
Q-GDEMAR: a general method for the identification of differentially expressed genes in microarrays with unbalanced groups†
Daniel V.
Guebel
ab,
Montserrat
Perera-Alberto
ac and
Néstor V.
Torres
*a
aSystems Biology and Mathematical Modelling Group, Science Faculty, University of La Laguna, Tenerife, Canary Islands, Spain. E-mail: ntorres@ull.edu.es
bBiotechnology Counselling Services, Buenos Aires, Argentina
cDepartment of Anatomy, Pathology, Histology and Physiology, University of La Laguna, Tenerife, Canary Islands, Spain
First published on 29th October 2015
Abstract
Microarray analysis is a powerful tool to simultaneously determine the pattern of transcription of large amounts of genes. For data post-processing distinct computational methods are currently used that, however, lead to different results regarding the genes expressed differentially. Herein, a new methodology for microarray data analysis named Q-GDEMAR is presented. It combines the quantile characterization of the entire distribution together with the Gaussian deconvolution of the central region of the microarray data distribution. Three discriminant variable variants are proposed that allow us to summarize data and compare groups even when their size is strongly unbalanced. In addition, a simple procedure to compute the false discovery rate (FDR) is also presented. The performance of the method is compared with that observed when using LIMMA (Linear Models Microarray) software as reference. In 58 out of 68 cases, Q-GDEMAR showed a higher sensitivity than LIMMA to detect differentially expressed genes (p = 1 × 10−10). The proposed method does not produce biased information, detecting genes with high sensitivity equally well at both tails of the distribution (p = 0.7428). Moreover, all detected genes were associated with very low levels of FDR (median value = 0.67%, interquartile range = 0.87%). Q-GDEMAR can be used as a general method for microarray analysis, but is particularly indicated when the conditions to be compared are unbalanced. The superior performance of Q-GEDEMAR is the consequence of its higher discriminative power and, the fact that it yields a univocal correspondence between the p-values and the values of the discriminating variable. Q-GDEMAR was tested only using Affymetrix microarrays. However, given that it operates after the step of data standardization, it can be used with the same quality features on any of the available mono- or dual-channel microarray platforms.
1 Introduction
cDNA- and oligonucleotide-based microarrays are routinely applied in the study of complex diseases in several biomedical fields, ranging from neurobiology and cancer to connective tissue or infectious pathologies.1–3 There are multiple protocols and software packages for the data analysis of microarrays.4–10 However, the meta-analysis of different microarray studies related to a same pathology usually yields very different results, no matter if the studies were carried out with the same or different analytical platforms,11–15 thus limiting the translation of the microarray findings to the clinical practice.16,17Although microarray discrepancies can be partially attributed to some unavoidable biological and analytical variability, the observed differences can also be the consequence of operational factors during the post-processing steps, once the data have been standardized as we discuss below.
Microarray data are routinely subjected to logarithmic transformation in order to “normalize” the distribution of such data. Normalization should increase the Gaussian character of the distribution (a feature required for t-Student, ANOVAs, and estimation of Bayesian probabilities). Hence, skewness and/or kurtosis should be minimized while the problem of heteroscedasticity of variances is alleviated.18 However, the efficiency of the logarithmic transformation in achieving an improved Gaussian character is not always guaranteed.19 An alternative to avoid these problems is the use of non-parametric methods, but these methods are far less powerful than the parametric ones when the size of the samples is not big enough.20
One of the main objectives pursued through the microarray studies is to identify “differentially expressed genes”, i.e., those genes whose level of expression changes under different conditions tested (e.g., age, sex, disease stage, treatments). The differentially expressed genes are recognized on the basis of their associated “statistical effects”, which usually requires the comparison of the “treatment groups” against a “control group” in terms of their mean values.
In order to discriminate the statistical effects of the tested conditions on the genes, there are two ways to summarize the mean comparisons: by “Fold-Change (FC)-Difference”21 or by log2(FC).22 Importantly, one or the other form leads to the identification of different sets of differentially expressed genes.23 In addition, neither of these forms account for the differences in the sizes of the groups. Optimal experimental designs require balanced groups. Otherwise, the means of the groups will have different reliabilities. Nevertheless, due to practical limitations the balance conditions cannot be fulfilled frequently. In these cases, the detection of differentially expressed genes poses additional problems.24
The statistical effects are assessed by ranking the values of the summarizing variable selected according to the significance of two magnitudes: the p-value (p = probability(error type I ≤ α) = probability(false positive)),25 and the false discovery rate (FDR). The latter is defined as the “expected” value of the ratio between the number of false positives and the total number of significant values declared. From the Bayesian perspective, and according to the type of assumptions considered, there are several forms to estimate the expected value of the ratio that defines the FDR.26–29 In any case, while the p-values give the significance of the statistical effect of a gene as if its expression were determined independently, the FDR protects against false inferences due to the problem of multiple comparisons involved when thousands of genes are being determined simultaneously.29
Another important question driving our search for an alternative procedure to compute the FDR values is our verification that current algorithms, such as Empirical-Bayes26 and Benjamini–Hochberg,27 actually do not provide a univocal correspondence between the p-values (or FDR) computed and the discriminating variable as should be expected (Fig. 1).
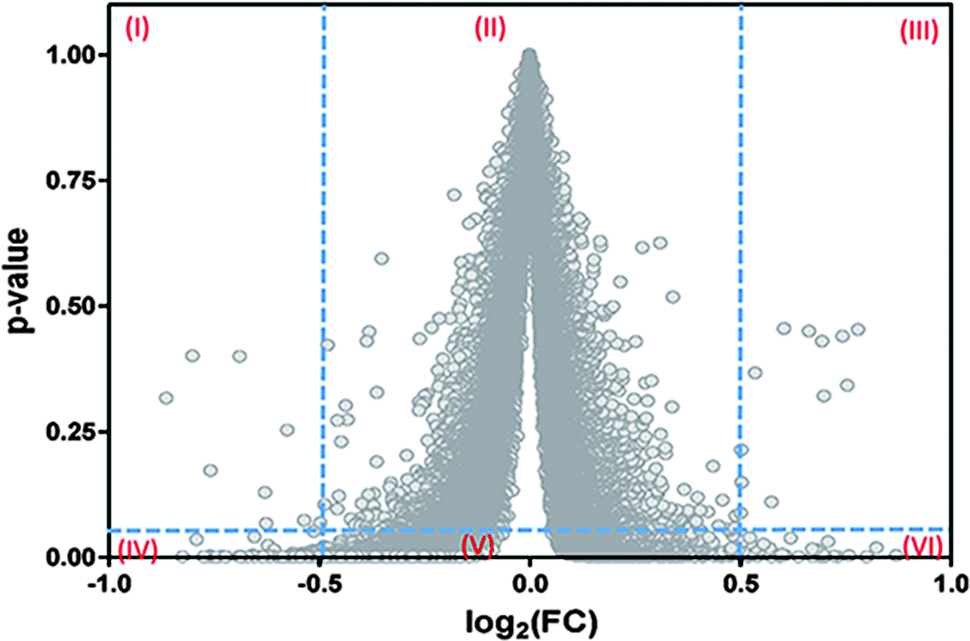 | ||
| Fig. 1 Failure of the current algorithms implemented in LIMMA to establish a univocal correspondence between the magnitudes of the fold-change (FC) and its associated probability level of significance (p-value). This analysis was performed on data from GSE4806030 but a similar result is obtained with data from other eleven microarrays. A similar profile of dispersion is also observed for the FDR values of this and other microarrays (results not shown). Roman numerals in the graph indicate the presence of six regions. The three upper regions differ in their significance with respect to the corresponding homologous at the bottom. Current algorithms as Empirical Bayes and Benjamini–Hochberg lead to a situation in which the same value of the FC admits multiple p-values (or FDR). Some of them can be interpreted as significant while others are interpreted as non-significant. | ||
A practical consequence of the absence of a univocal relationship between the p-values (or FDR values) and the fold-change (FC) in Fig. 1, is that this situation will contribute to some degree of misclassification of the genes in the microarray data. In fact, by taking two vertical lines at two feasible cut-off values of the discriminant variable at the x-axis (e.g., log2(FC) = ±0.5), and one horizontal line at a feasible limit of significance (e.g., p-value = 0.05), six regions are generated. The lack of unicity can be seen by comparing any of the juxtaposed regions (I and IV, II and V, III and VI) because the same value of FC appears associated with very low and very high p-values in all these cases.
Hence, while regions I and III are associated with potentially false negative cases, region V is associated with potentially false positive results. Note that due to the problem of multiple simultaneous comparisons, the horizontal line should actually be taken at values quite lower than p = 0.05. This last situation will improve the identification of the significant points located in the new, diminished regions IV–V–VI, but at the cost of worsening the reliability of the significance of the points located in the new, expanded regions I and III.
From Fig. 1 it can be concluded that of the six regions generated, only the regions named as IV and VI are suitable for the detection of differentially expressed genes. This explains why the current methods need to apply a double filtering criterion (FC and p-value) instead of a unique one, filtering either for FC or for p.31,32
Importantly, the absence of a univocal correspondence between the p-values and the fold-change values can explain the dissociation between the biological and statistical criteria observed with the interpretation of some microarrays.12,33 It is worth noting that if log(1/p) were considered instead of the p-values at the y-axis, Fig. 1 would turn into the well-known “volcano” plot.31,32
Likewise, when current algorithmic methods were tested for the reliability of the FDR values generated, we observed that sometimes none of the thousands of analysed genes were acceptable according to their FDR values, even when many of the same genes were associated with highly significant p-values. Moreover, in other instances the current algorithmic methods yield very significant p-values (p = 10−8 to p = 10−12) even when the empirical distribution of the experimental data cannot produce values beyond the order of p = 10−5 to p = 10−6.
Again, the excessive dispersion of the p-values for a given value of fold-change in Fig. 1 indicates that these values don't act as asymptotic limits to the p-values obtained empirically from the microarray samples as can be expected. This is a shortcoming of the method because the computation of the FDR by current algorithms depends strongly on the accuracy of the individual p-values obtained previously.33,34
In view of the methodological limitations previously commented, in the present paper we introduce a novel alternative for the post-processing analysis of the microarray data. At the same time, we introduce an alternative method to compute the FDR. Both methods are oriented to optimize the identification of differentially regulated genes. Finally, we will assess the performance of the proposed methodology by comparing it with LIMMA, a program included in the Bioconductor Project (http://https://www.bioconductor.org).
LIMMA is a particularly reliable reference for this comparison. First, it is a classic, almost standard method that is routinely updated.35,36 Also, for comparison we have to make combined use of its underlying algorithms (Empirical Bayes and Benjamini–Hochberg), which with slight variants are common to other programs.8 Finally, the core of LIMMA services can be additionally accessed by friendly interfaces, such the cases of the portal Babelomics9,37 and GEO2R (NIH, USA),38 both facilitating the operation of this program to any user.
In the Material and method section the rationale of Q-GDEMAR as well as the logic of the flux of operations implied in our algorithms and the definition of the discriminant variables used are presented. In the Results and discussion, we examine the efficiency of the current procedure of microarray normalization; we assess the overall performance of Q-GDEMAR when compared with LIMMA and characterize the relative efficiency of the discriminant variables used. Finally, we discuss the differences in Q-GDEMAR with respect to the Cauchy distribution. Note that detailed protocols for computing the p-values and FDR by our alternative approaches, as well as three tables containing data of interest for the analysis performed are provided in the ESI.†
2 Materials and methods
2.1 Compared microarrays
The following twelve sets of microarray data were downloaded from the NCBI Gene Expression Omnibus database (GEO): GSE48060,30 GSE54992,39 GSE35713,40 GSE5281,41 GSE1919,42 GSE36297,43 GSE48754,44 GSE28619,45 GSE34308,46 GSE1297,47 GSE11882,48 GSE46922,49 where GSE indicates the code of accession to the GEO data-sets (http://www.ncbi.nlm.nih.gov/gds). As a proof of concept, the data-sets were processed in parallel following the proposed method and the LIMMA,35 the latter being processed through the web interface GEO2R provided by NIH (USA, http://www.ncbi.nlm.nih.gov/geo/info/geo2r.html). It is worth mentioning that our study was designed to address a heterogeneous group of biomedical conditions. In addition, the data-sets cover an ample range of unbalanced situations regarding the number of control and treatment samples. These two experimental groups were subjected to three discriminant variables in Q-GDEMAR (see Table S1, ESI†), as well as tested using the standard method LIMMA. With the aim of gaining statistical power in the assessment of the methods, we decided to restrict the comparison of their performance to unique contrasts between the two experimental groups in each microarray, even when more complex analysis could also be possible in some cases.2.2 Q-GDEMAR: quantile characterization and Gaussian deconvolution of the central region of the MicroARray data distribution
Microarrays can determine the expression of up to fifty five thousand genes simultaneously. Each gene is monitored by specifically designed “probe-sets”, comprising several synthetic probes targeted to different segments of the gene sequence (Microarray Probing Mapping, available in: http://www.ensembl.org/info/genome/microarray_probe_set_mapping.html).The experimental evidence supports that most of the monitored genes usually do not vary their expression under the tested conditions, or if they do, only moderate stochastic fluctuations around a well-identifiable mean value are observed. In addition, two minor groups of genes that are up- and down-regulated can also be recognized. Hence, the problem of microarray analysis seems to lie on the deconvolution of three different populations of genes. However, given that the entire distribution of the data can be known, it is only necessary to identify precisely the genes whose expression does not vary in order to spot the occurrence of deregulated genes at the tails of the curve.
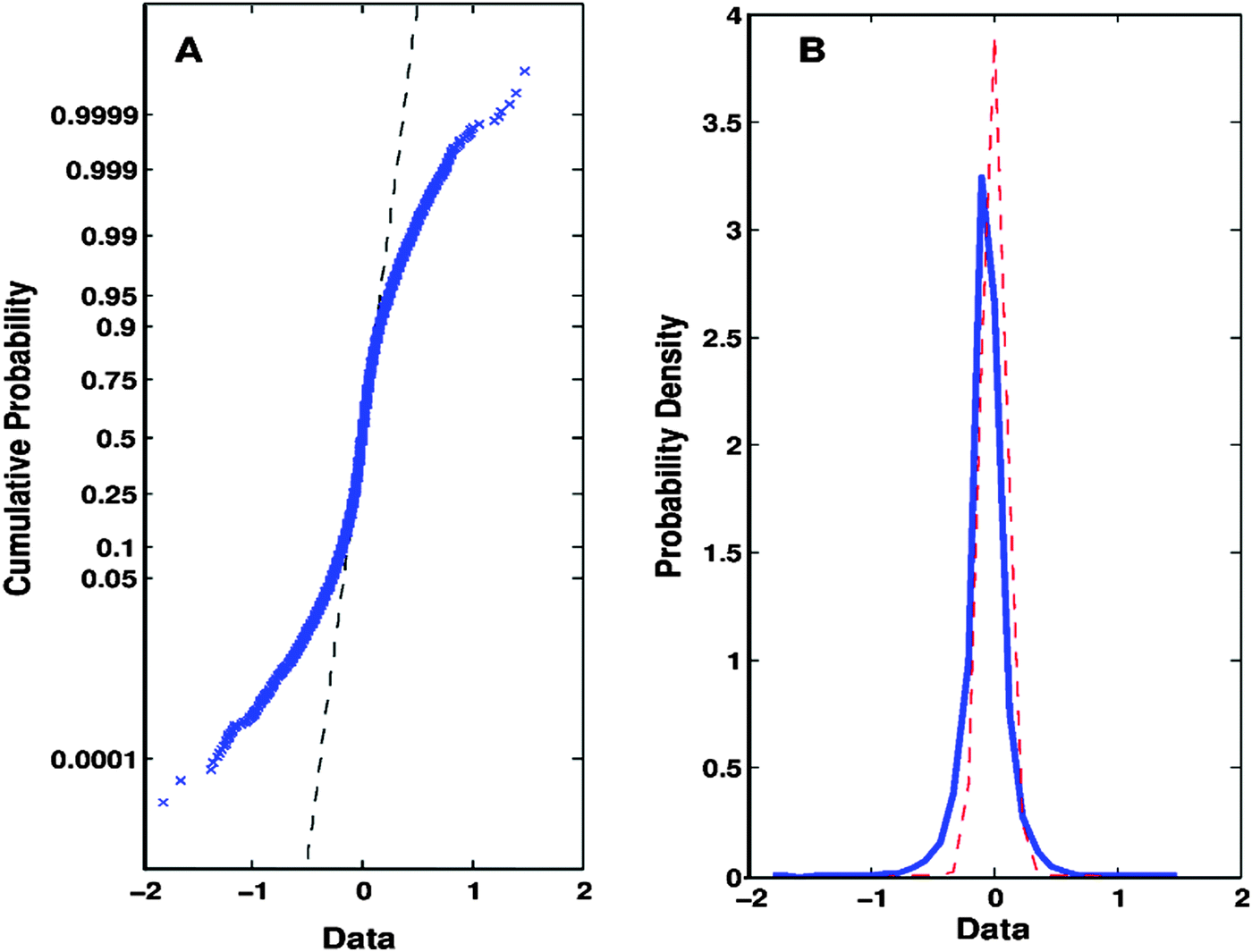 | ||
| Fig. 2 Evidences and principles that support the Q-GDEMAR method. (A) The intersection between the experimental data (crossed, blue markers) and the theoretical straight-line expected for a Gaussian distribution (dashed, black line) shows that the Gaussian model fits well in the central region of the data (i.e., the region of genes whose expression does not change significantly). (B) Consequently, it is possible to establish the variation in the probability profile of the entire set of experimental data (solid, blue curve) with respect to the Gaussian model fitted to the central region (dashed, red curve). Herein, “Data” refers to the expression of each gene as summarized by the difference Δ = median(log2(treatment group)) − median(log2(control group)). Original data were taken from GSE4806030 but the profiles depicted are also representative of the rest of the microarrays analysed. | ||
Note that in the case presented above, the “entire” data-set does not match a true Gaussian model (Fig. 2A) despite its bell-shaped distribution (Fig. 2B). However, the central region of the distribution complies with a Gaussian model in both plots (Fig. 2A and B). Importantly, the narrow dispersion of the Gaussian central region observed in Fig. 2B allows us to establish well-defined limits to the normal stochastic fluctuation for genes whose expression does not change significantly. This deconvolution process results in a gain of sensitivity in the determination of differentially expressed genes (significant genes are given by the difference of areas between the blue and the red curves at the tails of the entire distribution in Fig. 2B). At the same time, the risk of accepting a high number of false positive results is greatly diminished (note the reduced area under the tails of the red curve of the Gaussian central region in Fig. 2B). Hence, the value of the FDR would drastically reduce.
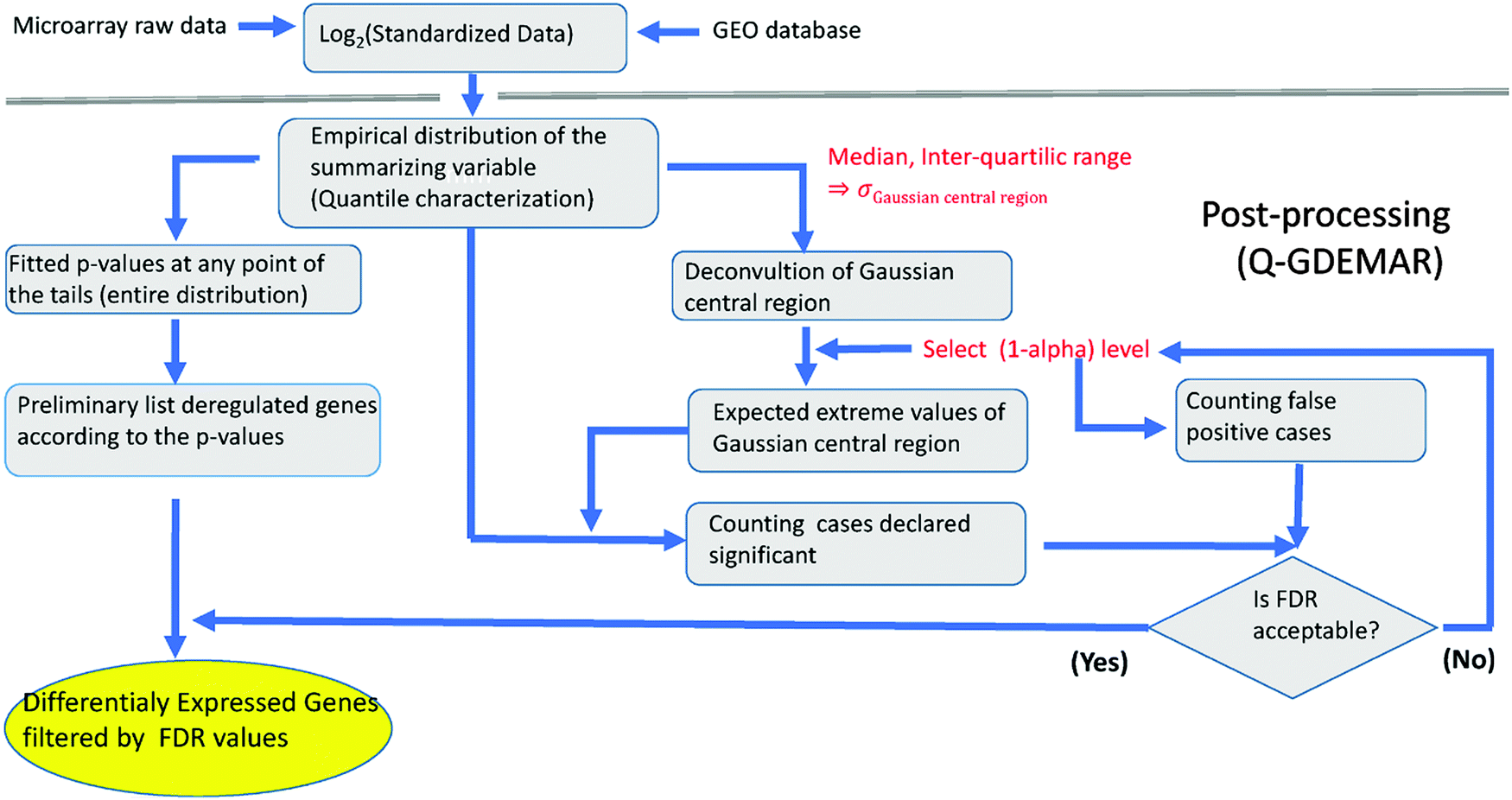 | ||
| Fig. 3 Flux diagram of the Q-GDEMAR method. See Appendices 1–2 (ESI†) for technical details. | ||
In microarray analysis, the variations between treatment conditions are determined by computing the “statistical effects”, i.e., the differences (or ratios) between the means of the groups compared. Means summarize the distribution of the experimental data in a unique, “representative” value but it is not the only possible parameter of centrality. In Q-GDEMAR the comparisons are implemented by using three types of discriminating variables: one is based on the difference between the means but corrected by their weighted average, and other two are direct variants of median-based comparisons.
Note that if non-balanced groups are compared in terms of means, each mean has a different degree of reliability and hence, some form of weighted average should be applied. In this regard, current methods have limitations.22 Instead, as used in the present method and shown in Fig. 2, the control group, with 6 replicates, and the treatment group, with 9 replicates, were compared on the basis of the difference of their log2(medians) along n = 43![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 742 genes without considering the actual number of replicates. Further details about the three types of discriminating variable used to deal with unbalanced groups are presented in Section 2.3.
742 genes without considering the actual number of replicates. Further details about the three types of discriminating variable used to deal with unbalanced groups are presented in Section 2.3.
In order to deal with the determination of the “significance” of the computed differences, in our method the deregulated genes are assessed by two complementary techniques. The first technique relies on the empirical modelling of the entire data distribution; the second is based on the identification of genes showing expression values beyond the extreme limits expected for the central Gaussian population of data.
The first approach is suitable to compute the significance of individual effects for each gene analysed. In this case, p-values will be computed based on the determination of some critical quantiles from the entire data distribution (see Appendix 1, ESI†). In doing so, the individual p-values are obtained without the need of assuming the Gaussian character of the overall data distribution. The second approach is suitable to compute the false discovery rate (FDR) (see Appendix 2, ESI†).
In contrast with current algorithms, such as that used by LIMMA, our method does not compute FDR for individual genes but for the set of differentially expressed genes on each tail of the overall distribution. Note that working together, the features of both combined approaches lead to a situation in which the identified groups of up- and down-regulated genes acquire different values of the FDR and comprise different number of genes.
2.3 Discriminating variables
In order to account directly or indirectly for the possible imbalance in size among the compared groups, we assayed three variants of discriminating variables. These variants are as follows.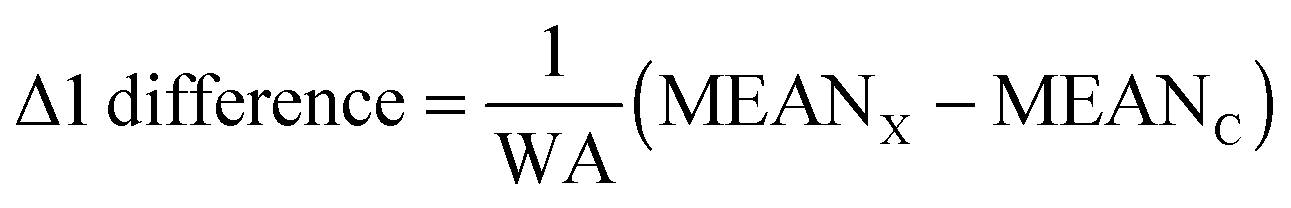 | (1) |
| MEANX = mean(log2(standardized valuesX)) | (2) |
| MEANC = mean(log2(standardized valuesC)) | (3) |
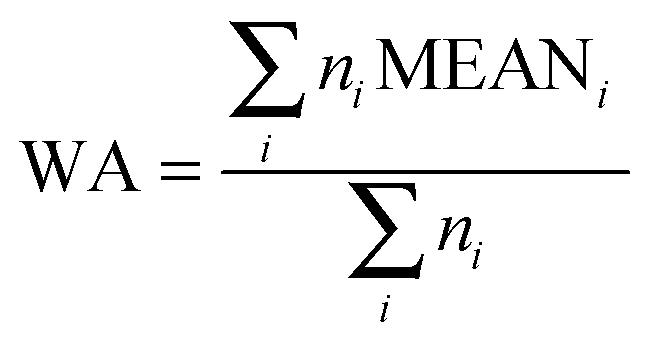 | (4) |
| Δ2 difference = median(log2(standardized valuesX)) − median(log2(standardized valuesC)) | (5) |
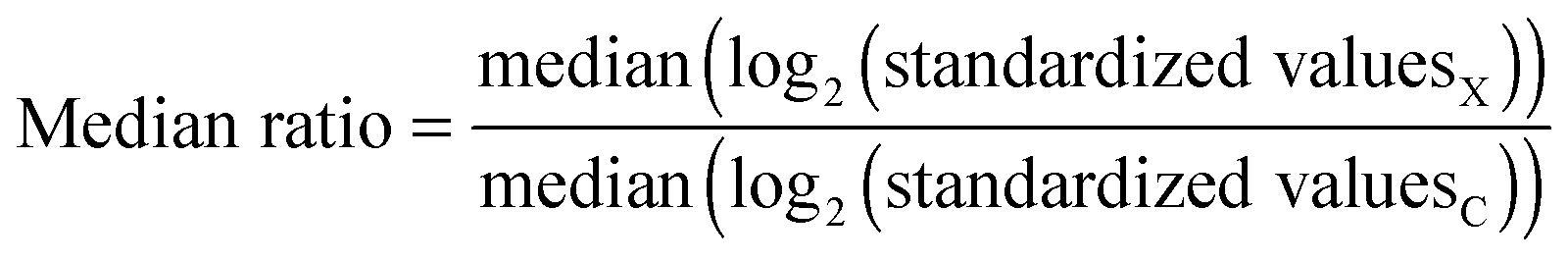 | (6) |
Even though the proposed median-based discriminatory variables only indirectly consider the fact that a median behaves itself as a random variable, this is not detrimental (see Section 3.5). In this median-based approach, the effects of the main comparisons (i.e., treatment vs. control) are “confounded” with the possible effects associated with the variation of the median with the size of the samples. We do not know about a law governing this type of variation, neither about some equivalent to central limit theorem, accounting for the distribution of replicated means, but for the case of medians since it could compensate the variation of the median value with the size of the sample.
Since median is a more robust parameter than the mean, it is expected that the performance of median values will be better than that obtained by the mean values. Particularly in the case of microarrays, the impact on median variation with the size of the sample might be lower than the impact due to other sources of variation present, such as the biological and technical variability. In fact, classic t-Student fails to deal with the low-expression genes, forcing current methods of microarray analysis to use modified forms of the t-statistic (regularized, penalized, or stabilized t-tests).8,28,32 From this “adapted” t-statistics, the p-values and the FDR are derived but neither of these forms are free of some strong assumptions.8,28 Hence, results obtained from the current algorithms are valid up to a given extent8,55 (see Section 4).
In the median-based approach, the implementation of the Gaussian-deconvolution operation imposes practical boundaries to the confounding effect. The analysis carried out on twelve real microarray data-sets, showing that the sensitivity of gene detection and the FDR of our median-based approach out-performed the one by LIMMA algorithms (see Section 2.3). Let us conclude that variation of the median with the size of the sample is not significant relative to other major sources of variation present in the system.
3 Results and discussion
3.1 How normalizing is the current normalization procedure?
As already commented, it is usually assumed that log-transformation is a universal recipe to get “normalized” data. However, an important residual trend between the variance with respect to the mean was verified when analysing the microarray data obtained from GSE54992, in both log2-transformed units (see Fig. 4A and B).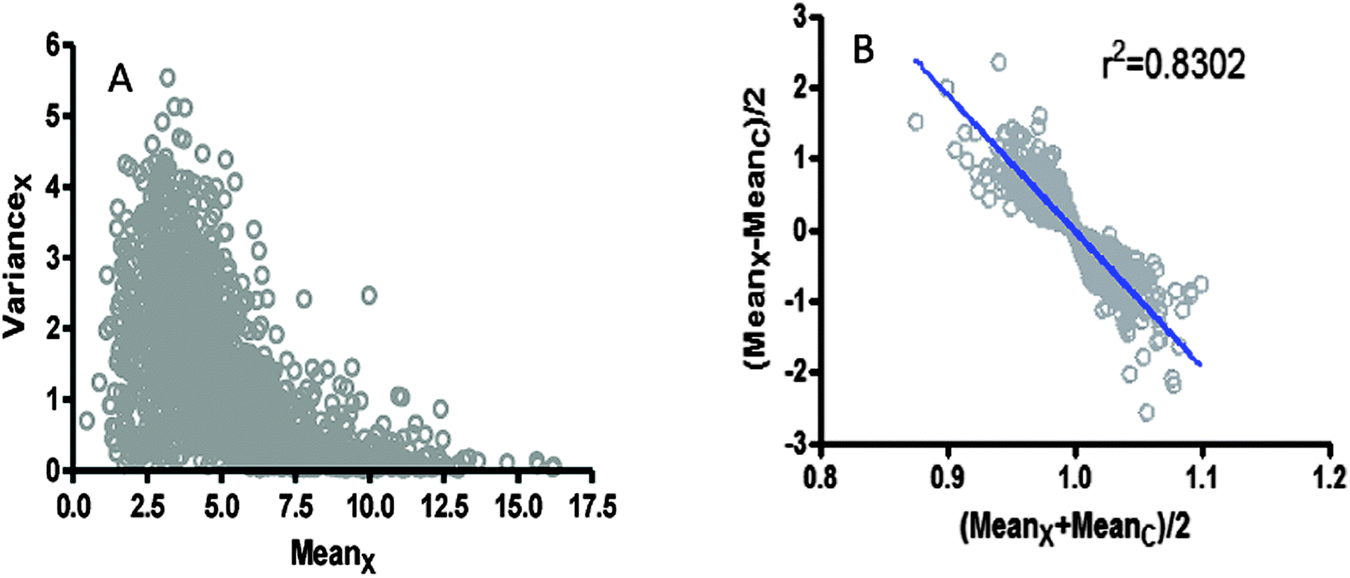 | ||
| Fig. 4 Pattern of dispersion in microarray data taken from GSE54992.39 (A) The variance is represented as a function of the mean value for the group with pathology. (B) The semi-distance between pathology and control groups is presented as a function of the average of expression in the same groups. | ||
Different degrees of residual trends also persist with the regression of the variance over the mean in the other microarrays analysed herein. For example, GSE48060 (potential function, R2 = 0.362), GSE46922 (polynomial 5th degree, R2 = 0.6359), and GSE11882 (linear trend, R2 = 0.8020), all of them showed significant patterns of residual trends of data after log-transformation (data not shown).
In order to avoid this residual trend, we assayed other types of data transformation. By doing so, we observed that sometimes the type of transformation needed depends on the combination of factors analysed (e.g., “old-female” could require a transformation different from that for “old-male”, and different from “young-female” or “young-male”). Therefore, it appears not to be recommendable to apply different data transformation when analysing different factors such as sex and gender simultaneously.
Note that equality of variances is a desirable requisite to be fulfilled when applying Empirical Bayes, which is the main method implemented when using LIMMA and many other programs. However, the presence of residual trends does not constitute a problem in the Q-GDEMAR method because it operates on the empirical distribution of the data, without any further assumption.
3.2 Overall method performance assessment
The biological interpretation of microarray results is influenced by the type of discriminant variable chosen as well as for the type of computational algorithm applied for data post-processing. To assess both effects, we tested the three discriminant variants of Q-GDEMAR (median ratio, Δ1-difference, Δ2-difference) (Section 2.3), and their performance was compared against the LIMMA program.To that purpose, twelve microarray data-sets (Section 2.1) were evaluated by the number of up- and down regulated genes detected (Section 2.2). We also computed the FDR associated with each microarray analysed by Q-GDEMAR, but using our own approach (Appendix 2, ESI†). In the case of LIMMA, the number of genes detected depends on the combined use that this program makes of the Empirical Bayes (for the estimation of the p-value),26 and the FDR (computed by Benjamini–Hochberg approach on the basis of the p-values).27
Actually, there is a unique definition for the FDR, independently of the way in which its value is estimated.26–29 Thus, the number of genes considered as differentially expressed in LIMMA was taken from the set of genes obtained by this program, but fulfilling the condition that their FDR values were lower than or equal to the FDR value determined by Q-GDEMAR. In this way, the comparisons between both methods were done at the same level of the FDR (see Fig. 5A–C).
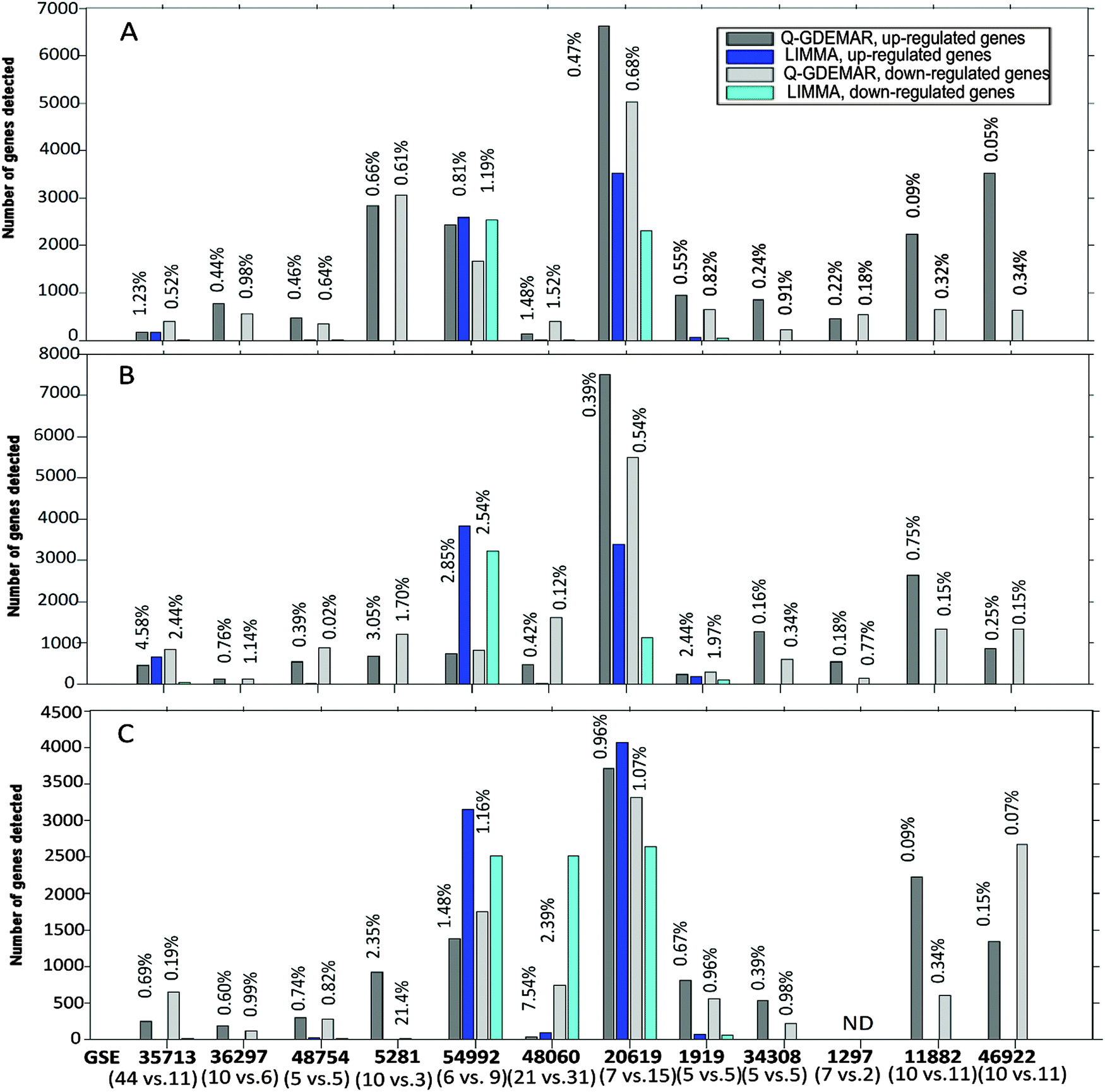 | ||
| Fig. 5 Comparative performance of Q-GDEMAR and LIMMA methods along twelve micro-array data sets according to the number of genes detected and the FDR achieved. Q-GDEMAR was evaluated through three discriminating variants: median ratio (top figure, A), Δ2-difference (middle figure, B), Δ1-difference (lower figure, C). The values above the bars indicate the percentage of FDR achieved in each case, at which comparison is done. The values between parentheses indicate the number of control and treatment samples which are being compared in each microarray. For details on the data, see Table S1 (ESI†). | ||
Fig. 5 shows some relevant features. First, LIMMA shows many gaps in the gene-number count. This indicates that LIMMA was able to detect just a few genes (or none at all) at the low level of the FDR. This is in strong contrast with the performance showed by Q-GDEMAR. Second, in 58 of the 68 comparisons done at the similar level of the FDR, Q-GDEMAR detected more genes than LIMMA did (see Table S1, ESI†). Finally, different non-parametric statistical tests (Wilcoxon's rank test, Sign test, and Fisher's exact test) confirmed the highly significant differences when Q-GDEMAR variants are overall compared against the LIMMA method (probability p ranges from 1 × 10−9 to 9.85 × 10−10 depending on the statistical method).
Also the finding that algorithms implemented through LIMMA are affected by a bias is important. In fact, the analysis of results from Table S1 (ESI†) shows that LIMMA detect systematically a higher number of up-regulated genes with respect to the number of down-regulated genes (Sign test, p = 2.74 × 10−4). Instead, this bias was not present in Q-GDEMAR even when different numbers of up- and down-regulated genes were always detected (Sign test, p = 0.7428).
Very often, researchers are forced to accept FDR values as high as 10–30% due to the difficulties found with their microarray data. In fact, in three of the twelve microarrays tested (GSE36297, GSE1297, and GSE46922), LIMMA showed a median value of the FDR ranging between 17% and 46%, even considering only those genes with extreme expression (absolute t-Student ≥4). Instead, with the combined use of Q-GDEMAR and our method to compute the FDR we can circumvent this limitation. In fact, we have obtained very low levels of the FDR (median value = 0.67%, interquartile range = 0.87%; see Fig. 5A–C). It is worth noting that our FDR evaluations included the detection of genes in three microarrays (the GSE 46922, GSE5281, and GSE1297) for which the LIMMA algorithms did not detect any significant gene at all (FDR varied between 88.5% and 98.9%, see Table S1, ESI†).
We thus conclude that Q-GDEMAR shows a superior performance with respect to LIMMA in sensitivity to detect differentially expressed genes. Moreover, it does this at very low levels of the false discovery rate (FDR).
3.3 Discriminating variants' performance assessment
Although Q-GDEMAR outperformed LIMMA in 85.3% of the analysed cases, a ranking of efficiencies for the individual variants can be established within each data-set (see Table S1, ESI†). The median ratio was the best discriminating variable in 50% of the cases analysed, Δ2-difference followed (33.33% cases), and lastly Δ1-difference (16.66% cases). This differential distribution is statistically significant (Chi2 = 12.04, p < 5 × 10−3), thus confirming that discriminating variants differ in the number of genes detected at a given instance.We have determined that the distribution curves of the three variants show quite similar profiles, but they do not keep a “strict” relation along their domains. Although each curve evolves monotonically, their mutual relationships change according to the number of genes detected (see Fig. 6).
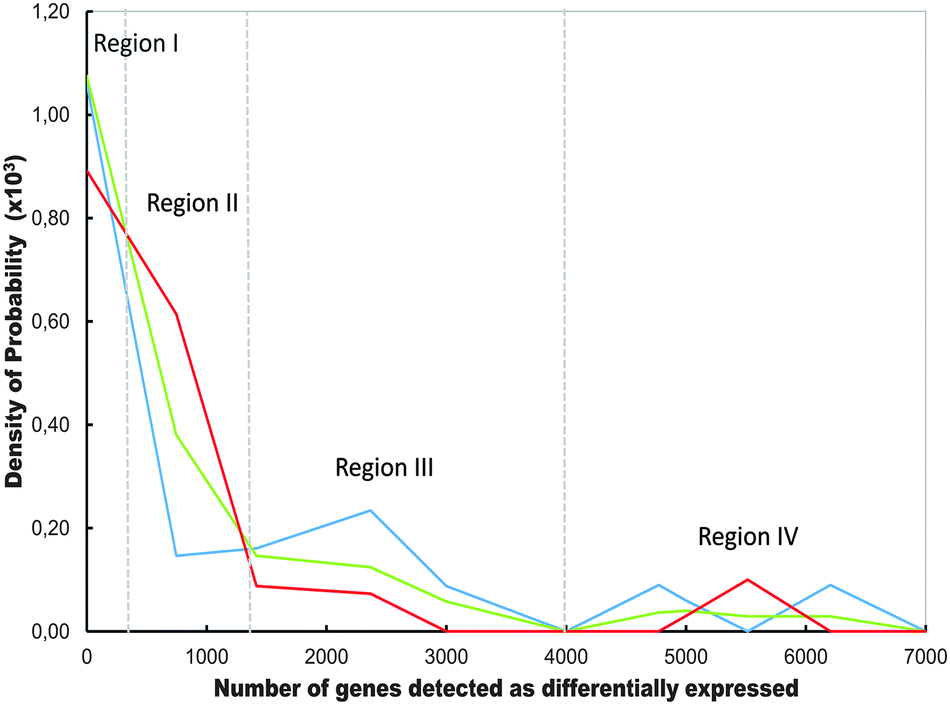 | ||
| Fig. 6 Comparative hierarchical order of the distributions curves corresponding to Q-GDEMAR variants as a function of the number of significant genes detected along the twelve microarrays analysed in Table S1 (ESI†). The discriminant variants compared are the median ratio (blue line), the Δ2-difference (red line) and, the Δ1-difference (green line). Regions (I−IV) are defined by the zones of the domain where the change in the hierarchical order takes place. | ||
The trends displayed in Fig. 6 can explain the data observed in Table S1 (ESI†), in the sense that the median ratio outperforms the Δ2- or Δ1-difference in some microarrays, but only within a given range of detected genes. The fact that the hierarchical order among the curves in Fig. 6 does not change from point to point, but along a few, discrete intervals suggests that the observed variations reflect true differences in the behaviour of the variants rather than mere noisy data.
Differences due to imbalance in size among the compared groups can also influence the performance of variants. This was observed in the GSE5281 microarray, where a poor correlation between the average and the median values (R2 = 0.625) is found in three samples of the treatment group. Nevertheless, the control group of the same microarray, which comprised ten samples, shows an excellent correlation (R2 = 0.999; data not shown). Hence, the median ratio variant out-performs the Δ1-difference in the case of GSE5281, because the better variant is based on median values, responding more accurately to the nature of the data (see Table S1, ESI†).
Moreover, “unbalanced sampling” could exert an “interaction effect” rather than a “fixed effect”. That is, the effect produced by size differences between the groups could vary depending on the high or low number of replications involved in a particular comparison. This is the case of the microarrays GSE35713 (44 control samples against 11 treatment samples) and GSE54992 (6 control samples against 9 treatment samples). In both cases, the Δ1-difference out-performed the median-based discriminating variants. This seems justified since in these cases, a high correlation between means and medians within each condition is guaranteed due to the high number of samples considered. In addition, the weight factor used as correction in Δ1-difference seems to be able to compensate the strongly unbalanced experimental design used. Besides, the number of genes detected in GSE35713 falls into region II of Fig. 6, providing an additional reason for the Δ1-difference variant out-performing the other discriminating variants.
In brief, given that the relative performance of each discriminant variant depends on the number of genes detected in each particular microarray, but the number of genes that will be detected is unknown beforehand, a practical consequence of Fig. 6 is that there is no a priori preferred form for the calculation of a suitable discriminating variable. The three variants have to be tested by Q-GDEMAR, and best selected for a given micro-array.
3.4 What determines the efficiency of the discriminant variants?
In order to quantify the efficiency of the discriminating variables we introduce the index Phi (Φ), defined as Φ = [1 − (σcentral/σglobal)], where σcentral is the standard deviation for the Gaussian central region of the data distribution (computed through the Gaussian deconvolution operation), and σglobal is the apparent standard deviation (computed on the entire data distribution).The index Φ characterizes how good is a given discriminant variant in separating the largest population of genes with invariant expression from those small groups of genes which are deregulated in a given condition. Note that if the Φ index is zero, there is no gained discrimination from the central region (i.e., σcentral = σglobal). In contrast, an ideal value of 1 would imply that the genes without significant variation would have a unique, constant value of expression (i.e., σcentral = 0). In this case, the discrimination between the genes belonging to the Gaussian central region and those de-regulated genes localized at the tails of the “entire” data distribution will be maximum. Otherwise, as in general could be expected, the Φ index will take some value comprised between zero and one (i.e., σcentral < σglobal).
Importantly, the “index Φ” shown to be significantly correlated with the “number of genes detected” by Q-GDEMAR variants (R-pearson = 0.806, p < 1 × 10−4), and this correlation persists even after being corrected by the kurtosis (partial correlation Rgenes_Φ(kurtosis) = 0.8418) or the skewness (partial correlation Rgenes_Φ(skewness) = 0.8194). Because other co-linear relations also occurred, the correlation matrix of system's variables (see Table S3, ESI†) was subjected to principal component analysis (see Fig. 7).
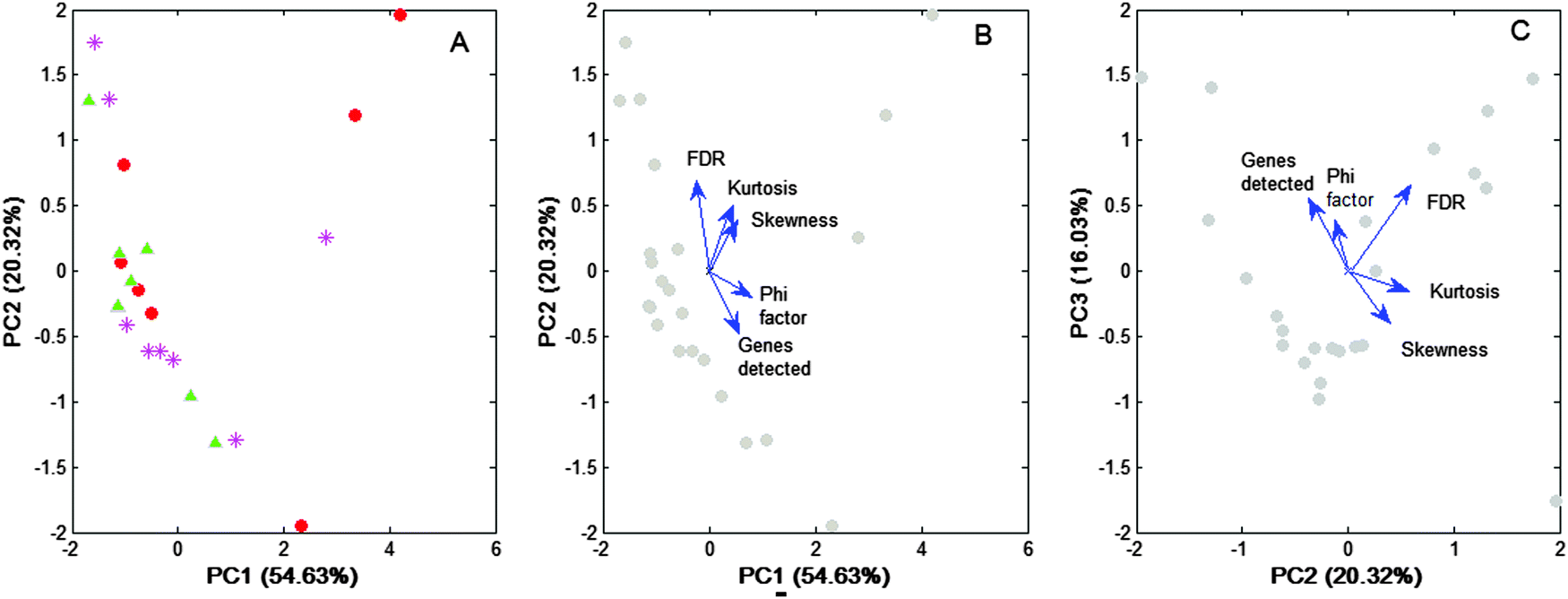 | ||
Fig. 7 Eigen-decomposition of the correlation matrix of Table S3 (ESI†). The correlation matrix (apparent dimension = 5) was projected on two bi-dimensional sub-spaces (PC1–PC2, PC2–PC3). (A) The scores of the projected points are disaggregated according to the type of discriminant variables used for processing: median ratio ( ), Δ2-difference ( ), Δ2-difference ( ), Δ1-difference ( ), Δ1-difference ( ). (B and C) Biplot representations of the points projected, but without disaggregation of the discriminating variants. The axes are the principal components (PC), and the values between parentheses give the percentage of variance explained by them. The relative position of the analysed variables in each sub-space is given by the eigenvectors of the correlation matrix (blue arrows). The closer the vectors, the higher the cosine of the angle comprised, and hence, the higher the correlation between the variables that they represent. ). (B and C) Biplot representations of the points projected, but without disaggregation of the discriminating variants. The axes are the principal components (PC), and the values between parentheses give the percentage of variance explained by them. The relative position of the analysed variables in each sub-space is given by the eigenvectors of the correlation matrix (blue arrows). The closer the vectors, the higher the cosine of the angle comprised, and hence, the higher the correlation between the variables that they represent. | ||
Fig. 7A shows that no particular pattern of the data is detected among the three discriminating variants used. This is in line with our previous finding concerning the “shape” similarity observed with the distribution profiles associated with each variant (Fig. 6). Importantly, data in Fig. 7A support the hypothesis that differences among the discriminant variants are not produced due to intrinsic differential features, but due to the fact that discriminant variants could share a common, still unidentified factor that is present at different levels under a given microarray condition.
Moreover the eigen-analysis performed confirms the trend of the “Φ index” to be positively correlated with the “number of genes detected” (see Fig. 7C and B). That is, the “Φ index” can be the unifying factor, shared by three discriminant variants, differing only in the level of efficiency achieved in each particular case.
Because PC1 together with PC2 explains 75% of the data variability, a main trend of the “FDR” to negatively correlate with the “number of genes detected” can be established (see Fig. 7B). However, a minor trend in which these variables appear in positive correlation is also observed (see Fig. 7C). Logically, index Φ correlated negatively with the FDR in the first case (see Fig. 7B), but positively in the second case (Fig. 7C). Anyway, from a practical point of view, it should be remembered that although a high value of the Φ index could increase the value of the FDR, in our method the FDR was usually very low (median value: 0.67%, interquartile range: 0.87%).
Concerning “kurtosis” and “skewness”, both show a main trend to worsen the FDR by increasing their value (see Fig. 7B), but some minor cases exist in which neither of these variables affected the FDR (see Fig. 7C). Moreover, skewness and kurtosis appeared as nearly orthogonal to the “Φ index” (see Fig. 7B), which is contrary to that shown in the matrix of correlations where these variables show significant correlations (see Table S2, ESI†).
The apparent discrepancies are explained because correlations in Table S2 (ESI†) were computed on the raw values of the variables, whereas in Fig. 7 the variables are Z-standardized for the principal component analysis given that they come from a correlation matrix. Hence, while in the first case correlations are dominated by the variables with higher variance, in the second case this influence is neutralized.
A near orthogonality was observed between the skewness–kurtosis pair with respect to the Φ index. This could mean either a non-linear relationship between the “shape” of the initial distribution of the data and the level of gain of discriminating efficiency achieved through the Gaussian deconvolution or that there is no relation at all between these variables. A method based on specific algorithms to deal in a complex way with skewness and kurtosis has been proposed to address the problem of (non-linear) interactions between the variables.10 However, in our approach, these structural features of the entire distribution are not treated separately, but subsumed in the operation of Gaussian deconvolution.
The occurrence of several main trends in the principal component analysis coexisting with other minor cases in which previous relationships appear inverted or relaxed denotes segmentation of the data. By cluster analysis, we verified this segmentation (results not shown). This opens the possibility of occurrence of “local” correlations rather than overall correlations. In spite of data segmentation, we have verified by using the R pearson correlation that the “Φ index” is strongly related to the “number of genes detected”. Analyses by partial correlation and principal component also confirm this relation. Although correlations do not necessarily imply causality, these findings strongly suggest that the efficiency of Gaussian deconvolution (i.e., the Φ index) effectively conditions the number of genes detected in the microarrays.
3.5 Is Cauchy distribution an alternative to the median ratio in Q-GDEMAR?
It has been proposed that genes differentially expressed can be determined by models based on the Cauchy distribution,53 a subtype of “alpha-stable” distributions.54 The Cauchy distribution is generated by the quotient of two normal, independent variables. Its main advantage is that median ratios of the samples converge to the true median ratio of the population while the tails of the distribution are “heavier” than that expected for a normal distribution, which is thus closer to the microarray reality.Our median ratio discriminating variant is also computed as the quotient of two independent variables. However, neither the control nor the treatment conditions followed a normal distribution in the microarray data-sets that we have analysed. Hence, it is not expected that the median ratio will follow a Cauchy distribution.
Moreover, Cauchy distributions are symmetric. Instead, in our median ratio distributions only a sub-set of microarrays are free of significant skewness. Interestingly, some particular subtypes of alpha-stable distributions allow some degree of restricted asymmetry.54 However, neither Cauchy nor alpha-stable distributions account for kurtosis, another prominent characteristic frequently observed in the median ratio distributions (see Table S2, ESI†). Therefore, it should not be expected that Cauchy or alpha-stable distributions can be used as a general method for microarray analysis.
As a practical example of the consequences generated when the requisites of Cauchy distributions are not fulfilled, we analysed the microarray GSE36297 using this method. The results are depicted in Fig. 8.
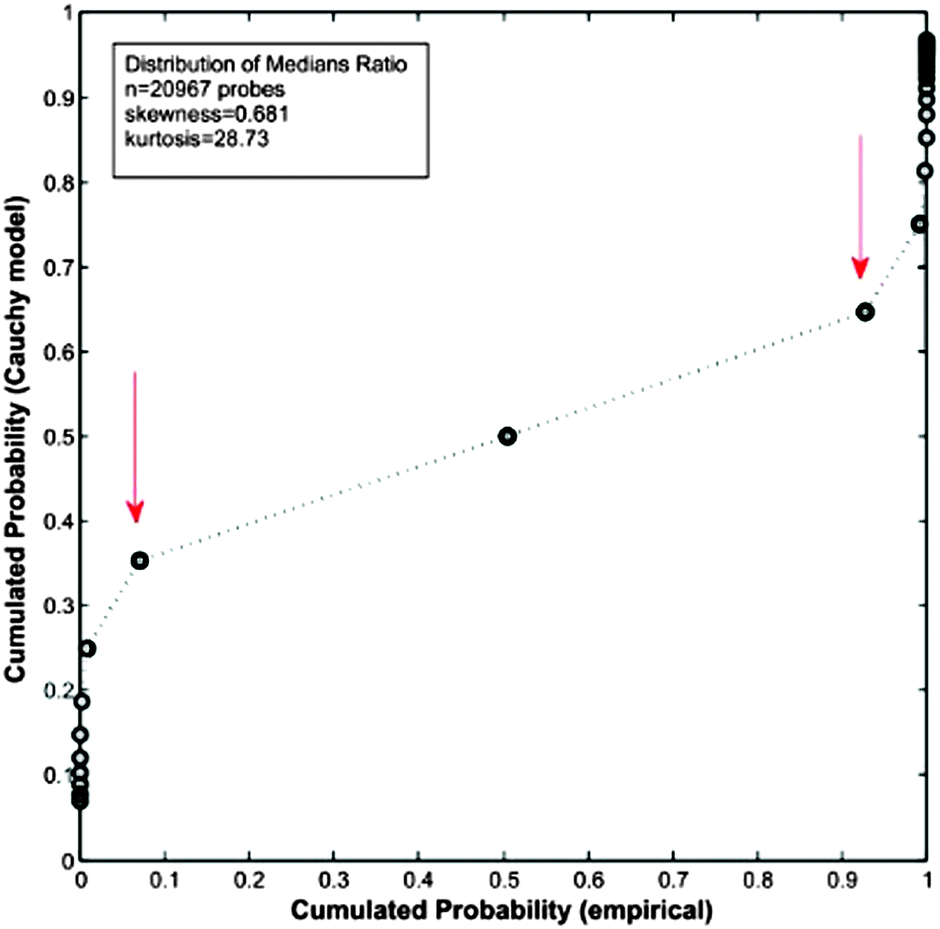 | ||
| Fig. 8 Q–Q plot between the “expected” cumulated probability distribution according to a Cauchy-based model and the “empirical” cumulated probability distribution. Computations were done on data from microarray GSE36297,43 which result in a Cauchy model with two parameters (location t = 0.9798 and scale s = 0.0389). | ||
As can be seen in Fig. 8, the fitting by a Cauchy distribution model is very poor. In fact, there is no quantile correspondence at the tails among of the compared distributions. The reason explaining the lack of fitting is the moderate level of kurtosis present in the data. This result is in contrast with the excellent performance obtained with the same microarray by Q-GDEMAR (see Tables S1 and S2, ESI†).
It should be noted that neither Q-GDEMAR nor Cauchy-distribution assumes that the overall data distribution is Gaussian. Hence, Q-GDEMAR also recognizes the problem of the “heavier tails”. But, Q-GDEMAR deals with “heavier tails” in a less restrictive and more accurate way than Cauchy-based models do.
4 Conclusions
The rigorous analysis of the microarray data is a hard and complex task. Its complexity arises from the convergence of multiple non-linear interacting factors. Some of them are analytical variability, biological variability inherent to the sampled subjects, the type of discriminating variable adopted, the absolute and the relative degree of imbalance between the compared groups. In order to fully rationalize these factors and to obtain an exhaustive explanation, a mathematical model where the multiple interrelations are represented is required.Because such a model is rarely available, other alternatives should be envisaged. Our proposal intends to fulfil this need. Being aware of its shortcomings and limitations, the proposed method has nonetheless some significant advantages. First, in 58 out of 68 cases compared Q-GDEMAR showed a higher sensitivity to detect differentially expressed genes (p = 1 × 10−10). Second, it allows the computation of two-tail FDR values in a simple straightforward way, which are associated with the sub-populations of up- and down regulated genes rather than to each individual gene. Third, in addition to its higher sensitivity, the procedure that we have developed allowed us to achieve very low levels of the FDR (median = 0.67%, interquartile range = 0.87%). Fourth, the method is statistically sound because univocal, well-defined p-values are assigned to the different values of the discriminant variable. Fifth, the outputs of the method, such as p-values and the FDR, are independent from any particular theoretical assumption and thus, the risk of under- or over-estimating the number of genes of interest is minimized.
Importantly, our method achieved high sensitivity for gene detection while maintaining very low levels of the FDR, in strong contrast with algorithms commonly applied. For example, simulation studies of Yang and co-workers' using five popular methods for microarray post-processing analysis showed that their power of detection ranges between 10 and 60% when the size of replication varies between 3 and 12 samples; a maximum power of 80% is achieved only if more than 25 samples are used, an experimental condition rarely fulfilled.8 On the other hand, simulation studies of Mc Carthy and Smyth practiced using six current methods for post-processing analysis showed that these methods can maintain low FDR values only if the number of differentially expressed genes considered is limited to less than 200 genes. If the list were increased to 400 genes, a FDR value as high as 33% would be generated.55
There are no rules about what should be considered an “acceptable” value of the FDR. A cost-benefit criterion based on practical considerations has to be used. The cut-off value of the FDR should be decided taking into account the aim of the analysis, such as the inherent level of biological variability, the analytical variability of the microarrays, and the cost of the polymerase chain reaction (PCR) validating the analysis. In any case, the FDR values condition the number of genes that can potentially be accepted as differentially expressed genes (the lower the FDR achieved, the higher is the number of identifiable deregulated genes). Many published microarray studies report a range from 10 to 500 top dysregulated genes. However, this criterion implicitly links the supposed biological importance exerted by one gene with the magnitude of the fold-change observed with it. However, it is a fact that many genes showing slight changes (∼15–20%) can also exert important effects. In addition, the consistency of the microarray findings can also be tested through network analysis and ontology analysis, besides being validated by PCR and/or proteomics. These two complementary procedures, being context-dependent techniques, would require lists of candidate genes as long as possible. Again, the importance of obtaining high sensitivity of detection together with low FDR values, both features of our method, makes our approach very valuable (Section 3.2).
Although Q-GDEMAR was primarily devised to deal with the detection of differentially expressed genes in unbalanced groups, actually it is a general method for the post-processing analysis of c-DNA and oligonucleotide-based microarrays. As such, its application can be extended to other experimental designs not included in the present study. Hence, Q-GDEMAR can deal with paired samples, (1-way) ANOVA, and factorial designs, time-regressions, dosage-regression, or survival (COX) analysis. Importantly, the post-processing analysis of the dual-channel microarrays2 can also be addressed using our method.
The common reason that justifies the ample scope of Q-GDEMAR is that in all the referred cases, the relation between treatments and control conditions finally converges in a unique, continuous, unimodal distribution to be analysed.
Q-GDEMAR and our procedure to compute the FDR rely on the fact that most of the genes within a microarray do not vary their expression significantly and follow a Gaussian distribution. This is not a supposition guided by mathematical convenience, but a fact supported by our analysis of the twelve microarray data-sets dealt with in this work (Fig. 2A, Section 2.1). What can change between different microarrays is not the presence of the Gaussian central region, but the extent of its dispersion. According to the features of each microarray distribution, the operation of deconvolution provides an estimation of its interquartile range (IQR = Q0.75 − Q0.25), which is related to the apparent SDGaussian_central_region through eqn (7) (Appendix 2, ESI†). Different SDGaussian_central_region values, through the interplay among eqn (7)–(14), lead to different levels of the FDR, and hence, to different number of genes detected as differentially expressed (Appendix 2, ESI†).
So, to compute the FDR either Q-GDEMAR or our procedure can function through the Gaussian deconvolution process, regardless of the profile of the overall data distribution of the microarray.
Authors contribution
DVG was responsible for the design and conception of the study, MPA was responsible for data acquisition and computing. All the authors contributed to the drafting of the manuscript, while NVT supervised the project and gave the final approval. All the authors agree on all the parts of the work.Acknowledgements
This work was funded by research grants from MINECO, Project BIO2014-54411-C2-2-R and the IMBRAIN project, REF. FP7-REGPOT-2012-CT2012-31637-IMBRAIN. DVG acknowledges the support given by the Campus Atlántico Tri-Continental (CEI-10/00018, Canary Islands, Spain). The authors thank Dr Catalina Feledi for her valuable collaboration.References
- N. Mah, A. Thelin, T. Lu, S. Nikolaus, T. Kühbacher, Y. Gurbuz, H. Eickhoff, G. Klöppel, H. Lehrach, B. Mellgård, C. M. Costello and S. Schreiber, A comparison of oligonucleotide and cDNA-based microarray systems, Physiol. Genomics, 2004, 16, 361–370 CrossRef CAS PubMed.
- Y. Guo, Q. Sheng, J. Li, F. Ye, D. C. Samuels and Y. Shyr, Large scale comparison of gene expression levels by microarrays and RNAseq using TCGA data, PLoS One, 2013, 8, e71462 CAS.
- K. J. Mantione, R. M. Kream, H. Kuzelova, R. Ptacek, J. Raboch, J. M. Samuel and G. B. Stefano, Comparing bioinformatic gene expression profiling methods, microarray and RNA-Seq, Med. Sci. Monit. Basic Res., 2014, 20, 138–142 CrossRef PubMed , review.
- R. C. Gentleman, V. J. Carey, D. M. Bate, B. Bolstadt and M. Detting, et al., Bioconductor, open software development for computational biology and bioinformatics, Genome Biol., 2004, 5, R80 CrossRef PubMed.
- G. R. Grant, E. Manduchi and C. J. Stoeckert, Analysis and management of microarray gene expression data, Current Protocols in Molecular Biology, 2007, ch. 19, unit 19.6, DOI:10.1002/0471142727.mb1906s77.
- S. K. Mohapatra and A. Krishnan, Microarray data analysis, Methods Mol. Biol., 2011, 678, 27–43 CAS , review.
- K. Owzar, W. T. Barry and S. H. Jung, Statistical considerations for analysis of microarray experiments, Clin. Transl. Sci., 2011, 4(6), 466–477 CrossRef CAS PubMed , review.
- D. Yang, R. S. Parrish and G. N. Brock, Empirical evaluation of consistency, accuracy of methods to detect differentially expressed genes based on microarray data, Comput. Biol. Med., 2014, 46, 1–10 CrossRef CAS PubMed.
- R. Alonso, F. Salavert, F. Garcia-Garcia, J. Carbonell-Caballero, M. Bleda and L. Garcia-Alonso, et al., Babelomics 5.0, functional interpretation for new generations of genomic data, Nucleic Acids Res., 2015, 43, W117–W121 CrossRef PubMed.
- J. Wang, M. Jia, L. Zhu, Z. Yuan, P. Li, C. Chang, J. Luo, M. Liu and T. Shi, Systematical Detection of Significant Genes in Microarray Data by Incorporating Gene Interaction Relationship in Biological Systems, PLoS One, 2010, 5(10), e13721 Search PubMed.
- J. P. Ioannidis, D. B. Allison, C. A. Ball, I. Coulibaly and X. Cui, et al., Repeatability of published microarray gene expression analyses, Nat. Genet., 2009, 41, 149–155 CrossRef CAS PubMed.
- A. Campain and Y. H. Yang, Comparison study of microarray meta-analysis methods, Bioinformatics, 2010, 11, 408–419 Search PubMed.
- Y. Tan and Y. Liu, Comparison of methods for identifying differentially expressed genes across multiple conditions from microarray data, Bioinformation, 2011, 7, 400–404 CrossRef PubMed.
- F. Hong and R. Breitling, A comparison of meta-analysis methods for detecting differentially expressed genes in microarray experiments, Bioinformatics, 2008, 24, 374–382 CrossRef CAS PubMed.
- L. C. Chang, H. M. Lin, E. Sibille and G. C. Tseng, Meta-analysis methods for combining multiple expression profiles, comparisons, statistical characterization and an application guideline, BMC Bioinf., 2013, 4, 368 CrossRef PubMed.
- P. Roepman, The future of diagnostic gene-expression microarrays, bridging the gap between bench and bedside, Bioanalysis, 2010, 2, 249–262 CrossRef CAS PubMed.
- M. Guarnaccia, G. Gentile, E. Alessi, C. Schneider, S. Petralia and S. Cavallaro, Is this the real time for genomics?, Genomics, 2014, 103, 177–182 CrossRef CAS PubMed.
- B. M. Bolstad, R. A. Irizarry, M. Astrand and T. P. Speed, A comparison of normalization methods for high density oligonucleotide array data based on variance and bias, Bioinformatics, 2003, 19, 185–193 CrossRef CAS PubMed.
- M. Raimers, Satistical analysis of microarray data, Addict. Biol., 2005, 10, 23–35 CrossRef PubMed.
- O. G. Troyanskaya, M. E. Garber, P. O. Brown, D. Botstein and R. B. Altman, Nonparametric methods for identifying differentially expressed genes in microarray data, Bioinformatics, 2002, 18, 1454–1461 CrossRef CAS PubMed.
- L. Guo, E. K. Lobenhofer, C. Wang, R. Shippy, S. C. Harris and L. Zhang, et al., Rat toxicogenomic study reveals analytical consistency across microarray platforms, Nat. Biotechnol., 2006, 24, 1162–1169 CrossRef CAS PubMed.
- V. G. Tusher, R. Tibshirani and G. Chu, Significance analysis of microarrays applied to the ionizing radiation response, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 5116–5121 CrossRef CAS PubMed.
- D. M. Witten and R. A. Tibshirani, A comparison of fold-change and the t-statistic for microarray data analysis, Standford University, 2007, available at http://statweb.stanford.edu/~tibs/ftp/daniela-fold.pdf Search PubMed.
- K. F. Kerr, Comments on the analysis of unbalanced microarray data, Bioinformatics, 2009, 25, 2035–2041 CrossRef CAS PubMed.
- D. B. Allison, X. Cui, G. P. Page and M. Sabripou, Microarray data analysis, from disarray to consolidation and consensus, Nat. Rev. Genet., 2006, 7, 55–65 CrossRef CAS PubMed.
- B. Efron, R. Tibshirani, J. D. Storey and V. Tusher, Empirical Bayes Analysis of a Microarray Experiment, J. Am. Stat. Assoc., 2001, 96, 1151–1160 CrossRef.
- Y. Benjamini and Y. Hochberg, Controlling the False Discovery Rate, A Practical and Powerful Approach to Multiple Testing, J. R. Stat. Soc. Ser. B, 1995, 57, 289–300 Search PubMed.
- E. Hansen and K. F. Kerr, A comparison of two classes of methods for estimating false discovery rates in microarray studies, Scientifica, 2012, 519394 Search PubMed , available at, http://www.hindawi.com/journals/scientifica/2012/519394/abs/.
- K. Shedden, W. Chen, R. Kuick, D. Ghosh and J. Macdonald, et al., Comparison of seven methods for producing Affymetrix expression scores based on False Discovery Rates in disease profiling data, BMC Bioinf., 2005, 6, 26 CrossRef PubMed.
- R. Suresh, X. Li, A. Chiriac, K. Goel, A. Terzic, C. Perez-Terzic and T. J. Nelson, Transcriptome from circulating cells suggest dysregulated pathways associated with long-term recurrent events following first-time myocardial infarction, J. Mol. Cell. Cardiol., 2014, 74, 13–21 CrossRef CAS PubMed.
- X. Cui and G. A. Churchill, Statistical tests for differential expression in cDNA microarray experiments, Genome Biol., 2003, 4(4), 210 CrossRef PubMed.
- W. Li, Volcano plots in analyzing differential expressions with mRNA microarrays, J. Bioinf. Comput. Biol., 2012, 10, 1231003 CrossRef PubMed.
- M. R. Dalman, A. Deeter, G. Nimishakavi and Z.-H. Duan, Fold change and p-value cutoffs significantly alter microarray interpretations, BMC Bioinf., 2012, 13, S11 CrossRef PubMed.
- E. Hansen and K. F. Kerr, A comparison of two classes of methods for estimating false discovery rates in microarray studies, Scientifica, 2012, 519394 Search PubMed , available in the web at, http://www.hindawi.com/journals/scientifica/2012/519394/.
- G. K. Smyth, Linear models and empirical Bayes methods for assessing differential expression in microarray experiments, Stat. Appl. Genet. Mol. Biol., 2004, 3(1), 3 Search PubMed.
- M. E. Ritchie, B. Phipson, D. Wu, Y. Hu, C. W. Law, W. Shi and G. K. Smyth, Limma powers differential expression analyses for RNA-sequencing and microarray studies, Nucleic Acids Res., 2015, 43, e47 CrossRef PubMed.
- I. Medina, J. Carbonell, L. Pulido, S. C. Madeira and S. Goetz, et al., Babelomics, an integrative platform for the analysis of transcriptomics, proteomics and genomic data with advanced functional profiling, Nucleic Acids Res., 2010, 38, W210–W213 CrossRef CAS PubMed.
- T. Barrett, S. E. Wilhite, P. Ledoux, C. Evangelista and I. F. Kim, et al., NCBI GEO, archive for functional genomics data sets update, Nucleic Acids Res., 2013, 41, D991–D995 CrossRef CAS PubMed.
- Y. Cai, Q. Yang, Y. Tang, M. Zhang and H. Liu, et al., Increased Complement C1q Level Marks Active Disease in Human Tuberculosis, PLoS One, 2014, 9, e92340 Search PubMed.
- H. Levy, X. Wang, M. Kaldunski, S. Jia and J. Kramer, et al., Transcriptional signatures as a disease-specific and predictive inflammatory biomarker for type 1 diabetes, Genes Immun., 2012, 13, 593–604 CrossRef CAS PubMed.
- W. S. Liang, T. Dunckley and T. G. Beach, et al., Gene expression profiles in anatomically and functionally distinct regions of the normal aged human brain, Physiol. Genomics, 2007, 28, 311–322 CrossRef CAS PubMed.
- U. Ungethuem, T. Haeupl, H. Witt, D. Koczan and V. Krenn, et al., Molecular signatures and new candidates to target the pathogenesis of rheumatoid arthritis, Physiol. Genomics, 2010, 42A, 267–282 CrossRef CAS PubMed.
- J. M. Kristensen, V. Skov, S. J. Petersson and N. Ørtenblad, et al., A PGC-1α- and muscle fibre type-related decrease in markers of mitochondrial oxidative metabolism in skeletal muscle of humans with inherited insulin resistance, Diabetologia, 2014, 57, 1006–1015 CrossRef CAS PubMed.
- D. R. Crooks, T. G. Natarajan, S. Y. Jeong and C. Chen, et al., Elevated FGF21 secretion, PGC-1α and ketogenic enzyme expression are hallmarks of iron–sulfur cluster depletion in human skeletal muscle, Hum. Mol. Genet., 2014, 23, 24–39 CrossRef CAS PubMed.
- S. Affò, M. Dominguez, J. J. Lozano and P. Sancho-Bru, et al., Transcriptome analysis identifies TNF superfamily receptors as potential therapeutic targets in alcoholic hepatitis, Gut, 2013, 62, 452–460 CrossRef PubMed.
- X. M. Wang, W. Y. Yik, P. Zhang and W. Lu, et al., The gene expression profiles of induced pluripotent stem cells from individuals with childhood cerebral adrenoleukodystrophy are consistent with proposed mechanisms of pathogenesis, Stem Cell Res. Ther., 2012, 3, 39 CrossRef PubMed.
- E. M. Blalock, J. W. Geddes, K. C. Chen and N. M. Porter, et al., Incipient Alzheimer's disease, microarray correlation analyses reveal major transcriptional and tumor suppressor responses, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 2173–2178 CrossRef CAS PubMed.
- N. C. Berchtold, D. H. Cribbs, P. D. Coleman and J. Rogers, et al., Gene expression changes in the course of normal brain aging are sexually dimorphic, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 15605–15610 CrossRef CAS PubMed.
- M. Jernås, Y. Hou, F. Strömberg Célind and L. Shao, et al., Differences in gene expression and cytokine levels between newly diagnosed and chronic pediatric ITP, Blood, 2013, 122, 1789–1792 CrossRef PubMed.
- T. M. Therneau and K. V. Ballman, What Does PLIER Really Do?, Cancer Inf., 2008, 6, 423–431 CAS.
- Z. Wu and R. A. Irizarry, Preprocessing of oligonucleotide array data, Nat. Biotechnol., 2004, 22, 656–658 CrossRef CAS PubMed.
- Z. Wu and R. A. Irizarry, Description of GCRMA package, 2014, Oct 13, http://available from, http://www.bioconductor.org Search PubMed.
- I. V. Yang, E. Chen, J. P. Hasseman, W. Liang and B. C. Frank, et al., Within the fold, assessing differential expression measures and reproducibility in microarray assays, Genome Biol., 2002, 3, research0062 Search PubMed.
- D. Salas-Gonzalez, E. E. Kuruoglu and D. P. Ruiz, A heavy-tailed empirical Bayes method for replicated microarray data, Comput. Stat. Data Anal., 2009, 53, 1535–1546 CrossRef.
- D. J. McCarthy and G. K. Smyth, Testing significance relative to a fold-change threshold is a TREAT, Bioinformatics, 2009, 25, 765–767 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5mb00541h |
| This journal is © The Royal Society of Chemistry 2016 |