
Identification of an inhibitor of the aminoglycoside 6′-N-acetyltransferase type Ib [AAC(6′)-Ib] by glide molecular docking†
Kevin
Chiem
a,
Saumya
Jani
a,
Brooke
Fuentes
a,
David L.
Lin
a,
Madeline E.
Rasche
b and
Marcelo E.
Tolmasky
*a
aCenter for Applied Biotechnology Studies, Department of Biological Science, College of Natural Sciences and Mathematics, California State University Fullerton, Fullerton, CA 92834-6850, USA. E-mail: mtolmasky@fullerton.edu
bCenter for Applied Biotechnology Studies, Department of Chemistry and Biochemistry, College of Natural Sciences and Mathematics, California State University Fullerton, Fullerton, CA 92834-6850, USA
First published on 16th November 2015
Abstract
The aminoglycoside 6′-N-acetyltransferase type Ib, AAC(6′)-Ib, confers resistance to clinically relevant aminoglycosides and is the most widely distributed enzyme among AAC(6′)-I-producing Gram-negative pathogens. An alternative to counter the action of this enzyme is the development of inhibitors. Glide is a computational strategy for rapidly docking ligands to protein sites and estimating their binding affinities. We docked a collection of 280![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compounds from 7 sub-libraries of the Chembridge library as ligands to the aminoglycoside binding site of AAC(6′)-Ib. We identified a compound, 1-[3-(2-aminoethyl)benzyl]-3-(piperidin-1-ylmethyl)pyrrolidin-3-ol (compound 1), that inhibited the acetylation of aminoglycosides in vitro with IC50 values of 39.7 and 34.9 μM when the aminoglycoside substrates assayed were kanamycin A or amikacin, respectively. The growth of an amikacin-resistant Acinetobacter baumannii clinical strain was inhibited in the presence of a combination of amikacin and compound 1.
000 compounds from 7 sub-libraries of the Chembridge library as ligands to the aminoglycoside binding site of AAC(6′)-Ib. We identified a compound, 1-[3-(2-aminoethyl)benzyl]-3-(piperidin-1-ylmethyl)pyrrolidin-3-ol (compound 1), that inhibited the acetylation of aminoglycosides in vitro with IC50 values of 39.7 and 34.9 μM when the aminoglycoside substrates assayed were kanamycin A or amikacin, respectively. The growth of an amikacin-resistant Acinetobacter baumannii clinical strain was inhibited in the presence of a combination of amikacin and compound 1.
Introduction
Aminoglycosides are bactericidal antibiotics that act by interfering with proper decoding of the mRNA during translation.1,2 Although not all aminoglycosides bind the 16S rRNA at identical sites, once bound they all produce a modification in the conformation of the A site that results in the loss of the ribosome proofreading capabilities.3–5 Aminoglycosides are used against Gram-negative bacteria such as Pseudomonas and those belonging to the Enterobacteriaceae family. They are also used to treat some infections caused by Gram-positives, but in this case they are usually combined with other antibiotics such as beta-lactams and vancomycin.6–9 Although widely effective for treatment of numerous severe infections, aminoglycosides present toxicity issues that must be considered when using them. The main adverse effects caused by these antibiotics are nephrotoxicity (generally reversible), ototoxicity (irreversible), and rarely neuromuscular toxicity.1,6,8 Another significant limitation in the use of aminoglycosides is the recent increase in resistance, which can occur through several mechanisms such as mutation or methylation of the 16S rRNA, reduced permeability, efflux, or enzymatic modification of the antibiotic molecule, the latter of which is the most prevalent in clinical settings.10–12 More than a hundred enzymes have been isolated from bacteria that catalyze the transfer of acetyl, phosphoryl, or nucleotidyl groups into –NH2 or –OH groups of aminoglycoside molecules leading to their loss of antibiotic activity.11–13 In spite of the existence of such a large number of modifying enzymes, a limited number of them are the most prevalent in clinical isolates. Within the group of the aminoglycoside acetyltransferases, which mediate transfer of an acetyl moiety to an –NH2 group of the 2-deoxystreptamine nucleus or the sugar moieties, the aminoglycoside 6′-N-acetyltransferase type Ib [AAC(6′)-Ib] is by far the most commonly found in the hospital.2,14–16 The gene coding for this enzyme can be hosted by most Gram-negative bacteria, usually as part of integrons and transposons within plasmids and chromosomes.14–18A strategy that proved effective in extending the useful life of β-lactams against resistance mediated by β-lactamases is the development of inhibitors that are used in combination with the antibiotic.19,20 In the past years there have been several efforts to apply this approach to contain the spread of resistance to aminoglycosides.10,11,21–34 Several kinds of inhibitors of aminoglycoside acetyltransferases have been identified. Early efforts permitted to identify a multisubstrate inhibitor of an AAC(3′).35 Bisubstrate and bisubstrate-like inhibitors were later designed to target AAC(6′) enzymes but most showed low efficacy in vivo.24,26–30,34,36,37 Antimicrobial peptides were also tested as inhibitors of aminoglycoside acetyltransefrases as well as aminoglycoside phosphotransferases.31 More recently cations such as zinc or copper complexed to ionophores showed promise as inhibitors of aminoglycoside acetyltransferase enzymes.22,23,25 Although numerous compounds that inhibit different aminoglycoside modifying enzymes have been found, none has yet reached the stage of application in therapeutic formulations.10,11 One strategy to identify compounds that are potential enzymatic inhibitors is molecular docking simulation, which has permitted the discovery of such compounds in numerous cases.38,39 We have recently identified compounds that inhibit the activity of AAC(6′)-Ib using AutoDock Vina40 but they did not show any activity in vivo.21 In this work we describe the identification of an AAC(6′)-Ib inhibitor by in silico molecular docking using Glide,41,42 a software that has been used to identify compounds that bind active sites and inhibit enzymatic activity.43,44
Results and discussion
Molecular docking was carried out using the X-ray crystal structure of AAC(6′)-Ib complexed with kanamycin C and acetyl CoA (Protein Data Bank code: 1V0C)45 and a collection of 280000 compounds from 7 sub-libraries of the Chembridge library.46 All compounds were analyzed by the ‘standard precision’ glide docking followed by applying the ‘extra precision’ glide docking to the top 10% ranking compounds. This procedure resulted in a ranking based on binding affinities, of which the top 78 were tested as inhibitors of AAC(6′)-Ib. Table S1† shows the compounds tested and the percent inhibition as determined by comparing the initial velocities of reactions taking place in the presence or absence of each tested compound. Only one compound, 1-[3-(2-aminoethyl)benzyl]-3-(piperidin-1-ylmethyl)pyrrolidin-3-ol, from here on referred to as compound 1 (Fig. 1), showed complete inhibition in these tests (Table S1†).
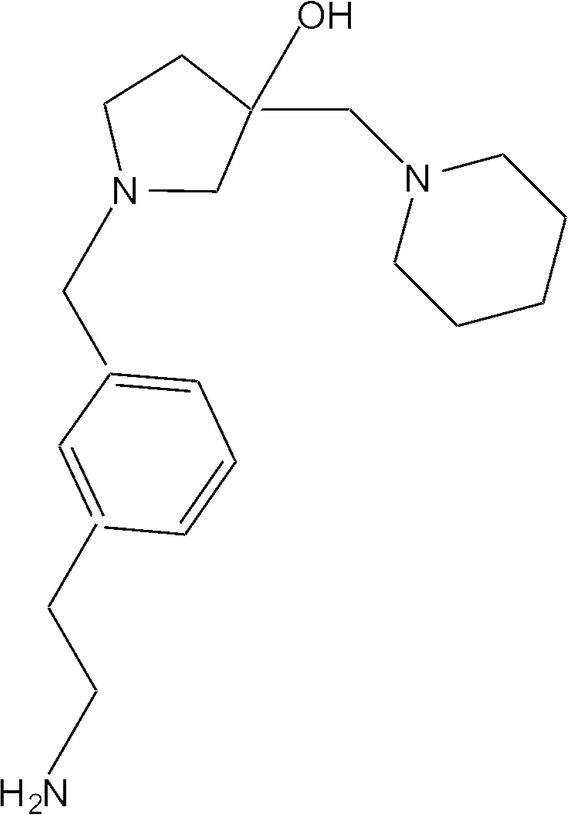 | ||
| Fig. 1 Chemical structure of 1-[3-(2-aminoethyl)benzyl]-3-(piperidin-1-ylmethyl)pyrrolidin-3-ol (compound 1). | ||
Since it is known that a variety of nonspecific compounds can form submicrometer aggregates and inhibit different enzymes,47 a reaction in the presence of 0.1% Triton X-100 was carried out to rule out non-specific protein aggregation as the cause of the observed inhibition. The level of inhibition of the acetylation reaction by compound 1 was similar in the presence and absence of Triton X-100. We then identified analogs of compound 1 using the “Show me analogs” function of the ZINC database46 at 80% identity and determined the inhibition activity of all 7 compounds identified that were not already in the first group of compounds selected by docking. Table S2† shows that although they show some inhibitory activity, none of them are more potent than compound 1. It was of interest that compound 26834434 (Table S2†) is quite similar to compound 1, yet it showed a much lower inhibitory activity. These results suggested that the substitutions at the pyrrolidine ring play an important role in the inhibitory capabilities of the compounds.
We subjected aminoglycoside molecules as well as inhibitors of aminoglycoside acetyltranferases previously described and compound 1 to extra precision docking and compared the XP GScores. Table S3† shows that all compounds assayed using this methodology could be docked to the kanamycin A binding site of AAC(6′)-Ib. All three aminoglycosides and a truncated aminoglycoside–coenzyme A bisubstrate analogue described by Gao et al.36 showed the highest binding affinity. Compound 1 exhibited the next highest affinity binding followed by chlorhexidine and several small molecules previously identified by Green et al.37 as inhibitors of the acetyltransferase Eis from Mycobacterium tuberculosis. These results validated the ability of the Glide docking program to identify potential compounds that have high binding affinity to the AAC(6′)-Ib region selected.
To determine the strength of compound 1 inhibition of the AAC(6′)-Ib enzyme we determined the half-maximal inhibitory concentration (IC50). The IC50 values determined using kanamycin A or amikacin as substrates were 39.7 and 34.9 μM, respectively (Fig. 2A and B). Kinetic analysis to determine the mode of inhibition showed uncompetitive inhibition with respect to acetyl CoA and mixed inhibition with a strong noncompetitive component with respect to kanamycin A (Fig. 2C and D). This later mechanism was confirmed by analyzing the data using the Michaelis–Menten equation variations for the different types of inhibition and determining the R2 values, an indication of goodness of fit. The highest values when the variable substrate was acetyl CoA corresponded to uncompetitive inhibition while the highest values in the case of kanamycin A were for mixed and non competitive inhibition, with mixed inhibition being a marginally better fit.
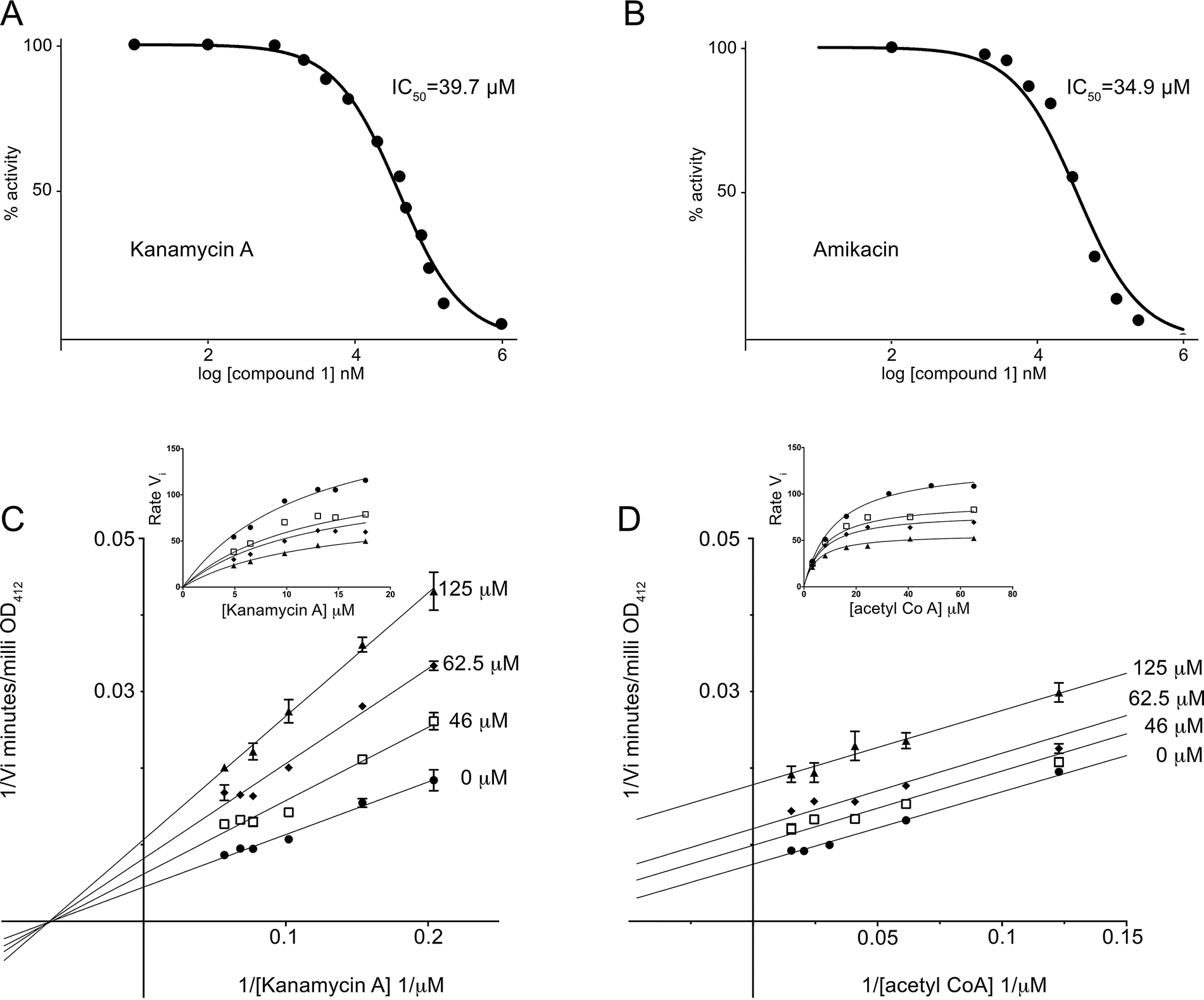 | ||
| Fig. 2 Inhibition of AAC(6′)-Ib. A and B. The percentage of acetylating activity of kanamycin A (A) and amikacin (B) was calculated for reaction mixtures containing different concentrations of compound 1. C and D. Lineweaver–Burk plots obtained carrying out reactions containing variable concentrations of kanamycin A (C) or acetyl CoA (D). Insets are non linear fitting with the Michaelis–Menten function modified for mixed inhibition (C) or uncompetitive inhibition (D). | ||
It has been previously reported that the acetylation reaction catalyzed by AAC(6′)-Ib occurs through an ordered sequential mechanism where addition of acetyl CoA is followed by the aminoglycoside substrate.45 Then, the inhibition patterns observed are consistent with a model in which following addition of acetyl CoA compound 1 binds the enzyme at one or more locations, not necessarily at the aminoglycoside binding site. These results were somewhat unexpected because the docking conditions to select potential inhibitors had been chosen to identify compounds with affinity to the kanamycin A binding site. Therefore, we extended the analysis to the complete protein structure to determine if there is another binding site for compound 1. We found one potential binding pocket outside of the kanamycin A and acetyl CoA binding sites. However, docking compound 1 to this site showed low predicted binding affinity. We do not know why compound 1, which acts as an inhibitor but seems to behave unexpectedly with respect to its binding characteristics, was identified by the Glide software. Further structural analysis may help clarify this apparent inconsistency.
The ability of compound 1 to inhibit growth of aac(6′)-Ib-harboring bacterial cells cultured in the presence of amikacin was tested in cultures of two clinical Klebsiella pneumoniae and Acinetobacter baumannii isolates. Addition of compound 1 to K. pneumoniae cultures containing amikacin did not change the growth pattern. However, A. baumannii A155 growth curves carried out in the presence of 6 or 8 μg ml−1 amikacin and 50 or 100 μM compound 1 showed that addition of 50 μM compound 1 resulted in complete growth inhibition when the amikacin concentration in the medium was 8 μg ml−1 (Fig. 3). Lowering the concentration of the antibiotic to 6 μg ml−1 required 100 μM compound 1 to inhibit growth (Fig. 3). Fig. 3 also shows that, as it has been observed before, there is a longer lag phase when amikacin is added to the growth medium but the same OD600 is reached at the stationary phase. We do not know why compound 1 did not show any effect when added to K. pneumoniae cultures. Possible explanations are that the compound cannot penetrate the capsule and/or cell wall and does not reach the cytoplasm at sufficient concentrations to exert the inhibitory effect or the presence of multiple copies of the gene, which in the tested strain is present in a high copy number plasmid, mediate synthesis of higher quantities of the enzyme that are enough to confer resistance in the presence of the inhibitor.
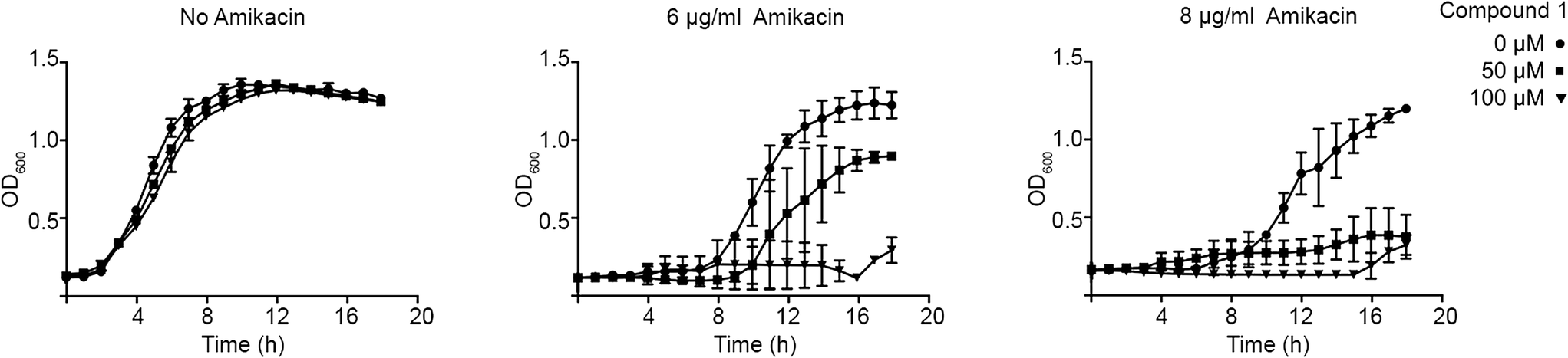 | ||
| Fig. 3 Effect of compound 1 on resistance to amikacin. A. baumannii A155 cells were cultured in 100 μl Mueller–Hinton broth in microtiter plates at 37 °C with the additions indicated in the figure, and the OD600 was periodically determined. | ||
Conclusions
In silico docking can be a viable strategy to identify enzyme inhibitors.21,48–52 We recently identified robust inhibitors of AAC(6′)-Ib using the molecular docking program Autodock Vina. These compounds are derivatives of barbituric acid that exhibit competitive inhibition against the aminoglycoside substrate but failed to inhibit the growth of resistant Gram-negative bacteria that harbor aac(6′)-Ib when added to cultures in combination with amikacin.21 This failure to enhance susceptibility of bacteria in culture could be due to deficient uptake or degradation inside the cells. In the search for other potential inhibitors, we analyzed a larger subset of compounds using Glide. As a result, we identified compound 1, which was able to inhibit the acetylation reaction in vitro. Furthermore, when compound 1 was combined with amikacin, it inhibited growth of an A. baumannii clinical isolate that harbors aac(6′)-Ib in its chromosome. Although the concentration required to inhibit growth was higher than one would desire, compound 1 could serve as a lead compound for designing more powerful inhibitors. Interestingly, compound 1, was unable to reverse resistance to amikacin of a K. pneumoniae strain that harbors the aac(6′)-Ib gene in a high-copy number plasmid. Whether this lack of activity is due to a permeability issue or the presence of high concentrations of enzyme inside the cells due to a gene dosage effect is being studied.Experimental section
Cloning, expression, and purification of AAC(6′)-Ib
The aac(6′)-Ib gene from pJHCMW1 (ref. 53) was amplified using the oligonucleotides CACCGCCGACATGATCC (nucleotides 6777–6793, accession number AF47977) and TCTAGAGTCGATACTTTCGCGTCACCGC (nucleotides 7983–8008 plus a termination codon to preclude a fusion to the tag included in the plasmid vector sequence) as primers. The fragment was inserted into the expression vector pBAD102 as recommended by the supplier (Life Technologies), generating a construct that codes for the AAC(6′)-Ib protein without a fusion to thioredoxin.54 Then, the aac(6′)-Ib gene was modified by site directed mutagenesis using the QuikChange II kit (Stratagene) to add a sequence coding for the carboxy-terminal hydrophilic extension of seven amino acids as described by Maurice et al.55 This modification increases solubility without changing the properties of the enzyme.55 The resulting recombinant plasmid, pBADMW131, was introduced in E. coli TOP10 and the gene was expressed by adding arabinose (0.2%) to log-phase cultures followed by incubation for 6 h at 30 °C. Cells were harvested by centrifugation, resuspended in Tris pH 7.5 buffer (50 mM), lysed by sonication, and the soluble lysate recovered after centrifugation was subjected to DEAE Sepharose ionic exchange chromatography eluting with a NaCl gradient (0–1 M) as described previously.21 This preparation was further purified by size exclusion chromatography using Bio-Gel P-30 polyacrylamide resins (Biorad) and eluting with a buffer containing Tris pH 7.5 buffer (50 mM) and NaCl (100 mM).Enzyme activity and kinetics
Enzymatic activity of AAC(6′)-Ib was determined using a method based on monitoring the increase in OD412 after 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) reacts with the CoA-SH released from acetyl CoA after acetylation of the aminoglycoside37 as described before.21 Briefly, the standard reaction mix contained acetyl CoA (150 μM), DTNB (0.2 mM), Tris-HCl pH 7.5 buffer (20 mM), and kanamycin A (18 μM), and when needed the testing compound at the indicated concentrations was incubated for 10 minutes at room temperature followed by addition of the enzyme. Acetylation was followed using a BioTek Synergy 2 plate reader monitoring absorbance at 412 nm. Initial velocities (Vi) were calculated using the Gen 5 software, version 2.01.13. Inhibition was assessed by comparison of initial velocities of acetylation reactions in the presence or absence of the compound (500 μM) to be tested. Apparent inhibition through protein aggregation was discarded by carrying out the reaction in the presence of Triton X-100 (0.1%). Results are averages of three separate experiments. Mode of inhibition was determined using Lineweaver–Burk plots and the Michaelis–Menten equation variations for the different types of inhibition using GraphPad Prism 6 software. Data were obtained by performing a series of reactions in the presence of a range of inhibitor concentrations adding one substrate at a constant excess concentration and the other at different concentrations as described before.21 When kanamycin A was the variable substrate the concentrations used ranged from 3 to 18 μM while acetyl CoA was present at 100 μM. When acetyl CoA was the variable substrate the concentrations ranged from 5 to 72 μM and kanamycin A was present at 25 μM.Molecular docking
Molecular docking was carried using the X-ray crystal structure of AAC(6′)-Ib complexed with kanamycin C and acetyl CoA retrieved from the Protein Data Bank (code: 1V0C),45 the molecular docking program Glide (grid-based ligand docking with energetics),41,42 and a collection of 280000 compounds from 7 sub-libraries (0, 2, 3, 5, 6, 9, and p1.0) of the Chembridge library.46 The structure of AAC(6′)-Ib bound to kanamycin C and acetyl CoA was retrieved and prepared for analysis by removing both substrates and all water molecules and assigning bond orders, creating zero-order bonds to metals, and creating disulfide bonds using the “protein prep” tool of the Glide program. Then, a grid box with coordinates X = 16, Y = 12, and Z = 13 was generated at the kanamycin A binding site. Before docking, the compounds of all 7 sub-libraries were prepared using the LigPrep and Epik tools of the Glide program. For docking compounds that were not present in the ZINC library such as inhibitors published previously (see Table S3†), the chemical structures were built in the program using the Build panel before using the LigPrep and Epik tools. The compounds were docked with the kanamycin A binding site using the ‘standard precision’ glide docking and ranked. The top 10% ranking compounds were then docked with the kanamycin A binding site using the ‘extra precision’ glide docking. The 78 top ranking compounds were tested as inhibitors of the AAC(6′)-Ib-mediated acetylation reaction. Other bindings sites outside of the region selected originally were identified using the ICM Pocket finder method on Molsoft.56 Compounds tested as potential inhibitors were purchased from ChemBridge Corp (San Diego, CA) and dissolved in 100% dimethyl sulfoxide (DMSO). Compound 1 is a mixture of the R and S enantiomers (only the –OH group has stereospecificity).
Acknowledgements
This work was supported by Public Health Service grant 2R15AI047115-04 from the National Institute of Allergy and Infectious Diseases, National Institutes of Health. We thank Christopher Meyer and Ali Saleh for helpful discussions on the mechanism of inhibition of compound 1.References
- P. Veyssier and A. Bryskier, in Antimicrobial Agents, ed. A. Bryskier, ASM Press, Washington DC, 2005, pp. 453–469 Search PubMed.
- S. B. Vakulenko and S. Mobashery, Clin. Microbiol. Rev., 2003, 16, 430–450 CrossRef CAS PubMed.
- S. Jana and J. K. Deb, Appl. Microbiol. Biotechnol., 2006, 70, 140–150 CrossRef CAS PubMed.
- J. M. Ogle and V. Ramakrishnan, Annu. Rev. Biochem., 2005, 74, 129–177 CrossRef CAS PubMed.
- H. S. Zaher and R. Green, Cell, 2009, 136, 746–762 CrossRef CAS PubMed.
- M. L. Avent, B. A. Rogers, A. C. Cheng and D. L. Paterson, Intern. Med. J., 2011, 41, 441–449 CrossRef CAS PubMed.
- J. Jackson, C. Chen and K. Buising, Curr. Opin. Infect. Dis., 2013, 26, 516–525 CrossRef CAS PubMed.
- J. Yao and R. Moellering, in Manual of Clinical Microbiology, ed. P. Murray, E. Baron, J. Jorgensen, M. Landry and M. Pfaller, ASM Press, Washington, DC, 2007, pp. 1007–1113 Search PubMed.
- P. Poulikakos and M. E. Falagas, Expert Opin. Pharmacother., 2013, 14, 1585–1597 CrossRef CAS PubMed.
- K. J. Labby and S. Garneau-Tsodikova, Future Med. Chem., 2013, 5, 1285–1309 CrossRef CAS PubMed.
- M. S. Ramirez and M. E. Tolmasky, Drug Resist. Updates, 2010, 13, 151–171 CrossRef CAS PubMed.
- K. J. Shaw, P. N. Rather, R. S. Hare and G. H. Miller, Microbiol. Rev., 1993, 57, 138–163 CAS.
- E. Azucena and S. Mobashery, Drug Resist. Updates, 2001, 4, 106–117 CrossRef CAS PubMed.
- M. S. Ramirez, N. Nikolaidis and M. E. Tolmasky, Front Microbiol., 2013, 4, 121 Search PubMed.
- M. E. Tolmasky, R. M. Chamorro, J. H. Crosa and P. M. Marini, Antimicrob. Agents Chemother., 1988, 32, 1416–1420 CrossRef CAS PubMed.
- M. Woloj, M. E. Tolmasky, M. C. Roberts and J. H. Crosa, Antimicrob. Agents Chemother., 1986, 29, 315–319 CrossRef CAS PubMed.
- M. E. Tolmasky and J. H. Crosa, Antimicrob. Agents Chemother., 1987, 31, 1955–1960 CrossRef CAS PubMed.
- G. Warburg, C. Hidalgo-Grass, S. R. Partridge, M. E. Tolmasky, V. Temper, A. E. Moses, C. Block and J. Strahilevitz, J. Antimicrob. Chemother., 2012, 67, 898–901 CrossRef CAS PubMed.
- S. M. Drawz, K. M. Papp-Wallace and R. A. Bonomo, Antimicrob. Agents Chemother., 2014, 58, 1835–1846 CrossRef PubMed.
- R. R. Watkins, K. M. Papp-Wallace, S. M. Drawz and R. A. Bonomo, Front Microbiol., 2013, 4, 392 Search PubMed.
- D. L. Lin, T. Tran, C. Adams, J. Y. Alam, S. R. Herron and M. E. Tolmasky, Bioorg. Med. Chem. Lett., 2013, 23, 5694–5698 CrossRef CAS PubMed.
- D. L. Lin, T. Tran, J. Y. Alam, S. R. Herron, M. S. Ramirez and M. E. Tolmasky, Antimicrob. Agents Chemother., 2014, 58, 4238–4241 CrossRef CAS PubMed.
- Y. Li, K. D. Green, B. R. Johnson and S. Garneau-Tsodikova, Antimicrob. Agents Chemother., 2015, 59, 4148–4156 CrossRef CAS PubMed.
- T. Lombes, G. Begis, F. Maurice, S. Turcaud, T. Lecourt, F. Dardel and L. Micouin, ChemBioChem, 2008, 9, 1368–1371 CrossRef CAS PubMed.
- K. Chiem, B. A. Fuentes, D. L. Lin, T. Tran, A. Jackson, M. S. Ramirez and M. E. Tolmasky, Antimicrob. Agents Chemother., 2015, 59, 5851–5853 CrossRef CAS PubMed.
- F. Gao, X. Yan, O. M. Baettig, A. M. Berghuis and K. Auclair, Angew. Chem., Int. Ed., 2005, 44, 6859–6862 CrossRef CAS PubMed.
- F. Gao, X. Yan, O. Zahr, A. Larsen, K. Vong and K. Auclair, Bioorg. Med. Chem. Lett., 2008, 18, 5518–5522 CrossRef CAS PubMed.
- F. Gao, X. Yan and K. Auclair, Chemistry, 2009, 15, 2064–2070 CrossRef CAS PubMed.
- K. Vong and K. Auclair, Med. Chem. Commun., 2012, 3, 397–407 RSC.
- K. Vong, I. S. Tam, X. Yan and K. Auclair, ACS Chem. Biol., 2012, 7, 470–475 CrossRef CAS PubMed.
- D. D. Boehr, K. A. Draker, K. Koteva, M. Bains, R. E. Hancock and G. D. Wright, Chem. Biol., 2003, 10, 189–196 CrossRef CAS PubMed.
- K. T. Welch, K. G. Virga, N. A. Whittemore, C. Ozen, E. Wright, C. L. Brown, R. E. Lee and E. H. Serpersu, Bioorg. Med. Chem., 2005, 13, 6252–6263 CrossRef CAS PubMed.
- M. Liu, J. Haddad, E. Azucena, L. P. Kotra, M. Kirzhner and S. Mobashery, J. Org. Chem., 2000, 65, 7422–7431 CrossRef CAS PubMed.
- M. L. Magalhaes, M. W. Vetting, F. Gao, L. Freiburger, K. Auclair and J. S. Blanchard, Biochemistry, 2008, 47, 579–584 CrossRef CAS PubMed.
- J. W. Williams and D. B. Northrop, J. Antibiot., 1979, 32, 1147–1154 CrossRef CAS PubMed.
- F. Gao, X. Yan, T. Shakya, O. M. Baettig, S. Ait-Mohand-Brunet, A. M. Berghuis, G. D. Wright and K. Auclair, J. Med. Chem., 2006, 49, 5273–5281 CrossRef CAS PubMed.
- K. D. Green, W. Chen and S. Garneau-Tsodikova, ChemMedChem, 2012, 7, 73–77 CrossRef CAS PubMed.
- G. Sliwoski, S. Kothiwale, J. Meiler and E. W. Lowe, Jr., Pharmacol. Rev., 2014, 66, 334–395 CrossRef PubMed.
- Y. C. Chen, Trends Pharmacol. Sci., 2015, 36, 78–95 CrossRef CAS PubMed.
- L. K. Wolf, Chem. Eng. News, 2009, 87, 48 Search PubMed.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P. Francis and P. S. Shenkin, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS PubMed.
- T. A. Halgren, R. B. Murphy, R. A. Friesner, H. S. Beard, L. L. Frye, W. T. Pollard and J. L. Banks, J. Med. Chem., 2004, 47, 1750–1759 CrossRef CAS PubMed.
- S. K. Tripathi, R. Muttineni and S. K. Singh, J. Theor. Biol., 2013, 334, 87–100 CrossRef CAS PubMed.
- G. Ozbuyukkaya, E. O. Olmez and K. O. Ulgen, J. Enzyme Inhib. Med. Chem., 2014, 29, 137–145 CrossRef CAS PubMed.
- M. W. Vetting, C. H. Park, S. S. Hegde, G. A. Jacoby, D. C. Hooper and J. S. Blanchard, Biochemistry, 2008, 47, 9825–9835 CrossRef CAS PubMed.
- J. J. Irwin, T. Sterling, M. M. Mysinger, E. S. Bolstad and R. G. Coleman, J. Chem. Inf. Model., 2012, 52, 1757–1768 CrossRef CAS PubMed.
- S. L. McGovern, B. T. Helfand, B. Feng and B. K. Shoichet, J. Med. Chem., 2003, 46, 4265–4272 CrossRef CAS PubMed.
- N. Dessalew and S. K. Singh, Med. Chem., 2008, 4, 313–321 CrossRef CAS.
- G. M. Morris, L. G. Green, Z. Radic, P. Taylor, K. B. Sharpless, A. J. Olson and F. Grynszpan, J. Chem. Inf. Model., 2013, 53, 898–906 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- S. Vadivelan, B. N. Sinha, S. J. Irudayam and S. A. Jagarlapudi, J. Chem. Inf. Model., 2007, 47, 1526–1535 CrossRef CAS PubMed.
- K. Yelekci, O. Karahan and M. Toprakci, J. Neural Transm., 2007, 114, 725–732 CrossRef CAS PubMed.
- R. Sarno, G. McGillivary, D. J. Sherratt, L. A. Actis and M. E. Tolmasky, Antimicrob. Agents Chemother., 2002, 46, 3422–3427 CrossRef CAS PubMed.
- A. Pourreza, M. Witherspoon, J. Fox, J. Newmark, D. Bui and M. E. Tolmasky, Antimicrob. Agents Chemother., 2005, 49, 2979–2982 CrossRef CAS PubMed.
- F. Maurice, I. Broutin, I. Podglajen, P. Benas, E. Collatz and F. Dardel, EMBO Rep., 2008, 9, 344–349 CrossRef CAS PubMed.
- J. An, M. Totrov and R. Abagyan, Mol. Cell. Proteomics, 2005, 4, 752–761 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5md00316d |
| This journal is © The Royal Society of Chemistry 2016 |