
De novo design of protein mimics of B-DNA†
Deniz
Yüksel
a,
Piero R.
Bianco
*b and
Krishna
Kumar
*ac
aDepartment of Chemistry, Tufts University, 62 Talbot Avenue, Medford, MA 02155, USA. E-mail: krishna.kumar@tufts.edu
bDepartment of Microbiology and Immunology, University at Buffalo, The State University of New York, Buffalo, NY 14214, USA. E-mail: pbianco@buffalo.edu
cCancer Center, Tufts Medical Center, Boston, MA 02111, USA
First published on 16th November 2015
Abstract
Structural mimicry of DNA is utilized in nature as a strategy to evade molecular defences mounted by host organisms. One such example is the protein Ocr – the first translation product to be expressed as the bacteriophage T7 infects E. coli. The structure of Ocr reveals an intricate and deliberate arrangement of negative charges that endows it with the ability to mimic ∼24 base pair stretches of B-DNA. This uncanny resemblance to DNA enables Ocr to compete in binding the type I restriction modification (R/M) system, and neutralizes the threat of hydrolytic cleavage of viral genomic material. Here, we report the de novo design and biophysical characterization of DNA mimicking peptides, and describe the inhibitory action of the designed helical bundles on a type I R/M enzyme, EcoR124I. This work validates the use of charge patterning as a design principle for creation of protein mimics of DNA, and serves as a starting point for development of therapeutic peptide inhibitors against human pathogens that employ molecular camouflage as part of their invasion stratagem.
Introduction
Dramatic examples of mimicry abound in the macroscopic visible world around us, and this strategy is also used by nature at the molecular level. Proteins imitate other proteins in instances where bacteria, viruses or parasites escape an immune response; agonists and antagonists bind to receptors; and autoimmune responses are evoked because a pathogen shares sequence similarities with the host's native proteins.1,2 Structural impersonation of the nucleic acids, DNA and RNA, by proteins is, however, relatively rare, and examples have only recently been identified.3–6Naturally occurring DNA mimicking proteins have been reported in a variety of organisms including prokaryotes (DinI in Escherichia coli BL21, HI1450 in Haemophilus influenza PittGG, MfpA in Mycobacterium tuberculosis, NuiA Nostoc sp., ArdA Enterococcus faecalis, CarS Myxococcus xanthus, DMP19 and DMP12 in Neisseria meningitidis, SAUGI in Staphylococcus aureus)7–14 bacteriophages (UGI in Bacillus subtilis bacteriophage PBS2 and p56 in Bacillus phage ϕ29, Ocr in enterobacteriophage T7, Gam in bacteriophage λ),15–18 eukaryotes (dTAFII230 (residues 11–77) Drosophila melanogaster, p53 transactivation domain (residues 33–60) in Homo Sapiens)19,20 and at least one eukaryotic virus (ICP11 in Nimaviridae whispovirus).21 The resemblance of such proteins to DNA endows each organism with the ability of controlling different aspects of protein–DNA interactions. The DNA double helix has unique structural features. It displays diverse chemistry in the major and minor grooves presenting either polar or hydrophobic environments. The most striking is the carefully arranged supramolecular display of negative charges on the sugar–phosphate backbone that links successive nucleotides.22 The crystal structures of the DNA mimics discovered so far revealed that they mimic the charge pattern and shape of DNA.23 Despite the inherent structural diversity of these DNA mimics, they share some common features. First, they have evolved to mimic a ‘bent’ or distorted form of DNA imitating the conformation DNA adopts upon binding target enzyme. Having already a bent shape makes them better binding partners for their targets. Second, they possess a hydrophobic ‘core’ to display the large number of negative surface charges, and still be able to stabilize their overall fold. Finally, they are small, compact, and architecturally robust.
In a particularly striking demonstration of DNA camouflage, the bacteriophage T7 evades the bacterial restriction–modification (R/M) system – responsible for recognizing and destroying foreign DNA – by injecting only a small portion of its genome during the first few minutes of infection.24 These early genes encode a transcript, ∼7000 nucleotides long, that contains the mRNA for gene 0.3, the product of which is an anti-restriction protein called Ocr (overcome classical restriction).24,25 The Ocr protein mimics B-DNA by displaying charges on its surface that mirror the pattern observed in the nucleic acid polymer.17,26,27 The R/M system is tricked into binding Ocr, thus preventing detection and destruction of phage DNA. We hypothesized therefore that if a structurally robust molecular template with the dimensions of DNA were available, molecular Trojan horses of the likes of Ocr could be assembled using charge patterning. We envisioned that coiled coils could satisfy a majority of the attributes required for engineering decoys that resemble DNA in displayed charge, and shape.
The most commonly observed coiled coils consist of α-helices tightly wound into a shallow left-handed super helix.28 These structures display a heptad repeat, usually denoted as [abcdefg]n, where the a and d residues are typically hydrophobic, lining the helical interface. The other positions b, c, e, f, g are solvent exposed and are occupied mostly by residues with polar or charged side chains at physiological pH.29,30 Because of this apparent simplicity, coiled coils have served as scaffolds in encoding novel structure and function.31–33 For example, core directed design strategies34,35 have been fruitful in building simple coiled coil structures where helix orientation36,37 and oligomerization states38 are readily controlled.
The influence of surface charge patterning, and the effect of electrostatic interactions on coiled coil stability have been reasonably well documented.39–41 One of the earliest rational design strategies based on charge patterning was the ‘Peptide Velcro’ where oppositely charged peptides with identical sequences (at the a–d and f positions; with a and d being leucine) – Acid-p1 containing glutamates, and Base-p1 containing lysines at the e and g positions formed stable heterodimeric structures.42 Others have also utilized a charge-patterning strategy especially at e and g positions to gain insight into the role of inter- and intra-helical electrostatic interactions in coiled coil assembly.39,43–45
We report here the design of DNA ‘look-alikes’ by exploitation of the well-packed hydrophobic core to dictate display of charged surface residues of coiled coils. Decoration of dimeric coiled coils with Asp and Glu residues on the solvent exposed faces gave structures resembling the negative charge pattern of the B-DNA double helix. We further demonstrate that the designed DNA mimics inhibit the restriction activity of a type I R/M enzyme and that the activity is correlated with structure. This work sets the stage for rationally designing new specific DNA mimics that can target desired protein–DNA interactions where charge mimicry is utilized, for instance by pathogens like M. tuberculosis.9
Results and discussion
Computational analysis and superimposition of B-DNA on dimeric coiled coils yielded DNA mimics (DMs)
The DNA duplex can be crudely approximated as a cylindrical structure. We stipulated that the designed mimic should fit within the cylinder dimensions so that its solvent exposed residues could be modified to resemble the charge pattern observed in DNA. We started with the well-characterized coiled coil domain (GCN4-p1) of a yeast transcription activator as the template.39–41,46,47 Molecular models of dimeric (pdb id: 2zta) and trimeric (pdb id: 1zij) coiled coils were superimposed on the Dickerson-Drew dodecamer B-DNA (pdb id: 1bna) using Macromodel (v 7.1, Schrödinger, LLC). This initial analysis demonstrated that the trimer was too large to fit inside the B-DNA cylinder, also eliminating higher order oligomers as possible candidates. Next, the B-DNA crystal structure was used to pattern the surface of the dimeric coiled coil. In order to fit the designed ensembles onto the DNA duplex, vectors were calculated for the DNA duplex such that they represented the axes of a cylinder that gave the minimum root mean square (rms) distance of the phosphorus atoms originating from the surface of the cylinder, and identified the residues closest to the phosphorus atoms in the side chains of the peptide ensemble. We sought to replace only the solvent exposed residues keeping the hydrophobic core intact with Val and Leu at a and d positions. Literature precedence suggests that this core composition with a single Asn residue at the a position enforces a dimeric structure (as opposed to a dimer–trimer equilibrium).38 The residues proximal (<8 Å) to phosphorous atoms in the DNA backbone were substituted with either glutamic or aspartic acid. Several iterations were carried out to select the pairs to provide the smallest rms distances (Table S1 in the ESI†). The initial fits were further improved by rotating the Glu and Asp side chains about their Cα–Cβ (χ1) and Cβ–Cγ (χ2) bonds. Rotamer libraries representing the most frequently populated dihedral angles in structural data banks for Glu and Asp residues were used to guide side chain conformations, while keeping the backbone rigid.48,49 After global optimization, rms distance fits by pairing 11 carboxyl groups of the peptides DM1 and DM2 with 11 phosphorous atoms on the B-DNA backbone were computed to be 2.3 Å and 2.2 Å, respectively (Fig. 1 and Tables S2 and S3 in the ESI†). A similar fitting procedure with Ocr reported an rms fit of 1.9 Å by pairing 12 carboxyl groups with 12 phosphorous atoms.17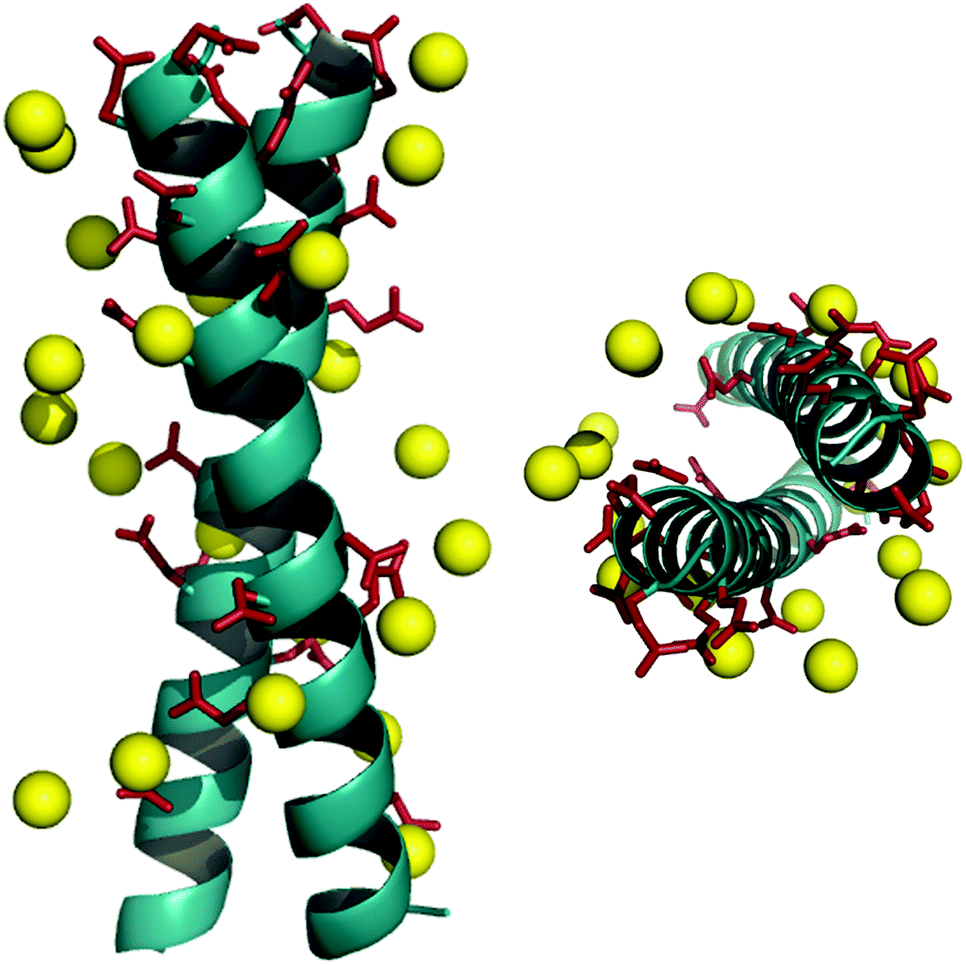 | ||
| Fig. 1 Superimposition of a DNA mimicking peptide on B-DNA backbone. Designed helical bundle (cyan) decorated with Asp and Glu residues (red sticks) is superimposed on B-DNA backbone (yellow spheres, only phosphorous atoms are shown) using Macromodel v.7.1. Front view (left), top view (right). | ||
Ocr exists as a dimer in solution. We introduced cysteine residues at either terminus to build constructs similar to Ocr in size. Glycine residues following Cys were used as spacers to mimic the dimerization interface of Ocr.17 This flexible spacer region could facilitate constructs to adopt a slightly bent conformation. We also positioned an asparagine residue in the hydrophobic core to ensure a parallel orientation, and restrict sundry oligomerization states.36,37 These considerations, and the accompanying computational effort led to synthesis of the DNA mimic (DM) peptides listed in Scheme 1A. In addition, two controls, DMscr and DMc, were also assembled. DMscr is a scrambled version of DM2 where the hydrophobic residues at a and d positions have been swapped with charged residues to abolish helical structure. DMc is designed to adopt random coil conformation while maintaining the same total charge as the other DMs.
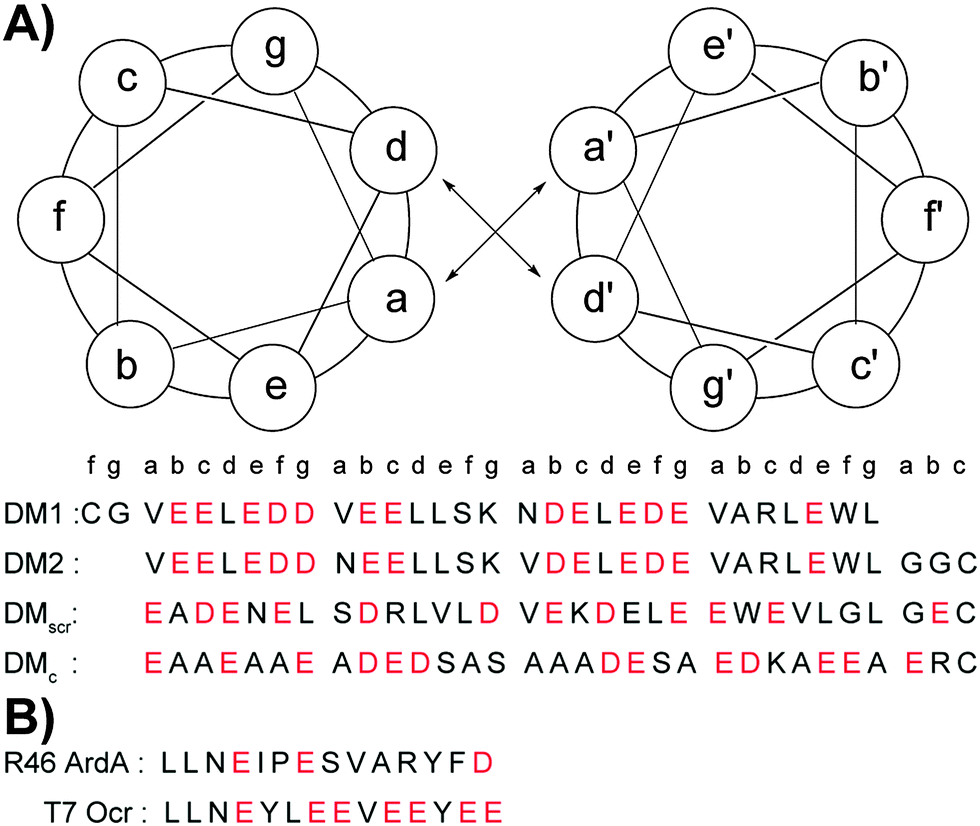 | ||
| Scheme 1 Helical wheel diagram and sequences of designed DMs (A) and control peptides that are known anti-restriction motifs (B). All peptides are acetylated at the N-terminus and amidated at the C-terminus. Corresponding position of each amino acid in the coiled coil heptad is denoted. Acidic residues are shown in red. | ||
DMs inhibit the ATPase activity of complex type I R/M enzyme, EcoR124I, in a coupled spectrophotometric ATPase assay
Bacterial R/M systems function by identifying and attacking non-self DNA.50–52 Typically, R/M systems have two distinct functions: the restriction endonucleases (REases) are responsible for recognition of unmethylated DNA sequences for cleavage and the methyltransferases (MTases) protect the genome by adding methyl groups on specific sites. Unlike type II R/M hydrolases, type I R/M enzymes recognize and bind specific, bipartite, asymmetric DNA sequences.50,51 After binding, the MTases remain bound to the recognition site whereas REases can translocate the adjacent DNA in a bidirectional manner, pulling the nucleic acid toward the bound complex. This results in formation of DNA loops in an ATP dependent process.53 Cleavage occurs when translocation is stalled either by topological strain on DNA54 or by a collision with another translocating complex.55Ocr is implicated as a broad-spectrum inhibitor of type I R/M enzymes and has been shown to inhibit EcoKI and EcoBI.56–58 These enzymes are motor proteins that couple ATP hydrolysis to DNA translocation and cleavage.50,51 In our previous studies, we utilized a coupled spectrophotometric ATPase assay to study ATP hydrolysis on supercoiled dsDNA (scDNA) containing a single recognition site for EcoR124I and demonstrated that ATP utilization is coupled directly to bidirectional, dsDNA translocation.59,60 Here, we used this assay to assess the inhibitory effects of designed DMs and Ocr mutants.
The inhibitory activity of Ocr against EcoR124I, a type IC R/M enzyme was tested first. The ATPase reactions were initiated by addition of scDNA following pre-incubation of EcoR124I with Ocr. The rate of ATP hydrolysis by EcoR124I was completely abolished at low concentrations of Ocr (Fig. S1B in the ESI†). Our designed DMs are highly negatively charged, but possess significantly fewer anionic side chain groups than Ocr. Therefore, we chose to compare the inhibitory activity of our peptides to Ocr99, a construct lacking 17 amino acids of the highly negatively charged C-terminal end of Ocr. Biophysical characterization of Ocr99 revealed that the removal of the highly negative C-terminus of Ocr does not affect its anti-restriction activity.26 This version of Ocr also retards ATP hydrolysis of EcoR124I albeit at modestly higher concentrations than Ocr itself (500 vs. 100 nM, respectively; Fig. S1A in the ESI†). We then performed the ATPase assay in the presence of various concentrations of DM1, DM1–SS–DM1, DM2–SS–DM2, and control peptides (Fig. 2 and Fig. S2 in the ESI†). The rate of ATP hydrolysis was calculated using eqn (1) (Experimental methods) and the results are summarized in Table 1. The DM1 peptide that lacks a N-terminal cysteine residue had the lowest ATPase50 value, 95.2 μM. However, when DM1 was allowed to disulphide bond, the ATPase50 concentration decreased to 22.5 μM. DM2–SS–DM2 peptide with 36.1 μM also had a similar ATPase50 value to DM1–SS–DM1. These results confirm that the size of the DM peptides play a crucial role in their inhibitory activity suggested by our computational design parameters. On the other hand, both control peptides DMscr–SS–DMscr and DMc–SS–DMc had much lower ATPase50 values, 182 and 237.3 μM, respectively suggesting that it is not only the total amount of charge but also how those anionic residues are positioned in three-dimensional space.
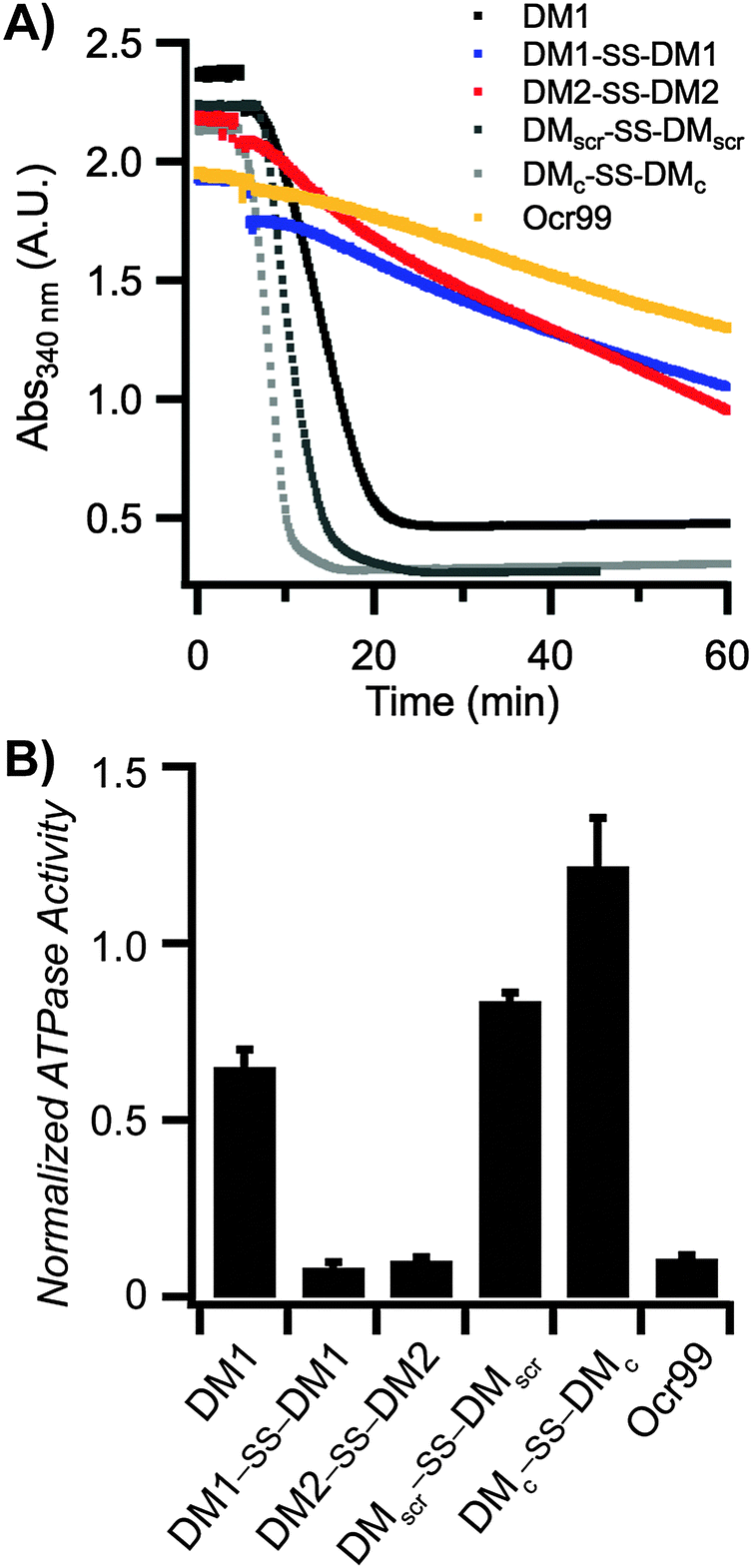 | ||
| Fig. 2 DMs inhibit ATPase activity of EcoR124I. (A) A representative plot of ATPase assay containing 50 μM of each indicated peptide and 500 nM of Ocr99. All reactions were initiated by addition of scDNA. [scDNA] = 5 nM, [EcoR124I] = 5 nM. (B) The rate of ATP hydrolysis was calculated at 50 μM peptide (or 500 nM Ocr99) concentration and normalized against control reactions without the indicated peptide within each experiment (n = 4 independent experiments). | ||
| Inhibitor | Residue/charge | ATPase50a (μM)/(relative activity) | scDNA50b (μM) |
|---|---|---|---|
| a Half maximal ATPase activity inhibition (ATPase50) represents the peptide concentration at which 50% of ATPase activity of the enzyme is inhibited. ATPase50 values were calculated by fitting a straight line to the average relative rate vs. peptide concentration data (n ≥ 3). b Peptide concentration at which 50% of total scDNA. scDNA50 was determined by fitting %scDNA vs. peptide concentration obtained from densitometric analysis of agarose gels (n ≥ 3). The data is represented as mean ± SEM. * No N-terminal cysteine residue. | |||
| DM1* | 29/−11 | 95.2 ± 7.0 (512) | 172.6 ± 3.9 |
| DM1–SS–DM1 | 60/−22 | 22.5 ± 0.2 (121) | 38.0 ± 0.9 |
| DM2–SS–DM2 | 62/−22 | 36.1 ± 0.2 (194) | 63.9 ± 0.7 |
| DMscr–SS–DMscr | 62/−22 | 182 ± 13.4 (978) | 612.0 ± 19.5 |
| DMc–SS–DMc | 62/−22 | 237.3 ± 13.4 (1276) | 245.5 ± 14.7 |
| Ocr99 | 99/−18 | 0.186 ± 0.004 (1) | 0.172 ± 0.011 |
| T7 Ocr | 14/−7 | Not active | Not active |
| R46 ArdA | 14/−2 | Not active | Not active |
DMs also inhibit the restriction activity of EcoR124I
As EcoR124I is a restriction endonuclease it conceivable that the decrease in the rate of ATP hydrolysis corresponds to the cleavage state of dsDNA. Hence, to determine whether the reduction in ATP hydrolysis rate correlates with inhibition of restriction activity of EcoR124I, samples from the ATPase assay were analyzed by agarose gel electrophoresis. We expected scDNA not to be nicked or linearized if the restriction activity of EcoR124I was also inhibited. Agarose gel analysis showed that scDNA with a single recognition site for EcoR124I was not restricted at concentrations of Ocr99 as low as 400 nM (Fig. 3B and Fig. S1C in the ESI†). In the presence of monomeric DM1, the DNA cleavage was impeded at higher concentrations, upwards of 100 μM. On the other hand, both disulphide bonded peptides DM1–SS–DM1 and DM2–SS–DM2 exhibited concentration dependent inhibition of restriction activity of EcoR124I starting at 20 and 50 μM, respectively (Fig. 3A). DMscr–SS–DMscr and DMc–SS–DMc control peptides were ineffective (at concentrations as high as 100 μM) as restriction enzyme inhibitors in accord with the design principles (Fig. 3B). To compare the potency of each peptide as restriction activity inhibitors, scDNA50 values were calculated from densitometric analysis of agarose gels (Table 1). The scDNA50 values and %scDNA remaining in the presence of 50 μM of peptide inhibitors also validated that DM1–SS–DM1 is the most effective restriction enzyme inhibitor in comparison to the control peptides (Table 1 and Fig. 3C). We further monitored DNA cleavage as a function of time in the presence of 40 μM DM1–SS–DM1 and demonstrated that our results are independent of ATPase assay conditions (Fig. S3 in the ESI†).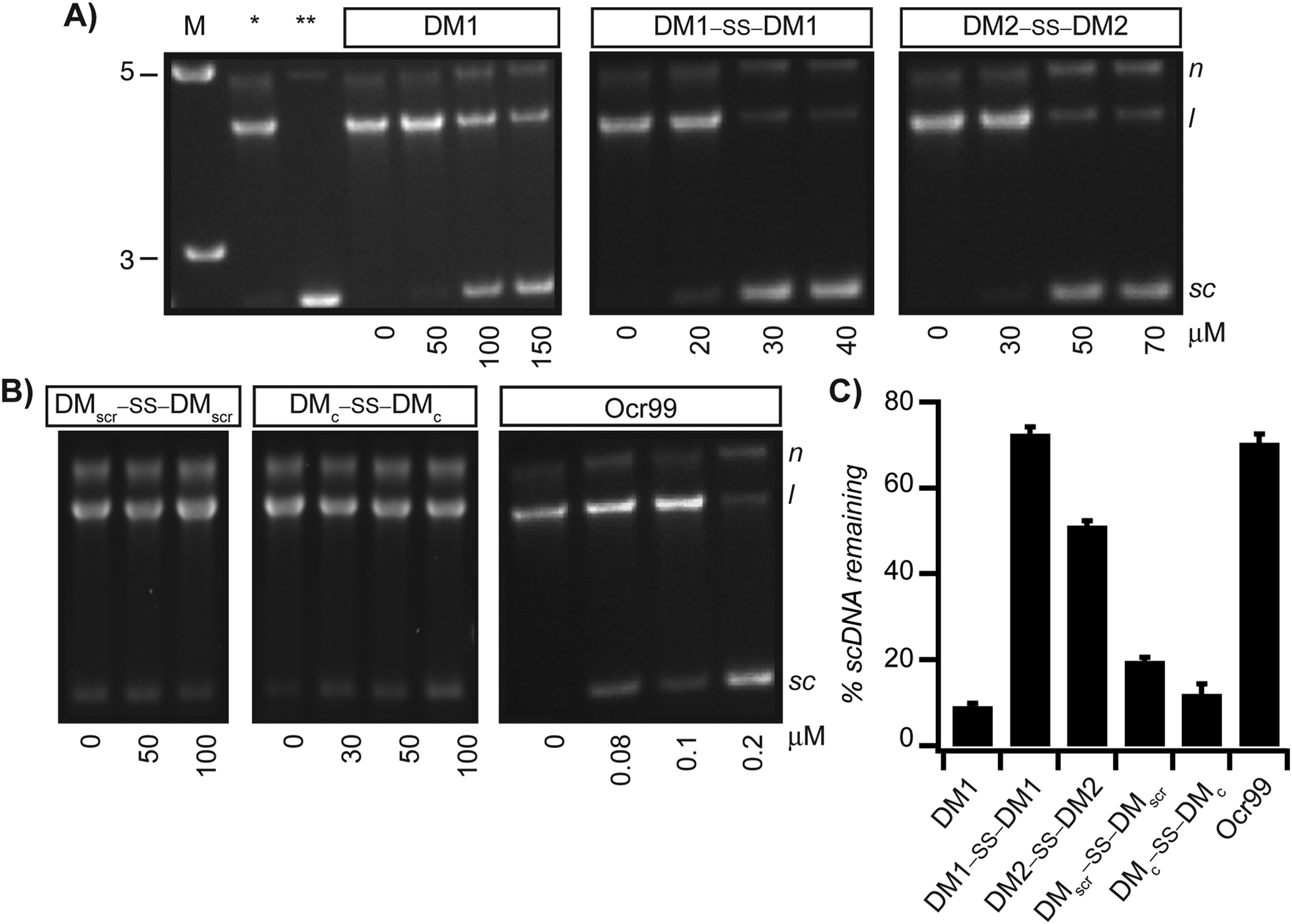 | ||
Fig. 3 DMs inhibit the restriction activity of EcoR124I. Reaction mixtures containing increasing concentrations of DM1, DM1–SS–DM1, DM2–SS–DM2 (A) and control peptides DMscr–SS–DMscr and DMc–SS–DMc and Ocr99 (B) from ATPase assays were analyzed by agarose gel electrophoresis to monitor DNA cleavage. All reactions were initiated by addition of scDNA. [scDNA] = 5 nM, [EcoR124I] = 5 nM. The sizes of the DNA molecular weight marker (M) are in kilobase-pairs. (C) The % of scDNA remaining in the presence of 50 μM peptide (except DM1–SS–DM1 at 40 μM and Ocr99 at 0.5 μM) was determined by densitometric analysis of agarose gels (n ≥ 4 independent experiments). *![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) scDNA digested in the absence of peptide inhibitor. **scDNA alone. N, nicked circular DNA; l, linear and sc, negatively supercoiled. scDNA digested in the absence of peptide inhibitor. **scDNA alone. N, nicked circular DNA; l, linear and sc, negatively supercoiled. | ||
We further compared the activity of designed peptides to two shorter peptides derived from conserved anti-restriction domain motifs. R46 ArdA is a conserved sequence isolated from ArdA family of proteins that are found in conjugative plasmids. The T7 Ocr sequence is identified as an anti-restriction motif in the in the highly charged C-terminus of Ocr.61 Both peptides were ineffective in inhibiting ATPase and restriction activities of EcoR124I at concentrations as high as 100 μM pointing to the importance of structure as prerequisite for activity (Fig. S4 in the ESI†).
Electrostatics and structural properties govern the interaction between DMs and EcoR124I
The Ocr monomer has a total charge of −28e, but it exists as a dimer in solution.17,26,27 Even though DMs are highly charged the total surface charge is still considerably less than Ocr (Table S4 in the ESI†). Hence, the inhibitory activities of the designed peptides were compared to Ocr99 where the total charge of the monomer is −18e. We observed that DM1 was not an effective inhibitor compared to DM1–SS–DM1 and DM2–SS–DM2 as the total charge displayed was half of the disulphide bonded peptides, −11e and −22e, respectively (Table 1). Similarly it has been shown that removal of 46% of the acidic residues in Ocr protein by chemical modification also leads to a 50-fold reduction in binding affinity for a type I R/M62 whereas single or double site mutations of Ocr does not affect its binding to M.EcoKI63 demonstrating the importance of electrostatics in inhibitory activity.We further argue that the structure of the scaffold that displays the charge pattern is pivotal in DNA mimics, and not just the total charge. Therefore, circular dichroism (CD) spectroscopy was used to probe the structure of DMs. DMs were unfolded at neutral and alkaline pH values likely due to repulsion between Glu and Asp residues (Fig. 4A). Jelesarov et al. have shown that kosmotropes (small anions of high charge density) can induce a strongly acidic 30-residue peptide to fold into a coiled coil by strengthening hydrophobic interactions.64 To demonstrate that DMs could also adopt helical structures when the negative charges are electrostatically shielded by a high dielectric medium, CD spectra were measured in the presence of kosmotropic salts. All DMs and Ocr99 were random coils in borate buffer alone (Fig. 4A and C). In contrast, the presence of 1.5 M (NH4)2SO4 induced coiled coil formation as judged by a random coil to helix transition observed in the CD spectra of DM1–SS–DM1, DM2–SS–DM2, and Ocr99 (Fig. 4B and C, Table S5 in the ESI†). On the other hand, DM1, DMscr–SS–DMscr and DMc–SS–DMc remained random coils under such conditions. These findings clearly establish that structure and activity are correlated.
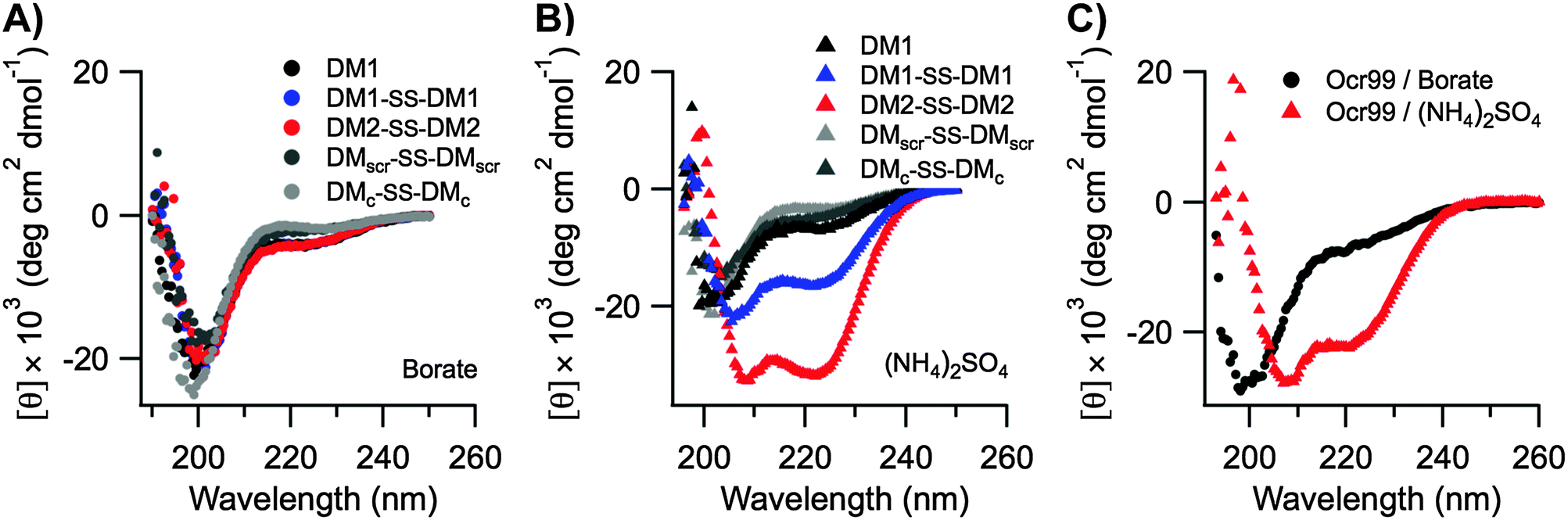 | ||
| Fig. 4 CD spectra of designed DM peptides and Ocr99. All DM peptides were random coils in borate buffer (A). A transition from random coil to coiled coil was observed in (NH4)2SO4 buffer for DM1–SS–DM1, DM2–SS–DM2 (B) and Ocr99 (C). All measurements were taken in 50 mM borate buffer pH 9.0 containing 1.5 M NH4(SO4)2 at 20 °C. [DM peptides] = 50 μM, [Ocr99] = 25 μM. Data represent the average of four scans. | ||
Our DMs were designed with an asparagine residue in the core so that a buried polar interaction could only be formed when the ensembles interacted in a parallel fashion. However, all DMs in Scheme 1 are in monomer–dimer equilibrium as judged by equilibrium sedimentation centrifugation (Table S6 in the ESI†). Ocr99, like Ocr itself, also exists as a dimer in solution.26 This difference in oligomeric states could also explain the moderately lower inhibitory effect of the DMs compared to Ocr99. It has also been reported that Ocr retains it anti-restriction activity as long as the mimicry of the electrostatics of the bend at the centre of the EcoKI DNA target sequence remains unchanged.65 Prior studies suggest that basic region leucine zipper peptides favour a DNA binding mechanism where monomers sequentially assemble into a dimer at the target DNA binding site.66 Similarly, we hypothesize that monomeric DMs are not unstructured in solution but they gain helicity and dimerize in the presence of EcoR124I. Based on prior estimates of the association constants of Ocr to DNA,26,58 the Kd of DM1–SS–DM1 binding to DNA can be approximated to be 100 nM or better. It is worth noting that through de novo design, we were able to attain activities and structure within two orders of magnitude of a natural product that has undergone extensive selection and has been optimized through eons of evolutionary pressures.
Experimental methods
Peptide synthesis, purification, and characterization
All peptides were synthesized on Advanced ChemTech 348 Ω synthesizer (Louisville, KY) using Rink amide NovaGel™ resin (resin substitution: 0.63 mmol g−1, 0.1 mmole scale synthesis) and 9-fluorenylmethoxycarbonyl (Fmoc) protection chemistry. Concentrations of peptide stock solutions were determined from UV-absorption of tryptophan (ε = 5690 M−1 cm−1) and tyrosine (ε = 1215 M−1 cm−1) residues at 280 and 276 nm, respectively in 6 M Gdn·HCl, 0.01 M phosphate buffer, pH 6.50.67 Details on synthesis, purification, and characterization are provided in the ESI.†Production of Ocr and Ocr99
E. coli strain BL21 (DE3) pLysS cells transformed with plasmids pAR2993, and pAR3790 were kind gifts from Prof. W. Studier (Brookhaven National Laboratory). Ocr and a truncated mutant of the protein lacking the C-terminal end, Ocr99, were expressed following literature protocols from plasmids pAR2993 and pAR3790, respectively.27,68,69 The concentration of Ocr and Ocr99 stocks were determined using ε = 31860 M−1 cm−1 and ε = 29430 M−1 cm−1, respectively at 280 nm.26
Purification of the EcoR124I holoenzyme
EcoR124I was purified exactly as described previously.59 The concentration of the holoenzyme was determined at 280 nm using ε = 366090 M−1 cm−1.
Plasmid production and purification
Plasmid pPB248 (4266 bp) with single binding site for EcoR124I was used for all the experiments. NovaBlue GigaSingles™ (Novagen) competent cells were used to transform the plasmids following the manufacturer's protocols. Cells were lysed and plasmids were purified using Qiagen plasmid maxi DNA purification kit (Qiagen). Purified DNA was stored at −20 °C in TE buffer (10 mM Tris·Cl, pH 8.5). The nucleotide concentration of dsDNA was determined by measuring the UV absorbance at 260 nm by a NanoDrop spectrophotometer (ND-1000, Thermo Scientific) and using ε = 6500 M−1 cm−1 (in nucleotides).Coupled spectrophotometric ATPase assay
A spectrophotometric assay that couples the hydrolysis of ATP to a decrease in absorbance of NADH was used to study inhibition of EcoR124I by designed peptides.59,70 The ATPase assay is based on the fact that upon hydrolysis of ATP to ADP by type I R/M system, one equivalent of phosphoenolpyruvate (PEP) is converted into pyruvate by pyruvate kinase. During this conversion pyruvate kinase uses PEP to regenerate ATP so that its concentration remains constant and ADP never accumulates. In a subsequent reaction, pyruvate is converted into lactate by lactate dehydrogenase (LDH) while one equivalent NADH is oxidized to NAD+. Oxidation of NADH results in a decrease in the absorption of NADH that is monitored at 340 nm. The rate of change in absorbance is directly correlated to rate of steady state ATP hydrolysis that is calculated by the following equation:| Rate of hydrolysis (μmol min−1 mL−1) = −dA/dt (min−1)/ε340 (NADH) | (1) |
400 M−1 cm−1 and stored in aliquots at −20 °C. NADH was dissolved in 10 mM Tris·Cl, pH 8.0, concentration was determined by UV measurements at 340 nm using ε = 6250 M−1 cm−1 and stored in small aliquots (sufficient for single assay) at −80 °C. All the reactions were performed at 23 °C unless otherwise noted and contained 20 mM Tris·OAc, pH 7.5, 100 μg mL−1 of bovine serum albumin, 5% glycerol, 7.5 mM phosphoenol pyruvate (PEP), 0.3 mM NADH, 20 U mL−1 pyruvate kinase (PK) and lactate dehydrogenase (LDH), 10 mM magnesium acetate, 1 mM ATP, 5 nM supercoiled pPB248, and 5 nM active EcoR124I holoenzyme. All inhibitors were solubilized in 20 mM Tris·Cl, pH 8.0 and were pre-incubated with the enzyme for 20 min while on ice. Reaction mixtures were allowed to equilibrate at 23 °C for 5 minutes and were initiated by addition of DNA. The absorbance data were collected using Varian Cary 50 spectrophotometer equipped with a 12-cell holder with a PCB150 peltier-controlled water bath. Reaction rates (in μM min−1) were calculated by fitting a straight line tangent to the data, and multiplying the slope by 160. All the experiments were performed in the absence of non-essential cofactor S-adenosyl methionine, which is not required for the ATPase and cleavage activities of EcoR124I.
DNA cleavage assay
DNA cleavage of supercoiled plasmid pPB248 by EcoR124I holoenzyme was monitored using agarose gel electrophoresis. The same reaction conditions as the ATPase assay were used except that reactions did not contain PK, LDH, NADH and PEP. Following DNA cleavage, time points were stopped by addition of 10× DNA loading buffer, containing ficoll (15% by w/v), 1% SDS, 10 mM EDTA, 0.25% bromophenol blue, 0.25% xylene cyanol FF. Samples were kept at RT until the last time point was collected and then were loaded onto 1% agarose gel and subjected to electrophoresis in TAE buffer (40 mM Tris·OAc, pH 9.0, 2 mM EDTA) at 35 V h−1 for 10 h. Gels were stained with ethidium bromide (0.5 μg mL−1) for 30 min and subsequently destained in water for 20 min. Destained gels were imaged using Molecular Imager GelDoc XR (Bio-rad, Hercules, CA) and analyzed using Quantity One software, v 4.5.1.Circular dichroism
Circular dichroism (CD) measurements were performed on a JASCO J-715 spectropolarimeter (Easton, MD) equipped with a JASCO PT-423S Peltier temperature controller using 1 mm pathlength quartz cuvettes. Three consecutive scans were taken per sample. A baseline was recorded for each condition and subtracted from the spectrum. Data were collected in 0.5 nm intervals, 4 s averaging, 1 nm bandwidth, and at a scanning speed of 10 nm min−1. Measurements were taken at several different temperature, and salt concentrations following literature protocols.42,64 Briefly, 10–50 μM of peptide was dissolved in 50 mM borate buffer, pH 9.0. Salt-induced folding was measured in 50 mM borate buffer and pH was adjusted to 9.0 after addition of the desired salt. Molar ellipticities were calculated using the relation:| [θ] = θobs × (MRW)/10 × l × c | (2) |
| Helical content (%) = [θ222] × 100/−40000 × [(1 − 2.5/n)] | (3) |
Analytical ultracentrifugation
Apparent molecular masses and oligomerization states of peptides were determined by sedimentation equilibrium on a Beckman ProteomeLab™ XL-I ultracentrifuge (Indianapolis, IN). Details on experimental set-up and data fitting are provided in the ESI.†Conclusions
The biological role of Ocr was elucidated more than three decades ago, but there are still only a handful of protein mimics of DNA known. Nevertheless, they have already found use in practical applications. For example, Hoffman and colleagues have utilized Ocr to increase the transformation efficiency of unmodified DNA in bacterial strains by inhibiting R/M enzymes.72 In another application, monomeric Ocr (Mocr) has been used as a universal affinity tag to purify several passenger proteins.73 MfpA, a DNA mimicking protein from M. tuberculosis, provides an ingenious resistance mechanism to fluoroquinolones by competing for the binding site of DNA gyrase.9 This protein is a potential target for clinical applications and the design of therapeutics.Inspired by these examples, we have used a charge patterning approach to create peptide mimics of B-DNA. We chose to use coiled coils as they are architecturally robust to allow surface amino acid substitution and tailor specificity. We have shown that with the correct surface charge distribution and size, the designed DMs inhibit a type I R/M enzyme. If the principles of binding of proteins to DNA are elucidated, this could provide a platform for the rational design of generic DNA mimics which might be able to target not only the bacterial R/M system but also other systems such as replication, repair and drug resistance. These synthetic mimics could eventually find diagnostic and therapeutic uses in the clinic. Further structural optimization in order to get the linker region to mimic the bent shape of B-DNA can be made by use of defined rigid scaffolds.74 Moreover, better control over oligomeric states of DMs can be achieved by incorporating fluorinated valine and leucine residues in the core. Based on our previous work34,75 we hypothesize that creating such a hydrophobic core will force highly charged monomers into dimers and hence improve the activity of DMs. Studies along these lines are underway in our laboratories.
Acknowledgements
This work was supported by the NIH (GM65500 to K. K; GM66831 to P. R. B.) and E. I. DuPont de Nemours & Co (to K. K.). The ESI-MS facility at Tufts is supported by the NSF (0320783). We thank F. W. Studier for providing plasmids (pAR2993, pAR3790) and D. Pamuk and S. T. Krishnaji for helpful discussions.Notes and references
- M. B. A. Oldstone, FASEB J., 1998, 12, 1255 CAS.
- J. M. Davies, Immunol. Cell Biol., 1997, 75, 113 CrossRef CAS PubMed.
- H. C. Wang, C. H. Ho, K. C. Hsu, J. M. Yang and A. H. J. Wang, Biochemistry, 2014, 53, 2865 CrossRef CAS PubMed.
- P. A. Tsonis and B. Dwivedi, Biochim. Biophys. Acta, Mol. Cell Res., 2008, 1783, 177 CrossRef CAS PubMed.
- D. T. F. Dryden and M. R. Tock, Biochem. Soc. Trans., 2006, 34, 317 CrossRef CAS PubMed.
- C. D. Putnam and J. A. Tainer, DNA Repair, 2005, 4, 1410 CrossRef CAS PubMed.
- O. N. Voloshin, B. E. Ramirez, A. Bax and R. D. Camerini-Otero, Genes Dev., 2001, 15, 415 CrossRef CAS PubMed.
- L. M. Parsons, D. C. Yeh and J. Orban, Proteins: Struct., Funct., Genet., 2004, 54, 375 CrossRef CAS PubMed.
- S. S. Hegde, M. W. Vetting, S. L. Roderick, L. A. Mitchenall, A. Maxwell, H. E. Takiff and J. S. Blanchard, Science, 2005, 308, 1480 CrossRef CAS PubMed.
- M. Ghosh, G. Meiss, A. M. Pingoud, R. E. London and L. C. Pedersen, J. Biol. Chem., 2007, 282, 5682 CrossRef CAS PubMed.
- P. M. Chilley and B. M. Wilkins, Microbiology, 1995, 141, 2157 CrossRef CAS PubMed.
- E. Leon, G. Navarro-Aviles, C. M. Santiveri, C. Flores-Flores, M. Rico, C. Gonzalez, F. J. Murillo, M. Elias-Arnanz, M. A. Jimenez and S. Padmanabhan, Nucleic Acids Res., 2010, 38, 5226 CrossRef CAS PubMed.
- H. C. Wang, T. P. Ko, M. L. Wu, S. C. Ku, H. J. Wu and A. H. J. Wang, Nucleic Acids Res., 2012, 40, 5718 CrossRef CAS PubMed.
- H. C. Wang, M. L. Wu, T. P. Ko and A. H. J. Wang, Nucleic Acids Res., 2013, 41, 5127 CrossRef CAS PubMed.
- C. D. Mol, A. S. Arvai, R. J. Sanderson, G. Slupphaug, B. Kavli, H. E. Krokan, D. W. Mosbaugh and J. A. Tainer, Cell, 1995, 82, 701 CrossRef CAS PubMed.
- J. I. Banos-Sanz, L. Mojardin, J. Sanz-Aparicio, J. M. Lazaro, L. Villar, G. Serrano-Heras, B. Gonzalez and M. Salas, Nucleic Acids Res., 2013, 41, 6761 CrossRef CAS PubMed.
- M. D. Walkinshaw, P. Taylor, S. S. Sturrock, C. Atanasiu, T. Berge, R. M. Henderson, J. M. Edwardson and D. T. F. Dryden, Mol. Cell, 2002, 9, 187 CrossRef CAS PubMed.
- R. Court, N. Cook, K. Saikrishnan and D. Wigley, J. Mol. Biol., 2007, 371, 25 CrossRef CAS PubMed.
- D. Liu, R. Ishima, K. I. Tong, S. Bagby, T. Kokubo, D. R. Muhandiram, L. E. Kay, Y. Nakatani and M. Ikura, Cell, 1998, 94, 573 CrossRef CAS PubMed.
- E. Bochkareva, L. Kaustov, A. Ayed, G. S. Yi, Y. Lu, A. Pineda-Lucena, J. C. C. Liao, A. L. Okorokov, J. Milner, C. H. Arrowsmith and A. Bochkarev, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15412 CrossRef CAS PubMed.
- H. C. Wang, T. P. Ko, Y. M. Lee, J. H. Leu, C. H. Ho, W. P. Huang, C. F. Lo and A. H. J. Wang, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20758 CrossRef CAS PubMed.
- J. D. Watson and F. H. C. Crick, Nature, 1953, 171, 737 CrossRef CAS PubMed.
- H. C. Wang, C. H. Ho, K. C. Hsu, J. M. Yang and A. H. Wang, Biochemistry, 2014, 53, 2865 CrossRef CAS PubMed.
- F. W. Studier, J. Mol. Biol., 1975, 94, 283 CrossRef CAS PubMed.
- F. W. Studier, J. Mol. Biol., 1973, 79, 237 CrossRef CAS PubMed.
- C. Atanasiu, O. Byron, H. McMiken, S. S. Sturrock and D. T. F. Dryden, Nucleic Acids Res., 2001, 29, 3059 CrossRef CAS PubMed.
- J. J. Blackstock, S. U. Egelhaaf, C. Atanasiu, D. T. F. Dryden and W. C. K. Poon, Biochemistry, 2001, 40, 9944 CrossRef CAS PubMed.
- A. Lupas, Trends Biochem. Sci., 1996, 21, 375 CrossRef CAS PubMed.
- J. M. Mason and K. M. Arndt, ChemBioChem, 2004, 5, 170 CrossRef CAS PubMed.
- F. H. C. Crick, Acta Crystallogr., 1953, 6, 689 CrossRef CAS.
- D. N. Woolfson, Adv. Protein Chem., 2005, 70, 79 CrossRef CAS PubMed.
- A. N. Lupas and M. Gruber, Adv. Protein Chem., 2005, 70, 37 CrossRef CAS PubMed.
- J. M. Mason and K. M. Arndt, ChemBioChem, 2004, 5, 170 CrossRef CAS PubMed.
- B. Bilgiçer and K. Kumar, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15324 CrossRef PubMed.
- D. N. Woolfson, Curr. Opin. Struct. Biol., 2001, 11, 464 CrossRef CAS PubMed.
- M. G. Oakley and P. S. Kim, Biochemistry, 1998, 37, 12603 CrossRef CAS PubMed.
- K. J. Lumb and P. S. Kim, Biochemistry, 1995, 34, 8642 CrossRef CAS PubMed.
- P. B. Harbury, T. Zhang, P. S. Kim and T. Alber, Science, 1993, 262, 1401 CAS.
- W. D. Kohn, C. M. Kay and R. S. Hodges, J. Pept. Sci., 1997, 3, 209 CrossRef CAS PubMed.
- P. Laigne, F. D. Sonnichsen, C. M. Kay, R. S. Hodges, K. J. Lumb and P. S. Kim, Science, 1996, 271, 1136 CrossRef.
- K. J. Lumb and P. S. Kim, Science, 1995, 268, 436 CAS.
- E. K. O'Shea, K. J. Lumb and P. S. Kim, Curr. Biol., 1993, 3, 658 CrossRef.
- D. N. Marti, I. Jelesarov and H. R. Bosshard, Biochemistry, 2000, 39, 12804 CrossRef CAS PubMed.
- W. D. Kohn, O. D. Monera, C. M. Kay and R. S. Hodges, J. Biol. Chem., 1995, 270, 25495 CrossRef CAS PubMed.
- J. R. Litowski and R. S. Hodges, J. Biol. Chem., 2002, 277, 37272 CrossRef CAS PubMed.
- D. Papapostolou, E. H. C. Bromley, C. Bano and D. N. Woolfson, J. Am. Chem. Soc., 2008, 130, 5124 CrossRef CAS PubMed.
- D. N. Woolfson and M. G. Ryadnov, Curr. Opin. Chem. Biol., 2006, 10, 559 CrossRef CAS PubMed.
- R. L. Dunbrack Jr and M. Karplus, J. Mol. Biol., 1993, 230, 543 CrossRef CAS PubMed.
- J. W. Ponder and F. M. Richards, J. Mol. Biol., 1987, 193, 775 CrossRef CAS PubMed.
- N. E. Murray, Microbiol. Mol. Biol. Rev., 2000, 64, 412 CrossRef CAS PubMed.
- A. A. Bourniquel and T. A. Bickle, Biochimie, 2002, 84, 1047 CrossRef CAS PubMed.
- N. E. Murray, Microbiology, 2002, 148, 3 CrossRef CAS PubMed.
- D. J. Ellis, D. T. F. Dryden, T. Berge, J. Michael Edwardson and R. M. Henderson, Nat. Struct. Biol., 1999, 6, 15 CrossRef CAS PubMed.
- E. Jindrova, S. Schmid-Nuoffer, F. Hamburger, P. Janscak and T. A. Bickle, Nucleic Acids Res., 2005, 33, 1760 CrossRef CAS PubMed.
- F. W. Studier and P. K. Bandyopadhyay, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 4677 CrossRef CAS.
- G. B. Zavilgelsky and S. M. Rastorguev, Mol. Biol., 2009, 43, 241 CrossRef CAS.
- C. Atanasiu, T. J. Su, S. S. Sturrock and D. T. F. Dryden, Nucleic Acids Res., 2002, 30, 3936 CrossRef CAS PubMed.
- P. K. Bandyopadhyay, F. W. Studier, D. L. Hamilton and R. Yuan, J. Mol. Biol., 1985, 182, 567 CrossRef CAS PubMed.
- P. R. Bianco and E. M. Hurley, J. Mol. Biol., 2005, 352, 837 CrossRef CAS PubMed.
- P. R. Bianco, C. Xu and M. Chi, Nucleic Acids Res., 2009, 37, 3377 CrossRef CAS PubMed.
- S. V. Nekrasov, O. V. Agafonova, N. G. Belogurova, E. P. Delver and A. A. Belogurov, J. Mol. Biol., 2007, 365, 284 CrossRef CAS PubMed.
- A. S. Stephanou, G. A. Roberts, L. P. Cooper, D. J. Clarke, A. R. Thomson, C. L. MacKay, M. Nutley, A. Cooper and D. T. F. Dryden, J. Mol. Biol., 2009, 391, 565 CrossRef CAS PubMed.
- A. S. Stephanou, G. A. Roberts, M. R. Tock, E. H. Pritchard, R. Turkington, M. Nutley, A. Cooper and D. T. Dryden, Biochem. Biophys. Res. Commun., 2009, 378, 129 CrossRef CAS PubMed.
- I. Jelesarov, E. Dürr, R. M. Thomas and H. R. Bosshard, Biochemistry, 1998, 37, 7539 CrossRef CAS PubMed.
- G. A. Roberts, A. S. Stephanou, N. Kanwar, A. Dawson, L. P. Cooper, K. Chen, M. Nutley, A. Cooper, G. W. Blakely and D. T. Dryden, Nucleic Acids Res., 2012, 40, 8129 CrossRef CAS PubMed.
- C. Park, J. L. Campbell and W. A. Goddard, J. Am. Chem. Soc., 1996, 118, 4235 CrossRef CAS.
- C. N. Pace, F. Vajdos, L. Fee, G. Grimsley and T. Gray, Protein Sci., 1995, 4, 2411 CrossRef CAS PubMed.
- S. S. Sturrock, D. T. F. Dryden, C. Atanasiu, J. Dornan, S. Bruce, A. Cronshaw, P. Taylor and M. D. Walkinshaw, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2001, 57, 1652 CrossRef CAS.
- K. K. Mark and F. W. Studier, J. Biol. Chem., 1981, 256, 2573 CAS.
- K. N. Kreuzer and C. V. Jongeneel, Methods Enzymol., 1983, 100, 144 CAS.
- Y. H. Chen, J. T. Yang and K. H. Chau, Biochemistry, 1974, 13, 3350 CrossRef CAS PubMed.
- L. M. Hoffman, D. J. Haskins and J. Jendrisak, EPICENTRE Forum 9,8, 2002, http://www.epicentre.com/typeone.asp Search PubMed.
- J. DelProposto, C. Y. Majmudar, J. L. Smith and W. C. Brown, Protein Expression Purif., 2009, 63, 40 CrossRef CAS PubMed.
- C. G. Levins and C. E. Schafmeister, J. Am. Chem. Soc., 2003, 125, 4702 CrossRef CAS PubMed.
- N. C. Yoder and K. Kumar, Chem. Soc. Rev., 2002, 31, 335 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, supplementary figures and tables. See DOI: 10.1039/c5mb00524h |
| This journal is © The Royal Society of Chemistry 2016 |