DOI:
10.1039/C5MB00718F
(Paper)
Mol. BioSyst., 2016,
12, 162-168
Molecular modeling approach to explore the role of cathepsin B from Hordeum vulgare in the degradation of Aβ peptides†
Received
25th October 2015
, Accepted 4th November 2015
First published on 4th November 2015
Abstract
The pathological hallmark of Alzheimer's disease is the accumulation of Aβ peptides in human brains. These Aβ peptides can be degraded by several enzymes such as hACE, hECE, hIDE and cathepsin B. Out of which cathepsin B also belongs to the papain super family and has been found in human brains, it has a role in Aβ peptide degradation through limited proteolysis. The Aβ concentrations are maintained properly by its production and clearance via receptor-mediated cellular uptake and direct enzymatic degradation. However, the reduced production of Aβ degrading enzymes as well as their Aβ degrading activity in human brains initiate the process of accumulation of Aβ peptides. So it becomes essential to investigate the molecular interactions involved in the process of Aβ degradation in detail at the atomic level. Hence, homology modeling, molecular docking and molecular dynamics simulation techniques have been used to explore the possible role of cathepsin B from Hordeum vulgare in the degradation of amyloid beta (Aβ) peptides. The homology model of cathepsin B from Hordeum vulgare shows good similarity with human cathepsin B. Molecular docking and MD simulation results revealed that the active site residues Cys32, HIS112, HIS113 are involved in the catalytic activity of cathepsin B. The sulfhydryl group of the Cys32 residue of cathepsin B from Hordeum vulgare cleaves the Aβ peptide from the carboxylic end of Glu11. Hence, this structural study might be helpful in designing alternative strategies for the treatment of AD.
Introduction
Cathepsin B belongs to the peptidase family and it has an important role in intracellular protein degradation. The proteins entering in the endolysosomal system are degraded by cathepsin B.1 Amyloid beta (Aβ) peptides play a central role in the pathogenesis of Alzheimer's disease, its accumulation in AD brains is due to the overproduction or insufficient clearance due to defects in the proteolytic degradation.2,3 The Aβ(1–42) peptide appears to be particularly critical in AD pathogenesis.4–6 The familial autosomal dominant (FAD) mutations and the defects in the proteolytic degradation results in an increase in the production or relative abundance of Aβ(1–42) in AD brains.7,8 Hence, degradation and clearance of Aβ peptides could be a promising therapeutic approach for the treatment of AD. Cathepsin B reduces Aβ levels by inducing C-terminal truncation of Aβ(1–42) which suggests that endogenous cathepsin B activity could reduce the levels of Aβ, especially Aβ1–42, and protect against AD-related deficits.9 ACE, ECE, IDE, neprilysin, plasmin, and MMP-9 are enzymes which have been implicated in cerebral Aβ clearance.10–14 Reducing inhibitor levels such as cystatin C can also enhance cathepsin B enzymatic activity.15 The people suffering from neurodegenerative disorders show abnormal distribution of cathepsin B and its endogenous inhibitors.16 Studies showed that the balance between cathepsin B and its endogenous inhibitors (secretory cystatin C and cystatin B) are important for the dissolution of amyloid plaques and the clearance of toxic oligomers.17 Human cathepsin B has certain limitations such as mutation, effect of inhibitors and direct oxidative damage which would lower cathepsin B activity.18 So there is a need to find the enzyme from a novel source which can be used to degrade Aβ peptides and control its deposition.
Hence, the structure of cathepsin B from the plant source Hordeum vulgare shows high homology with human cathepsin B19 and it could be useful to find out the degradation mechanism of Aβ peptides by plant cathepsin B which can be useful to develop new strategies against AD. So we developed a three-dimensional structure of cathepsin B from a plant source (Hordeum vulgare) using Modeller 9V7 software. The energy minimization of the predicted model was done using the steepest descent method to remove steric clashes. The structural validation was also done through various online servers which confirmed the good quality of the predicted structure. The sequence analysis and 3D model comparison between plant cathepsin B from Hordeum vulgare and human cathepsin B shows that they have high homology with each other. The molecular dynamic simulation confirms the stability of the predicted model. Molecular docking and MD simulation studies revealed a similar structural fold between plant cathepsin B and human cathepsin B. Hence, the predicted model of cathepsin B from the plant source could be important in the studies of Aβ peptide degradation.
Methods
Software used for the prediction and validation of three dimensional structures
The three dimensional model of plant cathepsin B from Hordeum vulgare was built by using SWISSMODEL,20 Geno3D,21 and MODELLER 9v7.22 Then the evaluation of the models was done by PROCHECK,23 PROSA24 and Verify-3D.25 The molecular docking study was performed with AutoDock 4.2.26 Structure visualization and analysis were carried out using Chimera.27
Sequence alignment, secondary structure analysis and homology modeling
The homology model of cathepsin B from Hordeum vulgare was built by using human cathepsin B as a template. The FASTA sequence of cathepsin B from Hordeum vulgare was retrieved from the NCBI protein sequence database (Accession No. BAK03491.1). The BLAST program was used to search for suitable templates available in the PDB. Finally cathepsin B from human (PDB ID: 2IPP) with a resolution of 2.15 Å was used as a template to build the model. The pairwise sequence alignment of the template sequence (2IPP) and the cathepsin B from Hordeum vulgare sequence was done using the EMBOSS program.28 Then the secondary structure analysis of the cathepsin B sequence from Hordeum vulgare was done by the “SOPMA” (Self-Optimized Prediction Method) program.29 Finally homology modeling of cathepsin B from Hordeum vulgare was performed by three homology modeling programs; SWISS-MODEL,20 Geno3D21 and Modeller9V7.22 The steepest descent energy minimization method30 was used to remove steric clashes.
Three dimensional model refinement and validation
The models obtained by SWISS-MODEL,20 Geno3D21 and MODELLER 9V722 were validated by inspecting the Ramachandran plot31 obtained from the PROCHECK analysis.23 After analysis of the models derived from SWISS-MODEL,20 Geno3D21 and MODELLER 9V722 the model constructed by MODELLER was finally chosen for further investigations on the basis of its geometry and 3D alignment with the template.
The PROSA24 server analysis is carried out to check the energy criteria in comparison with the potential of mean force derived from a large set of known protein structures. The second test was done to apply the energy criteria using a ProSA II energy plot to investigate whether the interaction energy of each residue with the remainder of the protein is negative. Furthermore, the compatibility of the model with its sequence was measured by Verify-3D.25 In Verify-3D,25 the vertical axis shows the average 3D–1D protein score for each amino acid in a 21 residue sliding window which would be useful to validate the predicted model. Furthermore, the Ramachandran plot was obtained through the PROCHECK analysis.23 The predicted model is compared with the template structure by using the PDBeFOLD server32 and also by using Chimera.27
Prediction of active site
The active site prediction in the refined model was done by automatic sequence alignment. We aligned the sequences of cathepsin B from Hordeum vulgare with the sequence of human cathepsin B (2IPP) for which the active site is known.19 The 3D alignment between cathepsin B from Hordeum vulgare and human cathepsin B (PDB ID: 2IPP) was done by Chimera27 to compare the active site pocket.
Molecular dynamic simulation of the predicted model of cathepsin B from Hordeum vulgare
The molecular dynamic simulation was performed using the Gromacs 4.0.4 program30 with an OPLS-AA force field.33,34 The homology model of cathepsin B from Hordeum vulgare built by MODELLER was used as a starting model for the MD. The complex was surrounded by a 0.9 Å layer of water and the system was neutralized with 14 Na+ ions. The solvated structure was minimized by the steepest descent method for 5730 steps at 300 K temperature and constant pressure followed by equilibration. After 1 ns equilibration of predicted model along with water molecules, the system was further simulated for period of 5 ns at 300k. The PME algorithms were used during simulation.30
Molecular docking of the simulated model of plant cathepsin B and Aβ peptide
The molecular docking was performed between the simulated model of plant cathepsin B and a patch of the Aβ peptide (10YDVHHNKLVFF20) from 1AML.pdb35 using AutoDock 4.0.26 The initial receptor and ligand structures were minimized by the steepest descent (SD) method using Gromacs 4.0.4 to remove the internal strain energy.30 The Lamarckian genetic algorithm (LGA) has been used in this study. The active site was defined using Auto Grid.26 The grid size was set to 48 × 44 × 54 points with a grid spacing of 0.375 Å centered on the selected flexible residues present in the active site of plant cathepsin B. The grid box contains the entire binding site of plant cathepsin B and provides enough space for translation and rotation of the Aβ peptide. A step size of 2 Å for translation and 50° for rotation were chosen and the maximum number of energy evaluations was set to 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500000. Thus, ten runs were performed. For each of the 10 independent runs, a maximum number of 27000 GA operations were generated on a single population of 150 individuals.
500000. Thus, ten runs were performed. For each of the 10 independent runs, a maximum number of 27000 GA operations were generated on a single population of 150 individuals.
Similarly, the whole Aβ peptide (1AML.pdb) was also docked with plant cathepsin B using AutoDock 4.0.26 The same protocol was followed as per the earlier docking between plant cathepsin B and a patch of the Aβ peptide. The same rigid and flexible files were used while the whole Aβ peptide was used as a ligand. The grid size was set to 50 × 50 × 50 points with a grid spacing of 0.375 Å centered on the selected flexible residues present in the active site of plant cathepsin B. The remaining parameters were kept similar to the earlier docking.
MD simulation of the docked complex of the simulated model of cathepsin B from Hordeum vulgare and amyloid beta peptide
Furthermore the docked complex of cathepsin B Hordeum vulgare and Aβ peptide 10YDVHHNKLVFF20 was simulated with the Gromacs 4.0.4 program30 using an OPLS-AA force field.33,34 The complex was surrounded with a 0.9 Å layer of water and the system was neutralized with 14 Na+ ions. The solvated structure was then minimized by the steepest descent method for 3704 steps at 300 K temperature and constant pressure, followed by equilibration. After 1 ns equilibration of docked complex, the whole system was further simulated for period of 50 ns at 300k. The PME algorithms were used during simulation.30 All of the MD simulation studies were performed on a HP Workstation. A structural comparison between the initial structure and final structure of the MD simulation was done using PDBeFOLD.32 All of the dynamic runs were then visualized using the VMD (Visual Molecular Dynamics) package.36 Images after simulation were generated using Chimera27 and PyMOL. Then the structure of cathepsin B from Hordeum vulgare and its docked complex with the Aβ peptide 10YDVHHNKLVFF20 was analyzed by various software, like “RasMol”37 “PyMOL” (http://PyMOL. sourceforge.net/) and “Chimera”.27
Results and discussion
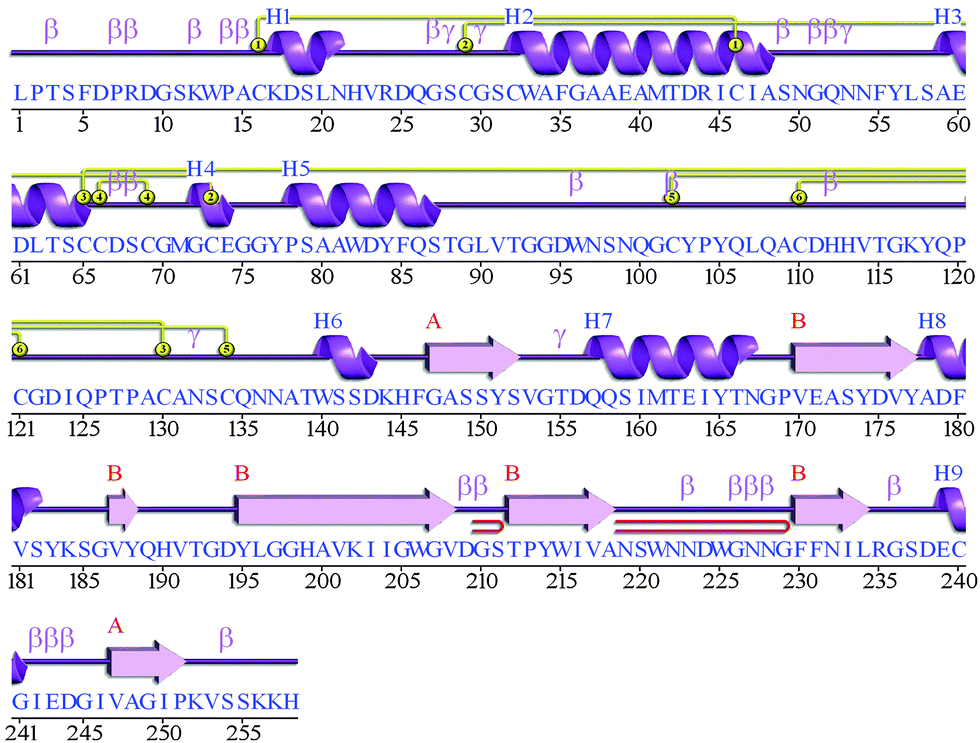
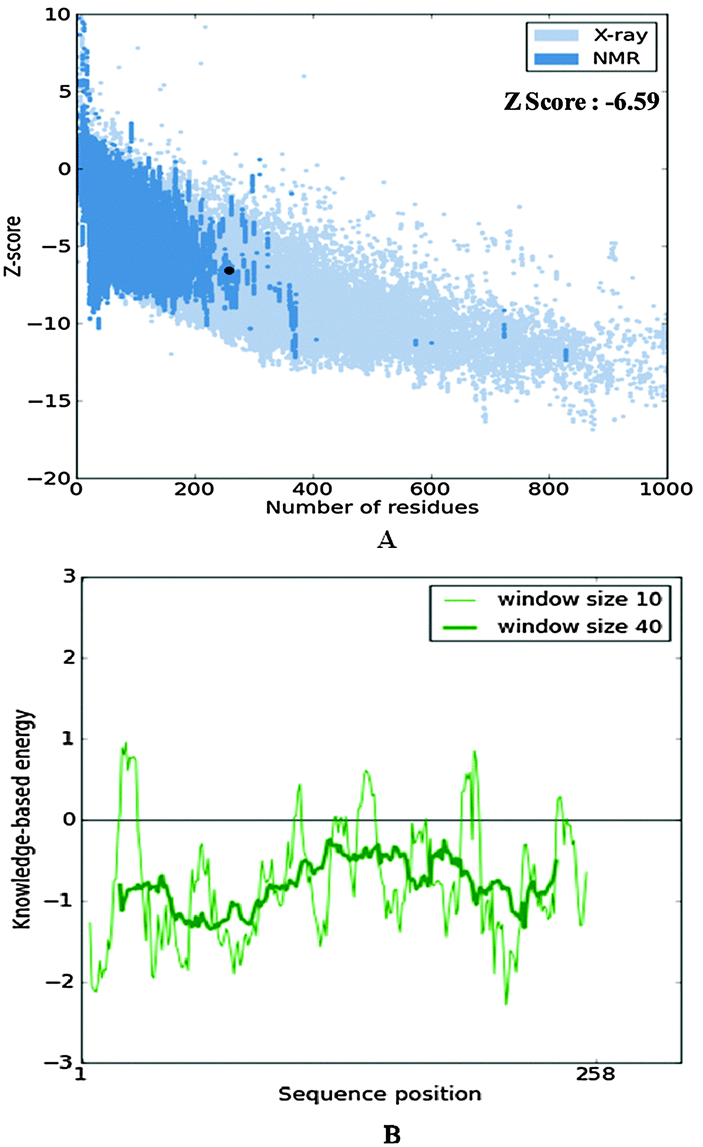
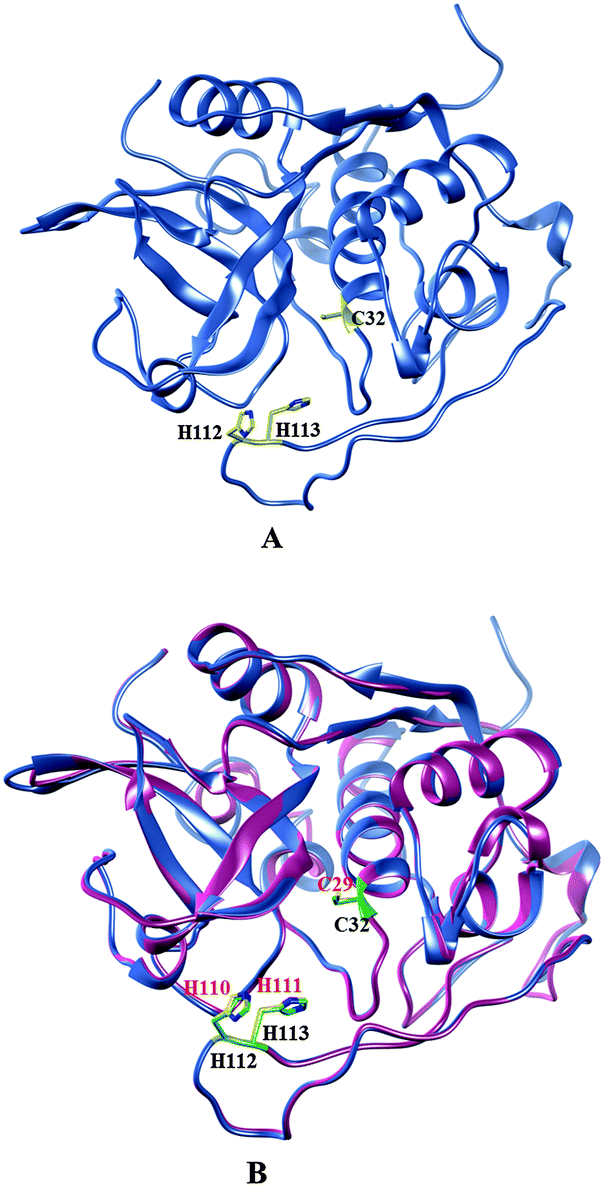
The secondary structure analysis of the cathepsin B sequence from Hordeum vulgare by the “SOPMA” method shows 18.22%, 54.65%, 17.44%, and 9.69% amino acids residues in the form of a helix, random coil, extended strand and beta turn respectively (Fig. S1, ESI†). The SOPMA analysis showed that the secondary structure element which occupied the largest part of the protein was the random coil followed by alpha helix, extended strand and beta turns for plant cathepsin B (Fig. S1, ESI†). The structure analysis was done by the PDBsum server38 which also showed that the random coil occupied the largest part of the protein followed by alpha helix, extended strand and beta turns (Fig. 1 and Fig. S2, ESI†). There was a 56% alignment (Fig. 2) between the target sequence of cathepsin B from Hordeum vulgare and the sequence of human cathepsin B (2IPP.pdb).19 The PROCHECK23 validation was done for all of the models of cathepsin B from Hordeum vulgare obtained by Geno3d (Table 1). Furthermore the three dimensional model of cathepsin B from Hordeum vulgare was predicted through the Swiss model server. The PROCHECK23 analysis was done for the model predicted by the Swiss model server (Table 1). Finally the three-dimensional structure of cathepsin B from Hordeum vulgare was modeled by comparative protein modeling methods using MODELLER 9V7 software.22 The stereochemical quality of the cathepsin B models was evaluated with the help of a Ramachandran plot using PROCHECK.23 Based on this plot the model contained 87.9% residues in the most favored regions, 10.7% of amino acids were present in allowed regions and only 1.4% of residues were in disallowed regions, a total of 98.6% of residues are present in favored regions (Fig. S3, ESI†). Hence, the PROCHECK23 analysis results are better for model predicted by MODELLER 9V722 rather than the SWISS MODEL20 and Geno3D.21 Another test ProSA24 was done for additional confirmation of the good quality of the cathepsin B model from Hordeum vulgare predicted by MODELLER 9V7.22 The Z score given by ProSA24 for cathepsin B from Hordeum vulgare predicted by MODELLER 9V722 was −6.59, which is in the range of native conformation and also the point given by the ProSA24 tool was found to be within the acceptable range of X-ray and NMR studies (Fig. 3A). The analysis of the cathepsin B model from Hordeum vulgare by PROSA24 showed negative interaction energy for maximum residues whereas very few residues displayed positive interaction energy (Fig. 3B). When comparing the structure of cathepsin B from Hordeum vulgare built by MODELLER22 with the template structure using PDBeFOLD32 and Chimera27 they showed good similarity (Fig. 4B). The sequence alignment and 3D comparison of cathepsin B from Hordeum vulgare with human cathepsin B (2IPP)19 shows that the active site residues (CYS32, HIS112, and HIS113) of cathepsin B from Hordeum vulgare are exactly the same to the active site residues (CYS29, HIS110, and HIS111) of human cathepsin B (Fig. 2 and 4B). The active site pocket of cathepsin B from Hordeum vulgare was also found to be similar to human cathepsin B (2IPP)19 (Fig. 4B). Therefore the residues CYS32, HIS112, and HIS113 might act as active site residues for cathepsin B from Hordeum vulgare.
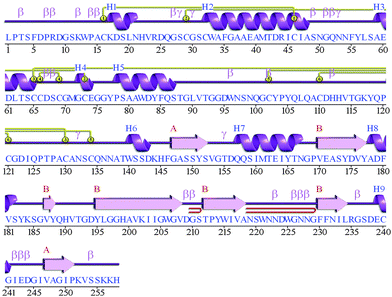 |
| | Fig. 1 Structural information obtained by the PDBsum server for the predicted model of cathepsin B from Hordeum vulgare. The letter H (blue) is for helices, strands are shown by A and B (red), beta and gamma turns are represented by β and γ respectively. Yellow circles show the disulphide bonds between residues. | |
 |
| | Fig. 2 Sequence alignment between cathepsin B from Hordeum vulgare and template (2IPP) catalytic residues are (red) indicated by arrows. | |
Table 1 The PROCHECK analysis for the models of plant cathepsin B obtained by Geno3D
| Model no. |
Residues in core region (%) |
Residues in allowed region (%) |
Residues in generously allowed region (%) |
Residues in disallowed region (%) |
| Model 1 |
65.5 |
27.4 |
4.2 |
3.0 |
| Model 2 |
63.1 |
31.5 |
4.2 |
1.2 |
| Model 3 |
65.5 |
30.4 |
3.6 |
0.6 |
| Model 4 |
65.5 |
29.8 |
3.0 |
1.8 |
| Model 5 |
62.5 |
31.5 |
4.8 |
1.2 |
| Swiss model |
77.6 |
21.2 |
0.6 |
0.6 |
| Modeler |
87.9 |
10.7 |
0.0 |
1.4 |
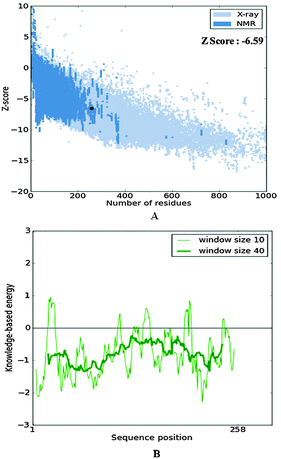 |
| | Fig. 3 The structure validation of the homology model of cathepsin B from Hordeum vulgare through ProSA. | |
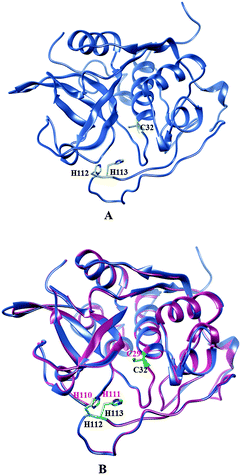 |
| | Fig. 4 (A) Active site residues (yellow) of plant cathepsin B. (B) Comparison of active site residues (yellow) of cathepsin B from Hordeum vulgare (cornflower blue) and active site residues (green) human cathepsin B (hot pink) (2IPP.pdb). | |
Molecular docking and MD simulation studies of the cathepsin B and Aβ peptide complex
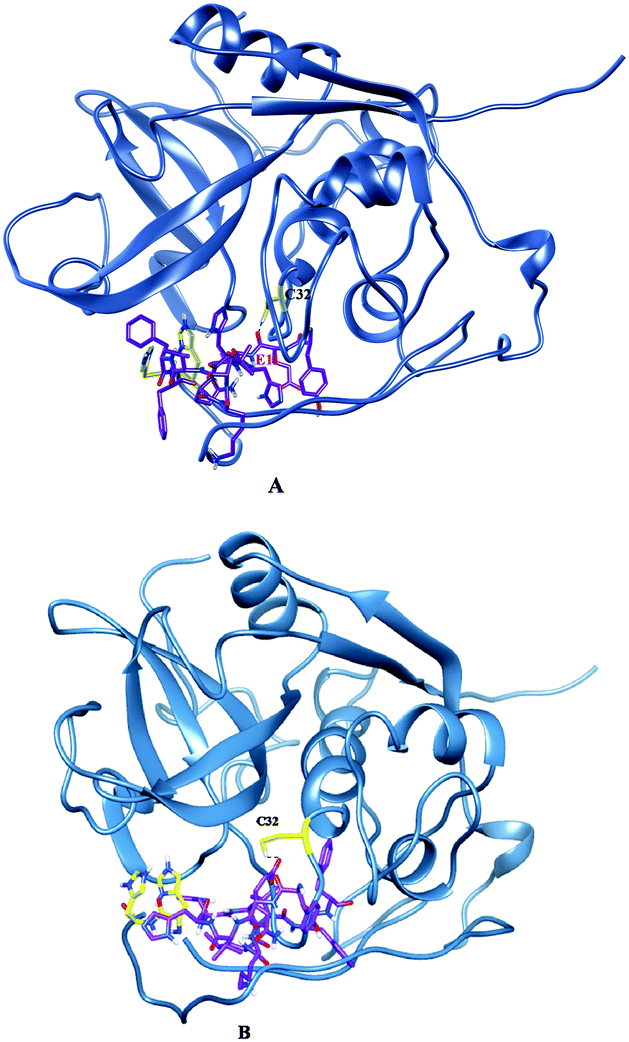
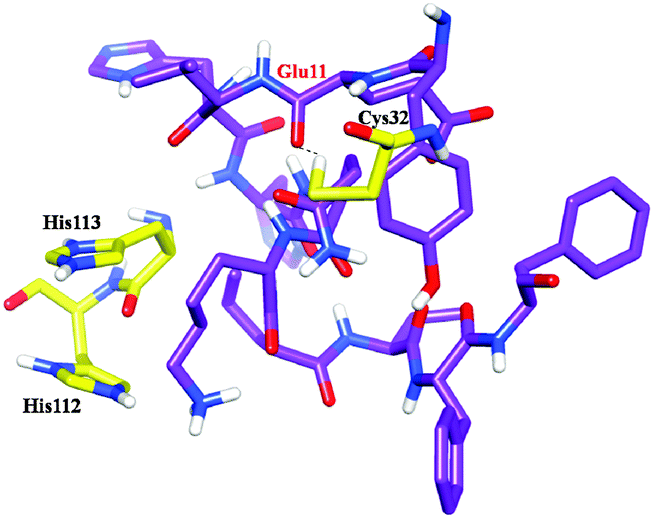
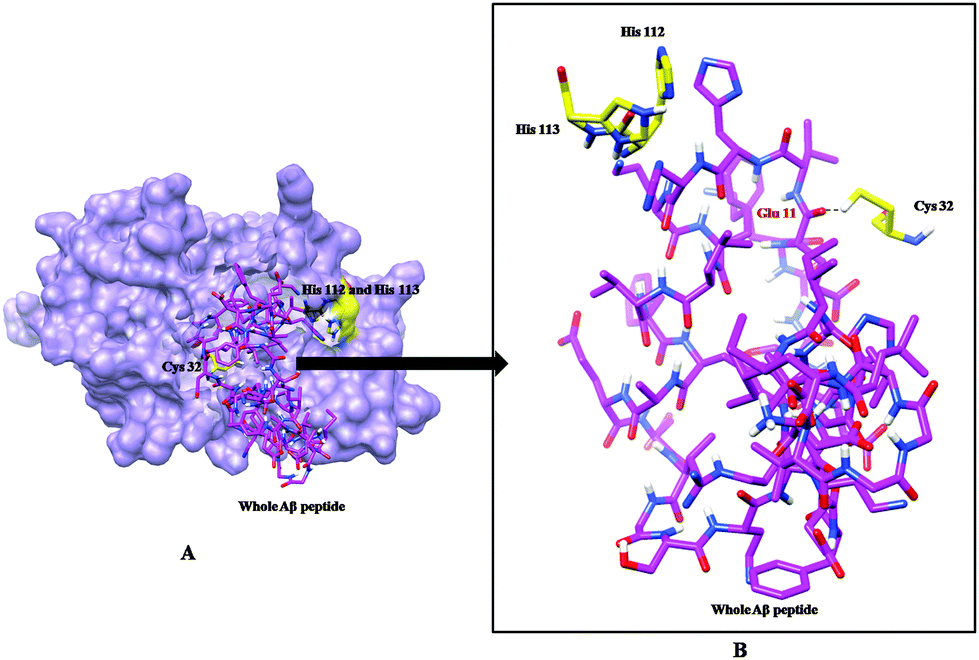
Molecular docking and MD simulation studies have been found to be useful in understanding protein and ligand interactions in detail at the molecular level.39–46 The residues such as (CYS32, HIS112, and HIS113) of cathepsin B from Hordeum vulgare were kept flexible and then docked with a patch of Aβ peptide (1AML.pdb) 10YDVHHNKLVFF20. The docked complex was analyzed before the MD which shows that the residue CYS32 of cathepsin B from Hordeum vulgare interacts with GLN11 from the patch of Aβ peptide (Aβ10–20) with an interatomic distance of 2.503 Å (Fig. 5A and Table 2). The docked complex of cathepsin B from Hordeum vulgare with the whole Aβ peptide (Aβ1–40) showed similar type of interactions as found in the docked complex of cathepsin B with the patch of the Aβ peptide (Aβ10–20) (Fig. 5A, 6 and 7). The CYS32 of cathepsin B from Hordeum vulgare interacts with GLN11 from the whole Aβ peptide (Aβ1–40) with an interatomic distance of 2.697 Å (Fig. 7 and Table 3).
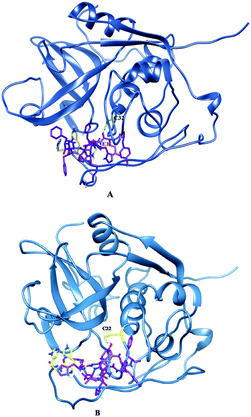 |
| | Fig. 5 Docked complex of cathepsin B from Hordeum vulgare and the patch of the Aβ peptide (A) before MD (B) after MD. | |
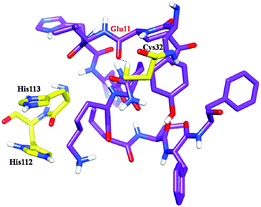 |
| | Fig. 6 Docked complex of cathepsin B from Hordeum vulgares showing the interactions between the active site residues (yellow) with the Aβ peptide (purple) after MD. | |
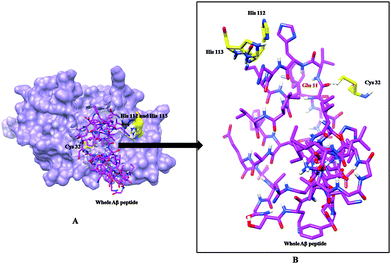 |
| | Fig. 7 (A) Docked complex of cathepsin B from Hordeum vulgare (cornflower blue) with the whole Aβ peptide (sticks have a magenta color) (1AML.pdb). (B) Interactions between the active site residues (yellow) of cathepsin B from Hordeum vulgare with the whole Aβ peptide (magenta). | |
Table 2 Interactions between the active site residues of cathepsin B from Hordeum vulgare with the patch of the Aβ peptide (Aβ10–20) before and after MD simulation
| Interaction between Aβ peptide and active site residues cathepsin B from Hordeum vulgare before MD |
Atomic distance in Å |
| GLU 11. O⋯CYS 32. HG: |
2.503 |
| Interaction between Aβ peptide and active site residues cathepsin B from Hordeum vulgare after MD |
Atomic distance in Å |
| GLU 11. O⋯CYS 32. HG: |
2.003 |
| GLU 11. H:⋯CYS 32. HG |
2.428 |
Table 3 Interactions between the active site residues of cathepsin B from Hordeum vulgare with the whole Aβ peptide (Aβ1–40)
| Interaction between whole Aβ peptide and active site residues cathepsin B |
Atomic distance in Å |
| TYR 10 O⋯CYS 32 HN2: |
2.549 |
| GLU 11 O⋯CYS 32 HN2: |
2.697 |
| CYS 32 HN2⋯GLU 11 H: |
2.949 |
| CYS 32 HN2⋯VAL 12 H: |
2.176 |
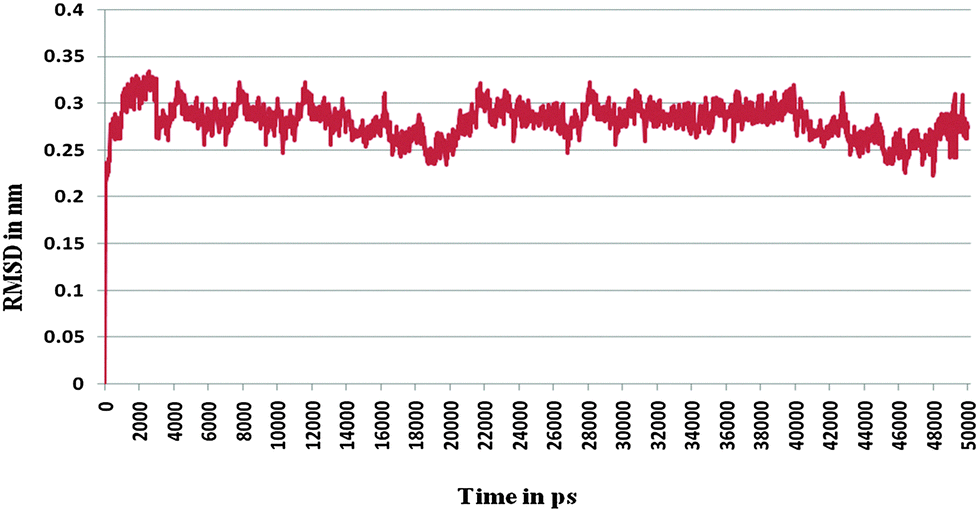
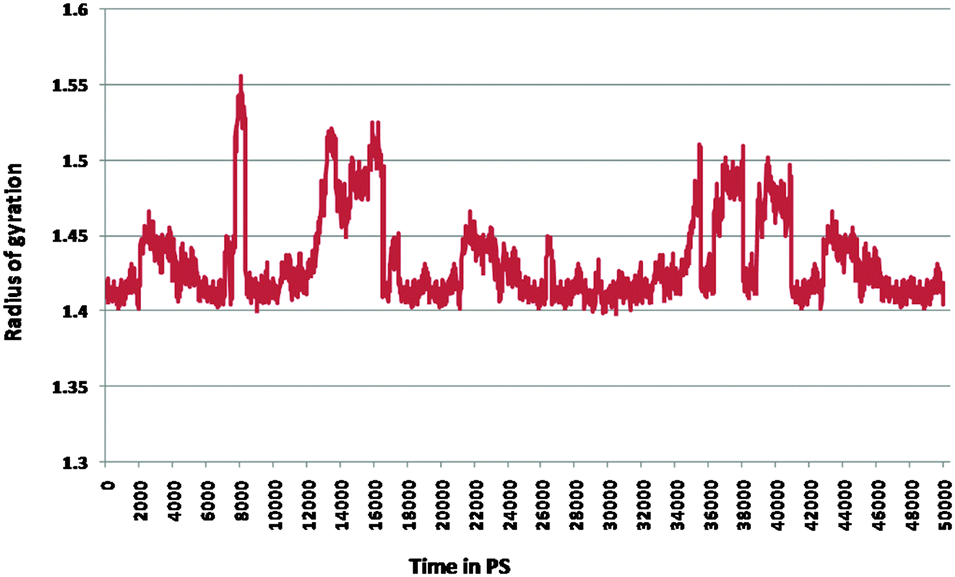
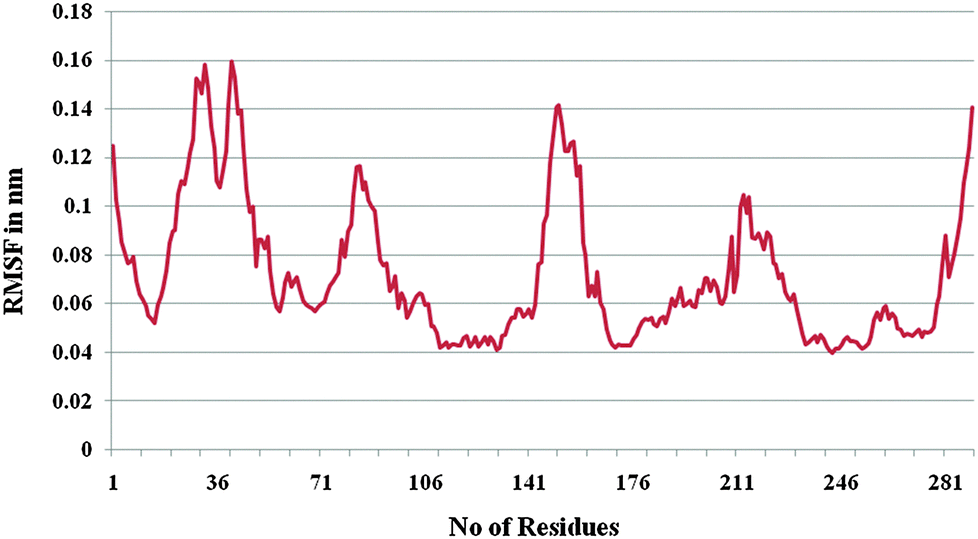
Furthermore, the docked complex of cathepsin B from Hordeum vulgare with the patch of the Aβ peptide was simulated for 50 ns. The average RMSD is 0.28 nm which shows the stable behaviour of the docked complex of cathepsin B from Hordeum vulgare and the patch of the Aβ peptide over the 50 ns simulation period (Fig. 8). The results for Rg (radius of gyration) also indicate stable behavior throughout the whole simulation time (Fig. 9). The dynamics of the residues can be identified by the RMSFs calculated for the C-alpha atoms (Fig. 10). The RMSF results show small fluctuations for the rigid structural elements and larger fluctuations for the ends and loops (Fig. 10). Generally these results indicate that the simulation was stable over the entire simulation time. Thus, the interaction between CYS32 of cathepsin B from Hordeum vulgare with GLN11 from the Aβ peptide was maintained throughout MD simulation trajectory (Fig. 5B, 6, 7 and Table 2) which suggests that cathepsin B from Hordeum vulgare might cleave the Aβ peptide at Glu11 which is similar to human cathepsin B.47
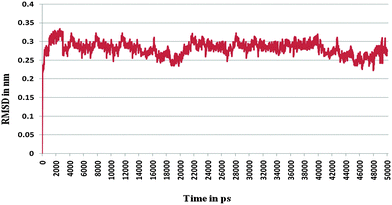 |
| | Fig. 8 RMSD of the docked complex of cathepsin B from Hordeum vulgares and Aβ peptide. | |
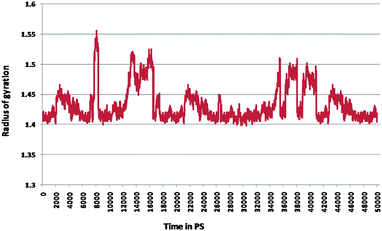 |
| | Fig. 9 Radius of gyration for the docked complex of cathepsin B from Hordeum vulgares and Aβ peptide. | |
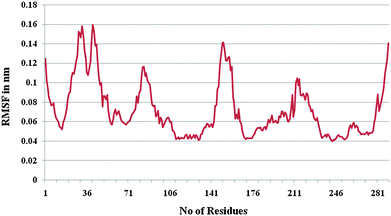 |
| | Fig. 10 RMSF of the docked complex of cathepsin B from Hordeum vulgares and Aβ peptide. | |
Conclusions
The three dimensional structure of cathepsin B from Hordeum vulgare was predicted by using a homology modeling technique. The sequence and structure analysis revealed that the active site residues of cathepsin B from Hordeum vulgare are exactly the same to human cathepsin B. The molecular docking results showed the proper interactions between the sulfhydryl group of Cys32 of cathepsin B with the backbone carboxyl oxygen atom of Glu11 from the Aβ peptide. This hydrogen bonding interaction between Cys32 of plant cathepsin B with the carboxylic end of Glu11 was maintained throughout the MD simulation which suggests its probable involvement in the cleavage of amyloid beta peptide. Hence, the structure of cathepsin B from Hordeum vulgare could be useful to study Aβ peptide degradation in detail at the atomic level. Thus, this study might be useful to understand amyloid beta peptide degradation as well as to design alternative strategies to control Alzheimer's disease.
Abbreviations
| AD | Alzheimer's disease |
| Aβ peptide | Amyloid beta peptide |
| Plant cathepsin B | Cathepsin B from Hordeum vulgare |
| MD | Molecular dynamics |
| RMSD | Root means square deviation |
Acknowledgements
KDS is thankful to University Grants Commission, New Delhi for providing financial support under UGC SAP DRS Phase-II programme sanctioned to Department of Biochemistry, Shivaji University, Kolhapur. MJD is thankful to Department of Science and Technology, New Delhi for providing fellowship as research assistance under the scheme DST-PURSE. Authors are thankful to Computer Centre, Shivaji University, Kolhapur for providing the computational facility.
References
- H. Braak and E. Braak, Acta Neuropathol., 1991, 82, 239–259 CrossRef CAS PubMed.
- J. Hardy and D. J. Selkoe, Science, 2002, 297, 353–356 CrossRef CAS PubMed.
- G. G. Glenner and C. W. Wong, Biochem. Biophys. Res. Commun., 1984, 120, 885–890 CrossRef CAS PubMed.
- G. K. Gouras, H. Xu, J. N. Jovanovic, J. D. Buxbaum, R. Wang, P. Greengard, N. R. Relkin and S. Gandy, J. Neurochem., 1998, 71, 1920–1925 CrossRef CAS PubMed.
- C. L. Masters, G. Simms, N. A. Weinman, G. Multhaup, B. L. McDonald and K. Beyreuther, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 4245–4249 CrossRef CAS.
- R. E. Tanzi and L. Bertram, Cell, 2005, 120, 545–555 CrossRef CAS PubMed.
- D. J. Selkoe, Neuron, 2001, 32, 177–180 CrossRef CAS PubMed.
- S. Mueller-Steiner, Y. Zhou, H. Arai, E. D. Roberson, B. Sun, J. Chen, X. Wang, G. Yu, L. Esposito, L. Mucke and L. Gan, Neuron, 2006, 51, 703–714 CrossRef CAS PubMed.
- J. S. Mort and D. J. Buttle, Int. J. Biochem. Cell Biol., 1997, 29, 715–720 CrossRef CAS PubMed.
- E. A. Eckman, D. K. Reed and C. B. Eckman, J. Biol. Chem., 2001, 276, 24540–24548 CrossRef CAS PubMed.
- N. Iwata, S. Tsubuki, Y. Takaki, K. Shirotani, B. Lu, N. P. Gerard, C. Gerard, E. Hama, H. J. Lee and T. C. Saido, Science, 2001, 292, 1550–1552 CrossRef CAS PubMed.
- W. Q. Qiu, D. M. Walsh, Z. Ye, K. Vekrellis, J. Zhang, M. B. Podlisny, M. R. Rosner, A. Safavi, L. B. Hersh and D. J. Selkoe, J. Biol. Chem., 1998, 273, 32730–32738 CrossRef CAS PubMed.
- H. M. Tucker, M. Kihiko, J. N. Caldwell, S. Wright, T. Kawarabayashi, D. Price, D. Walker, S. Scheff, J. P. McGillis, R. E. Rydel and S. Estus, J. Neurosci., 2000, 20, 3937–3946 CAS.
- K. J. Yin, J. R. Cirrito, P. Yan, X. Hu, Q. Xiao, X. Pan, R. Bateman, H. Song, F. F. Hsu and J. Turk, J. Neurosci., 2006, 26, 10939–10948 CrossRef CAS PubMed.
- N. Cimerman, M. T. Prebanda, B. Turk, T. Popovic, I. Dolenc and V. Turk, J. Enzyme Inhib., 1999, 14, 167–174 CrossRef CAS PubMed.
- K. Ii, H. Ito, E. Kominami and A. Hirano, Virchows Arch. A: Pathol. Anat. Histopathol., 1993, 423, 185–194 CrossRef CAS.
- B. Sun, Y. Zhou, B. Halabisky, I. Lo, S. H. Cho, S. Mueller-Steiner, X. Wang, A. Grubb and L. Gan, Neuron, 2008, 60(2), 247–257 CrossRef CAS PubMed.
- D. S. Wang, D. W. Dickson and J. Malter, J. Biomed. Biotechnol., 2006, 1–12 CrossRef PubMed.
- D. Musil, D. Zucic, D. Turk, R. A. Engh, I. Mayr, R. Huber, T. Popovic, V. Turk, T. Towatari, N. Katunuma and W. Bode, EMBO J., 1991, 9, 2321–2330 Search PubMed.
- T. Schwede, J. Kopp, N. Guex and M. C. Peitsch, Nucleic Acids Res., 2003, 31, 3381–3385 CrossRef CAS PubMed.
- C. Combet, M. Jambon, G. Deléage and C. Geourjon, Bioinformatics, 2002, 18, 213–214 CrossRef CAS PubMed.
- A. Sali and T. L. Blundell, J. Mol. Biol., 1993, 234, 779–815 CrossRef CAS PubMed.
- R. A. Laskowaski, M. W. McArther, D. S. Moss and J. M. Thornton, J. Appl. Crystallogr., 1993, 26, 283–291 CrossRef.
- M. Wiederstein and M. J. Sippl, Nucleic Acids Res., 2007, 35, W407–W410 CrossRef PubMed.
- D. Eisenberg, R. Luthy and J. U. Bowie, Methods Enzymol., 1997, 277, 396–404 CAS.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- P. Rice, I. Longden and A. Bleasby, Trends Genet., 2000, 16(6), 276–277 CrossRef CAS PubMed.
- C. Geourjon and G. Deléage, CABIOS, Comput. Appl. Biosci., 1995, 11(6), 681–684 CAS.
- V. D. Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. Berendsen, J. Comput. Chem., 2005, 26(16), 1701–1718 CrossRef PubMed.
- S. C. Lovell, I. W. Davis, W. B. Arendall, P. I. Bakker de, J. M. Word, M. G. Prisant, J. S. Richardson and D. C. Richardson, Proteins, 2002, 50, 437–450 CrossRef PubMed.
- E. Krissinel and K. Henrick, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2256–2268 CrossRef CAS PubMed.
- W. L. Jorgensen, D. S. Maxwell and J. TiradoRives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- G. A. Kaminski, R. A. Friesner, J. Tirado-Rives and W. L. Jorgensen, J. Phys. Chem. B, 2001, 105, 6474–6487 CrossRef CAS.
- H. Sticht, P. Bayer, D. Willbold, S. Dames, C. Hilbich, K. Beyreuther, R. W. Frank and P. Rosch, Eur. J. Biochem., 1995, 233, 293–298 CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- R. A. Sayle and E. J. Milner-White, Trends Biochem. Sci., 1995, 20(9), 374 CrossRef CAS PubMed.
- R. A. Laskowski, Nucleic Acids Res., 2009, 37, D355–D359 CrossRef CAS PubMed.
- G. Tseng, K. D. Sonawane, Y. V. Korolkova, M. Zhang, J. Liu, E. V. Grishin and R. H. Guy, Biophys. J., 2007, 92, 3524–3540 CrossRef CAS PubMed.
- C. B. Jalkute, S. H. Barage, M. J. Dhanavade and K. D. Sonawane, Protein J., 2013, 32, 356–364 CrossRef CAS PubMed.
- S. H. Barage and K. D. Sonawane, Protein Pept. Lett., 2013, 21, 140–152 CrossRef.
- M. J. Dhanavade, C. B. Jalkute, S. H. Barage and K. D. Sonawane, Comput. Biol. Med., 2013, 43, 2063–2070 CrossRef CAS PubMed.
- S. H. Barage, C. B. Jalkute, M. J. Dhanavade and K. D. Sonawane, Int. J. Pept. Res. Ther., 2014, 20, 409–420 CrossRef CAS.
- M. J. Dhanavade and K. D. Sonawane, Amino Acids, 2014, 46(8), 1853–1866 CrossRef CAS PubMed.
- R. S. Parulekar, S. H. Barage, C. B. Jalkute, M. J. Dhanavade and K. D. Sonawane, Protein J., 2013, 32, 467–476 CrossRef CAS PubMed.
- C. B. Jalkute, S. H. Barage, M. J. Dhanavade and K. D. Sonawane, Int. J. Pept. Res. Ther., 2014, 21, 107–115 CrossRef.
- E. A. Mackay, A. Ehrhard, M. Moniatte, C. Guenet, C. Tardif, C. Tarnus, O. Sorokine, B. Heintzelmann, C. Nay, J. M. Remy, J. Higaki, A. V. Dorsselaer, J. Wagner, C. Danzin and P. Mamont, Eur. J. Biochem., 1997, 244, 414–425 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5mb00718f |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.