DOI:
10.1039/C5GC02187A
(Tutorial Review)
Green Chem., 2016,
18, 76-104
Chemical and enzymatic modification of sophorolipids
Received
14th September 2015
, Accepted 26th October 2015
First published on 27th October 2015
Abstract
The significance of renewable resources within the chemical industry is constantly increasing. In the pursuit of sustainability, they serve as alternatives for fossil resources whose supply is limited and who have a major impact on the environment. Due to their complex structure and divergent biological activities, sophorolipids are interesting renewable resources. Unfortunately, industrial applications of natural sophorolipids are limited because of their high production cost. Therefore, chemical and enzymatic modifications provide an excellent tool to shift the application area of sophorolipids to high-added value sectors, in particular for the pharmaceutical sector. This review will give an overview of the modifications performed so far and their possible applications.
 Elisabeth Delbeke | Elisabeth Delbeke obtained her master's degree in Bioscience Engineering, Chemistry and Bioprocess Technology in 2012 at Ghent University. She is currently pursuing a PhD study at the Faculty of Bioscience Engineering, Ghent University supervised by Prof. Christian V. Stevens (SynBioC, Faculty of Bioscience Engineering) and Prof. Kevin Van Geem (LCT, Faculty of Engineering and Architecture). Her research focusses on the chemical modification of renewable resources, in particular sophorolipid biosurfactants, and evaluation of the biological properties of the new derivatives. |
 Marine Movsisyan | Marine Movsisyan (1990) was granted a Master's degree in Bioscience Engineering at Ghent University, Belgium, in 2013. She subsequently joined the SynBioC research group to pursue a PhD degree under the guidance of Prof. Christian V. Stevens at the Department of Sustainable Organic Chemistry and Technology. Marine's research interests lie in the synthesis of biobased building blocks using microreactor technology in corporation with an industrial partner. |
 Kevin Van Geem | Kevin Van Geem (associate professor) is member of the Laboratory for Chemical Technology of Ghent University. Thermochemical reaction engineering in general and in particular the transition from fossil to renewable resources are his main research interests. He is a former Fulbright Research Scholar of MIT and directs the Pilot plant for steam cracking and pyrolysis. He is the author of more than hundred scientific publications. He is involved in on-line and off-line analysis of complex petrochemical and biochemical samples using comprehensive two-dimensional gas chromatography. Direct experimental scale-up, detailed kinetic modeling, process modeling (Aspen, ProII), and the role of additives belong to his expertise. |
 Christian V. Stevens | Christian V. Stevens (1965) is full professor at the Department of Sustainable Organic Chemistry and Technology at the Faculty of Bioscience Engineering (Ghent University, Belgium). He graduated in 1988 and obtained a PhD in 1992 at Ghent University working with Prof. Norbert De Kimpe. He then performed post-doctoral work at the University of Florida with Prof. Alan Katritzky in 1992–93. In 2000, he became associate professor and full professor in 2008. C. Stevens published over 220 international peer reviewed papers and 14 patents. His research interest focuses on synthetic heterocyclic chemistry for agrochemical and medicinal applications, on chemical modification of renewable resources and on the use of flow chemistry to scale-up organic reactions that are difficult to scale-up in batch. |
Introduction
Sophorolipids are one of the most important classes of glycolipid biosurfactants. They consist of sophorose as a hydrophilic carbohydrate head and a hydrophobic lipid tail, which gives them an amphiphilic character. Sophorolipids lower the surface tension of water from 72.8 to 30–40 mN m−1 and have a critical micelle concentration of 40–100 mg L−1.1 They possess self-assembly properties due to the formation of nanostructures with supramolecular chirality.2 Their emulsifying properties can be applied for the recovery of oil and hydrocarbons and for the decontamination of soil and water.3 The most common natural derivatives are diacetylated sophorolipid lactone 1 and sophorolipid acid 2 (Scheme 1).
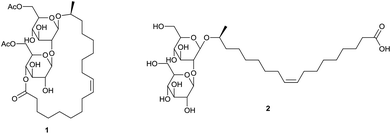 |
| | Scheme 1 Diacetylated sophorolipid lactone 1 and sophorolipid acid 2. | |
Interest in sophorolipids exponentially increased over the last 20 years, reaching over 200 citations in 2013 and 2014 (Fig. 1). Several reviews have been written on sophorolipids lately, with their focus on the microbial production of sophorolipids and application of these natural compounds.1,4 To date, natural sophorolipids are commercialized by different companies world-wide. The Japanese company Saraya and the Belgian company Ecover apply sophorolipids in their laundry, dishwashing and cleaning products while the former French company Soliance, now part of the Swiss company Givaudan, and the Korean company MG Intobio offer respectively the products Sopholiance S and Sopholine for cosmetic appplications.5 Other companies working on the industrial production of sophorolipids are Evonik, DSM and Croda.5,6 Despite these positive examples, application of natural sophorolipids is limited because of their high production cost. Van Bogaert et al. reported a production cost of 2 to 5 € per kg, but an up-to-date production price is not communicated by the companies.1
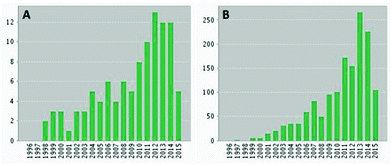 |
| | Fig. 1 Number of publications (A) and citations (B) per year on sophorolipids. | |
Modification of natural sophorolipids creates opportunities for their application in other high-added value sectors, in particular the pharmaceutical sector. Multiple modifications of natural sophorolipids have already been described, but were never combined in one review. In this review, it is aimed to provide an overview of all the chemical and enzymatic modifications performed on sophorolipids in the development of innovative high-added value compounds, in particular for the pharmaceutical sector.
Production of natural sophorolipids
Sophorolipids are produced by a selected number of yeast species.1,4a,7 They were first described by Gorin et al. in the early 1960s as an extracellular glycolipid produced by Candida apicola, formerly known as Torulopsis apicola. Other producing strains comprise Rhodotorula bogoriensis (formerly known as Candida bogoriensis), Starmerella bombicola (formerly known as Torulopsis bombicola and Candida bombicola) and Wickerhamiella domericqiae. Starmerella bombicola is the best known and preferred producing strain with a production yield of more than 400 g L−1. The sophorolipid biosynthesis starts with glucose as the hydrophilic carbon source and fatty acids, fatty acid methyl esters, triglycerides or alkanes as the hydrophobic carbon source (Scheme 2). All hydrophobic carbon sources are converted into fatty acids, which contain in general 16 or 18 carbon atoms with one or more double bonds. When no hydrophobic carbon source is present in the fermentation medium, fatty acids can be formed de novo via the acetyl-CoA pathway. The fatty acids are subsequently oxidized at the terminal (ω) or subterminal (ω − 1) position by a cytochrome P450 monooxygenase enzyme. The resulting hydroxylated fatty acids are β-glycosidically linked to a first glucose molecule at the 1′-position by a glycosyltransferase I. A second glucose molecule is linked to the 2′-position of the first one by a glycosyltransferase II, yielding sophorolipid acid 2. Subsequently, the carbohydrate head can be acetylated at the 6′- and/or 6′′-position by an acetyl-CoA dependent acetyl transferase. After excretion in the fermentation medium, lactonization can occur at the 4′′-position by an extracellular esterase.8 The major microbial product is the diacetylated, ω − 1 hydroxylated, mono-unsaturated sophorolipid lactone 1.
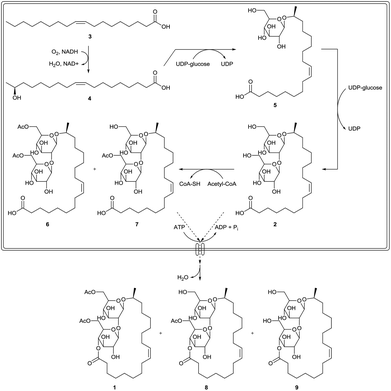 |
| | Scheme 2 Biosynthesis of sophorolipid derivatives.1 | |
Most sophorolipid fermentations are run at a temperature between 25 to 30 °C and a pH of 3.5. Sophorolipid synthesis starts in the stationary phase under nitrogen-limiting conditions and is very dependent on good aeration conditions. The highest yields are obtained when both a hydrophilic and a hydrophobic carbon source are present, but fermentation on only one type of carbon source is possible. After the fermentation, sophorolipids are extracted with ethyl acetate and washed with hexane to remove residual fatty acids. On larger scale, physical separation methods such as centrifugation are applied for the isolation. Lactonic sophorolipids were also purified by crystallization.9
The biggest bottleneck for the sophorolipid production process is the variability in the composition of the fermentation product. To optimize chemical modification procedures, one single compound is preferred as starting compound. If this is not the case, problems can arise along the modification pathway. For example, in the synthesis of sophorolipid alkyl esters via alkaline hydrolysis of di-acetylated sophorolipid lactone, the products are precipitated in water after reaction. In some cases, precipitation does not occur, possibly due to the presence of a considerable fraction of sophorolipid acids or residual fatty acids. Therefore, it is of utmost importance to have access to selective organisms for the fermentation process or suitable purification procedures to obtain the desired sophorolipid compounds in high purity.5,10
Physiological activity
Biodegradability and toxicity
The biodegradability of sophorolipids was determined via biological oxygen demand studies and the manometric respirometry method (OECD 301C and 301F method).4a,11 They can be classified as readily biodegradable since 61% of the product was degraded after only 8 days of cultivation. The aquatic toxicity is subdivided in acute and chronic toxicity. The acute toxicity is determined as the EC50 after 48 h on Daphnia magna, Tetrahymena terhmophila and Pseudokirchneriella subcapitata and proved to be ten times higher as compared to conventional surfactants.4a,12 The chronic toxicity was determined via a reproduction test on Daphnia magna. The no-observed-effect concentration (NOEC) was 11.3 mg L−1, which is also ten times higher as compared to conventional surfactants.12 The cytotoxicity was determined by the MTT method with human epidermal keratinocytes, displaying a lower cytoxicity for sophorolipids than for surfactin, arthrofactin, pluronic L31, sodium dodecyl sulfate and polyoxyethylene lauryl ether.11 The cytotoxicity was also evaluated on normal Chang liver cells, displaying an IC50 value of 81.9 mg mL−1 for crude sophorolipids and a higher cell viability for acidic sophorolipids compared to lactonic ones.13 Sophorolipids are not irritating to skin and eyes, have an oral safety level which is greater than or equal to 5 mL per kg body weight and cause no allergic reactions.14
Dermatological activity
Sophorolipids inhibit elastase activity, the enzyme which is responsible for the degradation of elastin fibres, at a concentration of 5% (w/v) and radical effects towards the hydroxy-radical at a concentration of 0.083% (w/v).14 Diacetylated sophorolipid lactones stimulate the metabolism of skin dermis fibroblast cells and hereby the in vitro neosynthesis of collagen.15 Therefore, they possess restructuring and tissue repairing activity which is higher as compared to crude sophorolipid mixtures. Besides moisturizing and skin-conditioning properties, sophorolipid lactones proved active to eliminate dandruff.16 Moreover, sophorolipids proved to possess desquamating, depigmenting and melanogenesis inhibiting activity.17 The desquamation is achieved by detachment of the corneocytes and offers opportunities for the use of sophorolipids against acne and as an anti-wrinkle product. Sophorolipids induce the secretion of leptin by adipocytes, hereby inducing the lipolysis of adipocytes and reducing the subcutaneous deposition of fat.18
Antimicrobial activity
Diacetylated sophorolipid lactones synthesized by Torulopsis bombicola inhibited the growth of Candida, Pichia, Debaryomyces, Saccharomycopsis and Lodderomyces yeasts on n-alkanes.19 Natural sophorolipids also inhibited the growth of Candida albicans, Candida Antarctica and Candida tropicalis with respectively 30, 32 and 25% at a concentration of 5 mg mL−1, while the inhibition with sophorolipid acids was respectively 24, 45 and 40%.20 Kulakovskaya et al. determined the minimum inhibitory concentrations (MIC) of sophorolipids against Filobasidiella neoformans, Candida tropicalis and Candida albicans as respectively 1, 15 and 15 mg mL−1.21 The antibacterial activity of sophorolipids against Rhodococcus erythropolis, Bacillus subtilis, Staphylococcus epidermidis, Streptococcus agalactiae, Moraxella sp., Pseudomonas putida, Enterobacter aerogenes and Escherichia coli was evaluated, displaying a higher activity against Gram-positive bacteria than Gram-negative bacteria.22 The activity was expressed as minimum lethal doses (MLD50) and was 98 μg mL−1 for R. erythropolis, B. subtilis, S. agalactiae and Moraxella sp., and >6250 μg mL−1 for S. epidermis, P. putida, E. aerogenes and E. coli. Kulakovskaya et al. determined the MIC values of sophorolipis against E. coli, Streptococcus salivarius and Micrococcus luteus as respectively 32, 23 and 23 mg mL−1.21 More antimicrobial activities against other species are described in a patent by Gross and Shah.23 Overall, sophorolipid lactones perform better than sophorolipid acids, with the lowest MIC values of 10 μg mL−1 for sophorolipid lactones against Bacillus subtilis and Rhodococcus rhodochrous. Sleiman et al. evaluated MIC values of different sophorolipid derivatives in sucrose and ethanol vehicles against Escherichia coli and Staphylococcus aureus.24 No significant inhibitory activity was observed for most derivatives at a concentration of 512 μg mL−1 and against S. aureus at a concentration of 218 μg mL−1 for two derivatives in an ethanol vehicle. The antibacterial activity of lauryl alcohol based sophorolipids was also evaluated against E. coli and S. aureus, respectively displaying a zero percent survival at 3000 μg mL−1 after 2 h and 600 μg mL−1 after 4 h.25 Both crude and acidic sophorolipids inhibited the growth of Cupriavidus necator and Bacillus subtilis at a concentration of 5% (v/v).26 They also disrupted biofilms of B. subtilis and a mixed culture of B. subtilis and S. aureus at a concentration of 5% (v/v). Synergistic effects were observed for administration of sophorolipids with the antibiotics tetracycline and cefaclor against respectively S. aureus and E. coli.27 Sophorolipids inhibited the motility of the harmful algae Alexandrium tamarense, Heterosigma akashiwo and Cochlodinium polykrikoides at concentrations of 10–20 μg mL−1.28 At these concentrations, sophorolipids have low adverse effects on the non-harmful microalgae Platymonas helgolandica var. tsingtaoensis, Isochrysis galbana and Nitzschia closterium f. minutissima.29 The EC50 for the zooplankton Strombidium sp., Calanus sinicus and Neomysis awatschensis was respectively 20, 50 and 150 μg mL−1 after 96 h. The EC50 for the zooplankton Artemia salina was 600 μg mL−1 after 24 h. The fish Lateolabrax japonicas and Paralichthys olivaceus displayed an EC50 of respectively 60 and 110 μg mL−1 and the relative clearance rate of the mussel Mytilus edulis decreased to 80% at a sophorolipid concentration of 20 μg mL−1. Synergistic effects were observed for the combination of sophorolipids and loess on the motility inhibition and removal of the harmful algae C. polykrikoides and A. tamarense.30 It can be concluded that sophorolipids display no significant antibacterial activity against clinically relevant bacteria, but are effective in the inhibition of harmful algae in concentrations which are not detrimental to most of the tested organisms.
Anticancer activity
Sophorolipids induced differentiation of human acute promyelocytic leukemia cell line HL60.31 At a concentration of 10 μg mL−1, the proliferation of the HL60 cells and the protein kinase C activity was inhibited after 2 days and differentiation into monocytes took place. For two other leukemic cell lines, the human myelogenous leukemia cell line K562 and the human basophilic leukemia cell line KU812, differentiation into megakaryocytes was also induced at a sophorolipid concentration of 15 μg mL−1. Chen et al. evaluated the anticancer activity of diacetylated sophorolipid lactone on liver cancer cell line H7402, lung cancer cell line A549 and leukemia cell lines HL60 and K562 via an MTT assay.32 The cell proliferation of all four cell lines was inhibited at concentrations ranging from 0 to 62.5 μg mL−1 after 2 days with a 0% cell viability at concentrations exceeding 62.5 μg mL−1. The mechanism of anticancer activity on the liver cancer cell line H7402 was identified as apoptosis.33 The condensation of chromatin, nuclear fragmentation and appearance of apoptotic bodies was observed. At a sophorolipid concentration of 50 μg mL−1, cell viability of both liver cancer cell line H7402 and lung cancer cell line A549 was completely inhibited while only a little decrease was observed for the normal liver cell lines HL7702 and Chang liver. The anticancer activity of diacetylated sophorolipid lactone and sophorolipid acid against human pancreatic cancer cells was also evaluated.34 A decreasing cytotoxicity of respectively 40.3 to 3.4% and 49 to 0% was observed with increasing concentration from 500 to 2000 μg mL−1. No cytotoxicity was observed against healthy peripheral blood mononuclear cells. Shao et al. described the anticancer activity of different sophorolipid derivatives against the esophageal cancer cell lines KYSE109 and KYSE450.35 Diacetylated sophorolipid lactone displayed the highest activity with complete inhibition of both cell lines at a concentration of 30 μg mL−1. The anticancer activity depended on the acetylation and unsaturation degree of the derivatives. No inhibition was observed for acidic sophorolipids. Evaluation of the anticancer activity on MDA-MB-321 breast cancer cell line demonstrated a higher activity for sophorolipid lactones compared to sophorolipid acids, with IC50 values of respectively 15–20 and 80 μg mL−1.36 Sophorolipids also induce differentiation in LN-229 glioma cell lines.37 Morphological changes were detected at concentrations of 10 μg mL−1 and 400 ng mL−1 for respectively oleic acid and linolenic acid based sophorolipids.
Immunoregulatory activity
Sophorolipids possess a pro-inflammatory activity by activating macrophages to induce the release of cytokines.17 They also promote wound healing due to the proliferative effect of these cytokines on the fibroblasts, the phagocytosis of bacteria, cells and cellular fragments which could obstruct the wound by macrophages and their fibrinolytic activity. Sophorolipids can reduce septic shock related mortality by inhibition of the nitrogen oxide production of macrophages and modulation of the inflammatory response.38 Experiments were performed in in vivo rat models with intravenous and intraperitoneal administration of Nembutal to induce septic shock. The survival rate after 36 h with and without addition of 5 mg kg−1 natural sophorolipid mixture was respectively 81.8 and 47.8% for the intervenous administration and 67 and 53% for the intraperitoneal administration. Macrophages which were in vitro cultured with lipopolysaccharides showed reduced nitrogen oxide production and induced expression of several cytokines. The viability of these macrophages also increased from 56 to 66% and from 23 to 36% after respectively 36 and 48 h.39 Moreover, sophorolipids at a concentration of 100 μg mL−1 decreased the IgE production in U266 myeloma cells with 63%.40 The effect of dosing was evaluated in the in vivo rat models.41 When a single dose was administered, survival increased with 28 and 42% after respectively 24 and 72 h while survival increased with 39 and 26% for sequential dosing after respectively 24 and 72 h. Increased mortality was observed for treatment with purified diacetylated sophorolipid lactones. Sophorolipids also reduced asthma severity in an in vivo asthma model with mice, which was demonstrated on the basis of decreased leukocytic infiltrate in the lungs and decreased levels of ovalbumin specific IgE in the bronchoalveolar lavage fluid.42
Spermicidal and antiviral activity
Sophorolipids are active as spermicidal and antiviral agents.43 Natural mixture sophorolipids and sophorolipid lactones displayed a minimum effective concentration of respectively 800 and 1000 μg mL−1 after 30 seconds of incubation. Sophorolipids at a concentration of 300 μg mL−1 immobilized spermatozoa completely and irreversibly after 2 minutes. Immobilization occurred faster than death and the mechanism of action is most likely membrane perturbation and disruption. Sophorolipids also displayed some degree of anti-HIV activity, with sophorolipid acids being more potent than sophorolipid lactones. Natural mixture sophorolipids and sophorolipid lactones exerted high cytotoxicity against human vaginal cells with a 50% effective concentration of around 15 μg mL−1, while sophorolipid acids displayed a 50% effective concentration higher than 100 μg mL−1. Natural mixture sophorolipids and sophorolipid lactones also induced the production of proinflammatory cytokines by vaginal epithelial cells. Sophorolipids also displayed anti-herpes virus activity for which the Epstein-Bar virus was used as a model organism.44 The EC50 values for diacetylated sophorolipid lactone and sophorolipid acid were respectively 25.8 and 49.2 μM.
Modifications in the context of structural characterization
The first modifications of sophorolipids produced by Torulopsis magnoliae are described by Gorin et al. in the context of the characterization of this class of biosurfactants (Scheme 3).45 In a first step, the crude sophorolipid product was transformed into unsaturated sophorolipid acid 2 and saturated sophorolipid acid 10 through removal of the acetyl groups with sodium methoxide. Acid methanolysis of sophorolipid acids 2 and 10 resulted in the cleavage of fatty acid methyl esters 11 and 12 which were separated via fractional crystallization. The saturated methyl ester 12 was oxidized with chromium trioxide into 17-oxo-stearic acid 13, which enabled the determination of the hydroxyl position. The unsaturated methyl ester 11 was oxidized with potassium permanganate and sodium periodate into azelaic acid 14 and 8-hydroxy-pelargonic acid 15, which enabled the determination of the double bond. Unsaturated sophorolipid acid 2, on the other hand, was oxidized with performic acid followed by alkaline hydrolysis into the threo-9,10-dihydroxy sophorolipid 16. Subsequent oxidation with lead(IV) acetate and reduction with sodium borohydride yielded sophorolipid alcohol 17. Partial acid hydrolysis furnished a mixture of 1,8-L-nonanediol 18, glucose 19, 1,8-L-nonanediol-β-D-glucopyranoside 20 and sophorose 21. The latter two were isolated by chromatography. Sophorolipid alcohol 17 and saturated sophorolipid acid 10 were methylated into respectively sophorolipid ether 22 and sophorolipid methyl ester 23. Both compounds were hydrolyzed into 2,3,4,6-tetra-O-methyl-D-glucose 24 and 3,4,6-tri-O-methyl-D-glucose 25 which confirmed the 1,2-glycosidic link between the two glucose units.
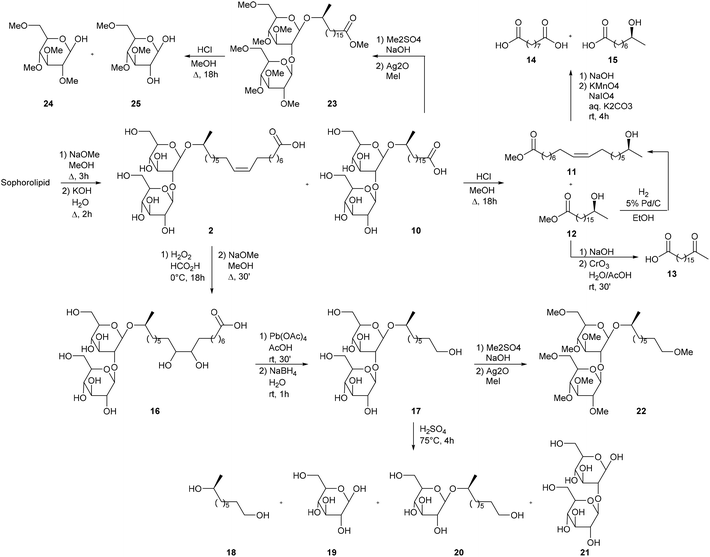 |
| | Scheme 3 Sophorolipid modifications in the context of the structure determination.45 | |
Similar modifications were performed by Tulloch et al. for the characterization of diacetylated sophorolipid 26 which is produced by Candida bogoriensis (Scheme 4).46 In a first step, sophorolipid 26 was transformed into diacetylated sophorolipid methyl ester 27 with diazomethane. Alkaline hydrolysis furnished the corresponding methyl ester 28. Acid methanolysis of sophorolipid acid 26 resulted in the cleavage of fatty acid methyl ester 29. Oxidation with chromium oxide at room temperature or 100 °C yielded respectively methyl 13-oxodocosanoate 30 or dodecanedioic acid 31 and tridecanedioic acid 32, which enabled the determination of the hydroxyl position. Diacetylated sophorolipid 17 was acetylated with acetic anhydride towards the peracetylated sophorolipid 33. Bromination towards α-acetobromosophorose 34 was followed by the synthesis of β-octaacetyl sophorose 35. Diacetylated sophorolipid 26 was further methylated towards sophorolipid methyl ester 36 and subsequently hydrolyzed into 2,3,4,6-tetra-O-methyl-D-glucose 24 and 3,4,6-tri-O-methyl-D-glucose 25 which confirmed the 1,2-glycosidic link between the two glucose units.
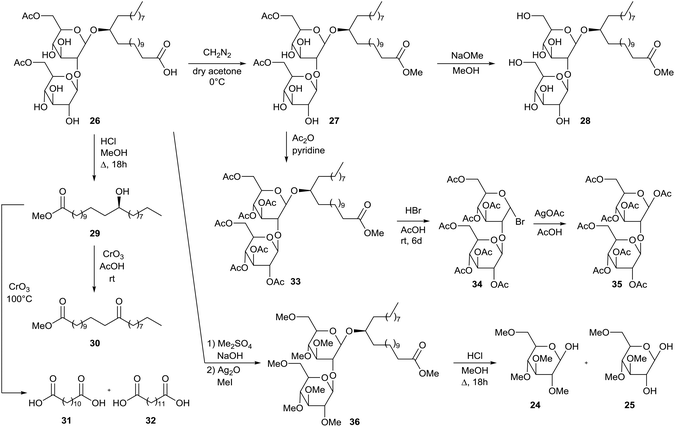 |
| | Scheme 4 Modifications for the structural determination of diacetylated sophorolipid 26.46 | |
Modifications starting from diacetylated sophorolipid lactone 37 are described by Tulloch et al. (Scheme 5).47 Diacetylated sophorolipid lactone 37 proved to be the major fermentation product and was obtained in pure form after crystallization and hydrogenation of the fermentation product followed by subsequent chromatography. Tetraphenylurethane sophorolipid 38 was synthesized via reaction with phenyl isocyanate. Acetylation of diacetylated sophorolipid lactone 37 was performed with acetic anhydride towards the hexaacetylated sophorolipid lactone 39 followed by brominolysis towards α-bromosophorose hexaacetate derivative 40. Subsequent treatment with silver acetate gave the corresponding β-bromosophorose hexaacetate derivative 41. The α-bromosophorose hexaacetate 40 was converted into β-methoxysophorose hexaacetate 42 followed by deacetylation towards β-methoxysophorose 43 and acetylation towards β-methoxysophorose heptaacetate 44. The latter compound was also prepared via brominolysis of β-sophorose octaacetate 35 towards β-bromosophorose heptaacetate 34 and subsequent reaction with methanol in the presence of silver carbonate. A third method for the synthesis of β-methoxysophorose heptaacetate 44 comprises the Koenigs–Knorr reaction of 3,4,6-tri-O-acetyl-β-D-glucopyranoside 45 and α-acetobromoglucose 46 in the presence of mercuric cyanide in nitromethane.
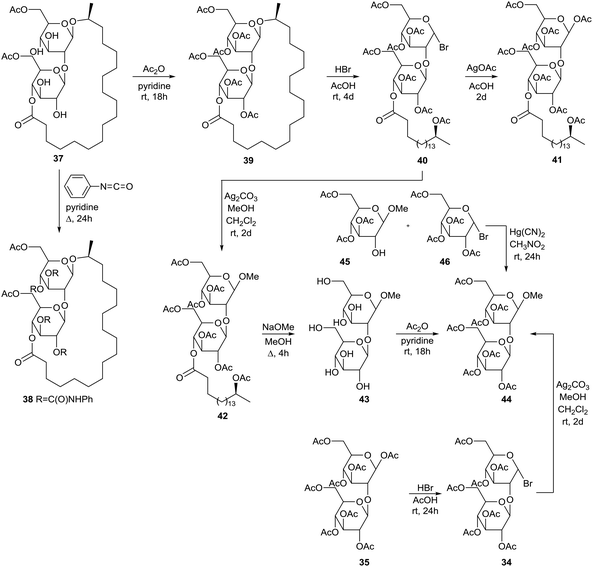 |
| | Scheme 5 Modifications of diacetylated sophorolipid lactone 37.47 | |
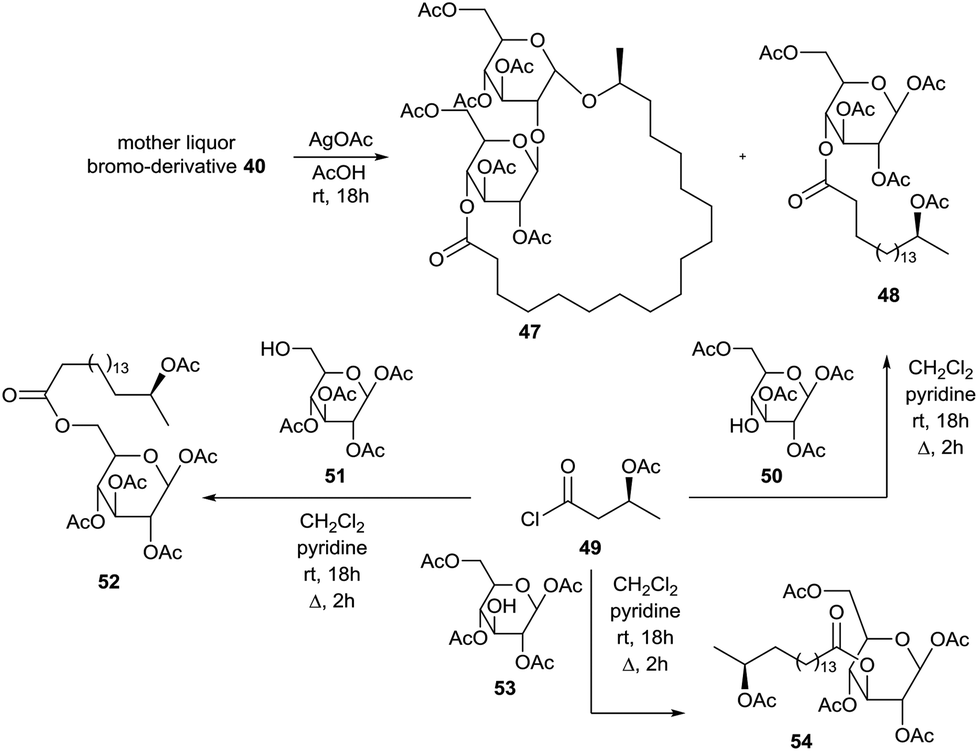
The brominolysis reaction of hexaacetylated sophorolipid lactone 39 also resulted in the cleavage of the linkage between the two glucose units and reversion of the anomeric centre. Treatment of the mother liquor after isolation of α-bromosophorose hexaacetate 40 with silver acetate and subsequent chromatography resulted in the isolation of hexaacetylated α-sophorolipid lactone 47 and β-glucose tetraacetate derivative 48 (Scheme 6). The structure of the latter compound was confirmed upon reaction of the acid chloride 49 synthesized from 17-L-acetoxyoctadecanoic acid and 1,2,3,6-tetra-O-acetyl-β-D-glucopyranose 50. In this context, acid chloride 49 was also coupled with 1,2,3,4-tetra-O-acetyl-β-D-glucopyranose 51 and 1,2,4,6-tetra-O-acetyl-β-D-glucopyranose 53 towards the β-glucose tetraacetate derivatives 52 and 54 respectively. Isolation and characterization of β-glucose tetraacetate derivative 48 demonstrated that the fatty acid tail is linked to the C4′′-position in the diacetylated sophorolipid lactone 37.
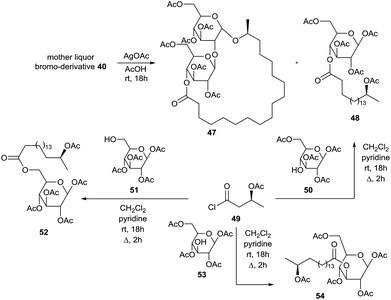 |
| | Scheme 6 Isolation of brominolysis side-products.47 | |
Next to diacetylated sophorolipid lactone 37, a second major fermentation product was present, i.e. diacetylated sophorolipid acid 55. Treatment of sophorolipid acid 55 with diazomethane on the one hand, and treatment of sophorolipid lactone 37 with very weak sodium methoxide on the other hand both resulted in the synthesis of diacetylated sophorolipid methyl ester 56 (Scheme 7). Subsequent deacetylation gave sophorolipid methyl ester 57, which was also prepared by deacetylation of sophorolipid lactone 37 and by treatment of sophorolipid acid 55 with a methanolic hydrogen chloride solution. The syntheses of sophorolipid methyl esters 56 and 57 confirmed that the structure of diacetylated sophorolipid acid 55 is similar to that of diacetylated sophorolipid lactone 37. Sophorolipid methyl ester 57 was acetylated towards heptaacetylated sophorolipid methyl ester 58 followed by brominolysis towards α-acetobromosophorose 34. When the reaction time was reduced to seven hours, heptaacetylated α-sophorolipid methyl ester 59 was obtained, which confirmed that brominolysis reaction conditions induced reversion of the anomeric centre. Deacetylation with sodium methoxide gave α-sophorolipid methyl ester 60.
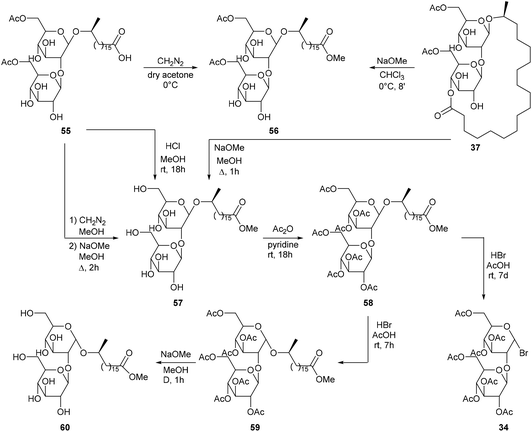 |
| | Scheme 7 Modifications of diacetylated sophorolipid acid 55.47 | |
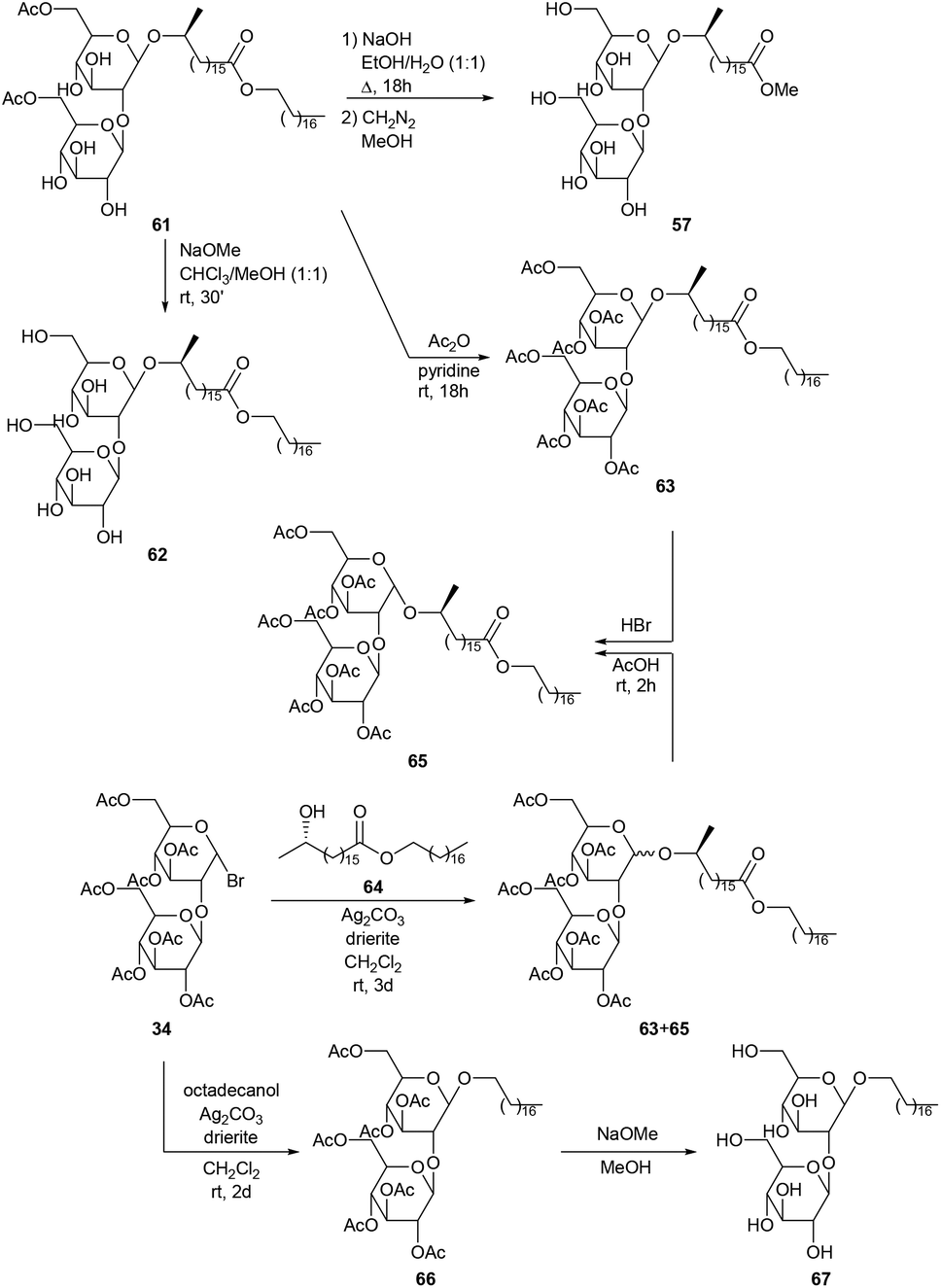
The unexpected production of diacetylated sophorolipid octadecyl ester 61via fermentation of octadecanol by Torulopsis bombicola and subsequent modifications is described by Tulloch and Spencer (Scheme 8).48 Alkaline hydrolysis with sodium hydroxide followed by reaction with diazomethane in methanol yielded sophorolipid methyl ester 57. Methanolysis with sodium methoxide gave sophorolipid octadecyl ester 62 and acetylation resulted in the synthesis of heptaacetylated sophorolipid octadecyl ester 63. To confirm the structure of diacetylated sophorolipid octadecyl ester 61, synthesis of heptaacetylated sophorolipid octadecyl ester 63via the Koenigs–Knorr reaction was attempted. Octadecyl 17-L-hydroxyoctadecanoate 64 was synthesized from 17-L-formyloxyoctadecanoyl chloride and octadecanol and subsequently reacted with α-acetobromosophorose 34. A mixture of α and β anomers 65 and 63 was obtained which was completely converted into heptaacetylated α-sophorolipid octadecyl ester 65 upon reaction with hydrogen bromide. Synthesis of the heptaacetylated derivative 66 of the desired fermentation product, i.e. an octadecyl β-sophoroside, was performed via the Koenigs–Knorr reaction of octadecanol with α-acetobromosophorose 34. Deacetylation gave octadecyl β-sophoroside 67.
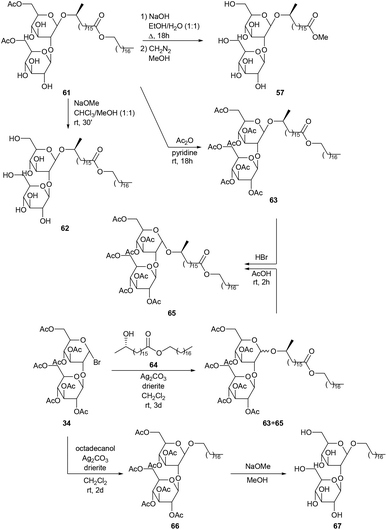 |
| | Scheme 8 Modifications of diacetylated sophorolipid octadecyl ester 61.48 | |
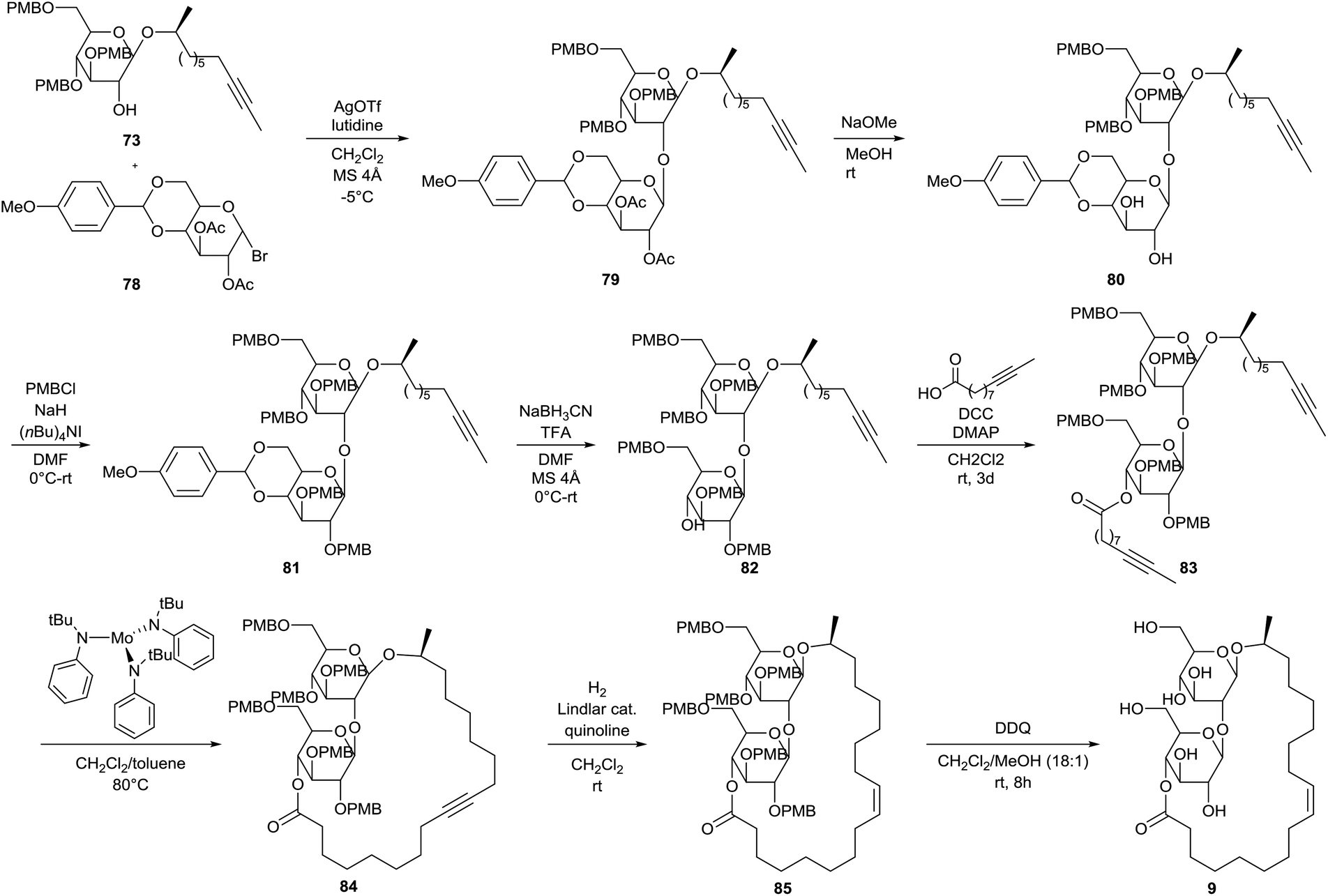
The first total synthesis of sophorolipid lactone 9 was described by Fürstner et al.49 In a first step, two major building blocks were synthesized (Scheme 9). The ring-opening reaction of (S)-propenoxide with Grignard reagent 68 yielded the enantiomerically pure alcohol 69. D-Glucal 70 was protected with p-methoxybenzyl chloride and the resulting tri-O-PMB ether 71 was subsequently treated with dimethyldioxirane to yield epoxide 72. Alcohol 59 was reacted with epoxide 72 into the first building block 73. On the other hand, D-glucose and p-methoxybenzaldehyde dimethylacetal 74 were coupled via a transacetalization reaction to yield 4,6-O-p-methoxybenzylidene acetal 75. Peracetylation towards acetal 76 was followed by selective deprotection of the anomeric centre yielding reducing sugar 77. Reaction with bromine delivered the second building block 78. The two building blocks were coupled via a glycosylation reaction under modified Koenigs–Knorr conditions which yielded the desired sophorose glycoside 79 (Scheme 10). Deacetylation into diol 80 was followed by protection with p-methoxybenzyl chloride. The resulting PMB ether derivative 81 was treated with sodium cyanoborohydride to remove the acetal protecting group. The p-methoxybenzylated sophorolipid 82 was esterified with 9-undecynoic acid towards diyne 83. A ring-closing metathesis reaction was performed which was catalyzed by a molybdenum catalyst to yield cycloalkyne 84. Protected sophorolipid lactone 85 was obtained via Lindlar hydrogenation and subsequent deprotection delivered the desired sophorolipid lactone 9.
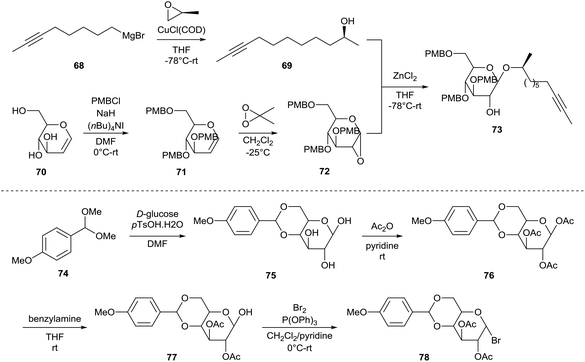 |
| | Scheme 9 Synthesis of building blocks 73 and 78.49 | |
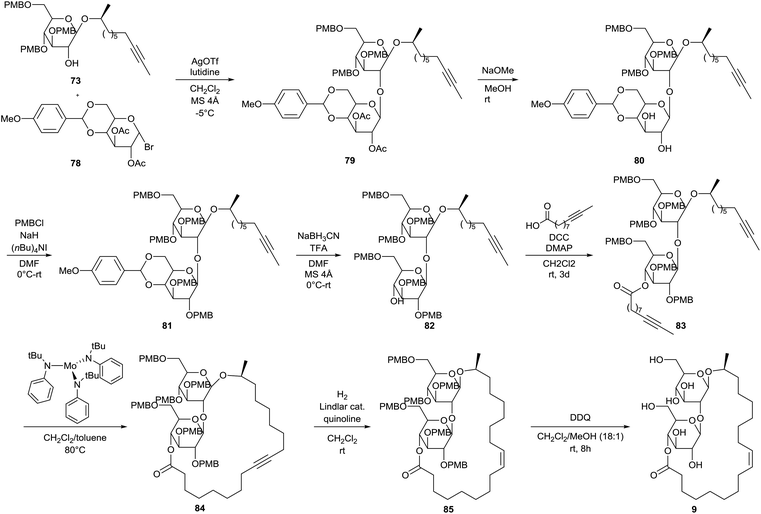 |
| | Scheme 10 First total synthesis of sophorolipid lactone 9.49 | |
Modifications towards new derivatives
Regioselective reactions at the sugar head group
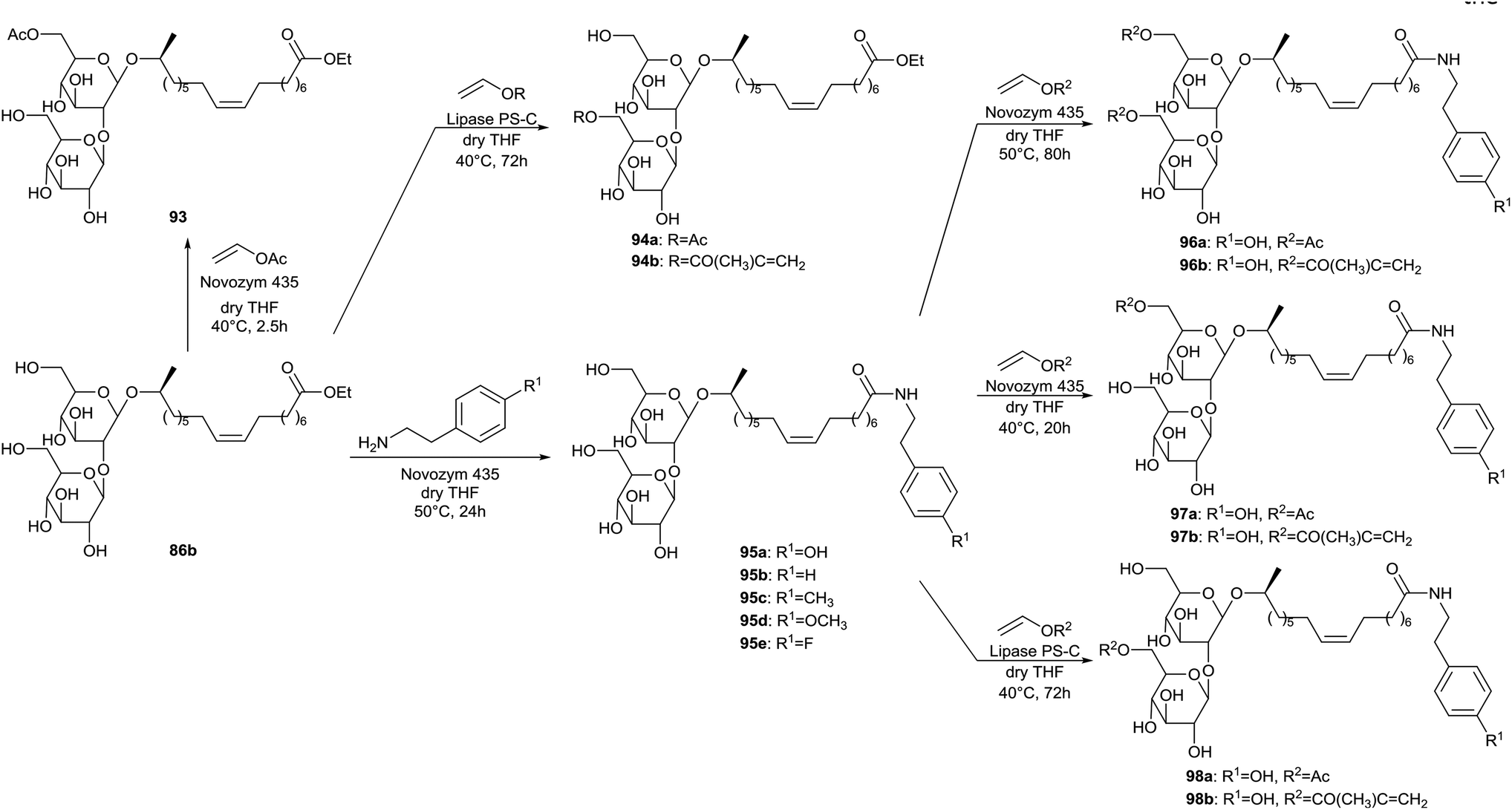
Regioselective acylations of the sophorolipid head group through the use of enzymes is described by Bisht et al. (Scheme 11).50 First, the crude sophorolipid fermentation product needed to be transformed into a single pure compound to enable the synthesis of well-defined sophorolipid analogues. Therefore, the starting product was subjected to alkaline hydrolysis with sodium alkoxides, yielding sophorolipid esters 86. Transformation of the starting product into sophorolipid acid was not useful, since this acid is only soluble in water and polar aprotic solvents which are not suitable for transesterification reactions with lipase enzymes. The sophorolipid esters 86 were subjected to lipase-catalyzed esterifications with an excess of vinyl acetate, vinyl acrylate or succinic anhydride. Multiple enzymes were evaluated and the highest conversion was obtained with Novozym 435. With this enzyme, selective acylations at both the 6′- and 6′′-position of the sugar head were obtained, yielding acylated sophorolipid esters 87. Acylation of only one of these positions was attempted through variation of the ratio sophorolipid ester/acylating agent. With a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 or less, 1,6′′-sophorolipid lactone 88 was formed which is an unnatural analogue of the 1,4′′-sophorolipid lactone. This lactone was subsequently used for the regioselective esterification at the 6′-position of the sugar head, yielding the monoacrylated sophorolipid lactone 89. Sophorolipid methyl ester 86a displayed a cytotoxicity of 63 ± 5% against human pancreatic cells at a concentration ranging from 500 to 2000 μg mL−1.34 Sophorolipid ethyl esters 86b and 87b displayed a lower cytotoxicity of respectively 23.6% at 1000 μg mL−1 and 42.6% at 500 μg mL−1. No cytotoxicity was observed against healthy peripheral blood mononuclear cells. Sophorolipid ethyl ester 87b also reduced septic shock related mortality in in vivo rat models with 23% and possessed high spermicidal and anti-HIV activity comparable to those of nonoxynol-9.41,43b Unfortunately, high cytotoxicity was exerted on human vaginal cells.
:1 or less, 1,6′′-sophorolipid lactone 88 was formed which is an unnatural analogue of the 1,4′′-sophorolipid lactone. This lactone was subsequently used for the regioselective esterification at the 6′-position of the sugar head, yielding the monoacrylated sophorolipid lactone 89. Sophorolipid methyl ester 86a displayed a cytotoxicity of 63 ± 5% against human pancreatic cells at a concentration ranging from 500 to 2000 μg mL−1.34 Sophorolipid ethyl esters 86b and 87b displayed a lower cytotoxicity of respectively 23.6% at 1000 μg mL−1 and 42.6% at 500 μg mL−1. No cytotoxicity was observed against healthy peripheral blood mononuclear cells. Sophorolipid ethyl ester 87b also reduced septic shock related mortality in in vivo rat models with 23% and possessed high spermicidal and anti-HIV activity comparable to those of nonoxynol-9.41,43b Unfortunately, high cytotoxicity was exerted on human vaginal cells.
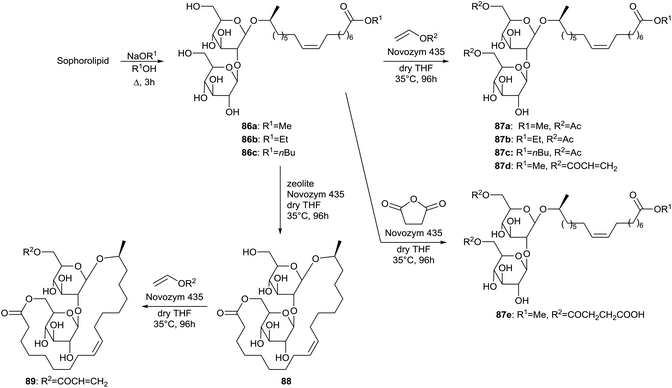 |
| | Scheme 11 Regioselective acylation of sophorolipid esters 86 at both the 6′- and 6′′-position.50 | |
More sophorolipid alkyl esters 86 were synthesized by Zhang et al. in order to evaluate the relationship between the length of the alkyl ester chain and the interfacial properties of the derivatives (Scheme 12).51 The surface tension was measured for all the derivatives. Critical micelle concentration (CMC) and the minimum surface tension decreased for increasing alkyl ester chain length. Adsorption at solid/liquid interfaces was studied as well, demonstrating adsorption on alumina but much less on silica. Hydrogen bonding was proposed to be the primary driving force for adsorption on alumina and the maximum adsorption density suggested bilayer formation at higher concentrations.
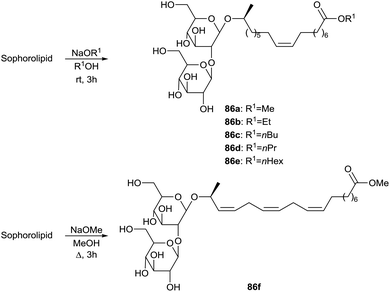 |
| | Scheme 12 Alkaline hydrolysis towards sophorolipid esters 86.51,52 | |
The transesterification of an α-linolenic acid based sophorolipid mixture into sophorolipid methyl ester 86f was described by Gupta & Prabhune (Scheme 12).52 Their minimum inhibitory concentrations against Bacillus subtilis, Escherichia coli and Pseudomonas aeruginosa were determined and proved to be respectively 20, 10 and 10 μg mL−1 for the fermentation product and respectively >20, 20 and 20 μg mL−1 for sophorolipid methyl ester 86f.53
Selective enzyme-catalyzed deacetylation of sophorolipid lactone 1 at the 6′-position was performed by Peng et al. with the lipase from Candida rugosa (Scheme 13).54 This mono-acetylated sophorolipid lactone 8 was subsequently hydrolysed into the non-acetylated sophorolipid lactone 9 with the cutinase from Humicola insolens. Methacrylate and butyrate groups were introduced at the 6′-position through an enzyme-catalyzed acylation of the mono-acetylated sophorolipid lactone 9 with the respective vinyl acylates. An azide group was introduced in two steps. First, a iodination was performed followed by reaction with sodium azide. Methacrylate and azide groups offer the opportunity to introduce bioactive groups through click reactions.
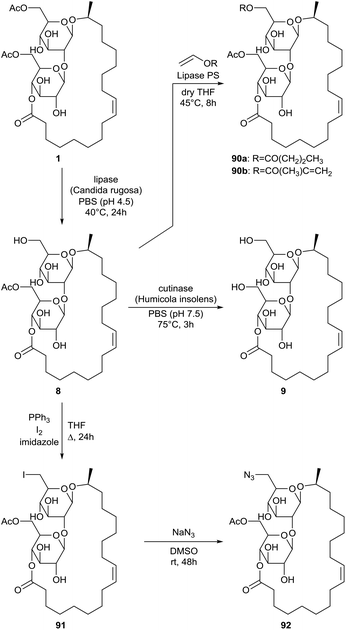 |
| | Scheme 13 Regioselective functionalization of sophorolipid lactone 1.54 | |
More enzyme-mediated regioselective acylations of sophorolipids at the 6′- and/or 6′′-position of the sugar head are described by Singh et al. (Scheme 14).55 Selective acylation at the 6′-position was accomplished through reaction of sophorolipid ethyl ester 86b with excess vinyl acetate and Novozym 435, yielding sophorolipid monoacetate 93. A small amount of the corresponding sophorolipid diacetate was formed as well. When Lipase PS is used as catalyst, selective acylation at the 6′′-postion is obtained. Vinyl acetate and vinyl methacrylate were used respectively for the synthesis of sophorolipid monoacetates 94a and 94b. Enzymatic catalysis was also evaluated for the amidation of sophorolipid ethyl ester 86b with a number of primary amines. Different enzymes were evaluated, of which only Novozym 435 was successful for the formation of sophorolipid amides 95 in high yield. The regioselective acylations with vinyl acetate and vinyl methacrylate at the 6′- and/or 6′′-postions of the sugar head were extended to the sophorolipid amines, yielding sophorolipid diacetate 96 and sophorolipid monoacetates 97 and 98. Also a one-pot synthesis of compound 96b was successfully accomplished.
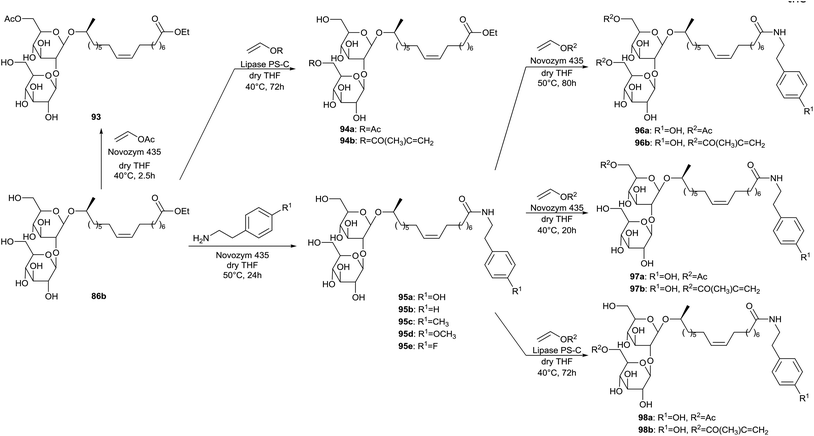 |
| | Scheme 14 Regioselective acylation and enzyme-catalyzed amidation of sophorolipid esters 86.55 | |
The modification of 2-dodecyl sophorolipid 99, prepared by fermentation with 2-dodecanol as substrate, was performed by Recke et al. (Scheme 15).56 The 2-dodecyl sophorolipid 99 was transformed into the 2-dodecyl glucolipid 100via the selective removal of a glucose unit with the β-glucuronidase enzyme. Subsequently, this glucolipid was acylated at the 6′-position with sebacic acid towards glucolipid 101. The free carboxylic acid function offers the opportunity for further derivatization towards polyesters. The 2-dodecyl sophorolipid 99 was also acylated at the 6′- and 6′′-position with the unusual fatty acids (R)-3-hydroxy decanoic acid and (S)-17-hydroxy stearic acid which are obtained via hydrolysis of respectively rhamnolipids and sophorolipids. Acylation with (R)-3-hydroxy decanoic acid resulted in the synthesis of one major product, i.e. the diacylated sophorolipid 102. On the other hand, acylation with (S)-17-hydroxy stearic acid was not complete after 48 hours, resulting in the formation of both the mono-acylated sophorolipid 103 and the di-acylated sophorolipid 104. Longer reaction times were however not evaluated to obtain full conversion. The emulsion stability, CMC values and reduction of the air–water surface tension (γ) and water-n-hexadecane interfacial tension (σ) of the derivatives were evaluated. A good stabilization of w/o emulsions was achieved with all derivatives. This was however not the case for o/w emulsions. CMC, γ- and σ-values were respectively 150–200 mg L−1, 27–50 mN m−1 and 3–7 mN m−1. Antimicrobial activities of the derivatives were also evaluated. All derivatives showed strong growth inhibition against the Gram-positive bacteria Bacillus megaterium and Bacillus subtilis, and against the fungus Candida magnolia. Only di-acylated sophorolipid 102 and the mixture of mono-acylated sophorolipid 103 and di-acylated sophorolipid 104 showed inhibition against the Gram-positive bacteria Staphylococcus capitis. No growth inhibition was observed against the Gram-negative bacteria Escherichia coli and Pseudomonas aeruginosa, the fungi Eurotium repens, Mycotypha microspora and Ustilago maydis, and against the alga Chlorella fusca. Finally, the anti-tumor promoting activity of the derivatives was evaluated via a short-term in vitro assay for Epstein-Barr virus activation in Raji cells induced by TPA using heptyl-galactosyl-glyceride as a reference compound. Concentrations were expressed as mol ratio of compound compared to TPA. At 1000 mol ratio/TPA, only 10–15% activation was observed for the derivatives. All compounds displayed a cell viability of 70% at this concentration, which corresponds to a weak cytotoxicity against the Raji cells.
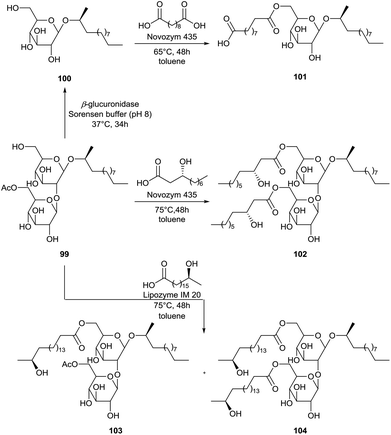 |
| | Scheme 15 Acylations at the 6′- and 6′′-position of sophorolipid 99 and glucolipid 100.56 | |
The enzymatic conversion of crude sophorolipid fermentation product into glucolipid 5 is described by Rau et al. (Scheme 16).57 In a first step, the crude fermentation product was transformed via alkaline hydrolysis into deacetylated sophorolipid acid 2. Different glycosidases were evaluated for the specific release of one glucose molecule and the best results were obtained with the hesperidinase and naringinase enzyme. A comparison was made for the interfacial activities of glucolipid 5 and sophorolipid lactone both at the air–water and water–n-hexadecane interface. In both cases, the interfacial activity of the sophorolipid lactone could not be matched by glucolipid 5.
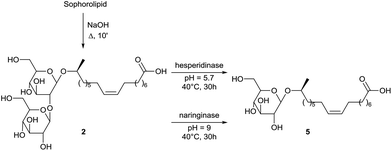 |
| | Scheme 16 Deglycosylation of sophorolipid acid 2.57 | |
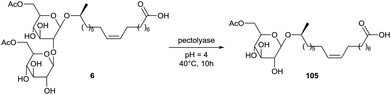
A similar deglycosylation of diacetylated sophorolipid acid 6 is described by Imura et al. (Scheme 17).58 For this acetylated substrate, no activity was observed with hesperidinase or naringinase as was the case for the non-acetylated compound. Selective cleavage of the β-1,2-glycosidic bond was obtained with invertase, pectinase solution, pectinase and pectolyase. The highest activity for the conversion towards acetylated glucolipid acid 105 was obtained with pectolyase. Evaluation of the surface active properties of sophorolipid acid 2, glucolipid acid 5, diacetylated sophorolipid acid 6 and acetylated glucolipid acid 105 showed little variation in CMC value for the different compounds.
 |
| | Scheme 17 Deglycosylation of diacetylated sophorolipid 6.58 | |
Non-selective reactions at the sugar head
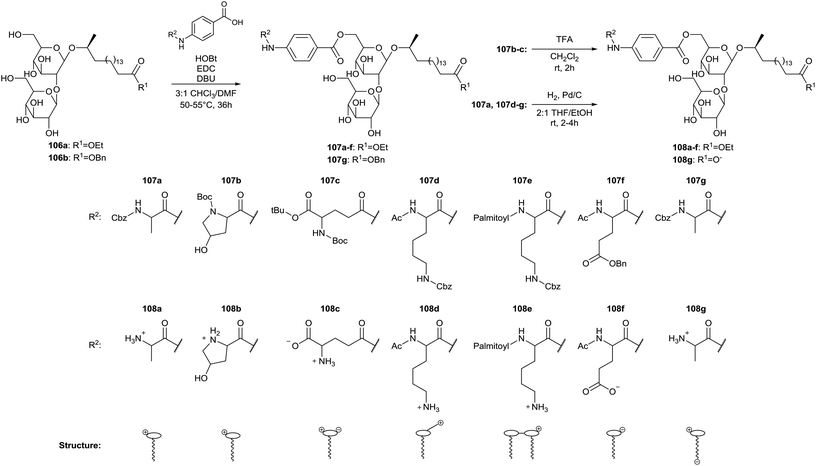
Modifications at the head group of stearic acid based sophorolipids are described by Zerkowski et al. (Scheme 18).59 The crude sophorolipid fermentation product is subjected to alkaline hydrolysis, yielding the sophorolipid free acid and the sophorolipid ethyl ester 106a. The sophorolipid free acid is transformed into the sophorolipid benzyl ester 106b through esterification with benzyl alcohol. Head group modified sophorolipid esters 107 were obtained through coupling of protected amino acids possessing a para-aminobenzoic acid linker (Paba) with the carbohydrate hydroxyl groups. The presence of a Paba-linker should avoid decoupling of the amino acids under alkaline conditions and an excess of sophorolipid ester is used to favor mono-acylation. Two major isomers are obtained for each derivative, most probably the 6′ and 6′′ adducts. Acidolytic or hydrogenolytic deprotection yields the water-soluble sophorolipids 108 with charged head groups. CMC values were determined for the derivatives at different pH-values and were 6–24 μM for comounds 108a–e and 51–110 μM for compounds 108f–g. The results show a significant impact of the modified head groups on the surface tension-lowering properties.
 |
| | Scheme 18 Modification at the head group with amino acids towards charged sophorolipid derivatives.59 | |
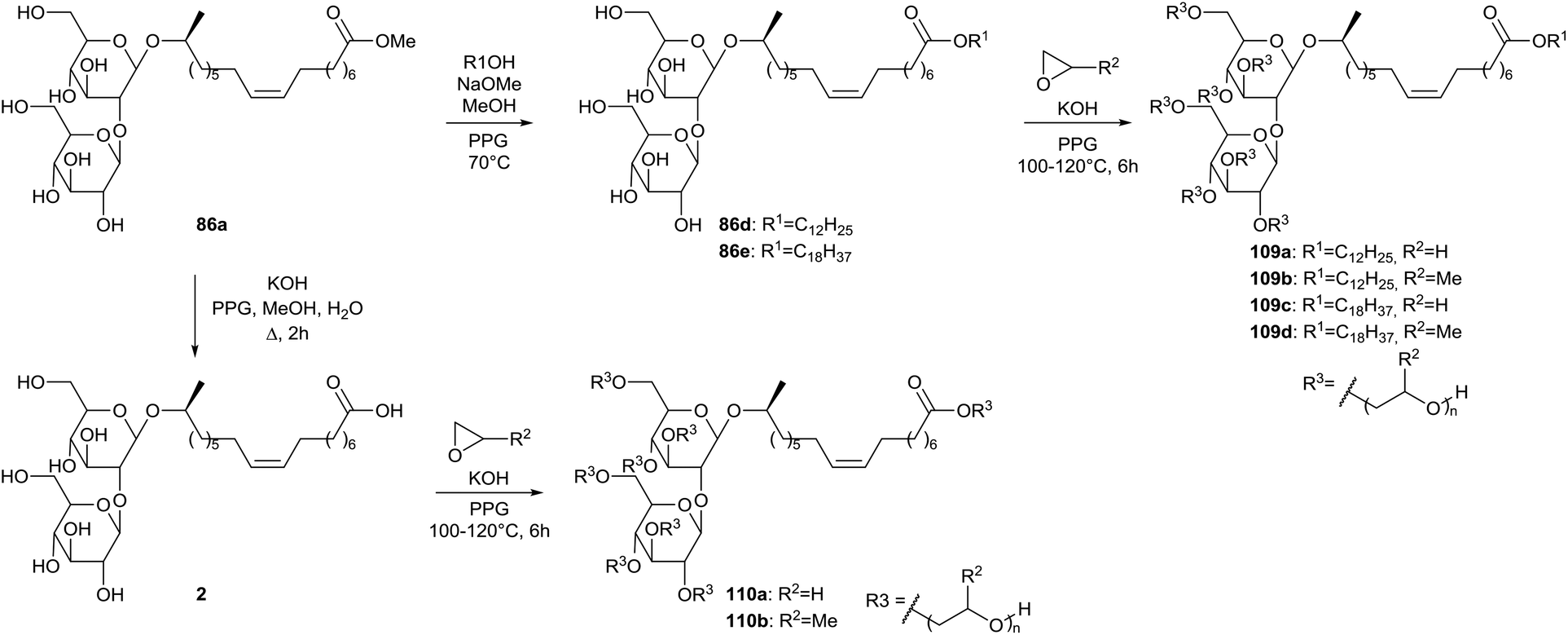
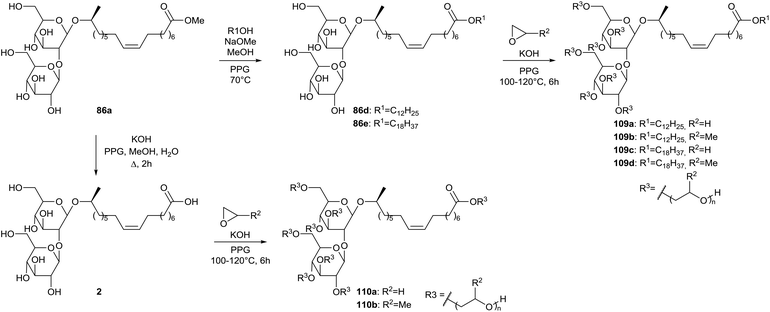
A patent was published by Inoue et al. on the synthesis of hydroxyalkyl-etherified sophorolipid esters (Scheme 19).60 The crude sophorolipid fermentation product was subjected to methanolysis to produce sophorolipid methyl ester 86a. After transformation towards sophorolipid esters 86d and 86e or sophorolipid acid 2, the hydroxyalkyl-etherified sophorolipid esters 109 and 110 were produced through reaction with alkylene oxides in the presence of an alkali catalyst. These hydroxyalkyl-etherified sophorolipid esters were incorporated in cosmetic compositions to prepare powdered compressed cosmetic materials such as face powder, cheek rouge, highlight or eye shadow or stick-shaped cosmetic materials such as stick-shaped rouge, lip cream, stick-shaped eye shadow or cosmetic pencil.61 They were also applied in cosmetics as moisture-retaining and moisturizing agents, giving a less sticky and better moisturizing feeling than conventional moisturizers such as glycerine, sorbitol and ethylene glycol.62 The hydroxyalkyl-etherified sophorolipid esters were formulated as moisturizers in hand cream, cleansing milk, skin lotion, facial pack and lip stick.
 |
| | Scheme 19 Synthesis of hydroxy-alkyl etherified sophorolipids 109 and 110.60 | |
Modifications at the end of the lipid tail
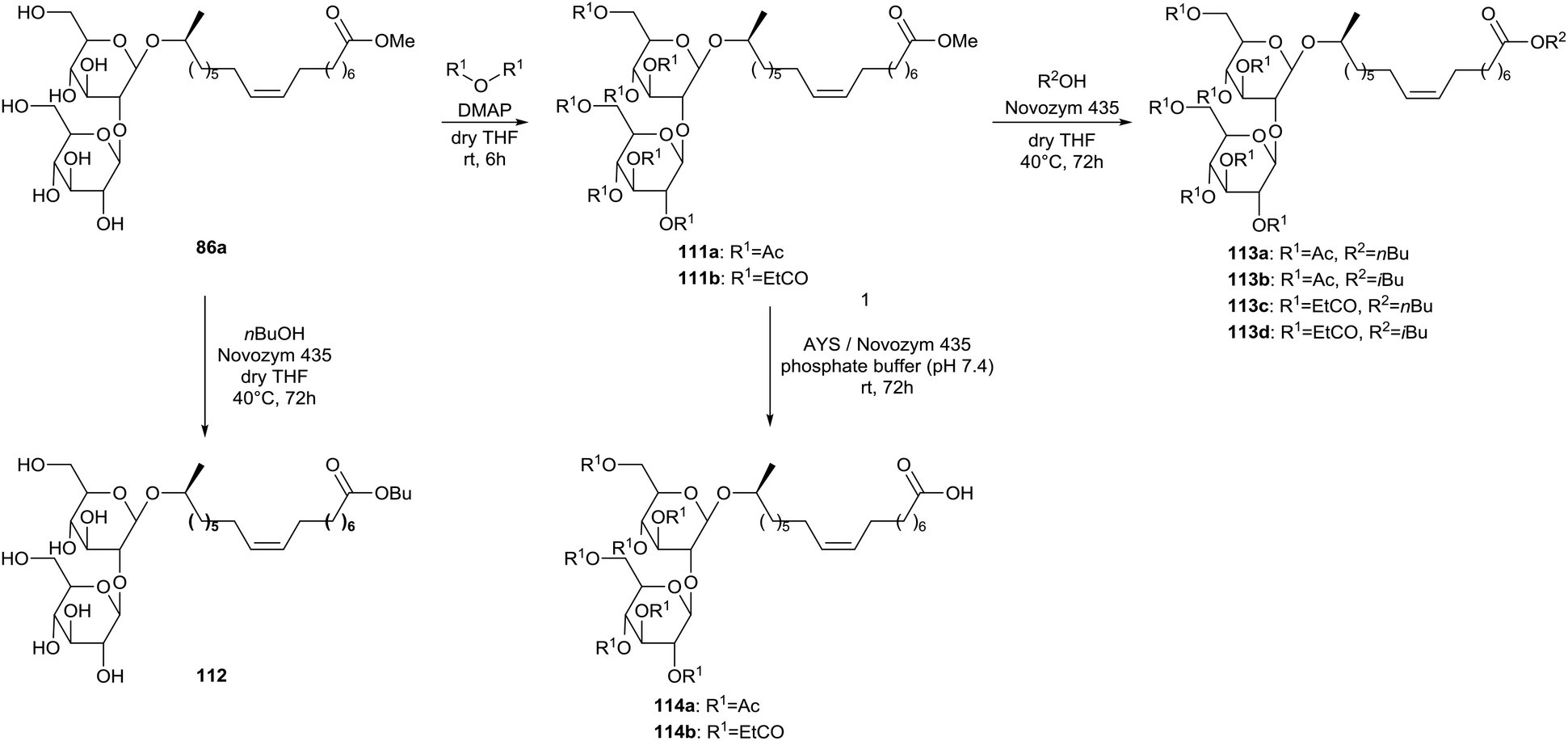
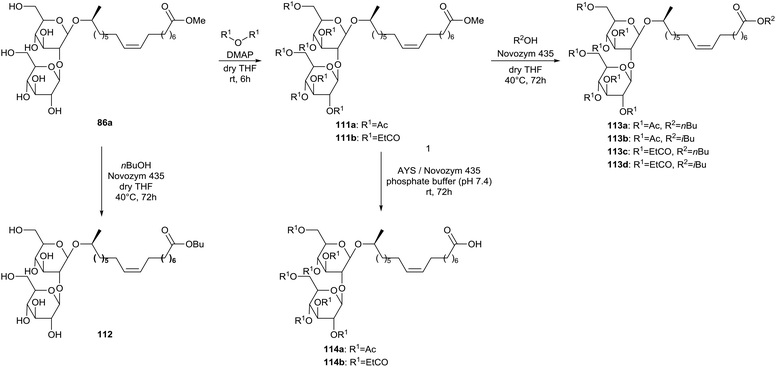
Enzyme-catalyzed transesterifications at the lipid tail are described by Carr & Bisht (Scheme 20).63 Sophorolipid methyl ester 86a was transformed into the corresponding peracylated sophorolipid methyl esters 111 through reaction with acetic and propionic anhydride in the presence of a catalytic amount of DMAP. Different enzymes were evaluated for the transesterification in presence of n-butanol and t-butanol. Only Novozym 435 proved to be successful for the transesterification of both methyl ester 86a and peracylated methyl esters 111 respectively, into sophorolipid butyl ester 112 and peracylated sophorolipid esters 113. Deacylation of the sugar head did not occur. It is suggested that the presence of the macrolactonic ring is necessary to fit the sugar head in the binding pocket of the lipase for deacylation to occur. A lipase catalyzed hydrolysis of the lipid tail was further performed with AYS and Novozym 435 in a phosphate buffer, yielding peracylated sophorolipid acids 114.
 |
| | Scheme 20 Enzyme-catalyzed transesterifications at the lipid tail.63 | |
Transesterification at the lipid tail was extended by Nuñez et al. for the coupling of a sugar monomer to the diacetylated sophorolipid methyl ester 87a (Scheme 21).64 A first attempt with glucose was not successful due to its poor solubility in organic solvents. Functionalization of the hydroxyl groups proved necessary where ketalization is favored for its selectivity. Galactose was used instead of glucose since the primary alcohol still has to be available for the transesterification. The transesterification reaction was performed with immobilized Novozym 435 and sophorolipid sugar esters 115 were formed which varied in the degree of acetylation. Deacetylation occured through the lipase-catalyzed transesterification of the acetylated primary alcohols and the functionalized galactose unit. Deacetylation at the 6′′-position resulted in the formation of 1,6′′-sophorolipid lactone 88 (9%) and its 6′-acetylated analogue (14%) (Scheme 11). Acid hydrolysis with HBF4 was performed for the deprotection of the acetyl and ketal groups towards sophorolipid galactopyranose ester 116.
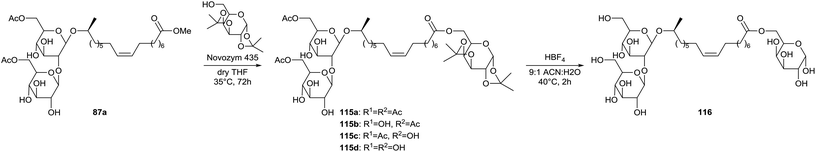 |
| | Scheme 21 Transesterification towards sophorolipid galactopyranose ester 116.64 | |
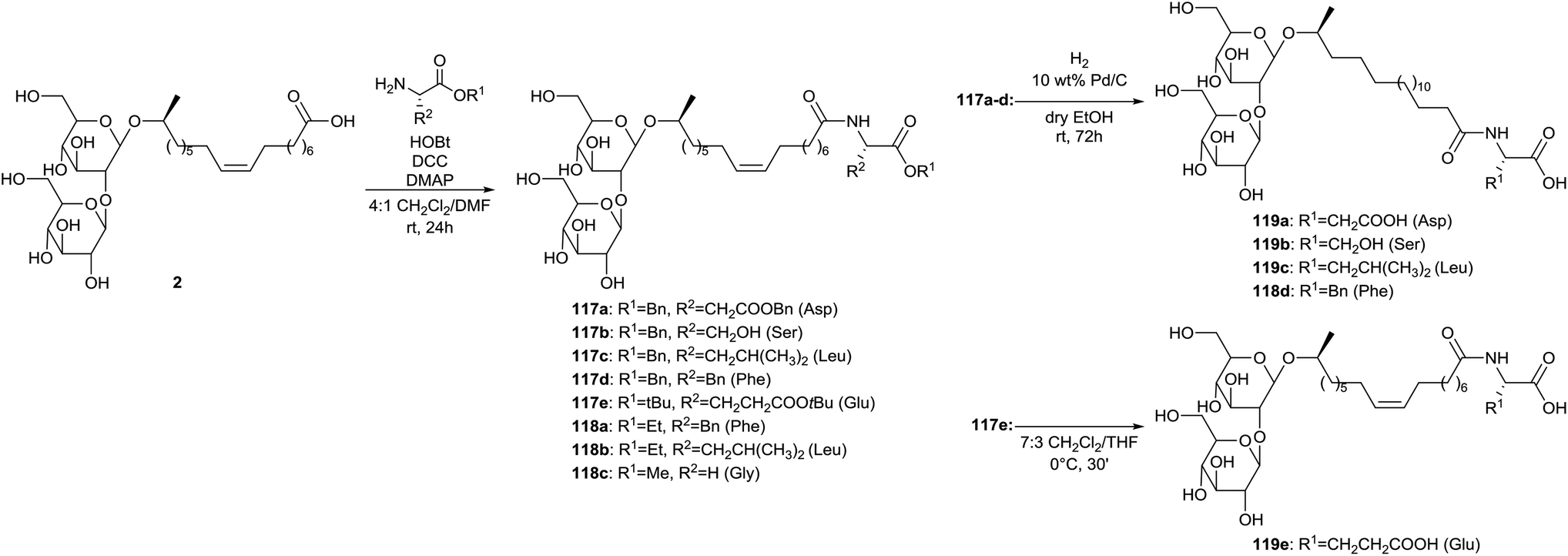
Coupling of sophorolipid acid 2 with different amino acids is described by Azim et al. (Scheme 22).65 The crude fermentation product was transformed into sophorolipid acid 2 through alkaline hydrolysis with NaOH. Protected amino acid conjugated sophorolipids 117 and amino acid conjugated sophorolipid alkyl esters 118 were synthesized using DCC coupling. Deprotection of the different protecting groups yielded amino acid conjugated sophorolipids 119. Deprotection of benzyl ester groups in compounds 117a–d was performed by hydrogenation on Pd/C which simultaneously resulted in the reduction of the double bond. Deprotection of t-butyl groups in compound 117e was accomplished under acid conditions. The antimicrobial, anti-HIV and spermicidal activity were evaluated for amino acid conjugated sophorolipid alkyl esters 118 and amino acid conjugated sophorolipids 119. All tested derivatives possessed antimicrobial activity against Gram-positive and Gram-negative organisms. The best results were obtained for leucine conjugated sophorolipid 119c, with MIC values of 1 and 2 μg mL−1 against respectively Moraxella sp. and S. sanguinis. Also the leucine conjugated sophorolipid ethyl ester 118b feautured high activity, especially against Moraxella sp. with a MIC value of 830 μg mL−1. Monoacetylated ethyl ester sophorolipid (MAEE) was also included in the antimicrobial screening and showed higher activity against all the organisms except for leucine conjugated sophorolipid ethyl ester 118b against Moraxella sp. All tested derivatives displayed virus inactivation with EC50 below 200 μg mL−1. The esterified derivatives 118 are more potent than the corresponding non-esterified compounds 119. Leucine conjugated sophorolipid 118c was the most potent and displayed even higher anti-HIV activity than the commercial spermicide nonoxynol-9 (EC50 ≈ 65 μg ml−1). None of the tested derivatives displayed significant spermicidal activity. Nonoxyl-9 and MAEE were used as positive controls.
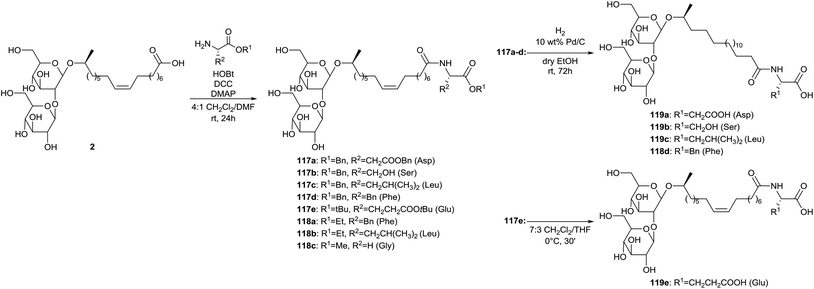 |
| | Scheme 22 Synthesis of amino acid conjugated sophorolipids.65 | |
Synthesis of sophorolipid amides 120 is described in a patent published by Schofield et al. (Scheme 23).66 Non-biogenic sophorolipid amides 120a–120i and biogenic sophorolipid amides 120j–120p were synthesized from sophorolipid methyl ester 86a through amidation with the corresponding amines. Sophorolipid N′,N′-dimethylethylamide 120b was quaternized with methyl iodide to the sophorolipid quaternary ammonium salt 121. This compound was also hydrogenated to the saturated sophorolipid N′,N′-dimethylethylamide 122 and subsequently quaternized to the saturated sophorolipid quaternary ammonium salt 123. Antimicrobial activities were evaluated against a wide variety of micro-organisms.
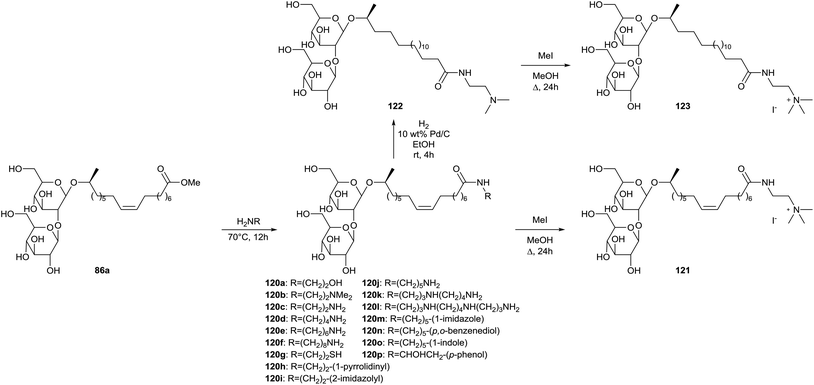 |
| | Scheme 23 Synthesis of non-biogenic and biogenic sophorolipid amides.66 | |
Hydrogenation of diacetylated sophorolipid lactone 1 and sophorolipid acid 2 was also performed, yielding respectively the saturated diacetylated sophorolipid lactone 37 and the saturated sophorolipid acid 10 (Scheme 24).66 Antimicrobial activities were evaluated against a wide variety of micro-organisms.
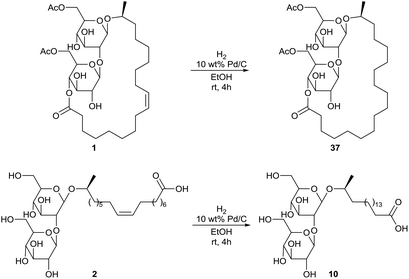 |
| | Scheme 24 Hydrogenation of diacetylated sophorolipid lactone 1 and sophorolipid acid 2.66 | |
Sophorolipid polymerization
The sophorolipid lactone 89 was used by Bisht et al. for the synthesis of polymers with amphiphilic properties (Scheme 25).67 Homopolymerisation was performed with AIBN as initiator, yielding sophorolipid homopolymer 124. This polymer is soluble in DMF and DMSO but not in water. To increase the hydrophilic character of the polymer, copolymers 125 were produced with acrylic acid and acrylamide. The copolymer composition was controlled by the monomer feed ratio. Copolymers 125a and 125b were synthesized with respectivily 3, 5, 10 and 50 mol% and 0.5, 1, 2, 3, 5 and 50 mol% sophorolipid lactone 89 in the feed. Copolymer 125b is soluble in water with less than 3 mol% sophorolipid lactone 89 in the feed.
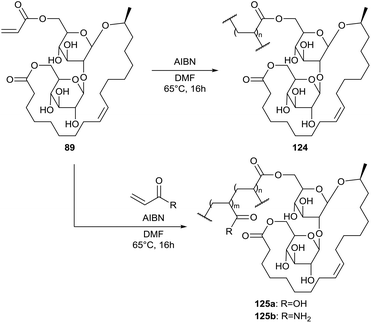 |
| | Scheme 25 Homo- and copolymerization of sophorolipid lactone 89.67 | |
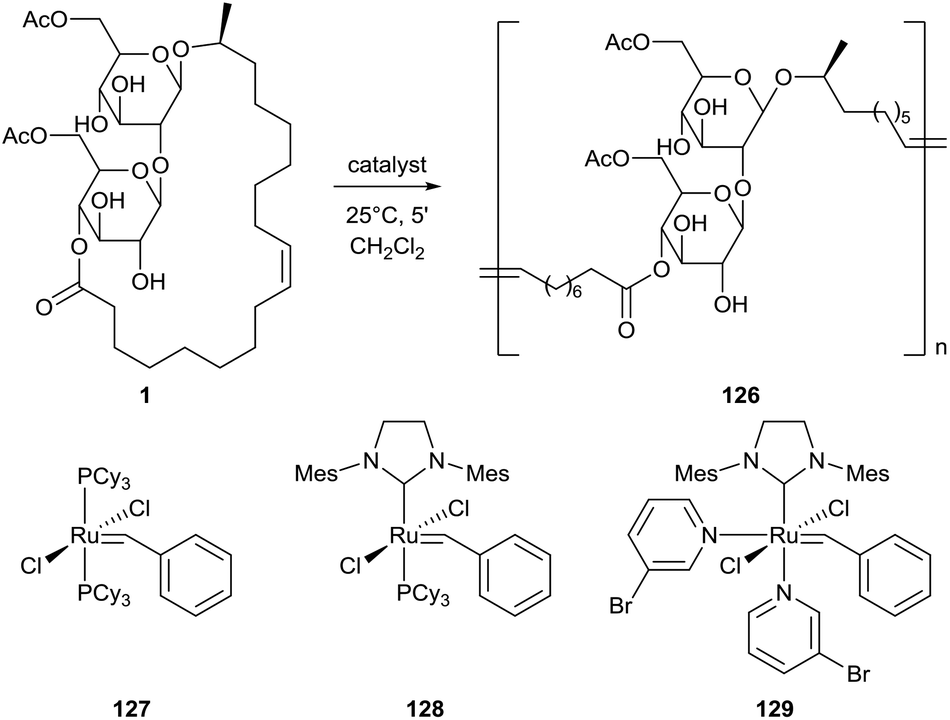
A ring-opening methatesis polymerization (ROMP) for the synthesis of a poly(sophorolipid) 126 from sophorolipid lactone 1 was first described by Gao et al. (Scheme 26).68 The presence of degradable links in the polymer structure makes these polymers promising bioresorbable materials. Three different ruthenium-based Grubbs catalysts were evaluated and the highest yields were obtained with catalyst 128. The solid-state properties of this poly(sophorolipid) were evaluated by Zini et al.69 Both cis and trans double bonds are present in the poly(sophorolipid), respectively in 90% and 10%. Thermogravimetric analysis demonstrated the presence of two main degradation steps. The first step around 350 °C may be associated with the degradation of the hydrophilic sugar moieties, the second step around 450 °C can be attributed to the thermal degradation of the hydrophobic fatty acid chain. The combined results of differential scanning calorimetry (DSC), X-ray diffraction (XRD) and temperature modulated DSC (TMDSC) suggest that the poly(sophorolipid) possesses crystalline domains which melt at 123 °C and has a glass transition at 61 °C. A kinetic study on the polymerization mechanism was performed by Peng et al., categorizing it as an enthalpy driven ROMP.70 The effect of catalyst loading, monomer concentration and solvent choice were evaluated for polymerization with Grubbs catalysts 128 and 129.
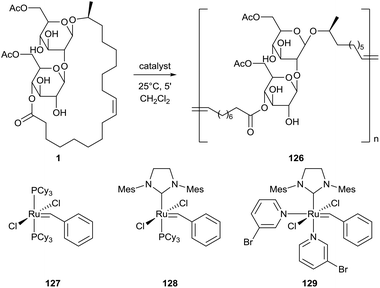 |
| | Scheme 26 ROMP towards poly(sophorolipid) 126.68 | |
The structural diversity of the poly(sophorolipid) was expanded by Peng et al. through the incorporation of modified sophorolipid lactone monomers in the polymer product (Scheme 27).54 The di-acetylated, mono-acetylated, non-acetylated and butyrated sophorolipid lactones 1, 8, 9 and 90a were homopolymerized according the optimal conditions which were determined in the kinetic study. TGA analysis showed that all polymers are stable up to 200 °C and DSC analysis was used to analyze their thermal behavior. No melting transitions were observed for the mono- and non-acetylated sophorolipid polymers, probably due to the absence of a crystalline phase caused by a restricted chain mobility. These polymers show significant potential as substrates for bone tissue engineering due to their solid-sate properties. Biological properties relevant for tissue engineering were determined for all polymers except for the non-acetylated sophorolipid polymer 130b due to stability problems. Cytotoxicity on human mesenchymal stem cells was evaluated via LDH and cell proliferation assays and a similar cytocompatibility was observed as the control tissue culture polystyrene (TCPS). All three polymers also displayed similar cell adhesion and spreading as the control CTPS. The capacity of the three polymers to support the differentiation of the mesenchymal stem cells to an osteoblast cell phenotype was evaluated, giving the best results for butyrated sophorolipid polymer 130c compared to the control TCPS. Finally, degradation of the polymers in an osteogenesis medium was determined. All three polymers underwent an appreciable molecular weight loss in fourteen days and the degradation rate was dependent on the substitution at the sugar moiety.
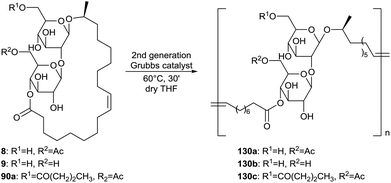 |
| | Scheme 27 Homopolymerization of sophorolipid lactone derivatives.54 | |
Copolymers of diacetylated sophorolipid lactone 1 and methacrylated or azidated sophorolipid compounds were prepared with control of the presence of the clickable functional groups (Scheme 28).54 Functionalization of the methacrylate containing copolymer 131avia a thiol–ene reaction was performed with mercaptoethanol as a model compound. Similarly, functionalization of the azide containing copolymer 131b was peformed with 3-butyn-1-ol via the azide–alkyne cycloaddition reaction. Copolymers containing both methacrylate and azide groups were also prepared.
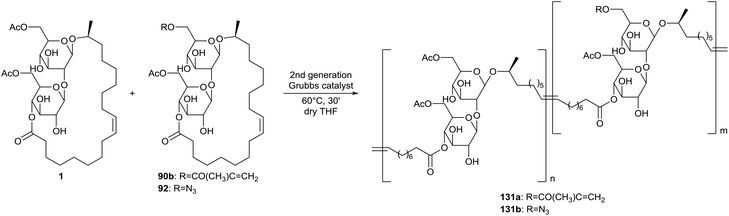 |
| | Scheme 28 Synthesis of copolymers with methacrylate and azide groups.54 | |
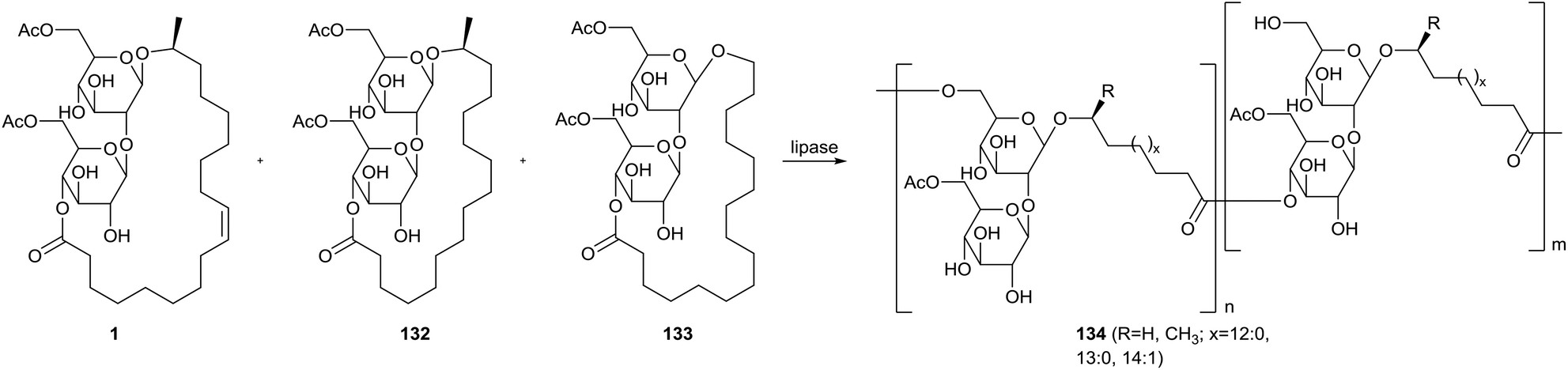
Ring-opening polymerization of sophorolipid lactones with lipases was explored by Hu & Ju (Scheme 29).71 The starting material for the polymerization was a mixture of ω − 1 C18:1 diacetylated sophorolipid 1, ω − 1 C16:0 diacetylated sophorolipid 132 and ω C16:0 diacetylated sophorolipid 133. Ethyl acetate, pyridine, isopropyl ether, toluene, cyclohexane and hexane were evaluated as solvents for polymerization with porcine pancreatic lipase (PPL), giving the highest conversion in isopropyl ether and toluene. An evaluation of the reaction mechanism revealed that the diacetylated sophorolipid lactones were selectively deacetylated at the 6′-position prior to the ring-opening polymerization. This results in linkage of the monomers via both the 6′-position and the 4′′-position at the sugar moiety. Four different lipases were used to evaluate the conversion efficiency in both acetonitrile and isopropyl ether, namely PPL, immobilized Mucor miehei lipase (MML), lyophilized Candida antarctica lipase (CAL-B) and lyophilized Pseudomonas sp. Lipase (PSL). The highest conversion was obtained with MML in isopropyl ether, but CAL-B proved to be the least solvent sensitive. Substrate selectivity dependeed on the temperature, with an increasing conversion at 60 °C but decreasing conversion at 50 °C for larger ring sizes. The overall polymerization proceeded the fastest at 60 °C.
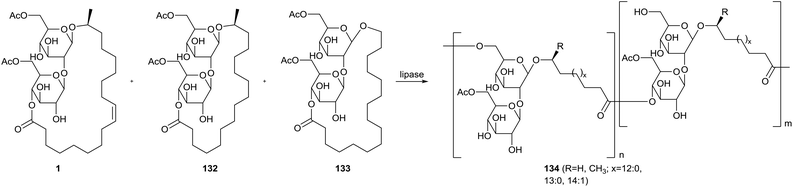 |
| | Scheme 29 Enzymatic ring-opening polymerization of sophorolipid lactones.71 | |
Modifications towards short-chained sophorolipids
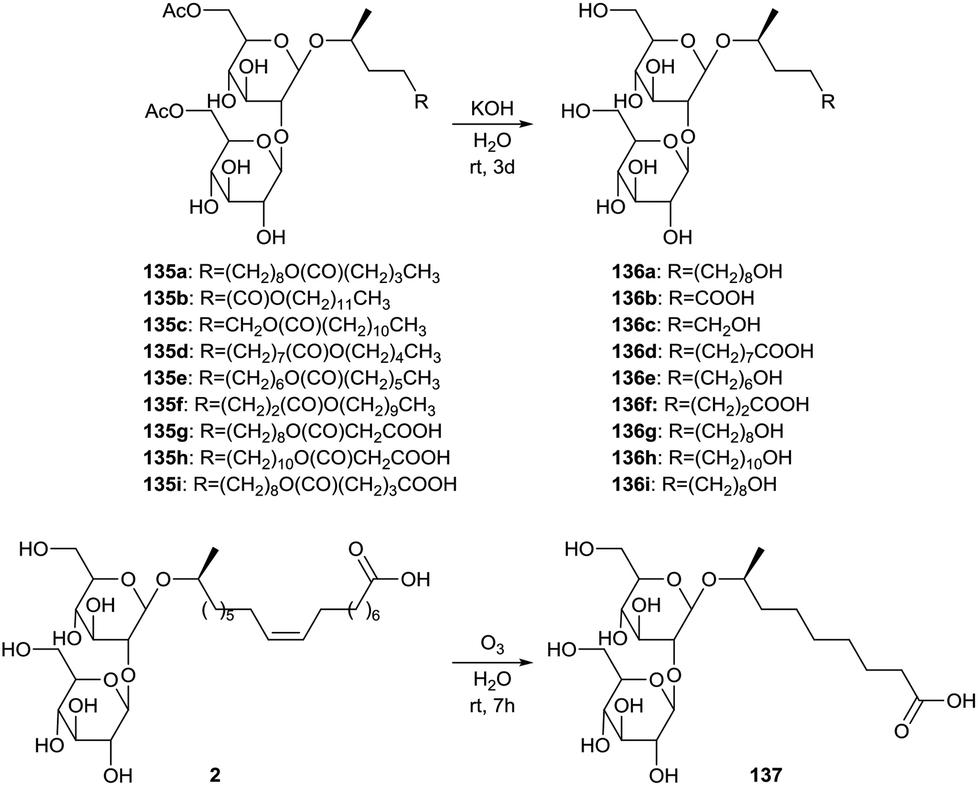
The synthesis of short-chained sophorolipids was described by Develter and Fleurackers, and by Van Bogaert et al. (Scheme 30).72 The esters dodecyl pentanoate, pentyl dodecanoate, decyl heptanoate, dodecyl malonate, myristyl malonate and dodecyl glutarate were used for the synthesis of sophorolipid esters 135 by Candida bombicola. These sophorolipid esters were subjected to alkaline hydrolysis, yielding short-chained sophorolipids 136. An alternative strategy for the production of short-chained derivatives implied the ozonolysis reaction of sophorolipid acid 2 in water towards C9 sophorolipid acid 137. The mixture of this short-chained sophorolipid acid and azelaic acid reduced the surface tension of water to 30 mN m−1 and dynamic surface tension measurements indicated that the reduction of the surface tension was faster compared to crude sophorolipids after fermentation. The mixture proved to be a better wetting agent than Simulsol AS48 and a comparable hydrotrope as Simulsol AS48 and crude sophorolipids.
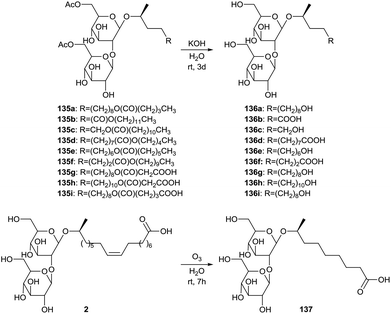 |
| | Scheme 30 Synthesis of short-chained sophorolipids.72 | |
The ozonolysis strategy was further exploited by Delbeke et al. for the synthesis of sophorolipid amines and sophorolipid quaternary ammonium salts (Scheme 31).73 Diacetylated sophorolipid lactone 1 was transformed into peracetylated sophorolipid methyl ester 111a according to procedures described earlier. A subsequent ozonolysis reaction with reductive work-up yielded sophorolipid aldehyde 138. Reductive amination with a variety of secondary amines furnished sophorolipid amines 139, which were subsequently quaternized and deprotected to yield sophorolipid quaternary ammonium salts 140 and 141. Eleven of the sophorolipid quaternary ammonium salts possessed antimicrobial activities against Gram-postive organisms. The best results were obtained for the derivatives with an octadecyl chain on the nitrogen atom.
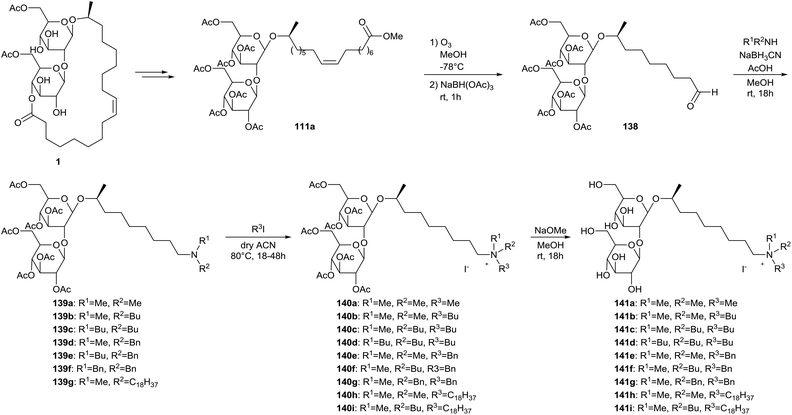 |
| | Scheme 31 Synthesis of sophorolipid amines 139 and sophorolipid quaternary ammonium salts 140 and 141.73 | |
Ring-opening cross-metathesis of sophorolipid lactone 1 was performed by Peng et al. for the synthesis of short-chained sophorolipid derivatives (Scheme 32).74 The cross-metathesis reactions with n-acrylates, trans-3-hexene, 1-hexene and ethylene were catalyzed with a second generation Grubbs catalyst 142. Cross-metathesis with n-acrylates resulted in the synthesis of three gemini type sophorolipids 143. Subsequent ethanolysis reaction yielded the corresponding ethyl ester 144. Cross-methathesis with trans-3-hexene, 1-hexene and ethylene followed by ethanolysis and optional hydrogenation yielded short-chain alkyl sophorolipids 146 and 147. Hydrogenation of unsaturated alkyl sophorolipid 146 was not performed since this terminal double bond is valuable for further functionalization. Critical micelle concentrations were determined via the Wilhelmy plate method and n-dodecyl-β-D-maltoside was used as a reference compound for comparison. Both the CMC and minimum surface tension (γmin) decrease with increasing hydrophobicity and are thus dependent on the chain length. The maximum surface adsorption density (Γm) and minimum surface coverage area per surfactant (Amin) were also calculated. Increasing the chain length results in a higher packing density and lower minimum surface area, especially from alkyl sophorolipid 146 to alkyl sophorolipid 147a. Sophorolipid ethyl ester 144 displayed the highest packing density and lowest minimum surface area despite its hydrophilic character. It is thought that the ester moiety induces additional lateral attraction which reduces the distance between the molecules at the air–water interface. Comparison of the values for n-dodecyl-β-D-maltoside and alkyl sophorolipid 147a demonstrates that sophorose occupies a larger effective area than maltose at the air–water interface. The influence of the chain length of alkyl sophorolipids on the CMC proved to be smaller than that of alkyl glucoside and maltoside sugar-based surfactants. This is likely due to the higher effective area occupied by the sophorose group.
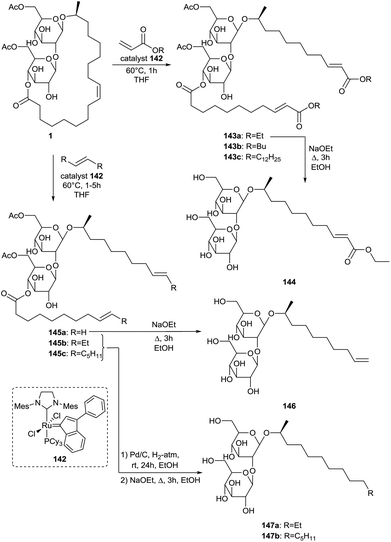 |
| | Scheme 32 Ring-opening cross metathesis towards short-chain sophorolipids.74 | |
Degradation of sophorolipids in smaller building blocks
Deacetylated sophorolipid acid 2 was converted into the ω − 1 hydroxy fatty acid 4 by Rau et al. via acid hydrolysis (Scheme 33).57a The ω − 1 hydroxy fatty acid 4 is a rare fatty acid which is difficult to prepare via organic synthesis. This fatty acid could be used as a building block for the synthesis of pharmaceutically valuable products and for polyester- and macrocyclic lactones.
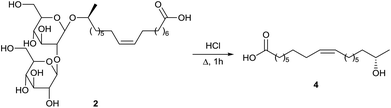 |
| | Scheme 33 Degradation towards 17-hydroxy oleic acid 4.57a | |
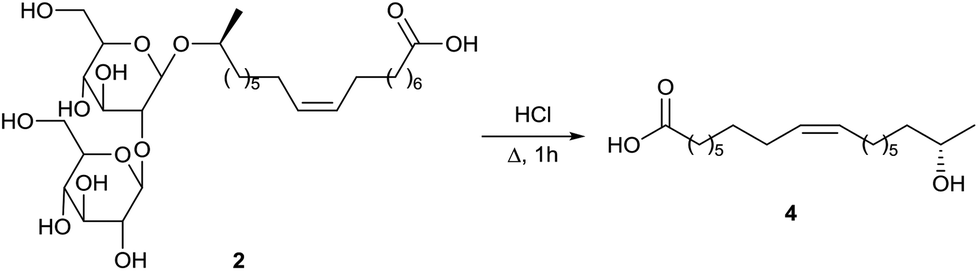
Zerkowski and Solaiman described the synthesis of polyfunctional fatty amines starting from sophorolipids (Scheme 34).75 In a first step, sophorolipid lactone 1 is subjected to acid hydrolysis to yield methyl 17-hydroxy oleate 148. An allylic substitution was used to introduce an amine function at the allylic position of the double bond. Methyl 17-hydroxy oleate 148 was reacted with N-bromosuccinimide and sodium azide towards azide 149 and was subsequently subjected to a Staudinger reaction. Protection of the amine function towards Boc-protected amine 150 was performed without intermediate isolation. Amine 148 was further subjected to a Mitsonobu reaction with phtalimide to yield diamine 151. After conversion of the ester function of amine 150 into a carboxylic acid function, a Curtius rearrangement with diphenylphosphorylazide and a Mitsonobu reaction were performed to yield triamine 153. Further modifications of methyl 17-hydroxy oleate 148 comprised a Mitsonobu reaction towards subterminal amine 154 with phtalmide and the synthesis of azide 155 followed by a Staudinger reaction and protection of the amine function towards Boc-protected amine 156 (Scheme 35). Conversion of the ester function into a carboxylic acid function and a subsequent Curtius rearrangement yielded diamine 157. Finally, methyl 17-hydroxy oleate 148 was converted into 17-hydroxy oleic acid 4 and was subsequently subjected to a Curtius rearrangement to yield amine 158 and isocyanate 159. The latter was converted into urea derivative 160 with mono-Boc piperazine. Amine 158 was transformed into azide 161 and subsequently subjected to a Staudinger reaction with protection of the amine function towards Ns-protected amine 162. Amine 158 was also reacted with N-bromosuccinimide and sodium azide towards azide 163 and additionally subjected to a Staudinger reaction and protection of the amine function towards Fmoc-protected amine 164.
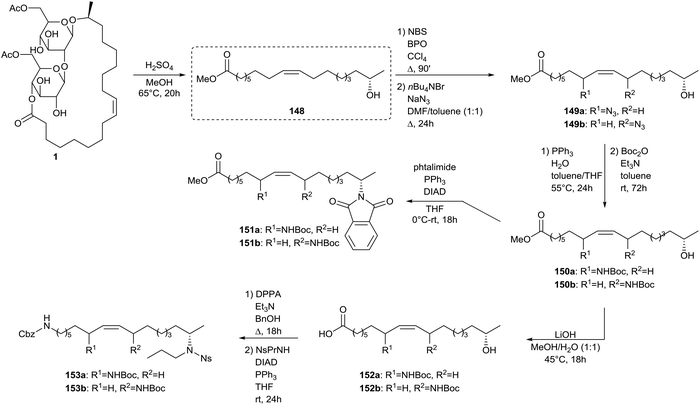 |
| | Scheme 34 Synthesis of polyfunctional fatty amines from sophorolipids (part 1).75 | |
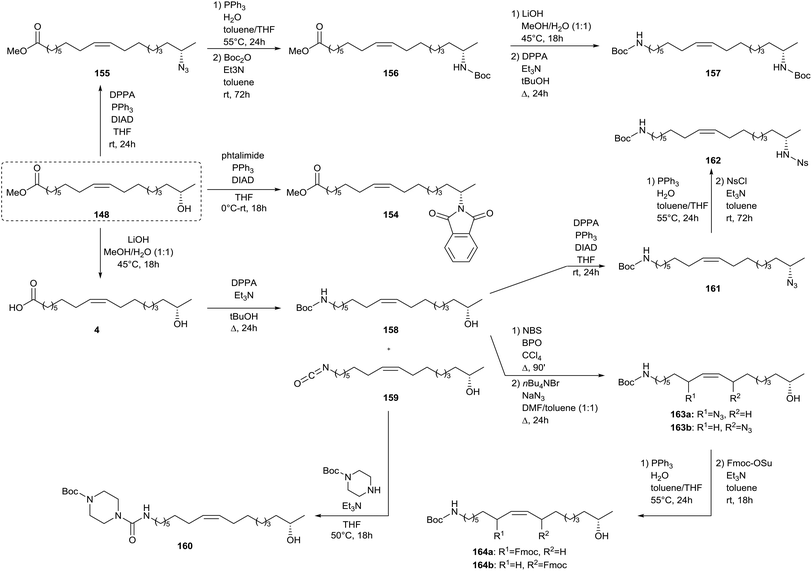 |
| | Scheme 35 Synthesis of polyfunctional fatty amines from sophorolipids (part 2).75 | |
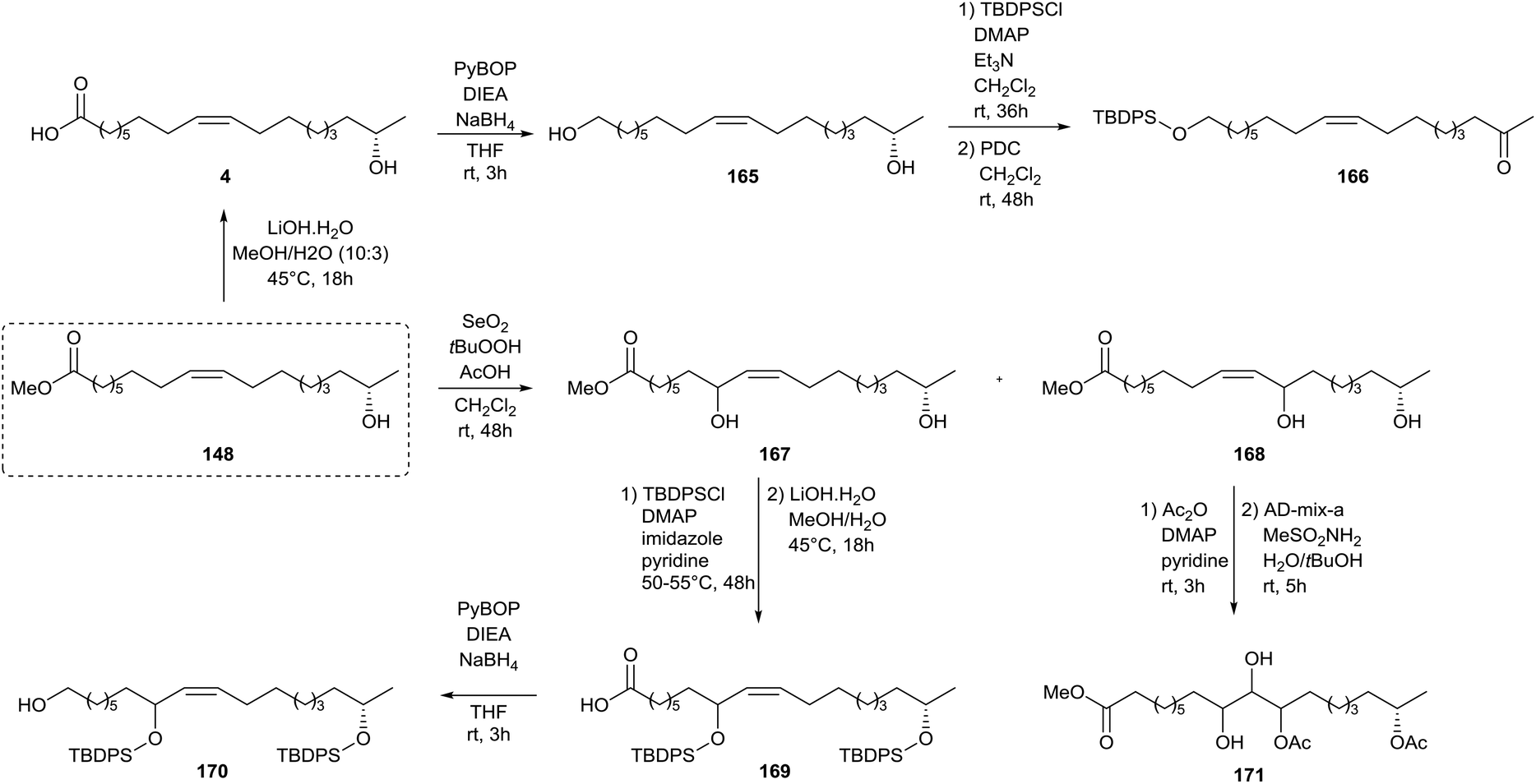
A similar strategy was adopted by Zerkowski and Solaiman for the synthesis of polyhydroxy fatty acids (Scheme 36).76 Methyl 17-hydroxy oleate 148 was converted into 17-hydroxy oleic acid 4 and subsequently reduced towards diol 165via an intermediate activated ester. The primary alcohol was selectively protected with t-butyldiphenylsilyl chloride after which the secondary alcohol was oxidized with pyridinium dichromate towards keton 166. Allylic hydroxylation with selenium dioxide of methyl 17-hydroxy oleate 148 yielded two isomers 167 and 168. The former was protected with t-butyldiphenylsilyl chloride, converted to carboxylic acid 169 and reduced to the primary alcohol 170. The latter was protected with acetic anhydride and subjected to the Sharpless’ asymmetric dihydroxylation towards methyl ester 171. Protection of methyl 17-hydroxy oleate 148 with t-butyldiphenylsilyl chloride followed by hydrolysis yielded carboxylic acid 172, which was subsequently reduced to alcohol 173 (Scheme 37). Sharpless’ asymmetric dihydroxylation and protection with 2,2-dimethoxypropane yielded alcohol 174. Olefin metathesis of methyl 17-hydroxy oleate 148 with a Grubbs’ first generation catalyst resulted in diol 175. Methyl 17-hydroxy oleate 148 was also directly subjected to Sharpless’ asymmetric dihydroxylation towards methyl ester 176 and subsequently protected with 2,2-dimethoxypropane towards methyl ester 177. Treatment with 1,2-dibromotetrachloroethane resulted in bromide 178. A new double bond was formed upon elimination with DBU towards methyl ester 179. Sharpless’ asymmetric dihydroxylation yielded methyl ester 180 whereas allylic hydroxylation with selenium dioxide resulted in three isomeric products 181.
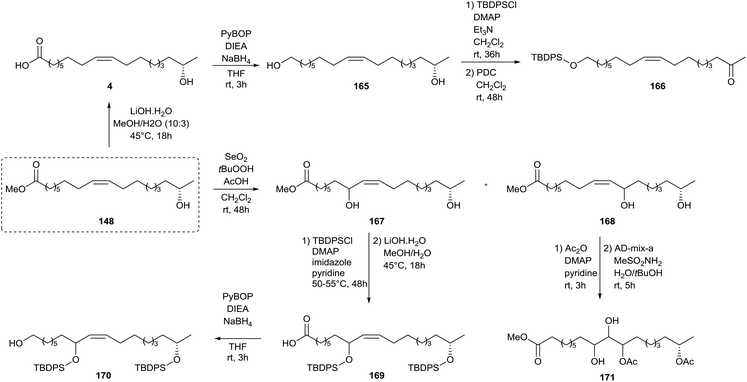 |
| | Scheme 36 Synthesis of polyhydroxy compounds from sophorolipids (part 1).76 | |
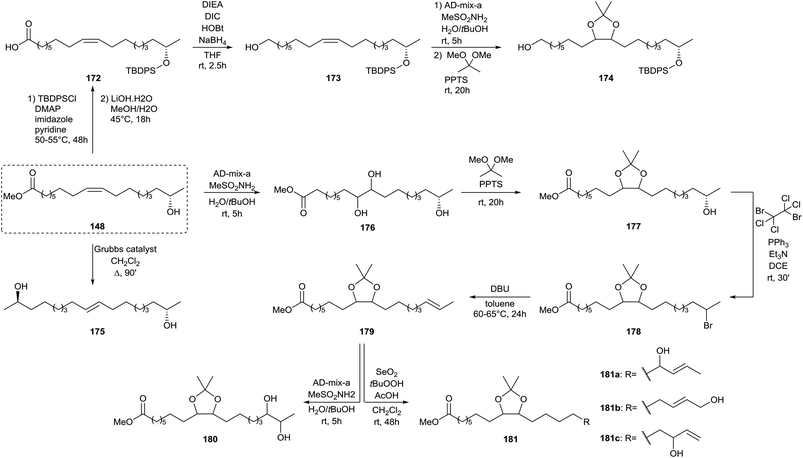 |
| | Scheme 37 Synthesis of polyhydroxy compounds from sophorolipids (part 2).76 | |
Sophorolipids were applied by Hoffmann et al. in the synthesis of the natural products Ebracteatoside C, Zizybeoside I and phenetyl glucoside (Scheme 38).77 The crude sophorolipid fermentation product was transformed into the sophorolipid methyl ester 86a. After protection of the carbohydrate hydroxyl groups and reduction of the double bond, sophorolipid methyl esters 58 and 183 were transformed into anomeric sophorose bromides 34 and 184. The acetylated sophorose bromide 34 was then treated under Koenigs–Knorr conditions with benzyl alcohol or 2-phenetylethanol to yield Zizybeoside 186 or phenetyl glycoside 187 after deprotection under Zemplén conditions. Sophorose bromides 34 and 184 were hydrolyzed and subsequently reacted with trichloroacetonitrile to form the trichloroacetimidates 189. Glycosylation of the acetate protected derivative 189a selectively led to α-anomer 190a. Benzoate protected derivative 189b was glycosylated with Pd(CH3CN)4(BF4)2 to selectively form the enriched β-anomer 190b and was subsequently hydrolyzed to furnish Ebracteatoside C 191.
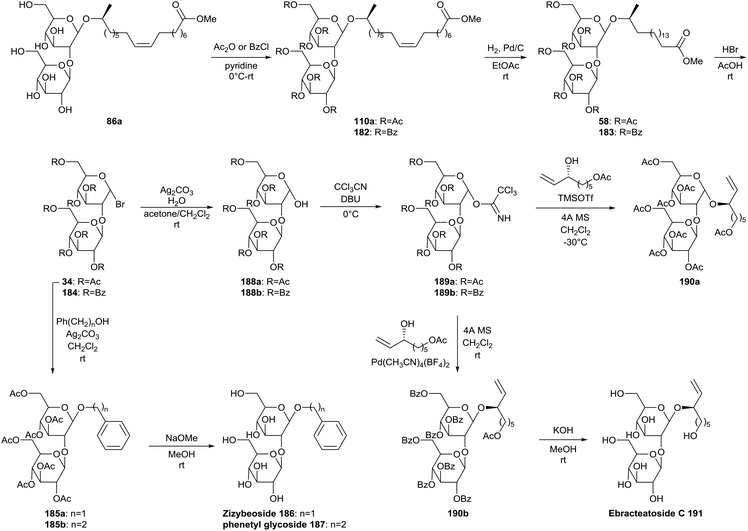 |
| | Scheme 38 Synthesis of natural products from sophorolipids.77 | |
Sophorolipid based nanoparticles
The application of sophorolipids for the formation of magnetic cobalt nanoparticles is described by Kasture et al. (Scheme 39 and 40).78 Sophorolipids have the advantage over oleic acid that they are better water soluble which resulted in the formation of more stable nanoparticles. The cobalt nanoparticles were synthesized by reduction of Co2+ with sodium borohydride using the sophorolipid acid 2 as capping agent. The nanoparticles were obtained from aqueous dispersions as a stable powder via simple centrifugation or magnetic separation and could easily be redispersed in water. Transmission electron microscopy (TEM) revealed that well separated, polydisperse particles were obtained with an average particle size of around 50 nm. The binding of the sophorolipids to the cobalt nanoparticle surface via the carboxylic acid function and the double bond was confirmed by Fourier transform infrared spectroscopy (FTIR). Room-temperature magnetization measurements were performed to demonstrate the good magnetic features of the sophorolipid-capped nanoparticles.
 |
| | Scheme 39 Sophorolipids as nanoparticle capping agents. | |
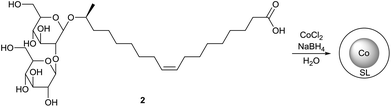 |
| | Scheme 40 Sophorolipid-capped cobalt nanoparticles.78 | |
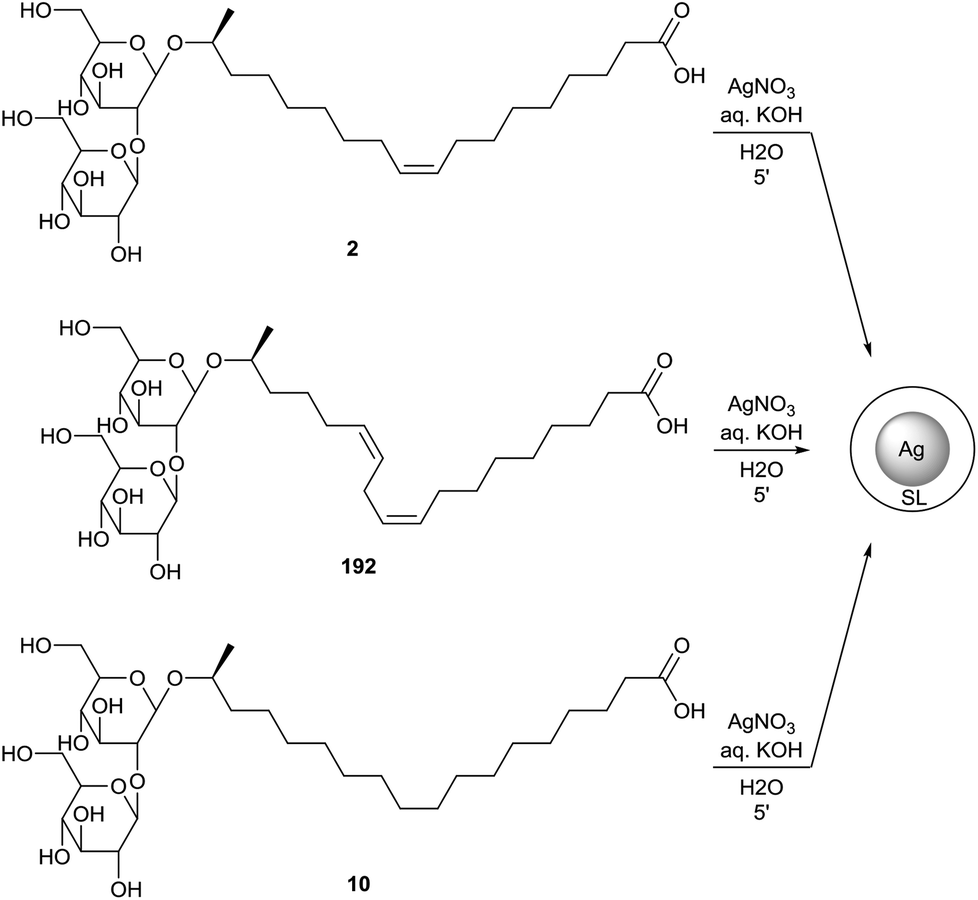
Kasture et al. also described the formation of sophorolipid based silver nanoparticles (Scheme 41).79 The sophorolipid acids 2 and 192 were used as reducing and capping agents in the synthesis of the nanoparticles with silver nitrate under alkaline conditions at different temperatures. The differences in size and dispersion for oleic acid sophorolipid-capped nanoparticles (OA) and linolenic acid sophorolipid-capped nanoparticles (LA) at different temperatures were evaluated. Particle size distribution was determined via both transmission electron microscopy (TEM) and dynamic light scattering measurements (DLS). It was observed that particle size decreased with increasing temperature and longer time is needed at low temperatures to obtain complete particle formation and growth. The nanoparticles were isolated as a stable powder via centrifugation and air-drying which could easily be redispersed in water.80 Just like for the cobalt nanoparticles, binding of the sophorolipids to the silver nanoparticle surface occurs via the carboxylic acid function and the double bond. Continuous flow synthesis of sophorolipid-capped silver nanoparticles is described by Kumar et al.81 Stearic acid based sophorolipid 10 was used as reducing and capping agent for the continuous flow experiments since the resulting nanoparticles were formed faster and featured a better size uniformity than those synthesized with oleic acid based sophorolipid 2. The influence of the flow rate on the average particle size and particle size distribution were evaluated. Higher flow rates led to better mixing but reduced residence time, which resulted in an incomplete reaction towards large polydisperse particles. At lower flow rates, the reaction was complete and small monodisperse spherical nanoparticles were obtained. The synthesis of sophorolipid-capped silver nanoparticles via segmented flow in a microreactor was performed by Kumar et al.82 Both liquid–liquid and gas–liquid segmented flows were applied with respectively kerosene and air as inert phase. The particle size was much smaller for gas–liquid flow than for liquid–liquid flow and the particle size distribution could be controlled by the choice of inert phase. Singh et al. investigated the antibacterial activities of these sophorolipid-capped silver nanoparticles.80 For sophorolipids as such, good inhibition was obtained against the Gram-positive bacteria Bacillus subtilis and Staphylococcus aureus while there was only a faint inhibition against the Gram-negative bacterium Pseudomonas aeruginosa. In case of the sophorolipid-capped nanoparticles, Bacillus subtilis and Staphylococcus aureus were exposed to concentrations ranging from 20 to 100 μg mL−1. The cell survival of Bacillus subtilis dropped to 0.4% after one hour, but no survival was observed for Pseudomonas aeruginosa. Sophorolipid-capped silver nanoparticles are thus more effective against Gram-negative bacteria. Silver proved to be the cause of the observed antibacterial effects via the induction of pore formation in the bacterial cell membrane through the formation of reactive oxygen species.
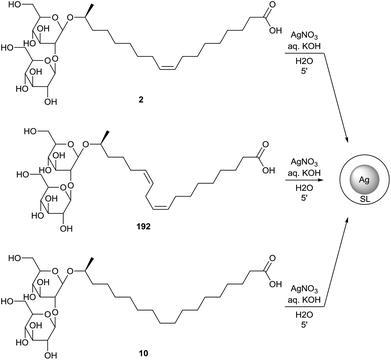 |
| | Scheme 41 Sophorolipid-capped silver nanoparticles.79,81 | |
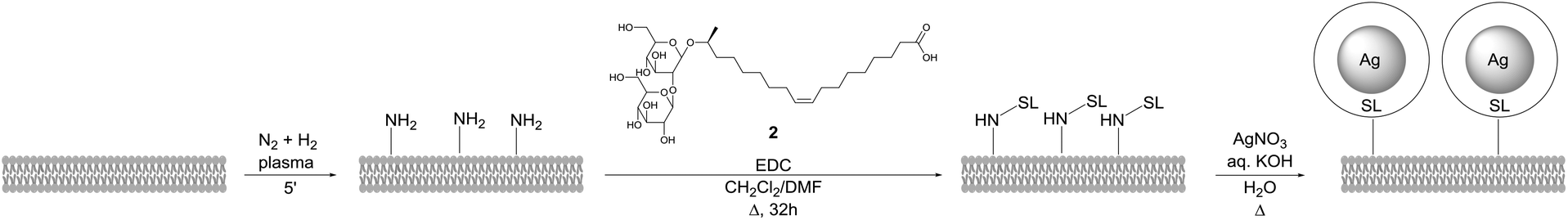
The same methodology was applied by D'Britto et al. for the synthesis of silver nanoparticle studded polyethylene scaffolds (Scheme 42).83 The polymer scaffold was first treated with in situ generated ammonia plasma to introduce amine groups. Sophorolipid 2 was covalently attached to the polymer surface via an amidation reaction followed by formation of silver nanoparticles at the polymer surface with silver nitrate. The size of these silver nanoparticles was approximately 60 to 70 nm. The antimicrobial activity of the modified polyethylene scaffolds against the Gram-positive bacteria Bacillus subtilis and Staphylococcus aureus, and the Gram-negative bacteria Pseudomonas aeruginosa and Escherichia coli was evaluated. Within six hours of exposure, the silver nanoparticle studded polyethylene scaffolds displayed a broad activity against both the Gram-positive and Gram-negative bacteria. The adhesion and proliferation of mammalian cells on the polymer scaffolds was also evaluated. This revealed that the growth of the mammalian cells was encouraged by the silver nanoparticle studded polyethylene scaffolds and that the cells exhibited good viability. These modified polymer scaffolds are good candidates for tissue-engineering and bio-implant applications.
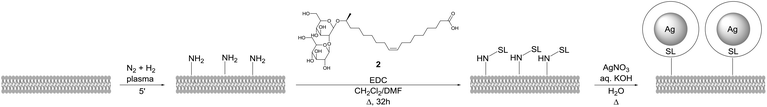 |
| | Scheme 42 Synthesis of silver nanoparticle studded polyethylene scaffolds.83 | |
Dhar et al. described the use of sophorolipids for the synthesis of biocompatible gold nanoparticles (Scheme 43).84 Gellan gum 193 was used as the primary reducing and capping agent in the formation of the gold nanoparticles with chloroauric acid, followed by conjugation of the nanoparticles with sophorolipid acid 2. The nanoparticles were further loaded with the anticancer drug doxorubicin hydrochloride 194. The average particle size was determined via transmission electron microscopy and amounted to 13 and 17 nm for respectively the gellan gum capped nanoparticles and the sophorolipid conjugated nanoparticles. After fluorescent labeling, the efficient cellular uptake of the nanoparticles by human glioma cell line LN-229 was demonstrated. The in vitro cytotoxicity of both sophorolipid-conjugated nanoparticles and doxorubicin chloride loaded nanoparticles were evaluated and compared to free doxorubicin chloride 194 against human glioma cell line LN-229 and human glioma stem cell line HNHC-2. The doxorubicin loaded nanoparticles performed better then both the sophorolipid-conjugated nanoparticles and the free doxorubicin chloride 194 against both cell lines. This synergistic effect could be explained by the better cell penetration of the doxorubicin loaded nanoparticles compared to free doxorubicin chloride 194.
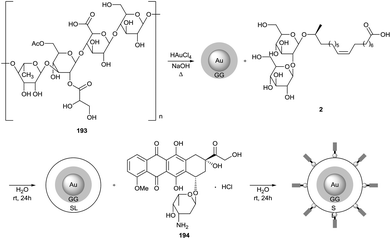 |
| | Scheme 43 Sophorolipid-conjugated gellan gum capped gold nanoparticles.84 | |
The formation of sophorolipid-capped iron oxide nanoparticles was executed by Baccile et al. (Scheme 44).85 The magnetic iron nanoparticles were synthesized with iron(II) chloride and iron(III) chloride in the presence of ammonia at room temperature and 80 °C. Both a two-step and one-step procedure were applied which resulted in nanoparticles with respectively a maghemite (γ-Fe2O3) or two-line ferrihydrite (Fe5HO8·4H2O) structure. Particle size distribution was determined via both transmission electron microscopy (TEM) and dynamic light scattering measurements (DLS). A very broad particle size distribution was obtained for the one-way synthesis pathway at room temperature via TEM, while monodisperse nanoparticles were obtained in all other cases. A narrow size distribution was obtained for all samples via DLS measurement in water and all average sizes are systematically higher than those measured by TEM. When DLS measurements were performed in an ethanol/water mixture (8:2), particle sizes increased compared to those measured in water, indicating the aggregation of the nanoparticles into large aggregates. The much higher increase of the average particle size for the nanoparticles synthesized at room temperature indicated that sophorolipids were more loosely bonded to the nanoparticle surface. FTIR analysis revealed that the sophorolipids are mainly attached to the nanoparticle surface via the carboxylic acid function. Adsorption studies were performed for the sophorolipid-capped iron oxide nanoparticles with two lectines, namely Concanavalin A and lectin from Bandeiraea simplicifolia. No specific interaction occurred with the lectin from Bandeiraea simplicifolia, but affinity for Concanavalin A was observed. Sophorolipid-capped iron oxide nanoparticles thus proved to be interesting as selective targets for sugar/protein interactions.
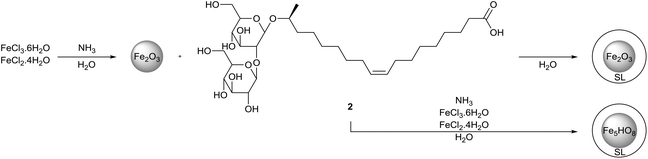 |
| | Scheme 44 Sophorolipid-capped iron oxide nanoparticles.85 | |
Singh et al. described the synthesis of sophorolipid self-assembled vesicular mesostructures via laser irradiation and their subsequent loading with iron oxide nanoparticles (Scheme 45).86 Laser irradiated sophorolipids proved to be highly fluorescent and in the case of diacetylated sophorolipid acid, uniform spherical microstructures were obtained. Upon irradiation, sheet-like structures are formed initially which are converted into fully developed spherical mesostructures after one hour. By adjusting the irradiation conditions, nanoparticles of approximately 100 nm could be obtained. After drying, the sophorolipid mesostructures could easily be redispersed in water. When Fe3O4 nanoparticles were added during the synthesis, sophorolipid mesostructures embedded with iron oxide nanoparticles were obtained. An MTT-assay on a HeLa derived cell line revealed that the sophorolipid mesostructures displayed no cytotoxicity against eukaryotic cells. It was also demonstrated that the sophorolipid mesostructures loaded with iron oxide nanoparticles could serve as effective hyperthermia agents.
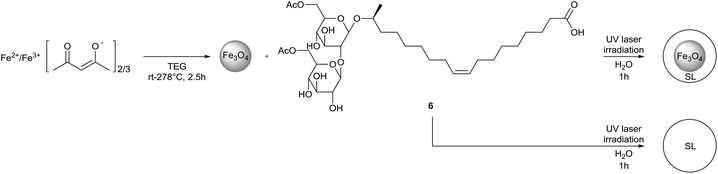 |
| | Scheme 45 Synthesis of sophorolipid mesostructures whether or not loaded with iron oxide nanoparticles.86 | |
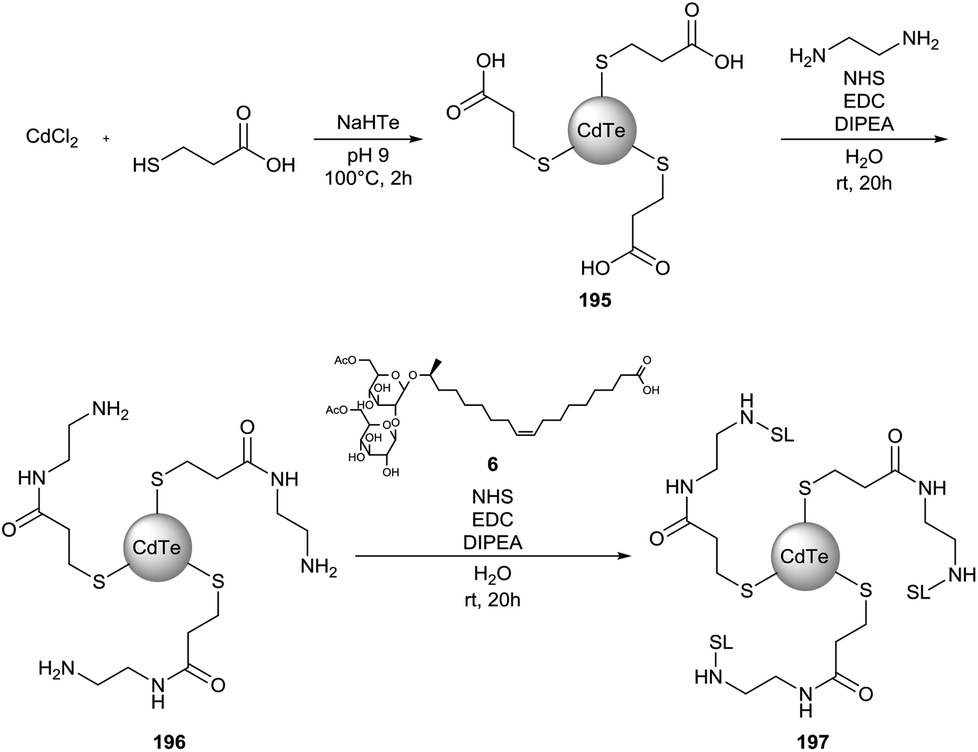
Sophorolipid conjugated cadmium telluride quantum dots (CdTe QD) were synthesized by Singh et al. (Scheme 46).87 In a first step, 3-mercaptopropionic acid was used as the capping agent in the synthesis of the CdTe QDs. Terminal amine groups were introduced via an EDC-mediated amidation reaction with ethylenediamine prior to the coupling of diacetylated sophorolipid acid 6. The obtained nanoparticles were nearly monodispersed with an average particle size of 5 nm for both CdTe QDs 195 and sophorolipid conjugated CdTe QDs 197via TEM analysis. DLS measurements indicated average particle sizes of 8 and 118 nm for respectively CdTe QDs 194 and sophorolipid conjugated CdTe QDs 197. Bioimaging studies revealed that sophorolipid conjugated CdTe QDs 197 were taken up in the cytosol of a cancer cell line. Cytotoxicity studies demonstrated a specific toxicity against the cancer cell line compared to a control cell line.
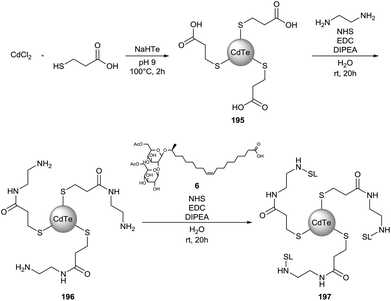 |
| | Scheme 46 Synthesis of sophorolipid conjugated cadmium telluride quantum dots.87 | |
Basak et al. described the synthesis of sophorolipid functionalized zinc oxide nanoparticles (Scheme 47).88 Diacetylated sophorolipid acid 6 was used for the nanoparticle synthesis in combination with zinc nitrate hexahydrate under alkaline conditions. The average particle size was calculated via the Debye–Scherrer formula and was found to be 6.55 and 6.02 nm for respectively naked and sophorolipid-capped zinc oxide nanoparticles. The antimicrobial activities of the zinc oxide nanoparticles were evaluated against the Gram-negative bacterium Salmonella enterica and the fungus Candida albicans. The sophorolipid-capped zinc oxide nanoparticles proved to exhibit a significant inhibitory effect on the growth of both strains at a concentration of 5 mg mL−1. Higher activities were observed for the sophorolipid-capped zinc oxide nanoparticles than for the naked zinc oxide nanoparticles, probably due to the better penetration of the former ones in the pathogenic cells. The mechanism of cell damage on S. enterica consisted of cell elongation followed by cell wall disruption. In the case of C. albicans, complete cell rupture occurred while cell elongation was not detected. A higher antimicrobial activity was observed against S. enterica compared to C. albicans.
 |
| | Scheme 47 Sophorolipid-capped zinc oxide nanoparticles.88 | |
Conclusions and perspectives
Sophorolipids are interesting renewable resources but to date, applications have only been commercialized for sectors in which they can hardly be competitive in terms of production cost. It was demonstrated that sophorolipids feature divergent biological activities which creates opportunities for application in high-added value sectors, in particular the pharmaceutical sector. Modification of these renewable feedstocks is an excellent strategy to improve their biological activity, but the purity of the sophorolipids is essential for the development of good synthesis pathways. The enzymatic sophorolipid modifications mostly enable selective transformations at the sugar head, whereas chemical modifications open up a whole new range of possible sophorolipid analogues. Natural sophorolipids were used for the synthesis of polymers, surface-active compounds, nanoparticles and as source of smaller, non-commercial building blocks. Future research should focus on optimizing fermentation processes towards pure natural sophorolipids in combination with developing selective modification pathways to obtain sophorolipid derivatives in high yields for high added value applications because competitiveness with the traditional base chemical industry will remain an issue.
Acknowledgements
Financial support from the Long Term Structural Methusalem Funding by the Flemish Government (grant number BOF09/01M00409) is gratefully acknowledged.
Notes and references
- I. N. A. Van Bogaert, K. Saerens, C. De Muynck, D. Develter, W. Soetaert and E. J. Vandamme, Appl. Microbiol. Biotechnol., 2007, 76, 23–34 CrossRef CAS PubMed.
-
(a) N. Baccile, N. Nassif, L. Malfatti, I. N. A. Van Bogaert, W. Soetaert, G. Pehau-Arnaudet and F. Babonneau, Green Chem., 2010, 12, 1564–1567 RSC;
(b) A. S. Cuvier, J. Berton, C. V. Stevens, G. C. Fadda, F. Babonneau, I. N. A. Van Bogaert, W. Soetaert, G. Pehau-Arnaudet and N. Baccile, Soft Matter, 2014, 10, 3950–3959 RSC;
(c) S. Q. Zhou, C. Xu, J. Wang, W. Gao, R. Akhverdiyeva, V. Shah and R. Gross, Langmuir, 2004, 20, 7926–7932 CrossRef CAS PubMed;
(d) N. Baccile, F. Babonneau, J. Jestin, G. Pehau-Arnaudet and I. Van Bogaert, ACS Nano, 2012, 6, 4763–4776 Search PubMed.
-
(a)
M. Baviere, D. Degouy and J. Lecourtier, Institut Français du Petrole, US Pat, 5326407, 1993 Search PubMed;
(b)
L. Pesce, Jeneil Biosurfactant Co., Idrabel Italia S.R.L., WO, 2002062495, 2001 Search PubMed;
(c)
J. Ducreux, D. Ballerini, M. Baviere, C. Bocard and N. Monin, Institut Français du Pétrole, US Pat, 5 654 192, 1995 Search PubMed;
(d) C. N. Mulligan, R. N. Yong and B. F. Gibbs, J. Hazard. Mater., 2001, 85, 111–125 Search PubMed;
(e) C. N. Mulligan, R. N. Yong and B. F. Gibbs, Eng. Geol., 2001, 60, 371–380 CrossRef;
(f) C. Schippers, K. Gessner, T. Muller and T. Scheper, J. Biotechnol., 2000, 83, 189–198 CrossRef CAS PubMed.
-
(a) I. N. A. Van Bogaert, J. X. Zhang and W. Soetaert, Process Biochem., 2011, 46, 821–833 CrossRef CAS;
(b) V. K. Morya, C. Ahn, S. Jeon and E. K. Kim, Mini-Rev. Med. Chem., 2013, 13, 1761–1768 CrossRef CAS PubMed;
(c) M. Borsanyiova, A. Patil, R. Mukherji, A. Prabhune and S. Bopegamage, Folia Microbiol., 2015 DOI:10.1007/s12223-015-0413-z;
(d)
I. N. A. Van Bogaert and W. Soetaert, in Biosurfactants: From Genes to Applications, ed. G. Soberón-Chávez, Springer, Berlin, Heidelberg, 2011, pp. 179–210 Search PubMed;
(e)
C. Söffing, Vom Biotensid zum Naturstoff und zurück, Düsseldorf University Press, Düsseldorf, 2012 Search PubMed;
(f)
V. K. Morya and E.-K. Kim, in Biosurfactants: Research Trends and Applications, ed. C. N. Mulligan, S. K. Sharma and A. Mudhoo, CRC Press, Boca Raton, 2014, pp. 105–124 Search PubMed;
(g)
I. N. A. Van Bogaert, K. Ciesielska, B. Devreese and W. Soetaert, in Biosurfactants: Production and Utilization – Processes, Technologies, and Economics, ed. N. Kosaric and F. V. Sukan, CRC Press, Boca Raton, 2015, vol. 159, pp. 19–36 Search PubMed;
(h)
D. W. G. Develter and S. J. J. Fleurackers, in Surfactants from Renewable Resources, ed. M. Kjellin and I. Johansson, John Wiley & Sons, Chichester, 2010, pp. 213–238 Search PubMed.
- S. L. K. W. Roelants, K. Ciesielska, S. L. De Maeseneire, B. Everaert, Q. Denon, H. Moens, B. Vanlerberghe, I. N. A. Van Bogaert, P. Van der Meeren, B. De Vreese and W. Soetaert, Biotechnol. Bioeng., 2015 DOI:10.1002/bit.25815.
-
K. V. Sajna, R. Höfer, R. K. Sukumaran, L. D. Gottumukkala and A. Pandey, in Industrial Biorefineries and White Biotechnology, ed. A. Pandey, R. Höfer, M. Taherzadeh, K. M. Nampoothiri and C. Larroche, Elsevier, Amsterdam, Oxford, Waltham, 2015, pp. 499–521 and other literature cited there Search PubMed.
- H. J. Asmer, S. Lang, F. Wagner and V. Wray, J. Am. Oil Chem. Soc., 1988, 65, 1460–1466 CrossRef CAS.
- K. Ciesielska, I. N. Van Bogaert, S. Chevineau, B. Li, S. Groeneboer, W. Soetaert, Y. Van de Peer and B. Devreese, J. Proteomics, 2014, 98, 159–174 CrossRef CAS PubMed.
- Y. M. Hu and L. K. Ju, J. Biotechnol., 2001, 87, 263–272 CrossRef CAS PubMed.
- N. Baccile, A. S. Cuvier, C. Valotteau and I. N. A. Van Bogaert, Eur. J. Lipid Sci. Technol., 2013, 115, 1404–1412 CrossRef CAS.
- Y. Hirata, M. Ryu, Y. Oda, K. Igarashi, A. Nagatsuka, T. Furuta and M. Sugiura, J. Biosci. Bioeng., 2009, 108, 142–146 CrossRef CAS PubMed.
- D. W. G. Develter and L. M. L. Lauryssen, Eur. J. Lipid Sci. Technol., 2010, 112, 628–638 CrossRef CAS.
- X. J. Ma, H. Li and X. Song, J. Colloid Interface Sci., 2012, 376, 165–172 CrossRef CAS PubMed.
-
G. Hillion, R. Marchal, C. Stoltz and F. Borzeix, Institut Françaic du Pétrole, Sophor S.A., US Pat, 5756471, 1995 Search PubMed.
-
F. Borzeix Concaix, Institut Françcais du Pétrole, Sophor S.A., US Pat, 6596265, 1999 Search PubMed.
-
H. Mager, R. Röthlisberger and F. Wagner, Wella Aktiengesellschaft, EP, 0209783, 1986 Search PubMed.
-
M. Maingault, Institut Français du Pétrole, US Pat, 5981497, 1996 Search PubMed.
-
P. Andre and F. Pellicier, LVMH Recherche, WO, 2004108063, 2004 Search PubMed.
- S. Ito, M. Kinta and S. Inoue, Agric. Biol. Chem., 1980, 44, 2221–2223 CrossRef CAS.
-
R. A. Gross and V. Shah, Synthezyme, LLC, US Pat, 8796228, 2009 Search PubMed.
- E. Kulakovskaya, B. Baskunov and A. Zvonarev, J. Oleo Sci., 2014, 63, 701–707 CrossRef CAS PubMed.
- V. Shah, D. Badia and P. Ratsep, Antimicrob. Agents Chemother., 2007, 51, 397–400 CrossRef CAS PubMed.
-
R. A. Gross and V. Shah, Polytechnic University, WO, 2004044216, 2002 Search PubMed.
- J. N. Sleiman, S. A. Kohlhoff, P. M. Roblin, S. Wallner, R. Gross, M. R. Hammerschlag, M. E. Zenilman and M. H. Bluth, Ann. Clin. Lab. Sci., 2009, 39, 60–63 CAS.
- V. D. Pulate, S. Bhagwat and A. Prabhune, J. Surfactants Deterg., 2013, 16, 173–181 CrossRef CAS.
- M. A. Diaz De Rienzo, I. M. Banat, B. Dolman, J. Winterburn and P. J. Martin, New Biotechnol., 2015, 32, 720–726 CrossRef CAS PubMed.
- K. Joshi-Navare and A. Prabhune, BioMed. Res. Int., 2013 DOI:10.1155/2013/512495.
- X. X. Sun, J. K. Choi and E. K. Kim, J. Exp. Mar. Biol. Ecol., 2004, 304, 35–49 CrossRef CAS.
- X. Sun, E. Kim and S. Sun, Chin. J. Oceanol. Limnol., 2010, 28, 1240–1247 CrossRef CAS.
- X. X. Sun, Y. J. Lee, J. K. Choi and E. K. Kim, Mar. Pollut. Bull., 2004, 48, 863–872 CrossRef CAS PubMed.
- H. Isoda, D. Kitamoto, H. Shinmoto, M. Matsumura and T. Nakahara, Biosci., Biotechnol., Biochem., 1997, 61, 609–614 CrossRef CAS PubMed.
- J. Chen, X. Song, H. Zhang and Y. Qu, Enzyme Microb. Technol., 2006, 39, 501–506 CrossRef CAS.
- J. Chen, X. Song, H. Zhang, Y. B. Qu and J. Y. Miao, Appl. Microbiol. Biotechnol., 2006, 72, 52–59 CrossRef CAS PubMed.
- S. L. Fu, S. R. Wallner, W. B. Bowne, M. D. Hagler, M. E. Zenilman, R. Gross and M. H. Bluth, J. Surg. Res., 2008, 148, 77–82 CrossRef CAS PubMed.
- L. J. Shao, X. Song, X. J. Ma, H. Li and Y. B. Qu, J. Surg. Res., 2012, 173, 286–291 CrossRef CAS PubMed.
- I. A. C. Ribeiro, C. M. C. Faustino, P. S. Guerreiro, R. F. M. Frade, M. R. Bronze, M. F. Castro and M. H. L. Ribeiro, J. Mol. Recognit., 2015, 28, 155–165 CrossRef CAS PubMed.
- K. Joshi-Navare, A. Shiras and A. Prabhune, Biotechnol. J., 2011, 6, 509–512 CrossRef CAS PubMed.
- M. H. Bluth, E. Kandil, C. M. Mueller, V. Shah, Y. Y. Lin, H. Zhang, L. Dresner, L. Lempert, M. Nowakowski, R. Gross, R. Schulze and M. E. Zenilman, Crit. Care Med., 2006, 34, 188–195 CrossRef CAS PubMed.
- S. L. Fu, C. Mueller, Y. Y. Lin, D. Viterbo, J. Pierre, V. Shah, R. Gross, R. Schulze and M. Zenilman, J. Am. Coll. Surgeons, 2007, 205, S44–S44 CrossRef.
- M. Hagler, T. A. Smith-Norowitz, S. Chicel, S. R. Wallner, D. Viterbo, C. M. Mueller, R. Gross, M. Nowakowski, R. Schulze, M. E. Zenilman and M. H. Bluth, J. Allergy Clin. Immunol., 2007, 119, S263–S263 CrossRef.
- R. Hardin, J. Pierre, R. Schulze, C. M. Mueller, S. L. Fu, S. R. Wallner, A. Stanek, V. Shah, R. A. Gross, J. Weedon, M. Nowakowski, M. E. Zenilman and M. H. Bluth, J. Surg. Res., 2007, 142, 314–319 CrossRef CAS PubMed.
- M. H. Bluth, S. L. Fu, A. Fu, A. Stanek, T. A. Smith-Norowitz, S. R. Wallner, R. A. Gross, M. Nowakowski and M. E. Zenilman, J. Allergy Clin. Immunol., 2008, 121, S2 CrossRef.
-
(a)
R. A. Gross, V. Shah and G. F. Doncel, US Pat, 2011/0223239, 2011 Search PubMed;
(b) V. Shah, G. F. Doncel, T. Seyoum, K. M. Eaton, I. Zalenskaya, R. Hagver, A. Azim and R. Gross, Antimicrob. Agents Chemother., 2005, 49, 4093–4100 CrossRef CAS PubMed.
-
R. A. Gross and V. Shah, Polytechnic University, WO, 2007130738, 2007 Search PubMed.
- P. A. Gorin, J. F. T. Spencer and A. P. Tulloch, Can. J. Chem., 1961, 39, 846–855 CrossRef CAS.
- A. P. Tulloch, J. F. T. Spencer and M. H. Deinema, Can. J. Chem., 1968, 46, 345–348 CrossRef CAS.
- A. P. Tulloch, A. Hill and J. F. T. Spencer, Can. J. Chem., 1968, 46, 3337–3351 CrossRef CAS.
- A. P. Tulloch and J. F. T. Spencer, J. Org. Chem., 1972, 37, 2868–2870 CrossRef CAS PubMed.
- A. Furstner, K. Radkowski, J. Grabowski, C. Wirtz and R. Mynott, J. Org. Chem., 2000, 65, 8758–8762 CrossRef CAS PubMed.
- K. S. Bisht, R. A. Gross and D. L. Kaplan, J. Org. Chem., 1999, 64, 780–789 CrossRef CAS PubMed.
- L. Zhang, P. Somasundaran, S. K. Singh, A. P. Felse and R. Gross, Colloids Surf., A, 2004, 240, 75–82 CrossRef CAS.
- R. Gupta and A. A. Prabhune, Biotechnol. Lett., 2012, 34, 701–707 CrossRef CAS PubMed.
- R. Gupta, S. K. Uma and A. Prabhune, Res. J. BioTechnol., 2012, 7, 40–45 Search PubMed.
- Y. F. Peng, D. J. Munoz-Pinto, M. T. Chen, J. Decatur, M. Hahn and R. A. Gross, Biomacromolecules, 2014, 15, 4214–4227 CrossRef CAS PubMed.
- S. K. Singh, A. P. Felse, A. Nunez, T. A. Foglia and R. A. Gross, J. Org. Chem., 2003, 68, 5466–5477 CrossRef CAS PubMed.
- V. K. Recke, M. Gerlitzki, R. Hausmann, C. Syldatk, V. Wray, H. Tokuda, N. Suzuki and S. Lang, Eur. J. Lipid Sci. Technol., 2013, 115, 452–463 CrossRef CAS.
-
(a) U. Rau, R. Heckmann, V. Wray and S. Lang, Biotechnol. Lett., 1999, 21, 973–977 CrossRef CAS;
(b) U. Rau, S. Hammen, R. Heckmann, V. Wray and S. Lang, Ind. Crops Prod., 2001, 13, 85–92 CrossRef CAS.
- T. Imura, Y. Masuda, H. Minamikawa, T. Fukuoka, M. Konishi, T. Morita, H. Sakai, M. Abe and D. Kitamoto, J. Oleo Sci., 2010, 59, 495–501 CrossRef CAS PubMed.
- J. A. Zerkowski, D. K. Y. Solaiman, R. D. Ashby and T. A. Foglia, J. Surfactants Deterg., 2006, 9, 57–60 CrossRef CAS.
-
S. Inoue, I. Kimura and M. Kinta, Kao Soap Co., Ltd, US Pat, 4195177, 1978 Search PubMed.
-
(a)
J. Kawano, T. Utsugi, S. Inoue and S. Hayashi, Kao Soap Co., Ltd, US Pat, 4305931, 1979 Search PubMed;
(b)
J. Kawano, T. Suzuki, S. Inoue and S. Hayashi, Kao Soap Co., Ltd., US Pat, 4305929, 1979 Search PubMed.
-
H. Tsutsumi, J. Kawano, S. Inoue and S. Hayashi, Koa Soap Co., Ltd, US Pat, 4305961, 1979 Search PubMed.
- J. A. Carr and K. S. Bisht, Tetrahedron, 2003, 59, 7713–7724 CrossRef CAS.
- A. Nunez, T. A. Foglia and R. Ashby, Biotechnol. Lett., 2003, 25, 1291–1297 CrossRef CAS PubMed.
- A. Azim, V. Shah, G. F. Doncel, N. Peterson, W. Gao and R. Gross, Bioconjugate Chem., 2006, 17, 1523–1529 CrossRef CAS PubMed.
-
M. H. Schofield, T. R. Thavasi and R. A. Gross, Polytechnic Institute of New York University, US Pat, 2013/0085067, 2012 Search PubMed.
- K. S. Bisht, W. Gao and R. A. Gross, Macromolecules, 2000, 33, 6208–6210 CrossRef CAS.
- W. Gao, R. Hagver, V. Shah, W. C. Xie, R. A. Gross, M. F. Ilker, C. Bell, K. A. Burke and E. B. Coughlin, Macromolecules, 2007, 40, 145–147 CrossRef CAS.
- E. Zini, M. Gazzano, M. Scandola, S. R. Wallner and R. A. Gross, Macromolecules, 2008, 41, 7463–7468 CrossRef CAS.
- Y. F. Peng, J. Decatur, M. A. R. Meier and R. A. Gross, Macromolecules, 2013, 46, 3293–3300 CrossRef CAS.
- Y. Hu and L.-K. Ju, Biotechnol. Prog., 2003, 19, 303–311 CrossRef CAS PubMed.
-
(a)
D. Develter and S. Fleurackers, Ecover N.V., EP, 1953237, 2007 Search PubMed;
(b) I. Van Bogaert, S. Fleurackers, S. Van Kerrebroeck, D. Develter and W. Soetaert, Biotechnol. Bioeng., 2011, 108, 734–741 CrossRef CAS PubMed.
- E. I. P. Delbeke, B. I. Roman, G. B. Marin, K. M. Van Geem and C. V. Stevens, Green Chem., 2015, 17, 3373–3377 RSC.
- Y. Peng, F. Totsingan, M. A. R. Meier, M. Steinmann, F. Wurm, A. Koh and R. A. Gross, Eur. J. Lipid Sci. Technol., 2015, 117, 217–228 CrossRef CAS.
- J. A. Zerkowski and D. K. Y. Solaiman, J. Am. Oil Chem. Soc., 2006, 83, 621–628 CrossRef CAS.
- J. A. Zerkowski and D. K. Y. Solaiman, J. Am. Oil Chem. Soc., 2007, 84, 463–471 CrossRef CAS.
- N. Hoffmann, J. Pietruszka and C. Soffing, Adv. Synth. Catal., 2012, 354, 959–963 CrossRef CAS.
- M. Kasture, S. Singh, P. Patel, P. A. Joy, A. A. Prabhune, C. V. Ramana and B. L. V. Prasad, Langmuir, 2007, 23, 11409–11412 CrossRef CAS PubMed.
- M. B. Kasture, P. Patel, A. A. Prabhune, C. V. Ramana, A. A. Kulkarni and B. L. V. Prasad, J. Chem. Sci., 2008, 120, 515–520 CrossRef CAS.
- S. Singh, P. Patel, S. Jaiswal, A. A. Prabhune, C. V. Ramana and B. L. V. Prasad, New J. Chem., 2009, 33, 646–652 RSC.
- D. V. R. Kumar, M. Kasture, A. A. Prabhune, C. V. Ramana, B. L. V. Prasad and A. A. Kulkarni, Green Chem., 2010, 12, 609–615 RSC.
- D. V. R. Kumar, B. L. V. Prasad and A. A. Kulkarni, Chem. Eng. J., 2012, 192, 357–368 CrossRef.
- V. D'Britto, H. Kapse, H. Babrekar, A. A. Prabhune, S. V. Bhoraskar, V. Premnath and B. L. V. Prasad, Nanoscale, 2011, 3, 2957–2963 RSC.
- S. Dhar, E. M. Reddy, A. Prabhune, V. Pokharkar, A. Shiras and B. L. V. Prasad, Nanoscale, 2011, 3, 575–580 RSC.
- N. Baccile, R. Noiville, L. Stievano and I. Van Bogaert, Phys. Chem. Chem. Phys., 2013, 15, 1606–1620 RSC.
- P. K. Singh, R. Mukherji, K. Joshi-Navare, A. Banerjee, R. Gokhale, S. Nagane, A. Prabhune and S. Ogale, Green Chem., 2013, 15, 943–953 RSC.
- P. Singh, K. Joshi, D. Guin and A. A. Prabhune, RSC Adv., 2013, 3, 22319–22325 RSC.
- G. Basak, D. Das and N. Das, J. Microbiol. Biotechnol., 2014, 24, 87–96 CrossRef CAS PubMed.
Footnote |
| † In equal contribution. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.