V. Voorhis, J. Am. Chem. Soc., 2010, 132, 11878–11880 CrossRef CAS PubMed.
- F. C. Jamieson, E. B. Domingo, T. McCarthy-Ward, M. Heeney, N. Stingelin and J. R. Durrant, Chem. Sci., 2012, 3, 485–492 RSC.
- S. Spencer, J. Cody, S. Misture, B. Cona, P. Heaphy, G. Rumbles, J. Andersen and C. Collison, J. Phys. Chem. C, 2014, 118, 14840–14847 CAS.
- J. R. Miller, L. T. Calcaterra and G. L. Closs, J. Am. Chem. Soc., 1984, 106, 3047–3049 CrossRef CAS.
- M. H. Lee, E. Geva and B. D. Dunietz, J. Phys. Chem. C, 2014, 118, 9780–9789 CAS.
- M. H. Lee, B. D. Dunietz and E. Geva, J. Phys. Chem. C, 2013, 117, 23391–23401 CAS.
- A. E. Johnson, N. E. Levinger, W. Jarzeba, R. E. Schlief, D. A. V. Kliner and P. F. Barbara, Chem. Phys., 1993, 176, 555–574 CrossRef CAS.
- F. Pöllinger, H. Heitele, M. E. Michel-Beyerle, C. Anders, M. Futscher and H. A. Staab, Chem. Phys. Lett., 1992, 198, 645–652 CrossRef.
- M. J. Weaver, J. Mol. Liq., 1994, 60, 57–71 CrossRef CAS.
- J. T. Hynes, J. Chem. Phys., 1986, 90, 3701–3706 CrossRef CAS.
- H. Sumi and R. A. Marcus, J. Chem. Phys., 1986, 84, 4894–4914 CrossRef CAS.
- Y. J. Yan, M. Sparpaglione and S. Mukamel, J. Phys. Chem., 1988, 92, 4842–4853 CrossRef CAS.
- I. Rips and J. Jortner, J. Chem. Phys., 1987, 87, 2090–2104 CrossRef CAS.
- B. Bagchi and G. R. Fleming, J. Phys. Chem., 1990, 94, 9–20 CrossRef CAS.
- J. Zhu and J. C. Rasaiah, J. Chem. Phys., 1991, 95, 3325–3340 CrossRef CAS.
- J. C. Rasaiah and J. Zhu, J. Chem. Phys., 1993, 98, 1213–1227 CrossRef CAS.
- K. V. Mikkelsen and M. A. Ratner, Chem. Rev., 1987, 87, 113–153 CrossRef CAS.
- J. Jortner, J. Chem. Phys., 1976, 64, 4860–4867 CrossRef CAS.
- K. K. Liang, A. M. Mebel, S. H. Lin, M. Hayashi, H. L. Selzle, E. W. Schlag and M. Tachiya, Phys. Chem. Chem. Phys., 2003, 5, 4656–4665 RSC.
- G. Nan, X. Yang, L. Wang, Z. Shuai and Y. Zhao, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 115203 CrossRef.
- Electron Transfer: From Isolated Molecules to Biomolecules, ed. M. Bixon and J. Jortner, Wiley, New York, 1999, vol. 106–107 Search PubMed.
- T. Liu, D. L. Cheung and A. Troisi, Phys. Chem. Chem. Phys., 2011, 13, 21461–21470 RSC.
- V. Lemaur, M. Steel, D. Beljonne, J.-L. Brédas and J. Cornil, J. Am. Chem. Soc., 2005, 127, 6077–6086 CrossRef CAS PubMed.
- C. Risko, M. D. McGehee and J.-L. Brédas, Chem. Sci., 2011, 2, 1200–1218 RSC.
- M. C. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger and C. J. Brabec, Adv. Mater., 2006, 18, 789–794 CrossRef CAS.
- S.-B. Li, Y.-A. Duan, Y. Geng, H.-Z. Gao, Y.-Q. Qiu and Z.-M. Su, RSC Adv., 2015, 5, 29041–29411 Search PubMed.
- T. Koopmans, Physica, 1934, 1, 104–113 CrossRef.
- Y. Kanai and J. C. Grossman, Nano Lett., 2007, 7, 1967–1972 CrossRef CAS PubMed.
- H. Ma and A. Troisi, J. Phys. Chem. C, 2014, 118, 27272–27280 CAS.
- H. Phillips, Z. Zheng, E. Geva and B. D. Dunietz, Org. Electron., 2014, 15, 1509–1520 CrossRef CAS.
- O. Gritsenko and E. J. Baerands, J. Chem. Phys., 2004, 121, 655–660 CrossRef CAS PubMed.
- W. Andreoni, Annu. Rev. Phys. Chem., 1998, 49, 405–439 CrossRef CAS PubMed.
- S. Few, J. M. Frost, J. Kirkpatrick and J. Nelson, J. Phys. Chem. C, 2014, 16, 8253–8261 Search PubMed.
- H.-D. Meyer, U. Manthe and L. S. Cederbaum, Chem. Phys. Lett., 1990, 165, 73–78 CrossRef CAS.
- M. H. Beck, A. Jäckle, G. A. Worth and H.-D. Meyer, Physical Reports, 2000, 324, 1–103 CrossRef CAS.
- H. Tamura, I. Burghardt and M. Tsukada, J. Phys. Chem. C, 2011, 115, 10205–10210 CAS.
- H. Wang and M. Thoss, J. Chem. Phys., 2003, 119, 1289–1299 CrossRef CAS.
- H.-D. Meyer and G. A. Worth, Theor. Chem. Acc., 2003, 109, 251–267 CrossRef CAS.
- U. Manthe, J. Chem. Phys., 2008, 128, 164116 CrossRef PubMed.
- U. Manthe, J. Chem. Phys., 2009, 130, 054109 CrossRef PubMed.
- O. Vendrell and H.-D. Meyer, J. Chem. Phys., 2011, 134, 044135 CrossRef PubMed.
- Q. Meng and H.-D. Meyer, J. Chem. Phys., 2013, 138, 014313 CrossRef PubMed.
- Q. Meng, S. Faraji, O. Vendrell and H.-D. Meyer, J. Chem. Phys., 2012, 137, 134302 CrossRef PubMed.
- B. Hartenstein, H. Bässler, S. Heun, P. Borsenberger, M. V. D. Auweraer and F. C. D. Schryver, Chem. Phys., 1995, 191, 321–332 CrossRef CAS.
- U. Albrecht and H. Bässler, Chem. Phys. Lett., 1995, 235, 389–393 CrossRef CAS.
- T. Offermans, S. C. J. Meskers and R. A. J. Janssen, J. Chem. Phys., 2003, 119, 10924 CrossRef CAS.
- C. Groves, Energy Environ. Sci., 2013, 6, 1546–1551 CAS.
- J. Nelson, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 155209 CrossRef.
- C. Deibel, T. Strobel and V. Dyakonov, Phys. Rev. Lett., 2009, 103, 036402 CrossRef PubMed.
- H. V. Eersel, R. A. J. Janssen and M. Kemerink, Adv. Funct. Mater., 2012, 22, 2700–2708 CrossRef.
- A. Miller and E. Abrahams, Phys. Rev., 1960, 120, 745–755 CrossRef CAS.
- N. Tessler, Y. Preezant, N. Rappaport and Y. Roichman, Adv. Mater., 2009, 21, 2741–2761 CrossRef CAS.
- B. G. Sumpter and M. Meunier, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 1071 CrossRef CAS.
- S. B. Darling, J. Phys. Chem. B, 2008, 112, 8891–8895 CrossRef CAS PubMed.
- D. M. Hinkens, Q. L. Chen, M. K. Siddiki, D. Gosztola, M. A. Tapsak, Q. Q. Qiao, M. Jeffries-EL and S. B. Darling, Polymer, 2013, 54, 3510–3520 CrossRef CAS.
- J. Niklas, K. L. Mardis, B. P. Banks, G. M. Grooms, A. Sperlich, V. Dyakonov, S. Beaupre, M. Leclerc, T. Xu, L. Yu and O. G. Poluektov, Phys. Chem. Chem. Phys., 2013, 15, 9562–9574 RSC.
- Q. Wu and T. V. Voorhis, J. Chem. Phys., 2006, 125, 164105 CrossRef PubMed.
- Q. Wu and T. V. Voorhis, J. Phys. Chem. A, 2006, 110, 9212 CrossRef CAS PubMed.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- M. Petersilka, U. J. Grossman and E. K. U. Gross, Phys. Rev. Lett., 1996, 76, 1212–1215 CrossRef CAS PubMed.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439–4448 CrossRef CAS.
- F. Furche and K. Burke, Annu. Rep. Comput. Chem., 2005, 1, 19–30 CAS.
- E. K. U. Gross, L. N. Oliveira and W. Kohn, Phys. Rev. A, 1988, 37, 2809–2820 CrossRef CAS.
- E. K. U. Gross, L. N. Oliveira and W. Kohn, Phys. Rev. A, 1988, 37, 2805–2808 CrossRef.
- M. Filatov, WIREs Comput. Mol. Sci., 2015, 5, 146–167 CrossRef CAS.
- A. Kazaryan, J. Heuver and M. Filatov, J. Phys. Chem. A, 2008, 112, 12980–12988 CrossRef CAS PubMed.
- M. Filatov, J. Chem. Theory Comput., 2013, 9, 4526–4541 CrossRef CAS PubMed.
- M. Huix-Rotllant, M. Filatov, S. Gozem, I. Schapiro, M. Olivucci and N. Ferré, J. Chem. Theory Comput., 2013, 9, 3917–3932 CrossRef CAS PubMed.
- M. Filatov and S. Shaik, Chem. Phys. Lett., 1999, 304, 429–437 CrossRef CAS.
- I. D. P. R. Moreira, R. Costa, M. Filatov and F. Illas, J. Chem. Theory Comput., 2007, 3, 764–774 CrossRef PubMed.
- B. P. Rand, D. Cheyns, K. Vasseur, N. C. Giebink, S. Mothy, Y. Yi, V. Coropceanu, D. Beljonne, J. Cornil, J.-L. Brédas and J. Genoe, Adv. Funct. Mater., 2012, 22, 2987–2995 CrossRef CAS.
- J. L. Brédas, J. P. Calbert, D. A. D. S. Filho and J. Cornil, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5804 CrossRef PubMed.
- A. Troisi and G. Orlandi, J. Phys. Chem. B, 2005, 109, 1849 CrossRef CAS PubMed.
- C. Groves, Energy Environ. Sci., 2013, 6, 3202–3217 CAS.
- M. Skompska and A. Szkurlat, Electrochim. Acta, 2001, 46, 4007–4015 CrossRef CAS.
- S. T. Turner, P. Pingel, R. Steyrleuthner, E. J. W. Crossland, S. Ludwigs and D. Neher, Adv. Funct. Mater., 2011, 21, 4640–4652 CrossRef CAS.
- T. S. Savenije, J. E. Kroeze, X. Yang and J. Loos, Thin Solid Films, 2006, 511–512, 2–6 CrossRef CAS.
- J. Stinchcombe, A. Pénicaud, P. Bhyrappa, P. D. W. Boyd and C. A. Reed, J. Am. Chem. Soc., 1993, 115, 5212–5217 CrossRef CAS.
- T. N. Truong and E. V. Stefanovich, Chem. Phys. Lett., 1995, 240, 253–260 CrossRef CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 5, 799–805 RSC.
- E. B. Isaacs, S. Sharifzadeh, B. Ma and J. B. Neaton, J. Phys. Chem. Lett., 2011, 2, 2531–2537 CrossRef CAS.
- D. L. Cheung and A. Troisi, J. Phys. Chem. C, 2010, 114, 20479–20488 CAS.
- J. A. Pople, D. L. Beveridge and P. A. Dobosh, J. Chem. Phys., 1967, 47, 2026–2033 CrossRef CAS.
- Y.-S. Huang, S. Westenhoff, I. Avilov, P. Sreearunothai, J. M. Hodgkiss, C. Deleener, R. H. Friend and D. Beljonne, Nat. Mater., 2008, 7, 483–489 CrossRef CAS PubMed.
- G. Raos, M. Casalegno and J. Idé, J. Chem. Theory Comput., 2014, 10, 364–372 CrossRef CAS PubMed.
- H. D. D. Gier, R. Broer and R. W. A. Havenith, Phys. Chem. Chem. Phys., 2014, 16, 12454–12461 RSC.
- A. Dreuw, J. L. Weisman and M. Head-Gordon, J. Chem. Phys., 2003, 119, 2943–2946 CrossRef CAS.
- D. J. Tozer, R. D. Amos, N. C. Handy, B. O. Roos and L. Serrano-Andrés, Mol. Phys., 1999, 97, 859–868 CrossRef CAS.
- Z.-L. Cai, K. Sendt and J. R. Reimers, J. Chem. Phys., 2002, 117, 5543–5549 CrossRef CAS.
- S. Grimme and M. Parac, ChemPhysChem, 2003, 3, 292–295 CrossRef PubMed.
- T. Stein, L. Kronik and R. Baer, J. Am. Chem. Soc., 2009, 131, 2818–2820 CrossRef CAS PubMed.
- A. Dreuw and M. Head-Gordon, J. Am. Chem. Soc., 2004, 126, 4007–4016 CrossRef CAS PubMed.
- D. J. Tozer, J. Chem. Phys., 2003, 119, 12697–12699 CrossRef CAS.
- J. Fabian, Theor. Chem. Acc., 2001, 106, 199–217 CrossRef CAS.
- M.-S. Liao, Y. Lu and S. Scheiner, J. Comput. Chem., 2003, 24, 623–631 CrossRef CAS PubMed.
- M. J. G. Peach, P. Benfield, T. Helgaker and D. J. Tozer, J. Chem. Phys., 2008, 128, 044118 CrossRef PubMed.
- N. T. Maitra, J. Chem. Phys., 2005, 122, 234104 CrossRef PubMed.
- J. Neugebauer, O. Gritsenko and E. J. Baerends, J. Chem. Phys., 2006, 124, 214102 CrossRef PubMed.
- S. L. Li and D. G. Truhlar, J. Chem. Phys., 2014, 141, 104106 CrossRef PubMed.
- D. Beljonne and J. Cornil, Multiscale Modeling of Organic and Hybrid Photovoltaics, Springer-Verlag, Berlin Heidelberg, 2014 Search PubMed.
- A. L. Sobolewski and W. Domcke, Chem. Phys., 2003, 294, 73–83 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, J. Chem. Phys., 2008, 128, 084106 CrossRef PubMed.
- L. Pandey, C. Doiron, J. S. Sears and J.-L. Brédas, Phys. Chem. Chem. Phys., 2012, 14, 14243–14248 RSC.
- I. T. Lima, C. Risko, S. G. Aziz, D. A. D. S. Filho and J.-L. Bredas, J. Mater. Chem. C, 2014, 2, 8873–8879 RSC.
- B. Yang, Y. Yi, C.-R. Zhang, S. G. Aziz, V. Coropceanu and J.-L. Brédas, J. Phys. Chem. C, 2014, 118, 27648–27656 CAS.
- M. A. Rohrdanz and J. M. Herbert, J. Chem. Phys., 2008, 129, 034107 CrossRef PubMed.
- T. Minami, I. Soichi and M. Nakano, Int. J. Quantum Chem., 2013, 113, 252–256 CrossRef CAS.
- J.-W. Song, T. Hirosawa, T. Tsuneda and K. Hirao, J. Chem. Phys., 2007, 126, 154105 CrossRef PubMed.
- D. Refaely-Abramson, R. Baer and L. Kronik, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 075144 CrossRef.
- A. V. Krukau, G. E. Scuseria, J. P. Perdew and A. Savin, J. Chem. Phys., 2008, 129, 124103 CrossRef PubMed.
- T. Körzdörfer, J. S. Sears, C. Sutton and J.-L. Brédas, J. Chem. Phys., 2011, 135, 204107 CrossRef PubMed.
- T. Stein, L. Kronik and R. Baer, J. Chem. Phys., 2009, 131, 244119 CrossRef PubMed.
- T. Stein, H. Eisenberg, L. Kronik and R. Baer, Phys. Rev. Lett., 2010, 105, 266802 CrossRef PubMed.
- N. Kuritz, T. Stein, R. Baer and L. Kronik, J. Chem. Theory Comput., 2011, 7, 2408–2415 CrossRef CAS PubMed.
- J. F. Janak, Phys. Rev. B: Condens. Matter Mater. Phys., 1978, 18, 7165–7168 CrossRef CAS.
- L. Kronik, T. Stein, S. Refaely-Abramson and R. Baer, J. Chem. Theory Comput., 2012, 8, 1515–1531 CrossRef CAS PubMed.
- T. Körzdörfer, R. M. Parrish, J. S. Sears, C. D. Sherrill and J.-L. Brédas, J. Chem. Phys., 2012, 137, 124305 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- O. A. Vydrov, J. Heyd, A. Krukau and G. E. Scuseria, J. Chem. Phys., 2006, 125, 074106 CrossRef PubMed.
- O. A. Vydrov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 234109 CrossRef PubMed.
- O. A. Vydrov, G. E. Scuseria and J. P. Perdew, J. Chem. Phys., 2007, 126, 154109 CrossRef PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1998, 108, 664–675 CrossRef CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- H. Iikura, T. Tsuneda, T. Yanai and K. Hirao, J. Chem. Phys., 2001, 115, 3540–3544 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- E. Livshits and R. Baer, Phys. Chem. Chem. Phys., 2007, 9, 2932–2941 RSC.
- R. Schueppel, K. Schmidt, C. Uhrich, K. Schulze, D. Wynands, J. L. Brédas, E. Brier, E. Reinold, H.-B. Bu, P. Baeuerle, B. Maennig, M. Pfeiffer and K. Leo, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 085311 CrossRef.
- J. C. Slater, Phys. Rev., 1951, 81, 385–390 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 110, 5121–5129 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2006, 110, 13126–13130 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- B. Hajgato, M. S. Deleuze, D. J. Tozer and F. D. Proft, J. Chem. Phys., 2008, 129, 084308 CrossRef CAS PubMed.
- G. D. Fletcher, J. Chem. Phys., 2015, 142, 134112 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2016 |