
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Template-assisted extrusion of biopolymer nanofibers under physiological conditions†
Mohammad
Raoufi
abc,
Neda
Aslankoohi‡
ab,
Christine
Mollenhauer
ab,
Heike
Boehm
abd,
Joachim P.
Spatz
ab and
Dorothea
Brüggemann
*abe
aDepartment of Biointerphase Science & Technology, Max Planck Institute for Medical Research, Jahnstraße 29, 69120 Heidelberg, Germany. E-mail: brueggemann@uni-bremen.de
bDepartment of Biophysical Chemistry, University of Heidelberg, INF 253, D-69120 Heidelberg, Germany
cNanotechnology Research Center, Faculty of Pharmacy, Tehran University of Medical Sciences, Tehran 1417614411, Iran
dCSF Biomaterials and Cellular Biophysics, Max Planck Institute for Intelligent Systems, Heisenbergstr. 3, D-70569 Stuttgart, Germany
eInstitute for Biophysics, University of Bremen, Otto-Hahn-Allee 1, 28359 Bremen, Germany
First published on 18th July 2016
Abstract
Biomedical applications ranging from tissue engineering to drug delivery systems require versatile biomaterials based on the scalable and tunable production of biopolymer nanofibers under physiological conditions. These requirements can be successfully met by a novel extrusion process through nanoporous aluminum oxide templates, which is presented in this study. With this simple method we are able to control the nanofiber diameter by chosing the size of the nanopores and the concentration of the biopolymer feed solution. Nanofiber assembly into different hierarchical fiber arrangements can be achieved with a wide variety of different proteins ranging from the intracellular proteins actin, α-actinin and myosin to the extracellular matrix components collagen, fibronectin, fibrinogen, elastin and laminin. The extrusion of nanofibers can even be applied to the polysaccharides hyaluronan, chitosan and chondroitin sulphate. Moreover, blends of different proteins or proteins and polysaccharides can be extruded into composite nanofibers. With these features our template-assisted extrusion process will lead to new avenues in the development of nanofibrous biomaterials.
Insight, innovation, integrationBiopolymer nanofibers with controllable characteristics have become increasingly important for the development of future biomaterials. In this study we present a novel extrusion approach to prepare nanofibers from a great variety of biopolymers under physiological buffer conditions. With this technique it is possible to extrude nanofibers with controllable diameters and different hierarchical arrangements. We have shown that this process can be applied to various intra- and extracellular proteins, polysaccharides and composites thereof. In future, the extrusion of such a new class of biopolymer nanofibers can be integrated into the development of novel biomaterials, implant surface coatings or tissue engineering scaffolds. |
Introduction
Among the large variety of synthetically prepared biomaterials protein nanofibers have become very important for biomedical applications as they can mimic the naturally occurring fibrous structure of the extracellular matrix (ECM) and many other natural protein assemblies.1,2 Such biopolymer nanofibers have a very high surface area combined with a small volume. Hence, the surface properties, which are important for chemical and biological interactions, often dominate over bulk properties.3 The mesh-like arrangement of nanofibers from ECM proteins and polysaccharides can benefit cellular adhesion and proliferation or tissue ingrowth in combination with mechanical strength.3 In the past, the tendency of cells to orient along fibers has often been exploited to design tissue engineering scaffolds4 since nanotopographical features in these synthetic ECM systems can regulate cell adhesion and other cellular functions.5Currently, three different techniques are established to fabricate polymeric nanofibers: self-assembly, phase separation and electrospinning.6,7 Self-assembly is based on the hydrophobic and ionic interactions of specially designed biopolymers.3,8 Temperature-induced phase separation requires very simple equipment and yields polymer nanofibers by freeze-drying of a solution with a polymer-poor and a polymer-rich phase.9,10 Both techniques allow the fabrication of biopolymer fibers with controlled functions and mechanical properties. Self-assembly even facilitates the preparation of nanofibers with very small diameters in the lowest ECM scale, i.e. 5 to 8 nm.7 Nevertheless, both self-assembly and phase separation are restricted to a few special polymers, lack scalability and are limited to laboratory scale use because of the low fiber yield, which is associated with these time-consuming procedures.7,11
Electrospinning is the most common nanofiber fabrication technique, being very versatile, scalable and able to produce fibers with a diameter range from 3 nm up to 10 μm.12 In a continuous process nanofibers with tailorable mechanical properties are fabricated through an electrically charged jet of polymer solution. Despite its use of simple equipment, electrospinning often is a time-consuming process unless parallel nozzles are used for simultaneous spinning of multiple nanofibers.13,14 Moreover, electrospinning of natural polymers like proteins is more difficult to control than for synthetic polymers as many biopolymers are not compatible with the large electric fields required for electrospinning.3 Water soluble proteins are particularly unstable during electrospinning due to weak mechanical properties,15 and the fabrication of nanofibrous protein networks with reproducible fiber diameters and porosity still remains challenging.16 Nevertheless, the main disadvantage of electrospun protein fibers is the use of organic solvents, which impedes the biological activity and functionality of many proteins.6,7
Recently, a flow processing technique was presented to prepare nanofibrous collagen scaffolds with aligned or randomly deposited nanofibers of approximately 30 to 45 nm in diameter.17,18 However, the fiber diameter could not be varied any further, and the hierarchical fiber assembly was restricted to dense fibril matrices with low porosity. In another study, composite fibrils containing collagen and calcium phosphate were formed by pH-driven self-assembly through track-etched polycarbonate nanopores in a gradient of pH 3 to pH 11. Depending on the nanopore geometry, the resulting mineralized collagen fibers exhibited large diameters between 120 and 760 nm and were several tens of micrometers long.19 Nevertheless, this study involved the use of non-physiological pH values, which affect the biological activity of various proteins or other biomolecules in the composite fibers and collagen was the only ECM protein to be assembled into nanofibers in this study.19
Biocompatible anodic aluminum oxide (AAO) is another nanoporous material, which is used in many biomedical applications ranging from filtration membranes,20 cell culture interfaces,21,22 drug delivery systems and implant coatings23 to biosensors.24 Nanoporous AAO membranes have highly ordered, self-organised nanochannels with regular pore size, uniform pore density and high porosity over a large scale.25,26 Pore diameters between approximately 5 nm and several hundred nanometers can be achieved using an efficient, low-cost anodisation process with polyprotic acids, such as sulphuric or oxalic acid.27 Beyond that, ordered AAO nanopores have become well-established template materials to prepare vertical nanowires and nanoparticle arrays from various materials such as metals, semiconductors or synthetic polymers.28–33 Recently, we introduced a novel extrusion approach based on nanoporous AAO membranes as a proof-of-concept to prepare fibronectin nanofibers in physiological buffers.34
In the present study we advanced this one-step technique to assemble nanofibers from manifold biopolymers for future biomedical applications. Here, we show for the first time that various intracellular and extracellular proteins as well as polysaccharides and composites thereof can be extruded into nanofibers with reproducible dimensions.
Materials and methods
Chemicals
Phosphate buffered saline (PBS) tablets were obtained from Life Technologies. Ethanol and acetic acid were purchased from Carl Roth. G-buffer with pH 7.5 was prepared from 2.0 mM Tris-HCl (Carl Roth, Karlsruhe, Germany), 0.2 mM CaCl2 (Carl Roth), 0.2 mM Adenosine-5′-triphosphate·Na2-salt (ATP, Serva), 0.02% NaN3 (Alfa Aesar, Karlsruhe, Germany) and 0.2 mM Dithiothreitol (Serva). D-buffer at pH 6.5 contained 0.6 mM KCl (Carl Roth) and 50 mM K2HPO4 (Carl Roth). A-buffer at pH 7.4 was prepared from 1 mM KHCO3 (AppliChem, Darmstadt, Germany) and 0.02% NaN3. Tris-buffered saline solution (TBS) at pH 7.5 was prepared from 150 mM NaCl (Roth) and 50 mM Tris-HCl. All solutions were prepared with nanopure, pH adjusted water from a TKA GenPure system (TKA, Germany).Fibrinogen from human plasma was provided from Calbiochem (San Diego, CA). Collagen type I from calf skin, elastin from bovine neck ligament, laminin from Engelbreth-Holm-Swarm murine sarcoma basement membrane, chondroitin sulfate sodium salt from shark cartilage, chitosan with medium molecular weight and hyaluronic acid sodium salt from Streptococcus equi were purchased from Sigma Aldrich (Munich, Germany). Collagen, fibrinogen, hyaluronan and chondroitin sulphate were stored in PBS, elastin was diluted in 0.02 M Tris buffer at pH 8.8, laminin was dissolved in TBS, and chitosan was diluted in 1% acetic acid. Paraformaldehyde (PFA) was provided by Serva Electrophoresis GmbH (Heidelberg, Germany).
Protein purification
Fibronectin was purified from human plasma by gel filtration and affinity chromatography over a Sepharose CL-4B column (Sigma), followed by a gelatin Sepharose column from GE Healthcare (Munich, Germany). Subsequently, fibronectin was eluted by 6 M urea (Sigma) in PBS and dialyzed against PBS before use.Actin was isolated from an acetone powder of rabbit skeletal muscle in G-buffer by modifying the protocol of Spudich and Watt.35 Actin was polymerized by adding 50 mM KCl and 2 mM MgCl2 (Carl Roth). Subsequently, KCl and MgCl2 were removed by dialyzation with G-buffer, and the depolymerized actin was purified by gel filtration with a Superdex 200 column (GE Healthcare) and stored in G-buffer. According to the protocol of Margossian and Lowey we also isolated myosin II from rabbit skeletal muscle using centrifugation and salting out.36 The purified myosin was diluted in D-buffer. α-Actinin was isolated from chicken gizzard following the protocol of Craig et al.37 After extraction with 1 mM KHCO3 α-actinin was salted out with (NH4)2SO4 (Carl Roth) and purified with ion exchange chromatography over a DEAE column (GE Healthcare) and gel filtration with a Superdex 200 column. Isolated α-actinin was stored in A-buffer.
Anodic alumina membranes
Nanoporous AAO membranes with pore diameters dAAO of 70 and 450 nm were prepared by anodization in a home-built setup according to our previous paper.38 Both sides open anodic alumina membrane were obtained by removing the underlying aluminum substrate (in a solution containing 3.5 g of CuCl2·H2O (Alfa Aesar), 100 ml of HCl (37 wt%, Carl Roth), and 100 ml of H2O) followed by chemical etching of the barrier layer (0.5 M aqueous phosphoric acid (Carl Roth) at 30 °C). Commercial Whatman® Anodisc membranes with diameters of 20 and 200 nm were purchased from Sigma.Extrusion of nanofibers
For the preparation of various nanofibers we designed a customized extrusion setup (see Fig. 1). A syringe containing the feed solution was placed in the hollow cylinder of the upper part. The AAO membrane was mounted below the syringe and sealed with an O-ring. A glass substrate (Gerhard Menzel GmbH, Braunschweig, Germany) was cleaned with ethanol and nanopure water, dried with nitrogen and placed in the bottom holder directly under the AAO membrane to collect the extruded fibers. Then we manually injected the feed solution through the AAO membrane. Protein, polysaccharide and protein composite solutions were prepared in different buffers with varying concentrations according to Tables 1, 2 and 3.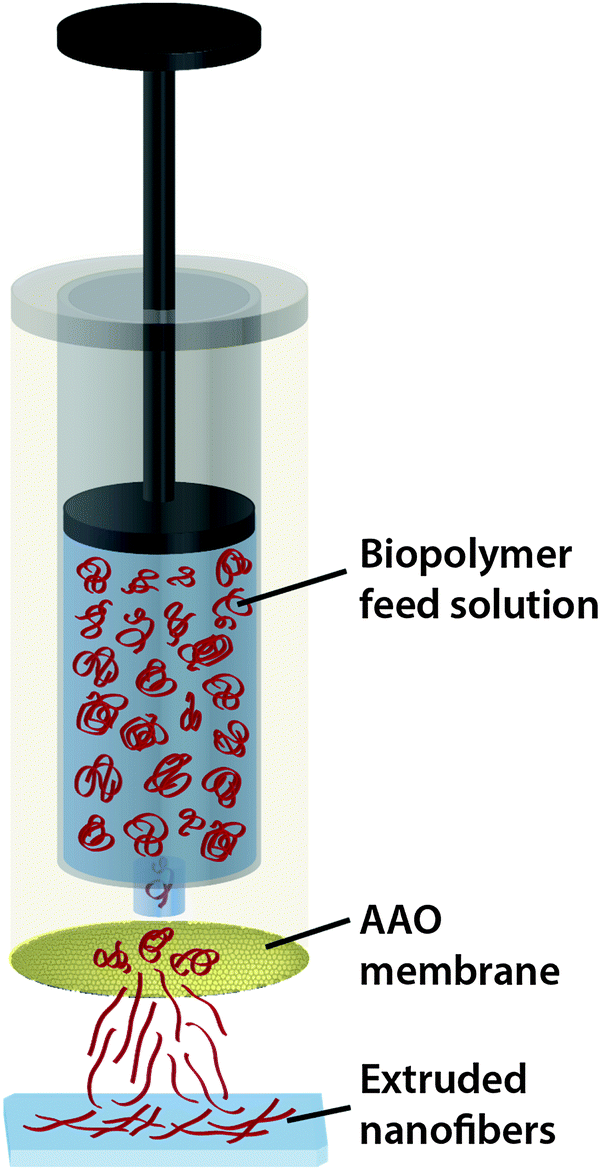 | ||
| Fig. 1 Schematic drawing of the extrusion of a biopolymer feed solution through a nanoporous AAO membrane. Extruded nanofibers are collected on a glass slide for further analysis. | ||
| Protein | Buffer | c (μg ml−1) | d AAO (nm) | d Fiber (nm) |
|---|---|---|---|---|
| Actin | G-buffer | 10 | 200 | 37 ± 8 |
| Actin | G-buffer | 100 | 450 | 64 ± 7 |
| Actin | G-buffer | 10 | 20 | 16 ± 3 |
| Myosin | D-buffer | 10 | 200 | 33 ± 7 |
| α-Actinin | A-buffer | 10 | 200 | 39 ± 5 |
| Protein | Buffer | d Fiber (nm) |
|---|---|---|
| Collagen | PBS | 29 ± 6 |
| Fibronectin | PBS | 33 ± 5 |
| Fibrinogen | PBS | 34 ± 3 |
| Elastin | Tris | 36 ± 3 |
| Laminin | TBS | 35 ± 7 |
| Blend | Buffer | d Fiber (nm) | |
|---|---|---|---|
| Protein/protein | Collagen/fibronectin | PBS | 32 ± 6 |
| Collagen/elastin | Tris/PBS | 38 ± 4 | |
| Myosin/actin | G-buffer/D-buffer | 35 ± 7 | |
| Protein/polysaccharide | Collagen/chondroitin sulphate | PBS | 33 ± 5 |
| Collagen/hyaluronan | PBS | 37 ± 5 | |
| Polysaccharide | Chondroitin sulphate | PBS | 28 ± 4 |
| Hyaluronan | PBS | 33 ± 8 |
To prepare the resulting fibers for SEM analysis they were collected in a drop of the cross-linking agent PFA in PBS (pH 7.4), which was deposited on a glass slide. After 1 hour of incubation, the fibers were rinsed with the respective buffer, followed by three rinsing steps with nanopure water and drying at room temperature. We also deposited fibers onto glass slides, which were incubated with 1% (w/v) poly-L-lysine (PLL, Sigma) in H2O for 10 minutes and subsequently dried with nitrogen.
Electron-microscopical analysis
After extrusion, the protein and composite fibers were dried and subsequently coated with an approximately 7 nm thick gold layer in a Baltec MED020 sputter system (Leica Microsystems GmbH, Wetzlar, Germany) and analyzed with scanning electron microscopy (SEM) using a Zeiss Ultra 55cv device (Zeiss, Oberkochen, Germany). All measurements were performed with an operation voltage of 3 to 5 kV. The software ImageJ (1.44p) was used to statistically analyze the average diameter and standard deviation of a minimum of 30 nanofibers per sample.Immunofluorescence staining and microscopy
Extruded composite nanofibers of collagen and fibronectin were fixed with 4% PFA in PBS for 10 min at room temperature. Following a 30 min blocking step with 0.5% BSA (Serva Electrophoresis GmbH) in PBS the nanofibers were incubated with primary antibodies in 0.1% BSA for 1 hour at room temperature. As primary antibodies we used monoclonal anti-collagen type I in mouse (Sigma, C2456) and polyclonal anti-fibronectin in rabbit (Sigma, F3648). For collagen nanofibers we employed anti-mouse secondary antibodies conjugated with Alexa 488 (Life Technologies, A21202) and for fibronectin fibers we used anti-rabbit Alexa 568 secondary antibodies (Life Technologies, A10042). The incubation time with secondary antibodies in 0.1% BSA was 30 min. Afterwards the substrates were mounted on microscope glass slides with Prolong Gold antifade reagent (Thermo Fisher). After 24 hours fluorescent imaging was carried out with an Axiovert 200 M microscope (Zeiss).Results and discussion
We have developed a novel method to fabricate nanofibers from a large variety of biopolymers under physiological conditions. Nanoporous aluminum oxide templates were used to extrude various intracellular and ECM proteins as well as polysaccharides and composites thereof into nanofibers with different hierarchical assemblies.Nanofibers of intracellular proteins
In its natural environment the intracellular protein actin assembles into filamentous structures, which are interconnected by α-actinin, thus forming a vital part of the cellular cytoskeleton. The actin-based cell motility is driven by myosin, a molecular motor, which binds to the actin filaments and converts ATP into mechanical energy.39 In order to further develop biomimetic model systems to study reconstructed cytoskeletal protein fibers in vitro we have analyzed the possibility to extrude intracellular proteins with our new extrusion approach.First, we extruded actin, α-actinin and myosin with a standard setting of 200 nm pore diameter and a protein concentration of 10 μg ml−1. This process reproducibly yielded nanofibrous assemblies for all of the above proteins with average nanofiber diameters ranging from 31 to 37 nm (see Table 1, Fig. 2A, C, E and Fig. S1, ESI†). When actin was extruded directly into a drop of PFA we also obtained fiber bundles of several micrometers in diameter, which reached a length in the millimeter range (see Fig. 2). For actin we also performed extrusions through 450 nm pores with 100 μg ml−1, which yielded a fiber diameter of 64 ± 7 nm (see Fig. 2D and Fig. S1, ESI†). When 10 μg ml−1 actin were extruded through 20 nm large pores the resulting nanofibers had a diameter of 16 ± 3 nm and were several micrometers long. These dimensions are close to natural actin filaments, which have diameters in the range of 7 nm with several micrometers in length.40 In future, extruded intracellular protein nanofibers may be a useful tool in recently developed synthetic cell systems41 or in mechanobiological studies, as they were previously performed, for instance using cantilever based techniques, micropipettes42 or optical tweezers.43 With the extrusion process the dimensions as well as the buffer conditions can be well controlled to mimic the natural environment of intracellular protein fibers more closely.
 | ||
| Fig. 2 SEM images of nanofibrous assemblies of intracellular proteins, which were extruded with different parameters and deposited onto glass slides with either PLL or PFA coating: (A) actin (c = 10 μg ml−1, dAAO = 200 nm, PLL), (B) actin fiber bundle with several millimeters length (c = 10 μg ml−1, dAAO = 200 nm, PFA), (C) α-actinin (c = 10 μg ml−1, dAAO = 200 nm, PLL), (D) myosin (c = 10 μg ml−1, dAAO = 200 nm, PFA), (E) actin (c = 100 μg ml−1, dAAO = 450 nm, PLL). | ||
ECM protein nanofibers
Using our customized setup we extruded several ECM proteins to explore the possibility to fabricate biomimetic ECM nanofibers in varying physiological buffer conditions (see Table 2). All proteins were concentrated at 10 μg ml−1 and extruded through nanopores with a diameter of 200 nm. Manual extrusion with this standard setting reproducibly yielded ECM protein nanofibers, which were deposited onto glass substrates with either PLL coating or into a drop of PFA solution (see Fig. 3A–F). The average diameter of single ECM nanofibers in both hierarchical assemblies was in the range of 29 to 36 nm (see Table 2 and Fig. S2, ESI†). This diameter range of ECM protein nanofibers conforms well to the above shown diameters of intracellular protein fibers obtained for our standard parameters. | ||
| Fig. 3 (A–F) SEM images of nanofibrous ECM protein structures, which were extruded with c = 10 μg ml−1 and dAAO = 200 nm and deposited onto glass slides with either PLL or PFA coating: (A) collagen on PLL, (B) fibronectin on PLL, (C) fibrinogen on PFA, (D) elastin on PLL, (E) laminin on PFA, (F) collagen on PLL, (G) fibronectin extruded onto PFA coated glass, (H) emerging collagen nanofibers from AAO nanopores after extrusion (c = 10 μg ml−1, dAAO = 70 nm, PFA). | ||
SEM analysis of the extruded ECM protein structures further revealed that the nanofibers also mainly assembled into two different hierarchical structures – like the intracellular proteins. We observed that proteins, which were extruded onto glass slides with PLL coating, mostly formed expanded two-dimensional nanofiber assemblies without any long-range order (see Fig. 3A, B, D and F). When the protein solutions were extruded into a drop of PFA solution, we primarily obtained highly aligned nanofiber bundles with several micrometers in diameter (see Fig. 3C, E and G). The extruded bundles of nanofibers often reached a length of several millimeters, which exceeds the previously reported length of protein nanofibers prepared by pH-driven nanofiber assembly by several orders of magnitude.19Fig. 3H shows collagen nanofibers emerging from AAO nanopores prior to the appearance of any of the two hierarchical fiber assemblies. We assume that the arrangement of protein nanofibers into either one of the observed hierarchical structures is a result of the substrate functionalization, for instance the positively charged PLL coating or a drop of the cross-linking agent PFA. Further experiments with different pH conditions to change the net charge of the protein nanofibers will be necessary to gain further insight into this assumption. Cross-linking of extruded protein nanofibers with PFA or other agents like carbodiimide or genipin might in future even increase the mechanical fiber properties, which can be beneficial for the development of novel durable biomaterials.44,45 Nevertheless, different crosslinking agents might affect the biological function of protein nanofibers.
Moreover, we analyzed for collagen and fibronectin how the diameter of extruded nanofibers depends on the concentration of the protein solution and the diameter of the AAO nanopores. Using pore diameters of 20 and 200 nm and varying the protein concentration between 10 and 1000 μg ml−1 we were able to reproducibly control the nanofiber dimensions (see Fig. 4). With concentrations below 10 μg ml−1 we could also prepare nanofibers, but the fiber yield was too low for subsequent SEM analysis. When a pore diameter of 20 nm was used for both, collagen and fibronectin, we could show that the nanofiber diameter increased from approximately 10 nm at 10 μg ml−1 to 17 and 16 nm at 1000 μg ml−1. The corresponding data points for collagen and fibronectin, which are indicated by black and red triangles in Fig. 4, are overlapping with each other. This trend indicates strongly resembling fiber diameters of both proteins for the respective extrusion parameters. With pore diameters of 200 nm the collagen and fibronectin fiber diameters increased from 29 and 32 nm at 10 μg ml−1 to 144 and 151 nm at 1000 μg ml−1, hence following the same trend for both ECM proteins. For low protein concentrations the fiber diameter stayed below the diameter of the template nanopore and reached the dimension of the pore diameter when the protein concentration was increased. These findings clearly indicate that the diameter of extruded nanofibers from different protein solutions can be tailored by adjusting the pore diameter and the protein concentration.
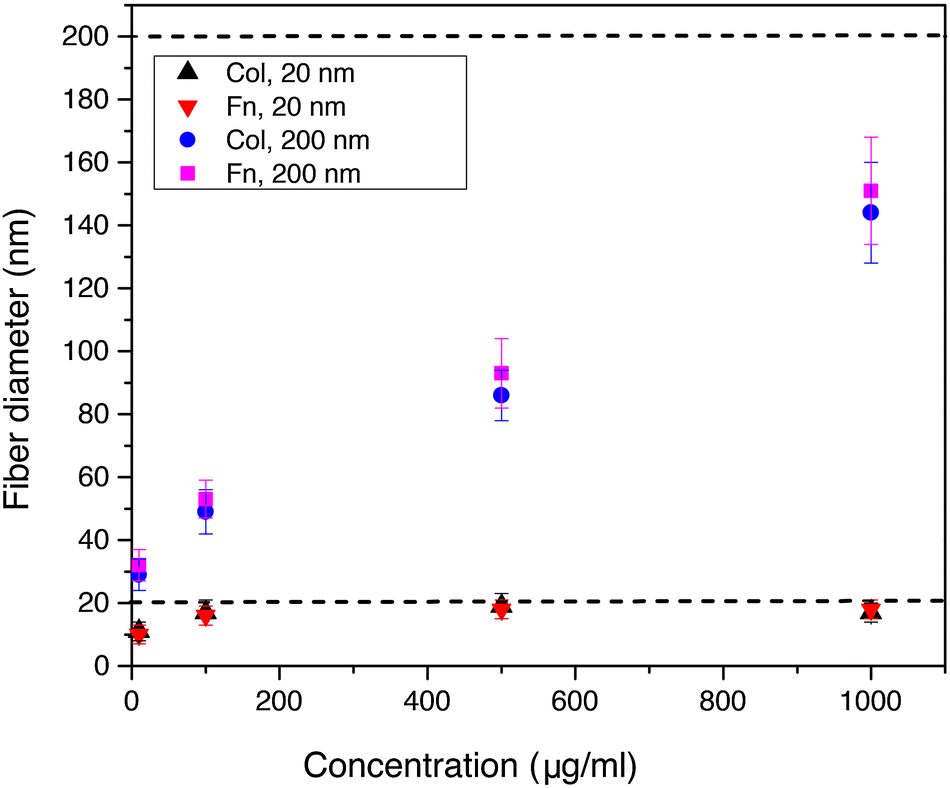 | ||
| Fig. 4 Dependence of the nanofiber diameter on the AAO pore diameter and the protein concentration measured for collagen and fibronectin. The dashed lines indicate the two different nanopore diameters of 20 nm and 200 nm, which were used for the extrusion experiments with varying concentration of the protein solution. With increasing protein concentration the nanofiber diameter approaches the size of the AAO nanopores. | ||
Considering the molecular level of the extrusion principle, we assume that single proteins get stretched when passing through the confined nanochannels of the AAO membrane as it was previously reported for protein translocation through various nanopores.46,47 In our recent FRET analysis of extruded fibronectin nanofibers we could support this assumption by showing that decreasing pore diameters yielded fibers, which were more stretched. It can be concluded that the molecules decreased their conformational entropy upon stretching in the spatially confined pores.34 Hence, we assume that temperature also plays an important role in our novel extrusion process and will focus on the underlying mechanisms in continuative studies. Furthermore, we previously found that an increase of protein concentration resulted in less stretched fibronectin nanofibers.34 This observation can be attributed to the occurrence of molecular crowding inside the pores, which diminishes the accessible volume of the protein molecules, i.e. the microscopic surface tension in the aqueous protein solution, thus counteracting the pore-induced stretching.48,49
In our novel extrusion approach the advantage of physiological buffers is combined with precise control of the nanofiber dimensions for a wide range of biopolymers, which could not be achieved with the previously presented flow processing technique, which also initiated fibrillogenesis in a physiological solution.17,18 Hence, the extrusion method holds great potential to prepare synthetic ECM systems with controlled nanotopography as future cell interfaces. Since the tuneable nanofiber diameters are correlated with varying stretching degrees within the fibers our extrusion technique might also facilitate the controlled unveiling of specific binding sites for tailored cellular interactions.
Polysaccharide and composite nanofibers
The natural ECM consists of nanofibers from various ECM proteins, which are surrounded by an aqueous solution of long-chain polysaccharides, such as hyaluronan and chondroitin sulphate. To design novel biomaterials, which mimic the natural extracellular environment more closely, we explored the possibility to prepare nanofibrous composites from different ECM proteins and polysaccharides. All solutions were extruded with a total biopolymer concentration of 10 μg ml−1 using 200 nm large pores. Thus, we prepared different nanofibrous arrangements with single fiber diameters ranging from 28 to 38 nm (see Table 3 and Fig. S3, ESI†), which is in good agreement with our previous extrusions of intracellular and ECM proteins.A blend of collagen and fibronectin was successfully extruded into micron-sized bundles of nanofibers (see Fig. 5A). When collagen and elastin were mixed and extruded we obtained a nanofibrous assembly shown in Fig. 5B, which strongly resembled the natural structure of explanted rat Achilles tendon sheaths.50 Subsequently, we blended collagen with the polysaccharides hyaluronan and chondroitin sulphate, respectively, and were able to extrude composite nanofibers, which were assembled into expanded assemblies (Fig. 5C and D). We also studied whether polysaccharides could be extruded on their own. Using hyaluronan, chitosan and chondroitin sulphate, respectively, we were able to produce pure polysaccharide nanofibers in vitro for the first time as exemplarily shown for a nanofibrous assembly of chondroitin sulphate in Fig. 5E. Like for ECM and intracellular proteins, we could also prepare pure polysaccharide fiber bundles with several millimeters in length. In future, nanofibrous composites containing different ECM proteins and/or polysaccharides could find application as tailored tissue engineering scaffolds, which closely mimic a specific tissue in vitro. Furthermore, extruding a blend of the intracellular proteins actin and myosin with our standard setting yielded two-dimensional arrangements of nanofibers (see Fig. 5F). Such assemblies of extruded intracellular protein nanofibers could be used in model systems to study biomimetic networks of filamentous and motor proteins.
 | ||
| Fig. 5 SEM images of protein composite nanofibers and polysaccharide nanofibers, which were extruded through pores with 200 nm diameter at a concentration of 10 μg ml−1: (A) fiber bundle extruded from a collagen–fibronectin blend, (B) fiber assembly extruded from a collagen–elastin blend, (C) nanofibrous assembly of an extruded collagen–hyaluronan blend, (D) extruded nanofibrous assembly of a collagen–chondroitin sulphate blend, (E) extruded chondroitin sulphate nanofibers, (F) extruded nanofibrous assembly of an actin–myosin blend. | ||
To investigate in more detail whether both components of a biopolymer blend are present in the extruded nanofibrous composite we exemplarily studied a mixture of collagen and fibronectin with immunofluorescence. The PBS feed solution in this experiment contained 500 μg ml−1 of each protein. After fixation and incubation of the extruded fibers with the corresponding primary and secondary antibodies the extruded nanofibers were analyzed via fluorescence microscopy. In Fig. 6 bundles of fluorescent composite nanofibers are shown. The fluorescent signal of Alexa 488 in Fig. 6A indicates the presence of collagen in the nanofibers, and with the Alexa 568 signal in Fig. 6B the presence of fibronectin in the extruded composite was shown. The superimposed fluorescent signals of both secondary antibodies in Fig. 6C confirm that both collagen and fibronectin were present in the extruded nanofibers.
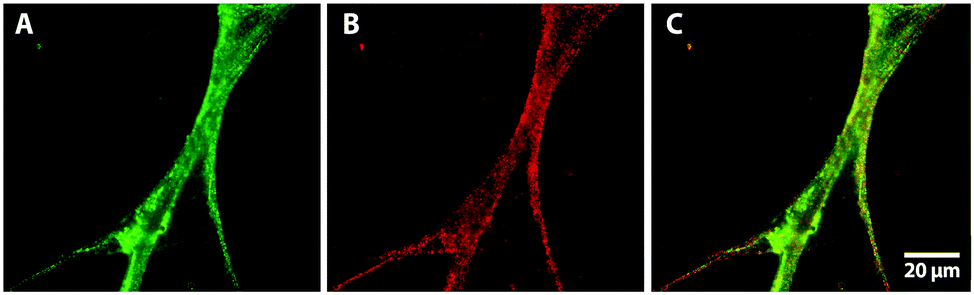 | ||
| Fig. 6 Immunofluorescence analysis of extruded nanofibers containing collagen and fibronectin confirms that both ECM proteins are present in the nanofibrous composite: (A) fluorescence signal of collagen with Alexa 488 secondary antibody, (B) fibronectin signal with Alexa 568 secondary antibody, (C) superimposed fluorescence signals of extruded collagen–fibronectin nanofibers. | ||
In future studies on nanofibrous composites it will be particularly exciting to extend the experimental scope towards super-resolution microscopy analysis to achieve a better understanding of the molecular distribution within the fibers. Such studies will also provide an important basis to tailor the biological functionality of extruded composite nanofibers for future biomedical applications.
Conclusions
We used our novel one-step extrusion approach through nanoporous membranes to prepare nanofibers of various biopolymers under physiological buffer conditions. Our results show that this technique holds great potential for the simple and reproducible fabrication of tailored protein and polysaccharide nanofibers as well as of various nanofibrous biopolymer composites. Different hierarchical nanofiber assemblies of ECM and intracellular proteins or polysaccharides could be prepared. Most importantly, we were able to control the diameter of our extruded nanofibers by adjusting the nanopore size of the extrusion membrane and the concentration of the biopolymer feed solution. Furthermore, with immunofluorescence analysis of blended collagen–fibronectin nanofibers we could show for the first time that both proteins were present in the extruded biopolymer composites.In future studies we will focus on controlling the hierarchical nanofiber assembly, for instance by regulating pH and flow conditions of the feed solution or by using different nanopore materials and substrate functionalizations. The next important step towards a new class of nanofibrous biomaterials will be to analyze the biological functionality of extruded nanofibers in selected cell culture test systems.
Acknowledgements
The authors wish to thank G. Gao and S. Young for their support in setting up the extrusion setup. The research leading to these results has received funding by the Max Planck Society, the European Research Council under the European Union's Seventh Framework Programme (FP/2007–2013)/ERC Grant Agreement no. 294852 and the Emmy Noether program of the German Research Council (BR 5043/1-1).References
- D. B. Khadka and D. T. Haynie, Nanomedicine, 2012, 8, 1242–1262 CAS.
- T. Scheibel, Curr. Opin. Biotechnol., 2005, 16, 427–433 CrossRef CAS PubMed.
- R. N. Palchesko, Y. Sun, L. Zhang, J. M. Szymanski, Q. Jallerat and A. W. Feinberg, Springer Handbook of Nanomaterials, Springer, Berlin, Heidelberg, 2013, pp. 977–1010 Search PubMed.
- A. Curtis and M. Riehle, Phys. Med. Biol., 2001, 46, R47–R65 CrossRef CAS PubMed.
- M. J. P. Biggs, R. G. Richards and M. J. Dalby, Nanomedicine, 2010, 6, 619–633 CAS.
- S. G. Kumbar, R. James, S. P. Nukavarapu and C. T. Laurencin, Biomed. Mater., 2008, 3, 034002 CrossRef CAS PubMed.
- C. P. Barnes, S. A. Sell, E. D. Boland, D. G. Simpson and G. L. Bowlin, Adv. Drug Delivery Rev., 2007, 59, 1413–1433 CrossRef CAS PubMed.
- C. A. E. Hauser and S. Zhang, Chem. Soc. Rev., 2010, 39, 2780–2790 RSC.
- Z. Ma, M. Kotaki, R. Inai and S. Ramakrishna, Tissue Eng., 2005, 11, 101–109 CrossRef CAS PubMed.
- R. Vasita and D. S. Katti, Int. J. Nanomed., 2006, 1, 15–30 CrossRef CAS PubMed.
- K. Jayaraman, M. Kotaki, Y. Zhang, X. Mo and S. Ramakrishna, J. Nanosci. Nanotechnol., 2004, 4, 1–2 CrossRef.
- Q. P. Pham, U. Sharma and A. G. Mikos, Tissue Eng., 2006, 12, 1197–1211 CrossRef CAS PubMed.
- L. A. Smith and P. X. Ma, Colloids Surf., B, 2004, 39, 125–131 CrossRef CAS PubMed.
- R. Nayak, R. Padhye, I. Kyratzis, Y. B. Truong and L. Arnold, Text. Res. J., 2012, 82, 129–147 CrossRef CAS.
- H. S. Yoo, T. G. Kim and T. G. Park, Adv. Drug Delivery Rev., 2009, 61, 1033–1042 CrossRef CAS PubMed.
- J. E. Plowman, S. Deb-Choudhury and J. M. Dyer, Protein Nanotechnology, Humana Press, Totowa, NJ, 2013, vol. 996, pp. 61–76 Search PubMed.
- E. S. Lai, C. M. Anderson and G. G. Fuller, Acta Biomater., 2011, 7, 2448–2456 CrossRef CAS PubMed.
- E. S. Lai, N. F. Huang, J. P. Cooke and G. G. Fuller, Regener. Med., 2012, 7, 649–661 CrossRef CAS PubMed.
- M. Maas, P. Guo, M. Keeney, F. Yang, T. M. Hsu, G. G. Fuller, C. R. Martin and R. N. Zare, Nano Lett., 2011, 11, 1383–1388 CrossRef CAS PubMed.
- A. Thormann, N. Teuscher, M. Pfannmöller, U. Rothe and A. Heilmann, Small, 2007, 3, 1032–1040 CrossRef CAS PubMed.
- D. Brüggemann, J. Nanomater., 2013, 2013, 1–18 CrossRef.
- N. Ferraz, J. Carlsson, J. Hong and M. K. Ott, J. Mater. Sci.: Mater. Med., 2008, 19, 3115–3121 CrossRef CAS PubMed.
- E. Gultepe, D. Nagesha, S. Sridhar and M. Amiji, Adv. Drug Delivery Rev., 2010, 62, 305–315 CrossRef CAS PubMed.
- M. Wesche, M. Hüske, A. Yakushenko, D. Brüggemann, D. Mayer, A. Offenhäusser and B. Wolfrum, Nanotechnology, 2012, 23, 495303–495310 CrossRef PubMed.
- G. E. J. Poinern, N. Ali and D. Fawcett, Materials, 2011, 4, 487–526 CrossRef.
- J. W. Diggle, T. C. Downie and C. W. Goulding, Chem. Rev., 1969, 69, 365–405 CrossRef CAS.
- A. Huczko, Appl. Phys. A: Mater. Sci. Process., 2000, 70, 365–376 CrossRef CAS.
- J. C. Hulteen and C. R. Martin, J. Mater. Chem., 1997, 7, 1075–1087 RSC.
- G. Schmid, J. Mater. Chem., 2002, 12, 1231–1238 RSC.
- F. Schröper, D. Brüggemann, Y. Mourzina, B. Wolfrum, A. Offenhäusser and D. Mayer, Electrochim. Acta, 2008, 53, 6265–6272 CrossRef.
- D. Brüggemann, K. E. Michael, B. Wolfrum and A. Offenhäusser, Int. J. Nano Biomater., 2012, 4, 108–127 CrossRef.
- D. Brüggemann, B. Wolfrum, V. Maybeck, Y. Mourzina, M. Jansen and A. Offenhausser, Nanotechnology, 2011, 22, 265104–265110 CrossRef PubMed.
- D. Weber, Y. Mourzina, D. Brüggemann and A. Offenhausser, J. Nanosci. Nanotechnol., 2011, 11, 1293–1296 CrossRef CAS PubMed.
- M. Raoufi, T. Das, I. Schoen, V. Vogel, D. Brüggemann and J. P. Spatz, Nano Lett., 2015, 15, 6357–6364 CrossRef CAS PubMed.
- J. A. Spudich and S. Watt, Int. J. Nano Biomater., 1971, 246, 4866–4871 CAS.
- S. S. Margossian and S. Lowey, Methods Enzymol., 1982, 85, 55–71 CAS.
- S. W. Craig, C. L. Lancashire and J. A. Cooper, Methods Enzymol., 1982, 85, 316–321 CAS.
- M. Raoufi, D. Tranchida and H. Schönherr, Langmuir, 2012, 28, 10091–10096 CrossRef CAS PubMed.
- D. Pantaloni, C. Le Clainche and M.-F. Carlier, Science, 2001, 292, 1502–1506 CrossRef CAS PubMed.
- P. B. Moore, H. E. Huxley and D. J. DeRosier, J. Mol. Biol., 1970, 50, 279–295 CrossRef CAS PubMed.
- D. Brüggemann, J. P. Frohnmayer and J. P. Spatz, Beilstein J. Nanotechnol., 2014, 5, 1193–1202 CrossRef PubMed.
- A. Kishino and T. Yanagida, Nature, 1988, 334, 74–76 CrossRef CAS PubMed.
- M. Streichfuss, F. Erbs, K. Uhrig, R. Kurre, A. E. M. Clemen, C. H. J. Boehm, T. Haraszti and J. P. Spatz, Nano Lett., 2011, 11, 3676–3680 CrossRef CAS PubMed.
- A. Sionkowska, Prog. Polym. Sci., 2011, 36, 1254–1276 CrossRef CAS.
- N. H. C. S. Silva, C. Vilela, I. M. Marrucho, C. S. R. Freire, C. Pascoal Neto and A. J. D. Silvestre, J. Mater. Chem. B, 2014, 2, 3715–3740 RSC.
- B. Cressiot, A. Oukhaled, L. Bacri and J. Pelta, J. Bionanosci., 2014, 4, 111–118 CrossRef.
- D. Rodriguez-Larrea and H. Bayley, Nat. Nanotechnol., 2013, 8, 288–295 CrossRef CAS PubMed.
- B. Martínez Haya and M. C. Gordillo, Macromol. Theory Simul., 2005, 14, 421–427 CrossRef.
- A. Nicholls, K. A. Sharp and B. Honig, Proteins: Struct., Funct., Bioinf., 1991, 11, 281–296 CrossRef CAS PubMed.
- M. Franchi, A. Trire, M. Quaranta, E. Orsini and V. Ottani, Sci. World J., 2007, 7, 404–420 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ib00045b |
| ‡ Present address: Helmholtz-Zentrum Geesthacht, Zentrum für Material- und Küstenforschung GmbH, Max-Planck-Straße 1, 21502 Geesthacht, Germany. |
| This journal is © The Royal Society of Chemistry 2016 |