
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew oligonucleotide derivatives as unreactive substrate analogues and potential inhibitors of human apurinic/apyrimidinic endonuclease APE1
Nikita A.
Kuznetsov
 ab,
Maxim S.
Kupryushkin
a,
Tatyana V.
Abramova
a,
Alexandra A.
Kuznetsova
a,
Anastasia D.
Miroshnikova
a,
Dmitry A.
Stetsenko
ab,
Dmitrii V.
Pyshnyi
ab and
Olga S.
Fedorova
*ab
ab,
Maxim S.
Kupryushkin
a,
Tatyana V.
Abramova
a,
Alexandra A.
Kuznetsova
a,
Anastasia D.
Miroshnikova
a,
Dmitry A.
Stetsenko
ab,
Dmitrii V.
Pyshnyi
ab and
Olga S.
Fedorova
*ab
aInstitute of Chemical Biology and Fundamental Medicine, Siberian Branch of the Russian Academy of Sciences, Novosibirsk 630090, Russia. E-mail: fedorova@niboch.nsc.ru
bDepartment of Natural Sciences, Novosibirsk State University, Novosibirsk 630090, Russia
First published on 29th October 2015
Abstract
Human apurinic/apyrimidinic endonuclease APE1 is one of the key enzymes of the base excision DNA repair system. The main biological function of APE1 is the hydrolysis of the phosphodiester bond on the 5′-side of an apurinic/apyrimidinic site (AP-site) to give the 5′-phosphate and 3′-hydroxyl group. It has long been known that AP-sites have mutagenic and cytotoxic effects and their accumulation in DNA is a potential hazard to the cell lifecycle. The structural and biochemical studies of APE1 are complicated by its high catalytic activity towards the AP-site and its cyclic or acyclic analogues. This work has focussed on the design, synthesis and analysis of oligonucleotide derivatives as potentially unreactive APE1 substrates. We have shown that the replacement of oxygen atoms in the phosphate group on the 5′-side from the AP-site analogue tetrahydrofuran (F) considerably decreases the rate of enzymatic hydrolysis of modified oligonucleotides. We have calculated that a N3′–P5′ phosphoramidate linkage is hydrolysed about 30 times slower than the native phosphodiester bond while phosphorothioate or primary phosphoramidate linkages are cleaved more than three orders of magnitude slower. The value of IC50 of the oligonucleotide duplex containing a primary phosphoramidate linkage is 2.5 × 10−7 M, which is in accordance with the APE1 association constant of DNA duplexes containing AP-sites. Thus, it is demonstrated that oligonucleotide duplexes with chemical modifications could be used as unreactive substrates and potential competitive inhibitors of APE1.
Introduction
Keeping DNA intact is the key to safeguarding genetic information and smooth running of all vital processes in the cell. In a mammalian genome of billions of nucleotides some 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 lesions per day may occur.1–4 Genotoxicity may result from a number of endogenous factors such as side effects of normal cell metabolites, and exogenous factors such as UV irradiation, ionising radiation and a variety of mutagenic chemical compounds.5,6
000 lesions per day may occur.1–4 Genotoxicity may result from a number of endogenous factors such as side effects of normal cell metabolites, and exogenous factors such as UV irradiation, ionising radiation and a variety of mutagenic chemical compounds.5,6
It is thought that a majority of nucleobase lesions are removed via the base excision repair (BER) pathway.7–9 This process is initiated by DNA glycosylases, which find lesions amongst all the undamaged bases and catalyse break-up of N-glycosidic bonds.10,11 After the action of DNA glycosylases the resulting apurinic/apyrimidinic site (AP-site) is targeted by apurinic/apyrimidinic endonuclease that cleaves the 5′-phosphate bond. The nick contains a hydroxyl group on the 3′-end and a 2′-deoxyribonucleoside-5′-phosphate on the 5′-end. Later stages of repair are performed by DNA polymerases and DNA ligases, which restore the initial DNA sequence.12,13
Disruptions in the work of DNA repair enzymes cause severe consequences such as premature aging, cancer and other diseases.14–17 It was shown that cells or animals in which some DNA glycosylase genes are knocked out become more sensitive to DNA-damaging factors.18–21 At the same time exclusion of the second player of the BER pathway AP-endonuclease results in cell death, which reveals a critical role APE1 plays in the restoration of the native DNA structure.22
Thus, the level of enzymatic activity of human AP-endonuclease APE1 has a considerable influence on the process of the removal of DNA lesions. APE1 is an extremely active enzyme,23,24 which is able to process many synthetic analogues of the AP-site such as tetrahydrofuran (F-site) or α,ω-alkanediols.24–26
Analysis of crystal structures of free APE127–29 and its complexes with DNA30–32 shows that currently there is still some uncertainty in our understanding of the multiple catalytic functions of the enzyme. Moreover, the stoichiometry of the divalent cations and their catalytic functions are still not clear.27–29,33,34 Therefore, the important aspects of the APE1-mediated catalytic mechanism remain unresolved due to the lack of convenient unreactive analogues of APE1 substrates.
The objective of this study was to prepare modified oligonucleotide derivatives and ascertain their utility in vitro as unreactive substrate analogues of human apurinic/apyrimidinic endonuclease APE1. Some of these derivatives may also have potential as inhibitors of APE1 in vivo. APE1 inhibition is expected to enhance genotoxicity of DNA lesions and accelerate cell death, which is the goal of radiation therapy and chemotherapy of cancer. On-going development of effective systems for cell delivery of DNA and RNA derivatives35 allows us to believe that those oligonucleotide inhibitors of APE1 may find their use in near future as effective and stable in vivo therapeutics.
Results and discussion
Rational design of oligonucleotide derivatives as potential APE1 inhibitors
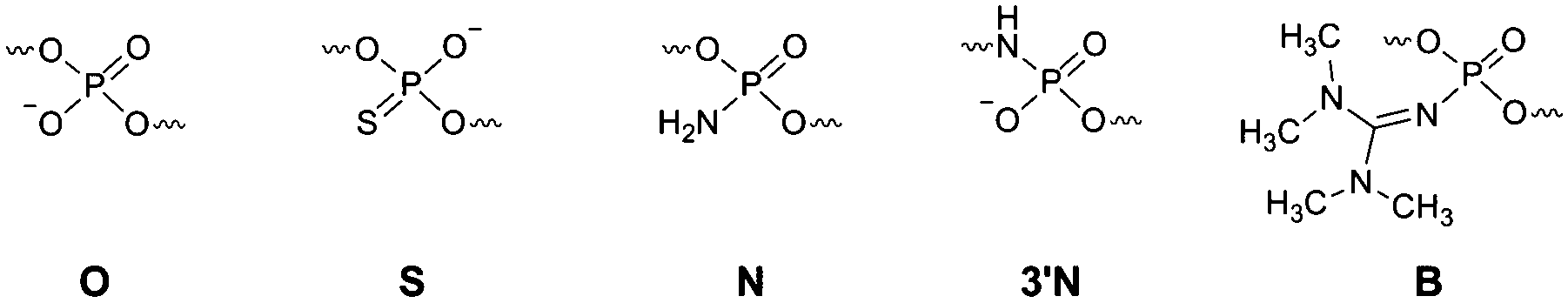
Information on the mechanism of the APE1-catalysed reaction25,28,30,36,37 prompted us to hypothesise that if we substitute the NH group for the 3′-bridging oxygen atom in the phosphate group of the nucleotide on the 5′-side from the AP-site to form an N3′–P5′ phosphoramidate, or sulphur S, an amino group NH2 or a 1,1,3,3-tetramethylguanidine (Tmg) group for any of the non-bridging oxygen atoms in the same phosphate group (Fig. 1), we disrupt its interactions with Mg2+ cations and amino acid residues in the active centre of the enzyme, which will likely decrease the rate of the catalytic reaction. Thus, in this work we set out to synthesise oligonucleotides incorporating those substitutions (Fig. 2 and Table 1). As APE1 exhibits also 3′–5′-exonuclease activity albeit less pronounced compared to its endonuclease activity, the 3′-terminal internucleotide phosphate group was chemically modified by the replacement of its oxygen atom by either a sulphur or a Tmg group to make the phosphate resistant to the exonuclease activity of the enzyme (Fig. 2 and Table 1).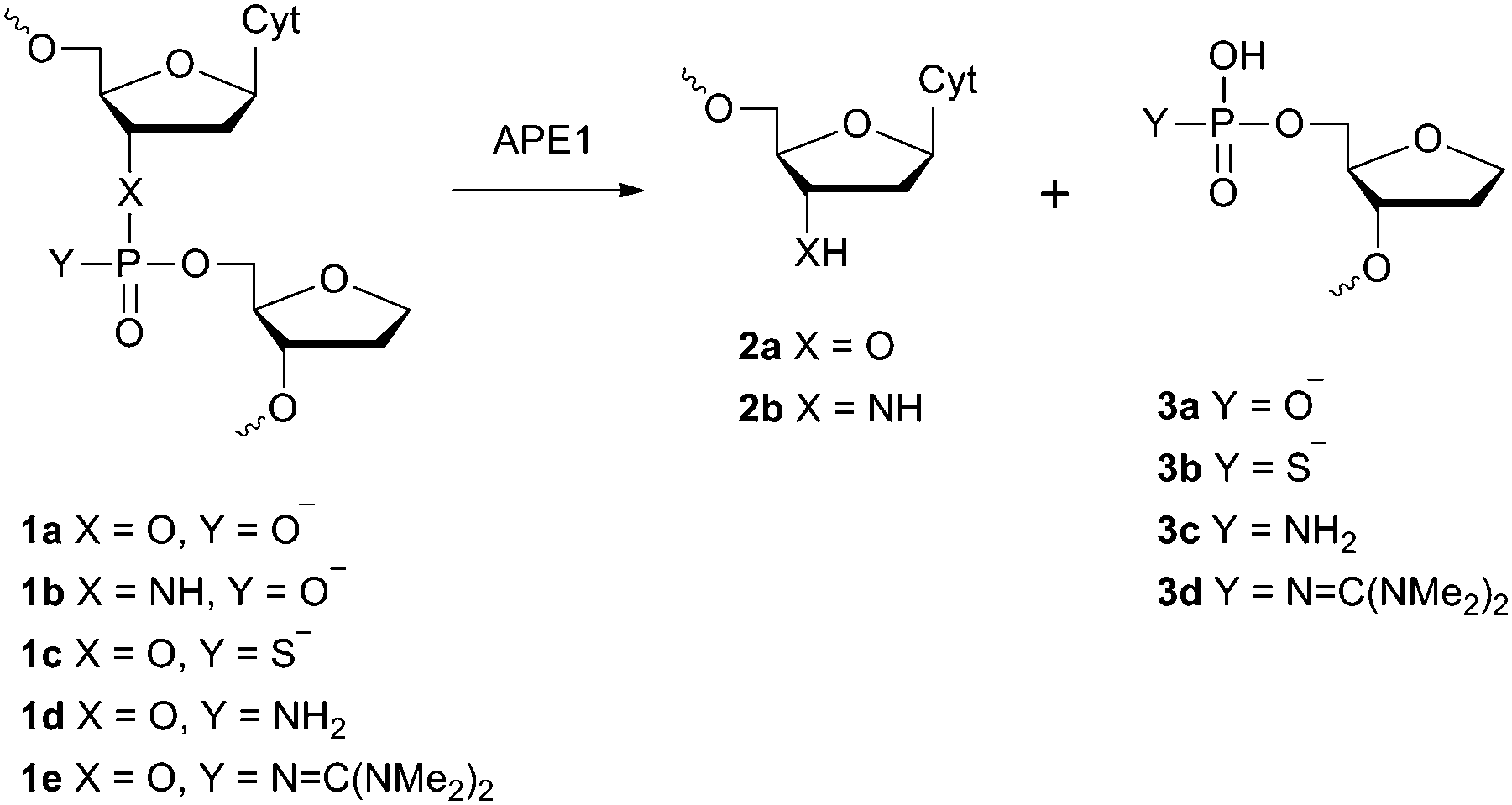 | ||
| Fig. 1 Structure of the F-site and chemical modifications of the phosphate group on its 5′-side that are resistant to APE1 hydrolysis. | ||
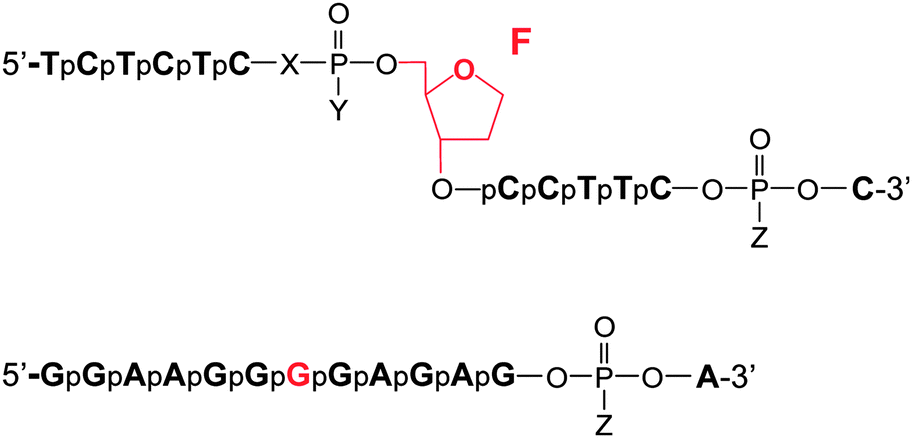 | ||
| Fig. 2 Oligonucleotide structures. Chemical modifications of phosphate groups are listed in Table 1. | ||
| Abbreviation | Chemical modification of the phosphate group |
|---|---|
a Tmg – 1,1,3,3-tetramethylguanidine group –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(NMe2)2. C(NMe2)2.
|
|
| FO | X = O, Y = O−, Z = O− |
| F3′N | X = NH, Y = O−, Z = O− |
| FS | X = O, Y = S−, Z = O− |
| FSS | X = O, Y = S−, Z = S− |
| FN | X = O, Y = NH2, Z = O− |
| FNB | X = O, Y = NH2, Z = Tmga |
| FBB | X = O, Y = Tmg, Z = Tmg |
| GO | Z = O− |
| GS | Z = S− |
| GB | Z = Tmg |
Instead of the natural AP-site, the oligonucleotides incorporated its chemically stable analogue (2R,3S)-2-(hydroxymethyl)-3-hydroxytetrahydrofuran (F-site). It is known that such a substitution scarcely affects the activity of APE1.24–26,38,39
Chemical synthesis of oligonucleotides with modified phosphate groups
An oligonucleotide with a N3′–P5′ phosphoramidate group was prepared using a dimer phosphoramidite I according to the approach described in ref. 40. Chemical synthesis of the dimer phosphoramidite I was accomplished as shown in Fig. 3. A known 5′-protected (2R,3S)-2-(hydroxymethyl)-3-hydroxy-tetrahydrofuran II41 was converted to 3′-protected compound III. After its 5′-phosphitylation and hydrolysis of the transient phosphoramidite H-phosphonate diester IV was obtained that was in turn reacted with protected 3′-deoxy-3′-aminocytidine V.40 After the selective removal of the levulinyl group and 3′-phosphitylation the desired dimer phosphoramidite I was obtained that was subsequently used in the synthesis of an oligonucleotide containing the AP-site mimic F and the N3′–P5′ phosphoramidate internucleotide linkage on the 5′-side of the F-site. To confirm that the P–N bond would be stable to the conditions of oligonucleotide synthesis, the protected dimer VI was unblocked by applying standard procedures of oligonucleotide deprotection, i.e. conc. aq. ammonia, 3 h at 55 °C, then 80% acetic acid, 30 min. The product was analysed by mass-spectroscopy. Its ESI-MS spectrum contained the only peak with m/z 405.10 that corresponded to the molecular ion [M − H]− of the fully unprotected dinucleotide VI.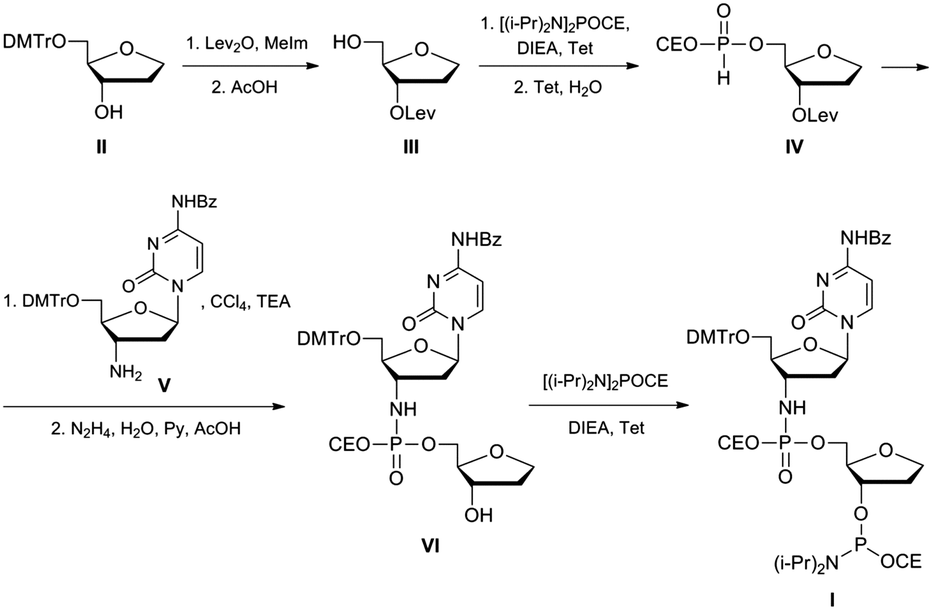 | ||
| Fig. 3 Synthesis of a dimer phosphoramidite I. | ||
Dimer phosphoramidite I was used in automated oligonucleotide synthesis together with regular nucleoside phosphoramidites to obtain the model oligonucleotides (Fig. 2 and Table 1).
Analysis of the rate of hydrolysis by APE1 of oligonucleotides with modified phosphate groups incorporated into DNA substrate duplexes
The efficiency of enzymatic hydrolysis by APE1 of double-stranded (ds) DNA substrates containing oligonucleotides with modified phosphate groups was ascertained by the separation of hydrolysis products by PAGE. Observed rate constants of chain scission for duplexes with substitutions only in the phosphate group on the 5′-side of the F-site were found to decrease in the order of FO/GO > F3′N/GO ≫ FN/GO > FS/GO (Fig. 4 and Table 2). Interestingly, the N3′–P5′ phosphoramidate linkage 1b in the DNA duplex F3′N/GO is hydrolysed only about 30 times slower than the native phosphodiester group 1a in the unmodified DNA duplex FO/GO. At the same time, oligonucleotides with phosphorothioate group 1c or primary phosphoramidate group 1d are hydrolysed more than three orders of magnitude slower than the unmodified duplex. Therefore, oligonucleotides containing those modifications could be potentially promising candidates for binding to and inhibiting the activity of APE1.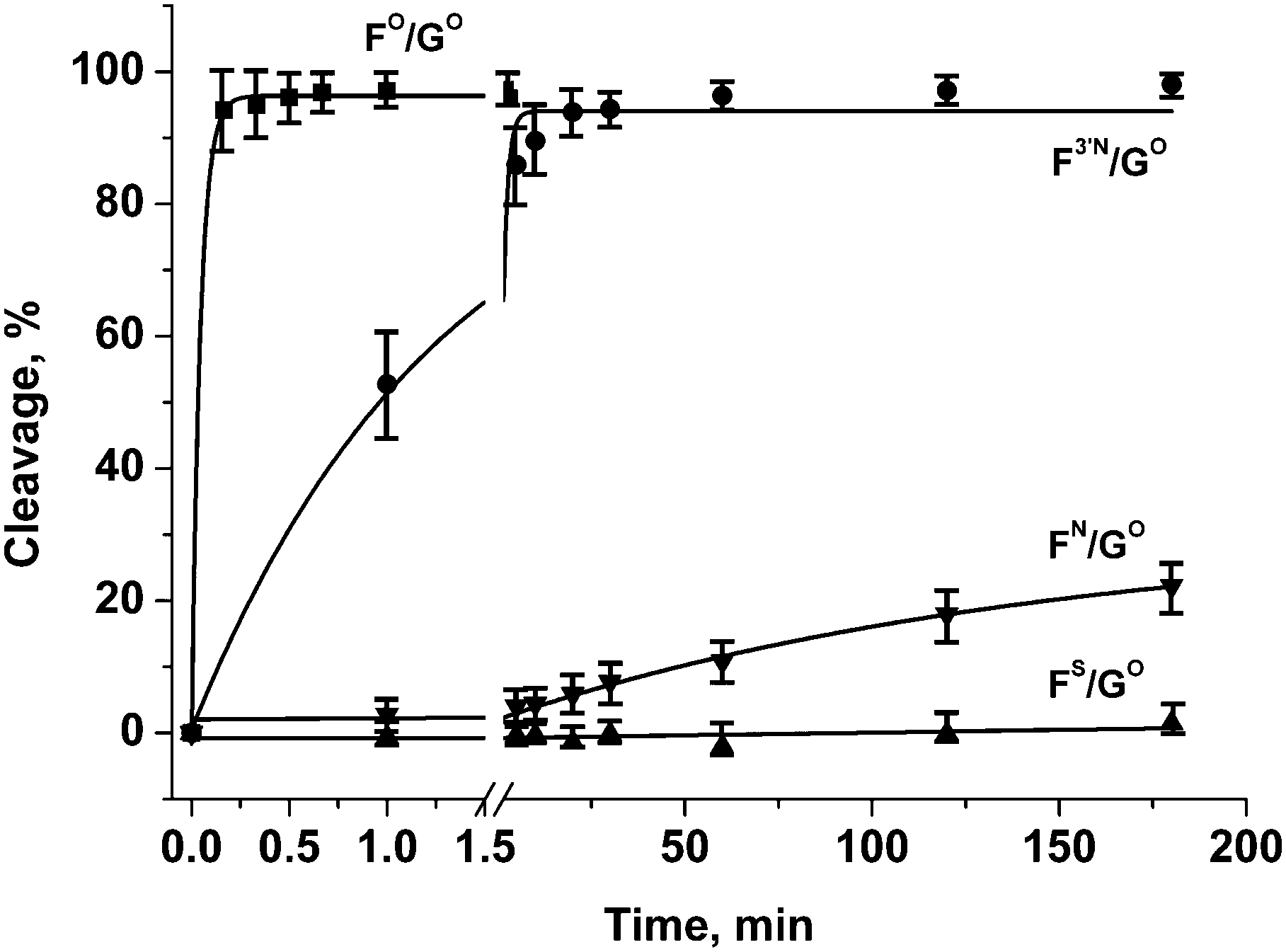 | ||
| Fig. 4 Endonuclease activity of APE1 towards DNA duplexes with modified phosphate groups on the 5′-side of the F-site. [APE1] = [dsDNA] = 1.0 μM. | ||
| DNA substrate | Observed rate constant kobs, min−1 | Dissociation constant Kd, Ma |
|---|---|---|
| a Obtained by fluorescence titration. | ||
| FO/GO | 22.4 ± 2.4 | ND |
| F3′N/GO | 0.8 ± 0.1 | (0.5 ± 0.4) × 10−7 |
| FN/GO | (6.8 ± 2.4) × 10−3 | (1.5 ± 0.5) × 10−7 |
| FS/GO | <1.0 × 10−4 | (1.1 ± 0.1) × 10−7 |
| FNB/GB | (1.9 ± 0.4) × 10−2 | ND |
| FBB/GB | <1.0 × 10−4 | ND |
As could be seen from the above data, the endonuclease activity of APE1 is considerably retarded for DNA duplexes containing phosphate modifications on the 5′-side of the F-site. However, analysis of the products of chain scission of those duplexes has revealed that some 3′–5′-exonuclease reaction that removes the 3′-terminal nucleotide has also occurred. To suppress the removal of the 3′-terminal nucleotide from the model oligonucleotides, their 3′-terminal internucleotide phosphate group was modified (Fig. 2 and Table 1). The tetramethyl phosphoryl guanidine group (Tmg) was employed as a nuclease-resistant phosphate group isostere.42 It was observed that such a modification blocks 3′–5′-exonuclease activity of APE1 for more than 12 h. The group was also tested as a protecting group for the phosphate group on the 5′-side of the F-site 1e (Fig. 1). The data obtained by PAGE analysis of reaction products have shown that the duplex containing the dinucleotide 1e was resistant to the endonuclease action of APE1 for at least 3 h (Fig. 5 and Table 2).
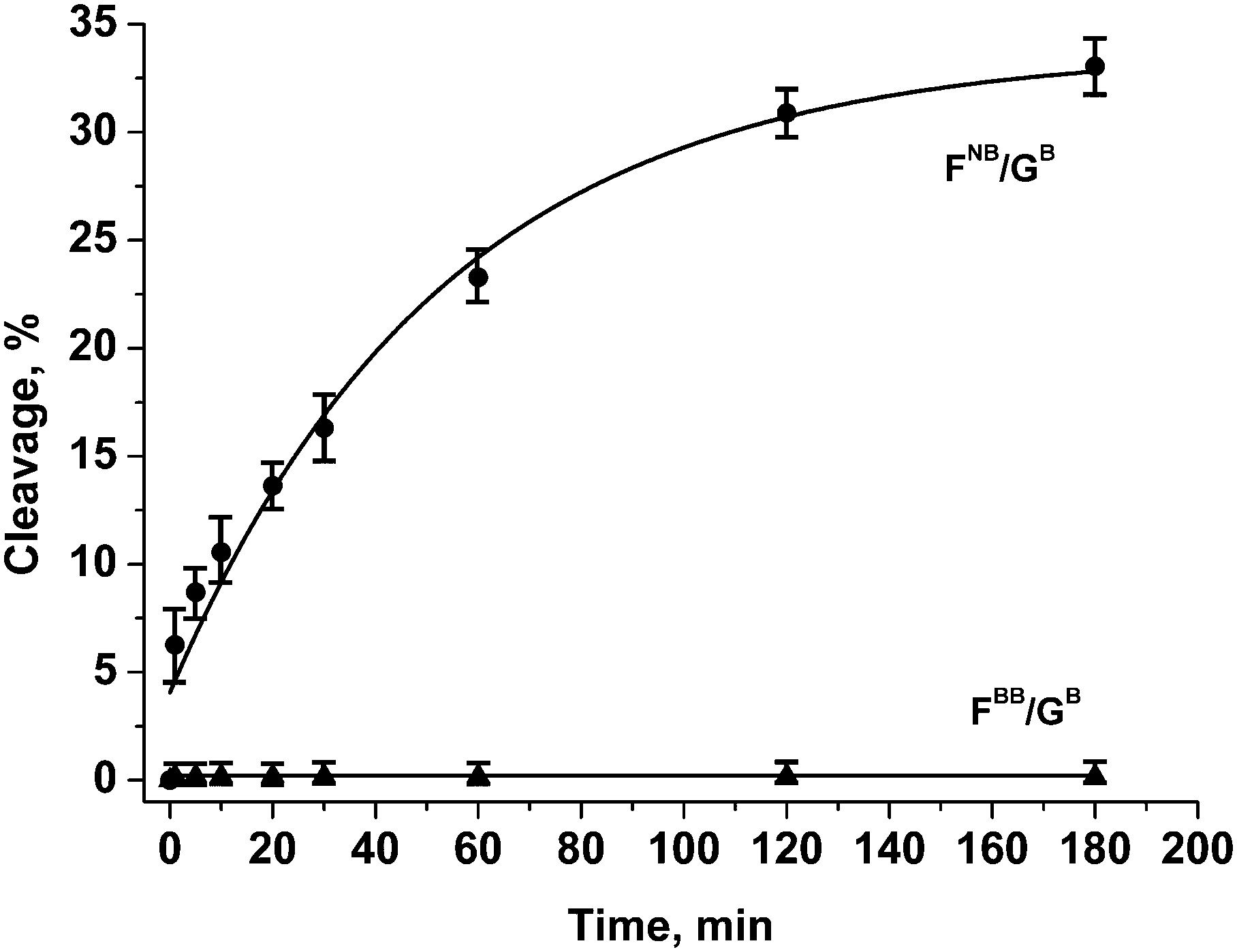 | ||
| Fig. 5 Endonuclease activity of APE1 towards DNA duplexes with modified phosphate groups on the 5′-side of the F-site and in the 3′-internucleotide position. [APE1] = [dsDNA] = 1.0 μM. | ||
Comparison of cleavage efficacy (Fig. 6) of the modified oligonucleotide derivatives has shown that the modifications reduce enzymatic activity of APE1 in the order FO/GO ≫ F3′N/GO ≫ FNB/GB > FN/GO > FS/GO ≈ FBB/GB.
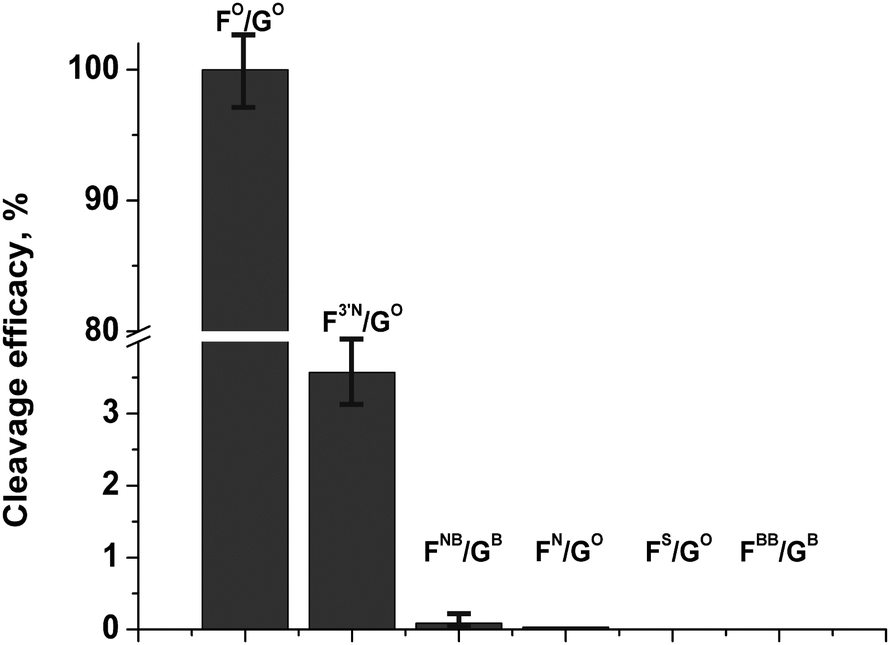 | ||
| Fig. 6 Comparison of cleavage efficacy of modified oligonucleotide derivatives. [APE1] = [dsDNA] = 1.0 μM. | ||
Binding of DNA duplexes to APE1
To ascertain the efficacy of APE1 binding to modified DNA duplexes, the kinetics of enzyme–substrate complex formation was studied by the “stopped-flow” method. The kinetic curve of the APE1 reaction with duplex FO/GO goes through characteristic phases (Fig. 7). The data for the APE1 interaction with duplexes incorporating the F-site that were published in ref. 38 and 39 prompted us to conclude that the initial (up until 0.1 seconds) decrease in fluorescence intensity could be attributed to the process of the formation of an enzyme–substrate complex that is responsible for the catalytic hydrolysis of the internucleotide phosphodiester bond. After that initial period within 0.1–10 seconds the complex dissociates with the concomitant release of the products of the reaction, which results in the increase in fluorescence intensity. As it is shown in Fig. 7, the interaction of APE1 with duplexes FN/GO and FNB/GB until 0.1 s was accompanied by only the decrease in fluorescence intensity that correlates with the formation of an enzyme–substrate complex. At the same time the interaction of APE1 with duplex FBB/GB did not result in any change in fluorescence intensity. This result may suggest that the presence of a bulky Tmg group on the 5′-side of the F-site has prevented the formation of an enzyme–substrate complex.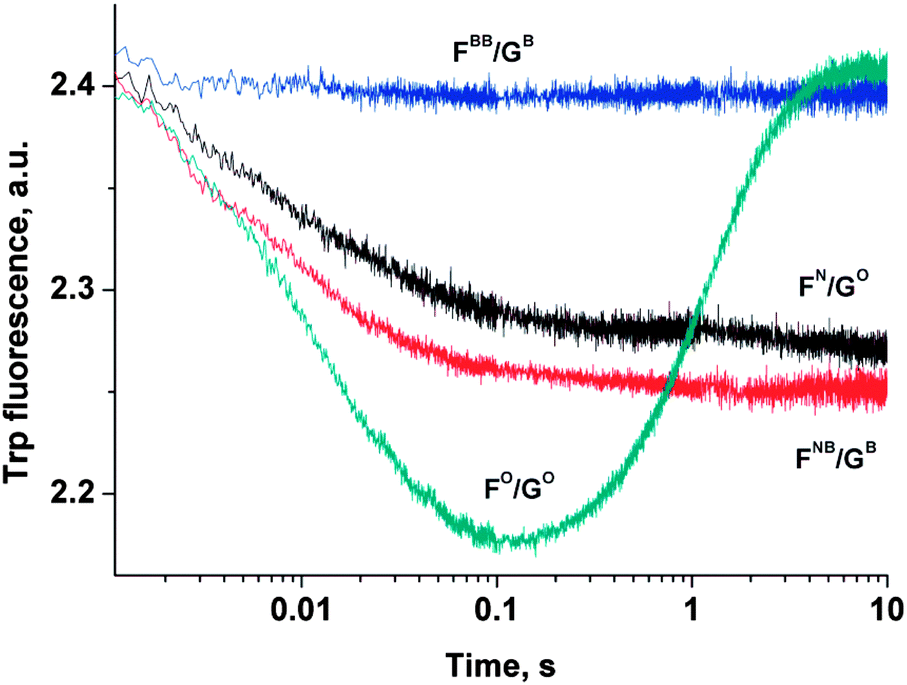 | ||
| Fig. 7 Kinetic curves of Trp fluorescence emission change during the APE1 interaction with modified DNA duplexes. [APE1] = [dsDNA] = 1.0 μM. | ||
To estimate the dissociation constant of the enzyme/inhibitor complex, APE1 was titrated with F3′N/GO, FN/GO and FS/GO duplexes. The change in APE1 protein fluorescence is shown in Fig. 8.
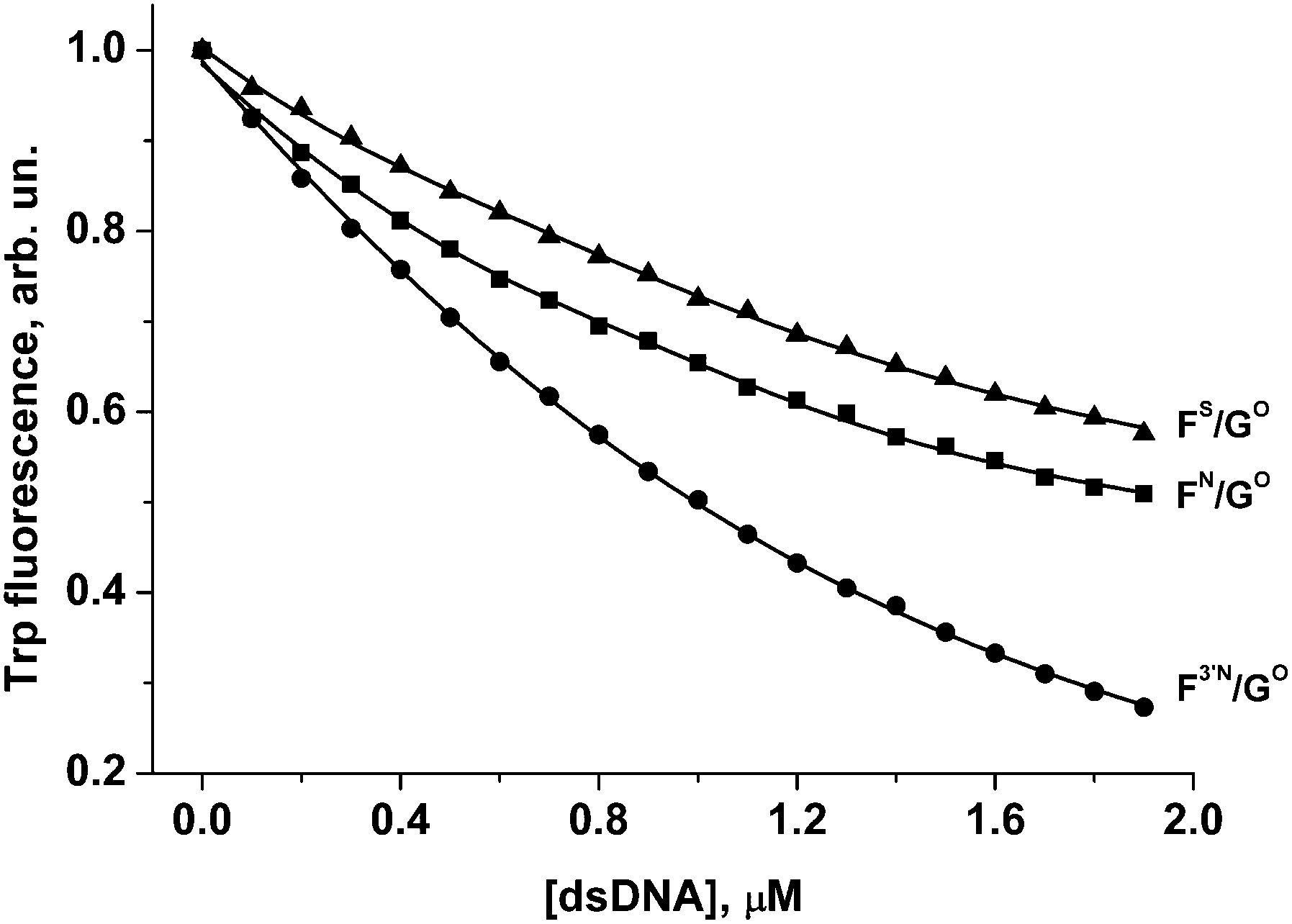 | ||
| Fig. 8 Change in APE1 fluorescence during titration with F3′N/GO, FN/GO and FS/GO duplexes. [APE1] = 1.0 μM. Triangle, FS/GO duplex; square, FN/GO duplex; circle, F3′N/GO duplex. | ||
Quenching of the Trp fluorescence was registered for all these DNA duplexes. The calculated dissociation constants Kd are listed in Table 2. It is worthy of note that values of Kd for FN/GO and FS/GO duplexes are approximately the same considering the standard deviation interval. It is also noteworthy that the F3′N/GO duplex is partially cleaved during the titration time. Probably, due to this fact the value of Kd for the F3′N/GO duplex is 2–3 times lower in comparison with FS/GO or FN/GO duplexes, respectively.
Assessment of inhibition efficacy
For one of the promising inhibitors of APE1, duplex FN/GO, we have assessed the inhibition effect for the reaction of hydrolysis of a DNA substrate FO/GO. For this, duplex FN/GO in various concentrations was added to the mixture of APE1 and FO/GO. As it could be seen in Fig. 9A, the rate of hydrolysis of a DNA substrate FO/GO was considerably decreased in the presence of FN/GO. The dependence of the observed reaction rate on the concentration of the inhibitor allowed us to estimate the value of IC50 = 2.5 × 10−7 M (Fig. 9C), which is in accordance with the value of APE1 association constants with F-containing DNA duplexes.38,39 Thus, the replacement of one of the non-bridging oxygen atoms in the phosphate group on the 5′-side from the F-site by the amino group to form a primary phosphoramidate did not significantly affect the affinity of APE1 for the DNA substrate. At the same time, the catalytic activity of APE1 has decreased considerably.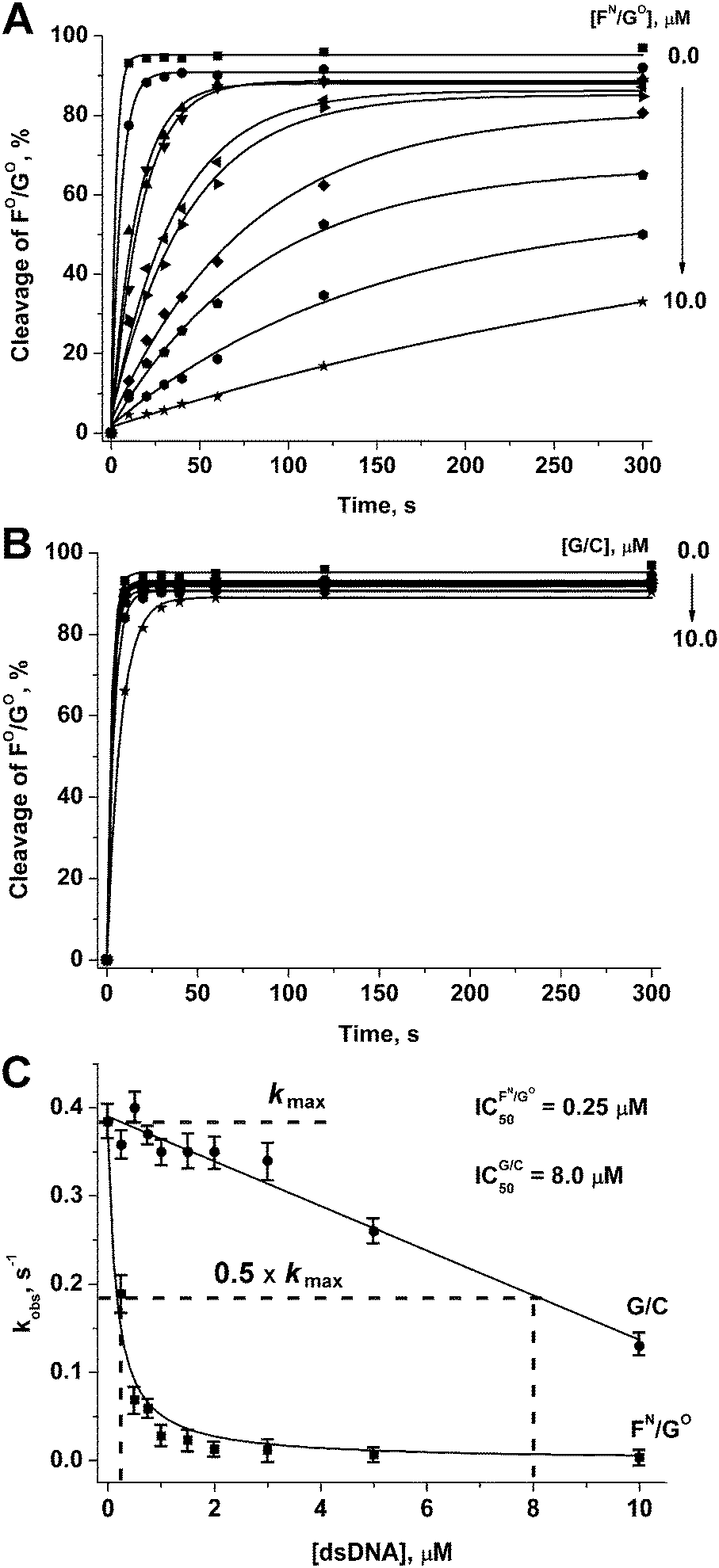 | ||
| Fig. 9 APE1-catalysed hydrolysis of a duplex FO/GO in the presence of duplexes FN/GO (A) and G/C (B). Estimation of the IC50 value (C). [APE1] = [FO/GO] = 1.0 μM. | ||
As a control, we have studied the kinetics of the interaction of APE1 with a DNA duplex 5′-d(CTCTCGCCTTCC)-3′/3′-d(GAGAGCGGAAGG)-5′ (G/C), which did not contain any of the modifications. As seen from Fig. 9B and C, duplex G/C slightly retards enzymatic activity of APE1 resulting in IC50 = 8.0 × 10−6 M. Thus, inhibitory activity of the duplex FN/GO is related to specific interactions of APE1 with the F-site.
It should be noted that IC50 for most of the reported small-molecule inhibitors of APE1 is in the 1.0–10.0 × 10−6 M range.43,44 One of the potent inhibitors of APE1 identified in the library of pharmacologically active compounds (LOPAC) was aurintricarboxylic acid (ATA), displaying an IC50 value of 5.5 × 10−8 M.45 The developments in oligonucleotide synthesis provided an opportunity for their use as therapeutic agents.46,47 Therefore, the compounds reported in the present work could be used not only for biochemical and structural studies of AP endonucleases but also could have potential for subsequent application for therapy.
Experimental
The following chemicals were used in this work: N,N,N′,N′-tetraisopropyl-2-cyanoethylphosphordiamidite, N,N-diisopropylethylamine (DIEA), 1H-tetrazole, levulinic acid and acetylacetone were from Aldrich (USA), 1-methylimidazole, N,N′-dicyclohexylcarbodiimide (DCC) and hydrazine hydrate were from Merck (FRG), the remainder were from “Reakhim” (Russia). All the reagents and solvents were used as purchased unless stated otherwise. Thin layer chromatography (TLC) was carried out on Kieselgel 60 F254 plates (Merck, FRG). The spots were visualized under UV light (254 nm) or by spraying with 0.25% cysteine solution in 30% H2SO4 and heating. Flash column chromatography was on Kieselgel 55–100 μm (Merck, FRG). NMR spectra were recorded on a Brücker AM-400 spectrometer (FRG). Chemical shifts δ are reported in ppm against tetramethylsilane as an internal standard for 1H and 13C NMR spectra, and 85% H3PO4 as an external standard for 31P NMR spectra. 13C and 31P NMR spectra were recorded with proton decoupling unless stated otherwise. Mass spectra were recorded on an Agilent ESI MSD XCT Ion Trap mass spectrometer (USA) in the SB RAS “Proteomics” Core Facility.1,4-Anhydro-2-deoxy-5-O-(4,4′-dimethoxytrityl)-D-ribitol (II) was prepared as described in ref. 41.
3′-Amino-N4-benzoyl-2′,3′-dideoxy-5′-O-(4,4′-dimethoxytrityl)-cytidine (V) was obtained according to ref. 40
1,4-Anhydro-2-deoxy-3-O-levulinyl-D-ribitol (III)
To a solution of levulinic acid (2.3 g, 19.5 mmol) in diethyl ether (50 ml) solid DCC (2.1 g, 10 mmol) was added in portions with stirring. After the reaction mixture was stirred for 3 h, the precipitate was filtered and washed by diethyl ether (10 ml), and the combined ether solution was concentrated in vacuo, the rest dissolved in pyridine (20 ml) and added to 1,4-anhydro-2-deoxy-5-O-(4,4′-dimethoxytrityl)-D-ribitol (II) (2.6 g, 6.5 mmol) and 1-methylimidazole (0.8 ml, 10 mmol). After 1 h, the reaction was quenched by 5% NaHCO3, concentrated in vacuo and partitioned between water and dichloromethane. The organic layer was dried over anhydrous Na2SO4, the solvent removed in vacuo, and the oily residue re-evaporated with toluene to remove traces of pyridine and dissolved in 80% aq. acetic acid (10 ml). The mixture was stirred for 3 h, then water (50 ml) was added, and the volatiles removed in vacuo (traces of water eliminated by co-evaporation with acetonitrile). The residue was separated by chromatography on a silica gel column eluted with a gradient of acetone in dichloromethane 0–30%. Yield of 1,4-anhydro-2-deoxy-3-O-levulinyl-D-ribitol (III) 0.85 g (60%). Rf 0.40 (CH2Cl2–EtOH 19:1). 1H NMR (CDCl3): 5.11 (1H, dt, J = 6.6, 2.3 Hz, H3), 4.06 (1H, dt, J = 8.3, 2.3 Hz, H4), 3.95–3.87 (2H, m, H1), 3.75 (1H, dd, J = 11.7, 4.0 Hz, H5), 3.68 (1H, dd, J = 11.7, 4.0 Hz, H5), 2.83–2.72 (2H, m, C(O)C![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) 2CH2C(O)CH3), 2.65–2.53 (2H, m, C(O)CH2C2C(O)CH3), 2.21 (1H, s, CH3), 2.19–2.13 (1H, m, H2), 2.06-2.00 (1H, m, H2); 13C NMR (CDCl3): 206.5, 172.7, 84.6, 76.3, 67.4, 62.7, 37.7, 32.9, 29.7, 27.9.
2CH2C(O)CH3), 2.65–2.53 (2H, m, C(O)CH2C2C(O)CH3), 2.21 (1H, s, CH3), 2.19–2.13 (1H, m, H2), 2.06-2.00 (1H, m, H2); 13C NMR (CDCl3): 206.5, 172.7, 84.6, 76.3, 67.4, 62.7, 37.7, 32.9, 29.7, 27.9.
1,4-Anhydro-2-deoxy-3-O-levulinyl-5-O-(2-cyanoethyl-H-phosphonyl)-D-ribitol (IV)
To a solution of compound III (0.68 g, 3.2 mmol) in dichloromethane (15 ml) DIEA (0.28 ml, 1.6 mmol) followed by 1H-tetrazole (0.1 g, 1.6 mmol) and N,N,N′,N′-tetraisopropyl-2-cyanoethylphosphordiamidite (1.0 ml, 0.35 mmol) were added. After 1 h of stirring, 0.4 M solution of 1H-tetrazole in 90% aq. MeCN (15 ml) was added, the mixture was stirred for another 1 h and partitioned between dichloromethane (30 ml) and brine (20 ml), the organic layer was washed by water (20 ml), separated, dried over Na2SO4 and evaporated to foam. Yield of crude IV 0.95 g (94%). Rf 0.13 (CH2Cl2–AcOEt–EtOH 17:17:2). 1H NMR (CDCl3): 6.93 (0.5H, d, J = 725.0 Hz, PH), 6.89 (0.5H, d, J = 725.0 Hz, PH), 5.13–5.05 (1H, m, H3), 4.35–3.79 (7H, m, H1, H4, H5, OC2C2CN), 3.75–3.60 (1H, m, H5), 3.54–3.30 (1H, m, OC2CH2CN), 2.82–2.67 (2H, m, C(O)C2CH2C(O)CH3), 2.61–2.49 (2H, m, C(O)CH2C2C(O)CH3), 2.17 (1H, s, CH3), 2.14–2.06 (1H, m, H2), 2.05–1.93 (1H, m, H2). 31P NMR (CDCl3): 8.97 (c), 8.35 (c). 31P NMR without P–H decoupling (CDCl3): 8.97 (d quint, J = 725.0, 9.8 Hz), 8.35 (d quint, J = 725.0, 9.8 Hz).
3′-N-[3′-Amino-N4-benzoyl-2′,3′-dideoxy-5′-O-(4,4′-dimethoxytrityl)-cytidyl]-(2-cyanoethyl)-phosphoryl-5-O-1,4-anhydro-2-deoxy-D-ribitol (VI)
H-Phosphonate IV (0.95 g, 3.0 mmol) was dissolved in a 1:1 mixture of pyridine and CCl4 (18 ml), then TEA (1.4 ml, 10 mmol) and aminonucleoside V (0.24 g, 0.38 mmol) were added with stirring. After 1 h of stirring, the solvent was removed in vacuo, the rest was dissolved in a 4:1 mixture of pyridine and acetic acid, and hydrazine hydrate (0.5 ml, 10 mmol) was added. After 20 min, the reaction was quenched by acetylacetone (2.0 ml, 20 mmol), diluted with dichloromethane (50 ml) and washed by water (2 × 30 ml). The organic layer was dried by Na2SO4, dichloromethane was removed in vacuo, the residue was dissolved in dichloromethane and chromatographed on a silica gel column eluted by a gradient of ethanol in CH2Cl2 0–20%. Fractions containing VI were pooled, evaporated to a small volume, and the product precipitated by 10 fold volume of petroleum ether. Yield of VI 0.13 g (40%). Rf 0.35 (CH2Cl2–EtOH 9:1). 31P NMR (CDCl3): 8.60, 8.34 (2 s, mixture of diastereomers). ESI MS: calculated for C45H48N5O11P 865.31, found m/z 864.20 [M − H]−.
Dimer phosphoramidite (I)
Phosphitylation was carried out as described in ref. 48. From 0.13 g of dimer VI 0.1 g of dimer phosphoramidite I were obtained (67% yield). Rf 0.50 (CH2Cl2–EtOH, 9:1). 31P NMR (CDCl3): 149.70, 149.66, 149.17, 149.02, 8.46, 7.88 (mixture of diastereomers).
Oligonucleotide synthesis
Automated synthesis of oligonucleotides was carried out on an ASM-800 DNA/RNA synthesiser (“Biosset”, Russia) using dimer phosphoramidite I together with standard commercial 2-cyanoethyl deoxynucleoside phosphoramidites and CPG solid supports from Glen Research (USA). Dimer phosphoramidite I was used as a 0.1 M solution in anhydrous acetonitrile. The volumes of I and activator 1H-tetrazole solutions were increased from 20 to 80 μl, and the reaction time was extended from 1 till 10 min. Phosphorothioate groups were introduced using sulfurizing reagent II (3-((dimethylaminomethylidene)amino)-3H-1,2,4-dithiazole-3-thione) from Glen Research, USA (0.1 M in pyridine–acetonitrile 3:2 v/v) according to the manufacturer's protocol. Oligonucleotides with internucleotide tetramethyl phosphoryl guanidine group (1e) were obtained as described in ref. 42. Oligonucleotides with an internucleotide primary phosphoramidate group (1d) were prepared as described in ref. 49
Native and modified oligonucleotides were isolated by reverse-phased HPLC on an Agilent 1200 HPLC system (USA) using a Zorbax SB-C18 5 μm column 4.6 × 150 mm in a linear gradient of acetonitrile 0–50% in 20 mM triethylammonium acetate, pH 7.0 in 30 min, a flow rate of 2 ml min−1. Fractions containing the desired product were pooled, concentrated in vacuo, dissolved in 0.1 ml of deionised water and precipitated by 2% LiClO4 in acetone. After centrifugation at 14500 rpm for 2 min, washing with acetone and drying on air for 20 min, oligonucleotide pellets were dissolved in deionised water and stored at −20 °C. Oligonucleotide homogeneity was assessed by 20% denaturing PAGE in 8 M urea, 0.1 M Tris-borate buffer, pH 8.3. The bands were visualised by staining with Stains-All (Sigma, USA).
Molecular masses of oligonucleotides were checked by LC-MS/MS ESI MS on an Agilent G6410A mass spectrometer (USA) in a negative ion mode. The samples were prepared by dissolving oligonucleotides in 20 mM triethylammonium acetate in 60% aq. acetonitrile until 0.1 mM concentration. The volume of the sample was 10 μl. Analysis was carried out using 80% aq. acetonitrile as an eluent, a flow rate of 0.1 ml min−1 and standard device settings. Molecular masses were calculated using experimental m/z values, obtained for each sample. The results are given in Table 3 and Fig. 8.
| Code | Oligonucleotide sequence, 5′–3′ | Molecular mass | |
|---|---|---|---|
| Calc. [M] | Exp. [M] | ||
| FS | TCTCTCSFCCTTCC | 3679.49 | 3679.54 |
| FSS | TCTCTCSFCCTTCSC | 3695.56 | 3695.50 |
| FN | TCTCTCNFCCTTCC | 3662.44 | 3662.5 |
| FNB | TCTCTCNFCCTTCBC | 3759.61 | 3759.32 |
| FBB | TCTCTCBFCCTTCBC | 3857.76 | 3857.34 |
| F3′N | TCTCTC3′NFCCTTCC | 3662.44 | 3662.45 |
| GS | GGAAGGGGAGAGSA | 4153.84 | 4154.14 |
| GB | GGAAGGGGAGAGBA | 4234.94 | 4234.84 |
|
|||
Enzymes and DNA duplexes
DNA duplexes that were used as substrates are given in Fig. 2. Oligonucleotides were purified by ion exchange HPLC on a Hamilton PRP-X500 12–30 μm 3.9 × 300 mm column followed by reverse-phased HPLC on a Nucleoprep 100-20 C18 10 × 250 mm column (Macherey-Nagel, FRG). Oligonucleotide homogeneity was checked by 20% denaturing PAGE.APE1 enzyme was isolated from the E. coli Rosetta2 cell line transformed by a plasmid pET11a carrying human APE1 gene.50
PAGE time-course experiments
5′-[32P]-Labelled oligonucleotides were used in experiments on separation of cleavage products by PAGE. The modified strands were 32P-labeled using [γ-32P]ATP and bacteriophage T4 polynucleotide kinase (New England Biolabs, Beverly, MA) according to the manufacturer's protocol.Single-turnover APE1 endonuclease assays were performed in the reaction buffer of 50 mM Tris-HCl, pH 7.5, 50 mM KCl, 0.1 mM EDTA, 5 mM MgCl2, 1 mM DTT, 9% glycerol at 25 °C.
To register the cleavage extent of modified oligonucleotide duplexes the reaction solution contained 1.0 μM APE1 and 1.0 μM dsDNA. Cleavage of the modified oligonucleotide duplexes was initiated by the addition of APE1. Aliquots of 2 μl of the reaction mixture were withdrawn at time intervals of 1, 5, 10, 20, 30, 60, 120 and 180 min, immediately quenched with 3 μl of gel-loading dye containing 7 M urea and 50 mM EDTA, and loaded on a 20% (w/v) polyacrylamide/7 M urea gel.
To register the cleavage extent of the FO/GO substrate in the presence of inhibitors the reaction solution contained 1.0 μM APE1 and 1.0 μM FO/GO and 0.0, 0.25, 0.5, 0.75, 1.0, 1.5, 2.0, 3.0, 5.0 or 10.0 μM FN/GO and G/C duplexes. The FO/GO substrate was initially incubated with FN/GO or G/C for 5 min. Cleavage of the FO/GO substrate was initiated by the addition of APE1. 2 μl aliquots of the reaction mixture were withdrawn at time intervals of 10, 20, 30, 40, 60, 120 and 300 s, immediately quenched with 3 μl of gel-loading dye containing 7 M urea and 50 mM EDTA, and loaded on a 20% (w/v) polyacrylamide/7 M urea gel.
The extent of cleavage was determined by radioautography and digital processing by Gel-pro Analyzer 4.0 software (Media Cybernetics, USA). The degree of cleavage was calculated as a ratio of the area of product peaks to the sum of the areas of the product and starting material peaks. All experiments were repeated in triplicate. The presented data points reflect the average value obtained from a set of repeats.
The fraction product was fitted by the single exponential curve using Origin software (Originlab Corp.) (eqn (1)).
| [product] = A × [1 − exp(−kobs × t)] | (1) |
Stopped-flow fluorescence measurements
Fluorescent kinetic curves were recorded on a stopped-flow spectrometer SX.18MV (Applied Photophysics, UK). For Trp fluorescence, excitation wavelength was 290 nm. The change in Trp fluorescence emission was observed on wavelengths above 320 nm (Schott filter WG 320). The dead time of the device was 1.38 ms, maximum recording time was 10 s. Each kinetic curve was the result of averaging of minimum three experimental curves. All the experiments were carried out in 50 mM Tris-HCl, pH 7.5, 50 mM KCl, 0.1 mM EDTA, 5 mM MgCl2, 1 mM DTT, 9% glycerol at 25 °C.Fluorescence titration of APE1 with oligodeoxyribonucleotide inhibitors
Dissociation constant Kd of the complex of APE1 with the inhibitors was determined by fluorescence titration. Each point on the fluorescence titration curves was obtained by the measurement of the fluorescence intensity of the reaction mixture (100 μl) containing APE1 (1.0 × 10−6 M) and an oligonucleotide inhibitor at the required concentration in a buffer (50 mM Tris-HCl, pH 7.5, 50 mM KCl, 0.1 mM EDTA, 5 mM MgCl2, 1 mM DTT, 9% glycerol). The mixtures were incubated at 25 °C for 2 min. Fluorescence spectra were measured at an excitation wavelength of 290 nm using a Varian Cary Eclipse spectrofluorimeter (Agilent Technologies, USA). Values of fluorescence intensity at the emission maximum (335 nm) were used to calculate the dissociation constants.The values of Kd were determined by non-linear regression analysis of data according to eqn (2), using program Origin software (Originlab Corp.).
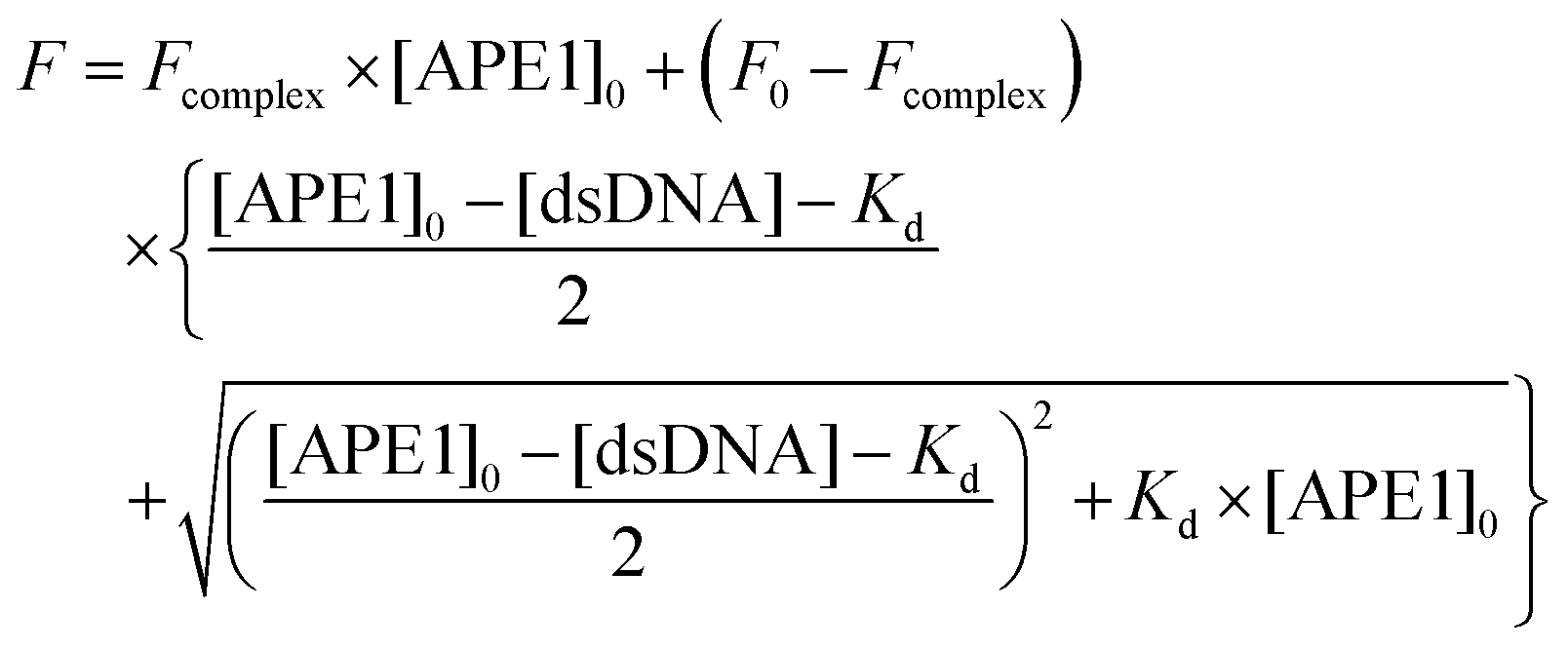 | (2) |
Conclusions
This work has focussed on the design, synthesis and analysis of oligodeoxyribonucleotide derivatives as unreactive substrate analogues of a key enzyme of the DNA repair, AP-endonuclease APE1. It was found that either S- or N-substitution of the oxygen atoms in the phosphate group on the 5′-side of the tetrahydrofuran analogue of the AP-site (F-site) leads to a significant decrease in the APE1 activity. The kinetic analysis demonstrated that these modified oligonucleotide duplexes could be used in vitro as specific uncleavable DNA substrate analogues of APE1 for biochemical or structural studies of AP-endonuclease mechanisms. Besides, the compounds in our opinion show promise for future cell and animal studies.Abbreviations
| APE1 | Human AP-endonuclease 1 |
| Bz | Benzoyl |
| CE | 2-Cyanoethyl |
| DCC | N,N′-Dicyclohexylcarbodiimide |
| DIPEA | N,N-Diisopropylethylamine |
| DMTr | 4,4′-Dimethoxytrityl |
| DTT | Dithiothreitol |
| Lev | Levulinyl |
| MeIm | 1-Methylimidazole |
| Py | Pyridine |
| TEA | Triethylamine |
| Tet | 1H-Tetrazole |
| Tmg | 1,1,3,3-Tetramethylguanidine |
| AP-site | Apurinic/apyrimidinic site |
| F | (2R,3S)-2-(Hydroxymethyl)-3-hydroxytetrahydrofuran |
Acknowledgements
The work was supported by the Presidential Grant for Leading Scientific Schools NS-1205.2014.4; Russian Foundation for Basic Research Grants No. 13-04-00013 to O. S. F., No. 14-04-01018 to T. V. A., No. 15-34-20121 to A. A. K., No. 15-04-00467 to N. A. K., No. 13-04-01176 to D. V. P., No. 15-04-06331 and 15-29-01334 to D. A. S.; bursary of the President of the Russian Federation (SP-3752.2015.4) to M. S. K. Part of the work with Trp fluorescence detection combined with stopped-flow kinetics was specifically funded by Russian Science Foundation grant No. 14-14-00063 to N. A. K. Synthesis of modified oligonucleotides was partly supported by the Ministry of Education and Science of the Russian Federation (Agreement No. 14.B25.31.0028).Notes and references
- T. Lindahl and B. Nyberg, Biochemistry, 1972, 11, 3610–3618 CrossRef CAS PubMed.
- J. H. Hoeijmakers, N. Engl. J. Med., 2009, 361, 1475–1485 CrossRef CAS PubMed.
- T. Iyama and D. M. Wilson, 3rd, DNA Repair, 2013, 12, 620–636 CrossRef CAS PubMed.
- S. Wolters and B. Schumacher, Front. Genet., 2013, 4, 19 Search PubMed.
- L. A. Loeb and C. C. Harris, Cancer Res., 2008, 68, 6863–6872 CrossRef CAS PubMed.
- L. H. Hurley, Nat. Rev. Cancer, 2002, 2, 188–200 CrossRef CAS PubMed.
- D. O. Zharkov, Cell. Mol. Life Sci., 2008, 65, 1544–1565 CrossRef CAS PubMed.
- A. B. Robertson, A. Klungland, T. Rognes and I. Leiros, Cell. Mol. Life Sci., 2009, 66, 981–993 CrossRef CAS PubMed.
- D. M. Wilson, 3rd and L. H. Thompson, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 12754–12757 CrossRef.
- R. P. Cunningham, Mutat. Res., 1997, 383, 189–196 CAS.
- S. C. Brooks, S. Adhikary, E. H. Rubinson and B. F. Eichman, Biochim. Biophys. Acta, 2013, 1834, 247–271 CrossRef CAS PubMed.
- B. Pascucci, G. Maga, U. Hubscher, M. Bjoras, E. Seeberg, I. D. Hickson, G. Villani, C. Giordano, L. Cellai and E. Dogliotti, Nucleic Acids Res., 2002, 30, 2124–2130 CrossRef CAS PubMed.
- J. L. Parsons and G. L. Dianov, DNA Repair, 2013, 12, 326–333 CrossRef CAS PubMed.
- E. Trushina and C. T. McMurray, Neuroscience, 2007, 145, 1233–1248 CrossRef CAS PubMed.
- M. S. Cooke, M. D. Evans, M. Dizdaroglu and J. Lunec, FASEB J., 2003, 17, 1195–1214 CrossRef CAS PubMed.
- M. D. Evans, M. Dizdaroglu and M. S. Cooke, Mutat. Res., 2004, 567, 1–61 CAS.
- M. W. Germann, C. N. Johnson and A. M. Spring, Med. Res. Rev., 2012, 32, 659–683 CrossRef CAS PubMed.
- B. J. Glassner, G. Weeda, J. M. Allan, J. L. Broekhof, N. H. Carls, I. Donker, B. P. Engelward, R. J. Hampson, R. Hersmus, M. J. Hickman, R. B. Roth, H. B. Warren, M. M. Wu, J. H. Hoeijmakers and L. D. Samson, Mutagenesis, 1999, 14, 339–347 CrossRef CAS PubMed.
- A. Klungland, I. Rosewell, S. Hollenbach, E. Larsen, G. Daly, B. Epe, E. Seeberg, T. Lindahl and D. E. Barnes, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 13300–13305 CrossRef CAS.
- J. L. Parsons and R. H. Elder, Mutat. Res., 2003, 531, 165–175 CrossRef CAS PubMed.
- H. Nilsen, I. Rosewell, P. Robins, C. F. Skjelbred, S. Andersen, G. Slupphaug, G. Daly, H. E. Krokan, T. Lindahl and D. E. Barnes, Mol. Cell, 2000, 5, 1059–1065 CrossRef CAS PubMed.
- H. Fung and B. Demple, Mol. Cell, 2005, 17, 463–470 CrossRef CAS PubMed.
- R. L. Maher and L. B. Bloom, J. Biol. Chem., 2007, 282, 30577–30585 CrossRef CAS PubMed.
- K. M. Schermerhorn and S. Delaney, Biochemistry, 2013, 52, 7669–7677 CrossRef CAS PubMed.
- D. M. Wilson, 3rd, M. Takeshita, A. P. Grollman and B. Demple, J. Biol. Chem., 1995, 270, 16002–16007 CrossRef.
- L. Y. Kanazhevskaya, V. V. Koval, Y. N. Vorobjev and O. S. Fedorova, Biochemistry, 2012, 51, 1306–1321 CrossRef CAS PubMed.
- M. A. Gorman, S. Morera, D. G. Rothwell, E. de La Fortelle, C. D. Mol, J. A. Tainer, I. D. Hickson and P. S. Freemont, EMBO J., 1997, 16, 6548–6558 CrossRef CAS PubMed.
- P. T. Beernink, B. W. Segelke, M. Z. Hadi, J. P. Erzberger, D. M. Wilson, 3rd and B. Rupp, J. Mol. Biol., 2001, 307, 1023–1034 CrossRef CAS PubMed.
- B. A. Manvilla, E. Pozharski, E. A. Toth and A. C. Drohat, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 2555–2562 CrossRef CAS PubMed.
- C. D. Mol, T. Izumi, S. Mitra and J. A. Tainer, Nature, 2000, 403, 451–456 CrossRef CAS PubMed.
- C. D. Mol, D. J. Hosfield and J. A. Tainer, Mutat. Res., 2000, 460, 211–229 CAS.
- S. E. Tsutakawa, D. S. Shin, C. D. Mol, T. Izumi, A. S. Arvai, A. K. Mantha, B. Szczesny, I. N. Ivanov, D. J. Hosfield, B. Maiti, M. E. Pique, K. A. Frankel, K. Hitomi, R. P. Cunningham, S. Mitra and J. A. Tainer, J. Biol. Chem., 2013, 288, 8445–8455 CrossRef CAS PubMed.
- A. S. Lipton, R. W. Heck, S. Primak, D. R. McNeill, D. M. Wilson, 3rd and P. D. Ellis, J. Am. Chem. Soc., 2008, 130, 9332–9341 CrossRef CAS PubMed.
- N. Oezguen, C. H. Schein, S. R. Peddi, T. D. Power, T. Izumi and W. Braun, Proteins, 2007, 68, 313–323 CrossRef CAS PubMed.
- E. Wickstrom, Adv. Drug Delivery Rev., 2015, 87, 25–34 CrossRef CAS PubMed.
- D. M. Wilson III and D. Barsky, Mutat. Res., 2001, 485, 283–307 Search PubMed.
- S. S. David and S. D. Williams, Chem. Rev., 1998, 98, 1221–1261 CrossRef CAS PubMed.
- N. A. Timofeyeva, V. V. Koval, D. G. Knorre, D. O. Zharkov, M. K. Saparbaev, A. A. Ishchenko and O. S. Fedorova, J. Biomol. Struct. Dyn., 2009, 26, 637–652 CAS.
- L. Y. Kanazhevskaya, V. V. Koval, D. O. Zharkov, P. R. Strauss and O. S. Fedorova, Biochemistry, 2010, 49, 6451–6461 CrossRef CAS PubMed.
- S. M. Gryaznov and R. L. Letsinger, Nucleic Acids Res., 1992, 20, 3403–3409 CrossRef CAS PubMed.
- R. Eritja, P. A. Walker, S. K. Randall, M. F. Goodman and B. E. Kaplan, Nucleosides Nucleotides, 1987, 6, 803–814 CAS.
- M. S. Kupryushkin, D. V. Pyshnyi and D. A. Stetsenko, Acta naturae, 2014, 6, 116–118 CAS.
- D. M. Wilson, 3rd and A. Simeonov, Cell. Mol. Life Sci., 2010, 67, 3621–3631 CrossRef PubMed.
- R. I. Al-Safi, S. Odde, Y. Shabaik and N. Neamati, Curr. Mol. Pharmacol., 2012, 5, 14–35 CrossRef CAS PubMed.
- A. Simeonov, A. Kulkarni, D. Dorjsuren, A. Jadhav, M. Shen, D. R. McNeill, C. P. Austin and D. M. Wilson, PLoS One, 2009, 4, e5740, DOI:10.1371/journal.pone.0005740.
- V. Wacheck and U. Zangemeister-Wittke, Critical reviews in oncology/hematology, 2006, 59, 65–73 CrossRef CAS PubMed.
- D. Castanotto and C. A. Stein, Curr. Opin. Oncol., 2014, 26, 584–589 CrossRef CAS PubMed.
- S. V. Vasil'eva, T. V. Abramova, T. M. Ivanova, G. V. Shishkin and V. N. Silnikov, Bioorg. Khim., 2004, 30, 264–272 Search PubMed.
- D. A. Stetsenko, M. S. Kupryushkin and D. V. Pyshnyi, Russian Pat., 2014117293, 2014 Search PubMed.
- S. Daviet, S. Couve-Privat, L. Gros, K. Shinozuka, H. Ide, M. Saparbaev and A. A. Ishchenko, DNA Repair, 2007, 6, 8–18 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |