
Ionic liquid gel materials: applications in green and sustainable chemistry
Patricia C.
Marr
* and
Andrew C.
Marr
Queens University Ionic liquid Laboratories (QUILL) and The School of Chemistry and Chemical Engineering, Queen's University Belfast, The David Keir Building, Stranmillis Road, Belfast, Northern Ireland BT9 5AG, UK. E-mail: p.marr@qub.ac.uk
First published on 9th November 2015
Abstract
Ionic liquid gel materials offer a way to further utilise ionic liquids in technological applications. Combining the controlled and directed assembly of gels, with the diverse applications of ionic liquids, enables the design of a heady combination of functional tailored materials, leading to the development of task specific/functional ionic liquid gels. This review introduces gels and gel classification, focusing on ionic liquid gels and their potential roles in a more sustainable future. Ionic liquid gels provide the ability to build functionality at every level, the solid component, the ionic liquid, and any incorporated active functional agents. This allows materials to be custom designed for a vast assortment of applications. This diverse class of materials has the potential to yield functional materials for green and sustainable chemistry, energy, electronics, medicine, food, cosmetics, and more. The discussion of the development of ionic liquid gel materials for applications in green and sustainable chemistry centres on uses of ionic liquid gels in catalysis and energy.
 Patricia C. Marr | Dr Patricia C. Marr's interest in materials, started while studying for a masters in materials chemistry at the University of Aberdeen. An interest in gels in particular followed during her PhD in St. Andrews. Working with materials and supercritical fluids at the University of Nottingham launched a special interest in green chemistry. This in combination with working on colloids, polymers and interfaces at the University of Bristol and ionic liquids with QUILL, has gelled to form a special research interest in ionic liquids, materials, and ionic liquid gels. Dr P. C. Marr is currently at the School of Chemistry and Chemical Engineering, Queen's University Belfast. |
 Andrew C. Marr | Dr Andrew C. Marr's interests in organometallic chemistry and catalysis initiated at the University of Aberdeen and sped up while studying for a PhD at the University of St Andrews. With a special interest in green chemistry from his time working at the University of Nottingham, Dr Marr has focussed on bio and chemo catalytic chemical transformations for green chemistry. Dr A. C. Marr is currently at QUILL and the School of Chemistry and Chemical engineering, Queen's University Belfast. |
1. Introduction
The gel state: gel classification
A substance is described as a gel when there is a liquid phase dispersed in a continuous solid phase. A gel is a colloid, and as such is one of the eight combinations of solid, liquid and gas as defined by the term colloidal system, dispersion or solution.1The term gel is thought to come from a contraction of the word gelatine and is often attributed to Thomas Graham, a 19th century Scottish chemist recognised as the founder of colloid chemistry.2,3 Gels are often difficult to classify because a gel can be soft, solid or liquid-like. In order to qualify as a gel the material must be crafted from two or more components, one of which is a liquid, present in a considerable quantity.4,5
The term gel is used ubiquitously in daily life to describe a material with behaviour lying somewhere between that of a solid and a liquid, and this rather general description has led to considerable confusion as to what comprises a gel state.6 There are some fundamental criteria that have to be met before a material can be considered a gel.
• Heterogeneous – a two (or more) component system composed of a liquid and solid (solid component not visible to the naked eye).
• Solid-like properties – sustain shear stress.
• The presence of a 3D network.
In addition to these qualifying properties often a liquid to gel transition is observed. A gel therefore can be recognised as a two phase heterogeneous mixture of a solid and a liquid which through self-assembly/bond formation (physical or chemical) is organised into a liquid dispersed phase and a solid continuous phase. The continuous solid phase can be a variety of solids from thread-like to spherical. The microstructure of a gel arises from attractive forces between at least one of its components forming a percolated network.7 Gels can be classified in many ways, the most common ones being classification according to the nature of the interactions, physical and mechanical properties (rheology), nature of the solvent, and nature of the solid.
Classification of gels
 | ||
| Fig. 1 Classisification of gels by nature of interactions. | ||
 | ||
| Fig. 2 Gel classification by nature of the continuous (solid) phase. | ||
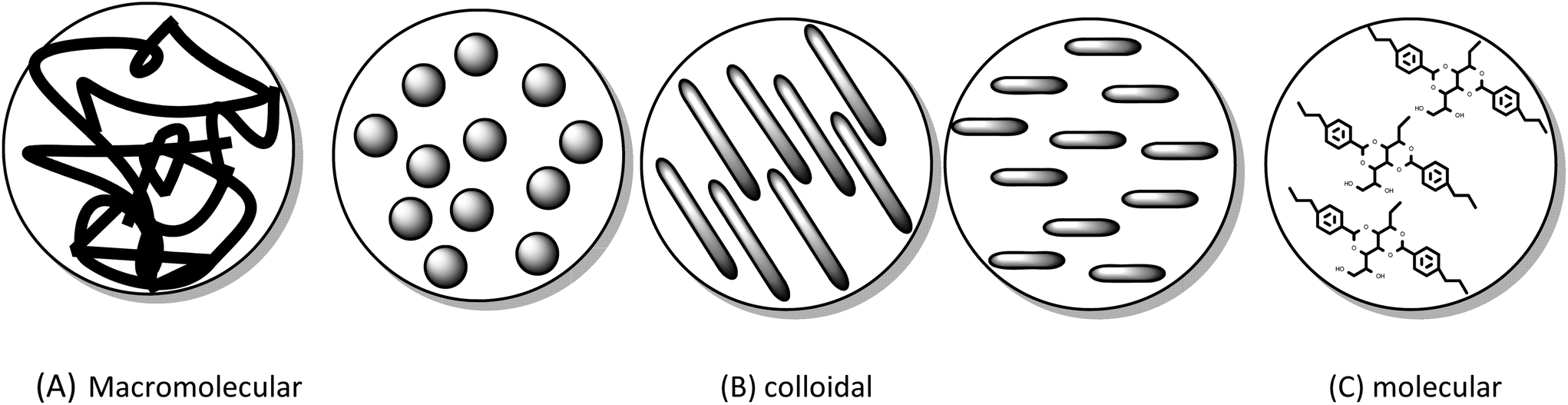 | ||
| Fig. 3 Types of continuous phase (A) macromolecular (B) colloidal (spheres, rods, discs) (C) molecular. | ||
Many initial observations of molecular and low molecular mass gelators were based on chance observations on heating and cooling, but gels of this type are now being studied in a more methodical approach. The design of molecules that can act as gelators and the addition of functionality to existing gelators are exciting directions and challenges. LMMG/LMOGs are being found that can gel solvents under other conditions such as photo irradiation, sonication, chemical or electrical stimuli.29
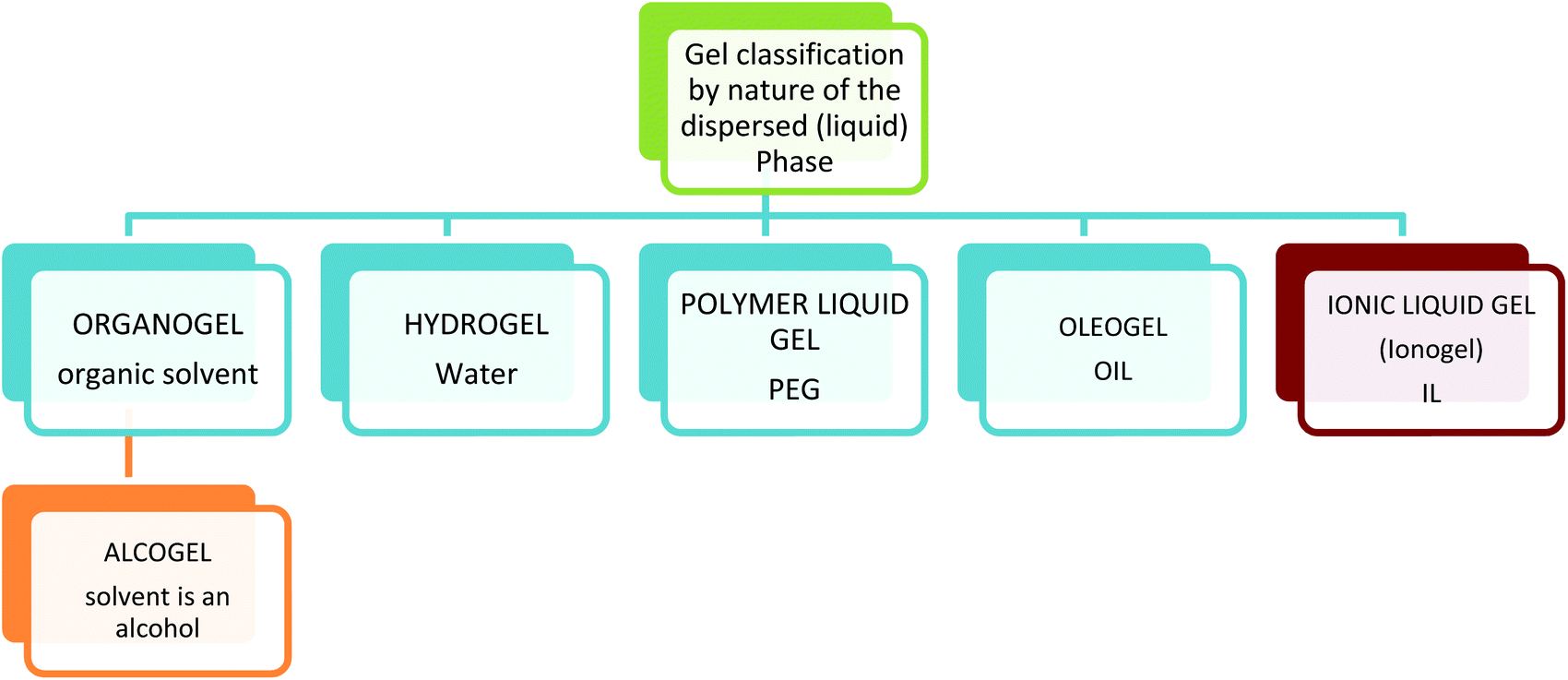 | ||
| Fig. 4 Gel classification by nature of the dispersed (liquid) Phase. | ||
Gels are termed organogels when the dispersed phase (liquid trapped) is an organic solvent, oleogels when the dispersed phase is oil, polymer liquid gels when the liquid phase is a polymer and hydrogels when the dispersed phase is aqueous. Most hydrogels are formed by polymer networks and crosslinked polymer networks, usually surface modified to enhance hydrogen bonding interactions, thus trapping water in the matrix. These are the most common gel type found in everyday life such as food, cosmetics, and nappies. So ubiquitous are the presence of these materials in daily life that the word gel has become synonymous with polymer hydrogels. More and more hydrogels are being designed and discovered and the level of complexity is increasing.31 Hydrogels can range in properties from low to high mechanical strength,32 from low tack to highly adhesive,33 have shape memory,34 and are capable of converting chemical energy into mechanical energy.35
Sometimes the solid phase is the polymer and sometimes the liquid phase is the polymer, this is a good example of how tricky the classification of gel systems can be. For example a poly(ethyleneglycol) (PEG) hydrogel is where the PEG polymer is the solid continuous phase, and water is the dispersed phase, but a PEG gel can also be where PEG is used as a viscous liquid and solvent for the sol–gel processing of silica,36 (Fig. 5) in this case it is the dispersed liquid phase.37–39
 | ||
| Fig. 5 Poly(ethyleneglycol) (PEG) silica gels. Materials made with PEG as the liquid (dispersed) phase and silica as the solid (continuous) phase, (sol–gel preparation of TEOS with PEG as the solvent and template). Photo PCM 2004. | ||
 | ||
| Fig. 6 Structures formed on solvent removal (A) gel, (B) aerogel, (C) xerogel, and (D) ceramic/dense closed structure. | ||
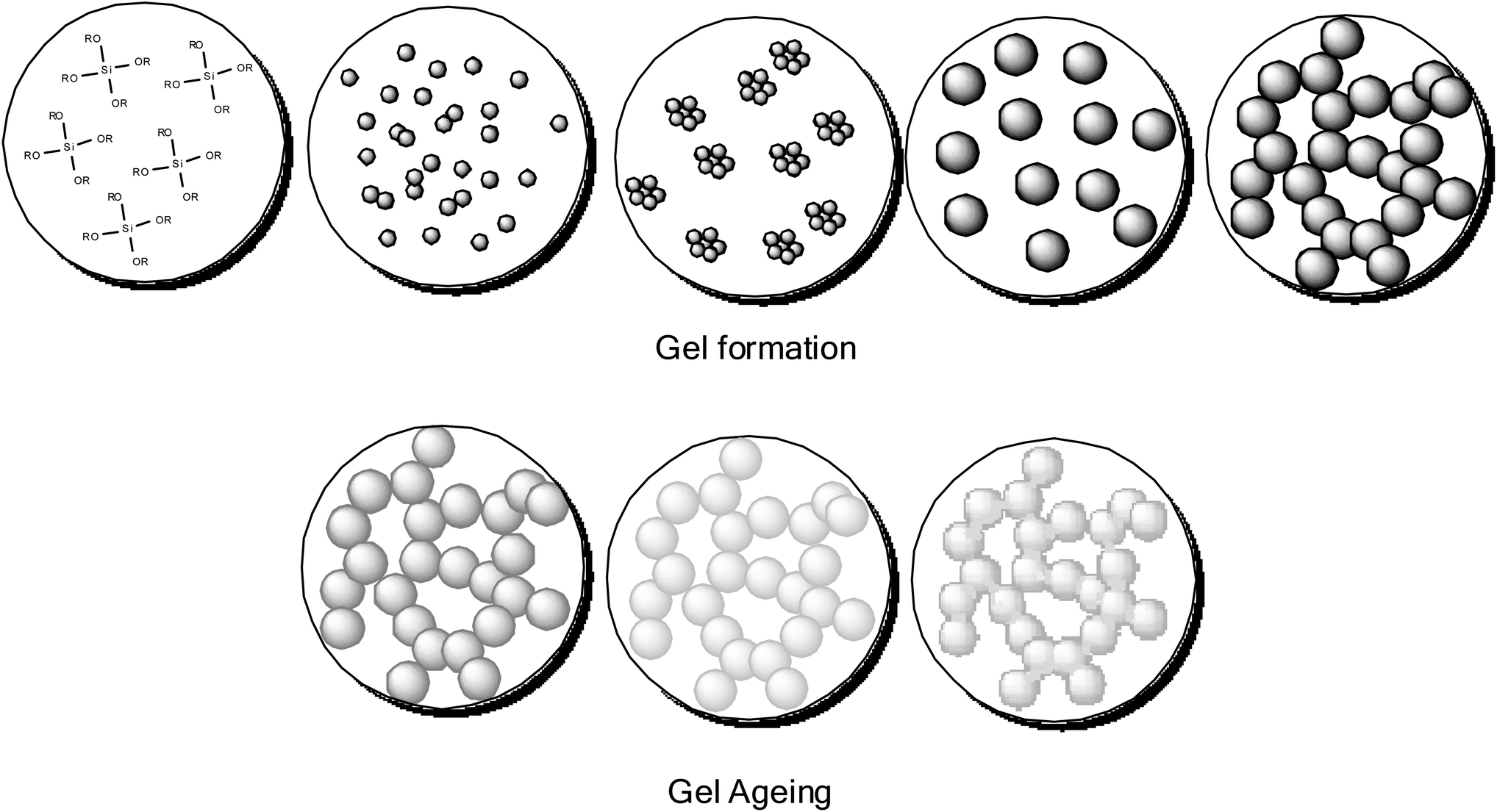 | ||
| Fig. 7 Sol–gel process (silicon alkoxide precursor) gel formation and gel aging. | ||
For example a typical traditional preparation of a silica gel using this method can be achieved by the dissolution of a silicon alkoxide (e.g. tetraethyl orthosilicate, TEOS) in alcohol (solvent), upon addition of a catalyst (either acid or base), forming a silica sol in alcohol. The sol aggregates and assembles into a gel trapping in the solvent. A gel in this state is an alcogel. The structure and size of the pores of the gel are determined by the solvent used, and the relative concentration of the solid and liquid components. Removal of the alcohol by evaporation leads to the formation of a xerogel. In this state some of the open porous structure is lost and the material becomes denser. Removal of the alcohol by supercritical drying with CO2 forms an aerogel, as the material retains its porosity. This is possible as drying supercritically allows the removal of the alcohol with minimum capillary forces acting on the internal structure of the solid network, allowing the porosity of the network to remain intact (Fig. 6). Irrespective of the starting alkoxide, the sol–gel process creates colloidal powders with controlled morphology, these colloidal particles exhibit the ability to aggregate to form a gel in a solvent. This low temperature approach to materials preparation is a versatile method and has been applied in many diverse applications. Processing from the sol state can produce many material forms, from fibres through to dense ceramics and aerogels. Percolation theory, and the closely related fractal theory are used to explain the growth and style of gels formed.41
2. Ionic liquid gels
From the most basic viewpoint ionic liquid gel materials comprise two components, an ionic liquid, and a continuous solid phase.Ionic liquids
Ionic liquids (ILs) are salts that are liquid. Just like common everyday salts they consist of positive (cation) and negative (anion) charges bound together by electrostatic interactions. However unlike solid salts that pack neatly together to form a crystalline structure, ionic liquids are composed of ions that do not pack well together, allowing a freedom of movement, and thus allowing the salt to take on the property of flow. The ionic liquid can be considered as a sea of charges incapable of organising into a highly regular structure, while still maintaining strong associations with each other (Fig. 8). | ||
| Fig. 8 Common ionic liquid cation and anions. | ||
A large combination of salts (1018 ionic liquids are theoretically possible)46 can be combined and functionality can be introduced on the anion or the cation, thus allowing tailored functional ionic liquids. The wide range of possible cations and anions means that the design of ionic liquids for functional applications is a dynamic area.
The beauty of ionic liquids is that virtually anything is possible. Many functional ionic liquids are being developed with a wide array of possible applications in mind. In the literature one can find a diverse range of ionic liquids and ionic liquid applications. Ionic liquids can be tailored to exhibit extremes of behaviour: to support cells and enzymes47,48 or kill microbes,49 have functional pharmaceutical therapeutic ability50,51 and be used in drug delivery,52 or have toxic tendencies,53 facilitate the cleanup and removal of environmental pollutants such as CO2,54–56 for metal extraction57–61 but with misuse can have the potential to cause environmental damage,62 designed to be biodegradable63–65 made from renewable resources such as amino acids66 or biomass,67 or made to be durable and long lasting. Ionic liquids can be hydrophobic, hydrophilic, acidic or basic. Research in to ionic liquids has led to the design of task specific/functionalised ionic liquids61 tailored explicitly for many applications, catalysis68–70 electrochemistry,71 conductivity,72 and perfume delivery.73 The use of ionic liquids in green chemistry was described in 200074 and since then the area has expanded in many directions, both in academia and in industry.75,76 New ionic liquids are designed daily, and many new directions will continue to be discovered. However liquid is not always the most desirable phase for a given application. The use of ionic liquids in some applications is dependent on the ionic liquid being a liquid, however there are applications which would benefit from the tailored functionality of an ionic liquid, but require that the functional component be a solid. Gel materials allow the functionality of the ionic liquid to be utilized by providing a more solid/semisolid structure (the continuous solid phase) whilst retaining liquid mobility within the dispersed liquid phase. What is now being understood is that the confinement of ionic liquids within pores or on thin films has a direct influence on the ionic liquid. Ionic liquid gels can offer the design of materials with controlled material strength from soft solid to solid and with varying pore sizes/shapes and surface interactions. The mobility of the liquid within the pores, and ordering of the ionic liquid have led to enhancements seen in catalytic activity,77 charge transport,78 and electrical conductivity.79,80 When in the gel state ionic liquids can be compelled to order when subjected to a confined space and/or an electric field80–82 and show bulk liquid dynamics83 or enhanced charge transport in nanoconfinement.84,85 Confinement in silica can also influence the melting point of the IL.83 This has been proposed to occur because the pore size changes the anion cation distance.86 Ionic liquids can show order in the liquid phase, ionic liquids are so varied that all types of liquid structure can exist. Influences such as the strength of charge between the cation and the anion, polarity of the molecules, the nature of any substituent groups, and whether the molecule is amphiphilic or nonamphiphilic, all contribute to the manifestation or absence of structure. Other influences such as temperature, and presence of solvent, can also change the self-assembly in the bulk phase. Ionic liquids can show surfactant behaviour, co-surfactant behaviour and liquid crystallinity,87 and show structures from bicontinuous, and micellar to liquid crystal, and transitions from micelles to vesicles or ribbons have been observed.88 A recent review on structure and nanostructure in ionic liquids discussed known observed morphologies.89 Interaction of an ionic liquid with a charged surface has been observed to further influence the self-assembly of the ionic liquid, by both experimental and theoretical study.80,90 Ionic liquid gel materials can provide a myriad of ways to confine/entrap and study the organisation of the ionic liquids when confined, and their study will offer new possibilities for ILs in catalysis, energy, and sustainability as these structure function relationships are better understood and controlled.
Ionic liquid gel: classification
Ionic liquid gel materials offer a symbiosis between the dispersed ionic liquid phase and solid continuous phase. The liquid dispersed/trapped inside a solid retains the function of the ionic liquid and therefore the gel can retain the favourable properties of ionic liquid, such as thermal stability and negligible vapour pressure, combined with the practicality of a solid. Generally these confined ionic liquids retain or display enhanced functional properties when contained within a gel material. The ionic liquids can play both a structural and functional role.This section will overview the different types of gel based ionic liquid material seen in the literature. The papers have been chosen to highlight examples of historical significance, and papers of special interest to catalysis and energy.
Ionic liquid gel: nomenclature
Using the current classification of gels by nature of the solvent, (Fig. 4) gels within which the liquid component is made up entirely of ions (an ionic liquid), can be classified as ionic liquid gels or ion gels.In the field of ionic liquid materials many naming systems are being adopted simultaneously to refer to ionic liquid gels, and these differences in terminology make this a difficult subject to reference. In many cases these terminologies suffer from the further complication that the same name can be used to describe materials that contain an ionic liquid, irrespective of whether the ionic liquid is immobilised by post treatment of a pre formed solid with an ionic liquid,91 or contained within a network (gel).92 In this review it is our intention to cover as many differently named variants as possible, ionic liquid and gel, ion gel(s), ionogel(s), ion-gel(s), ionic gel(s), confined ionic liquid(s), supported nanoliquid(s), ionic liquid templated material(s), ion jellys and bucky gels, to get a fuller picture of what is being reported. Further complexity comes from the fact that sometimes the newer term ionic liquid is interchanged with the wider term molten salt.93,94 The use of other closely related materials to ionic liquids such as deep eutectic mixtures/deep eutectic solvents (DES)95,96 zwitterionic liquids (ZIL)s and alkali metal ionic liquids (AMILs)97 can make finding the full range of gel materials made from ions difficult.
The most frequently used term is ionogel, commonly used to describe silica gels containing ionic liquid made by the sol–gel process.98 Sol–gel derived ionic liquid gels in which a large percentage (>10%) of the IL remains in the material are termed ionogels.99 However the term ionogel is also used more generally to describe any ionic liquid containing material, irrespective of the nature of the gelator,98 (colloidal, polymeric or supramolecular). In yet further usages ‘ionogel’ has been used extensively to describe materials that contain an ionic liquid confined within a polymer matrix (polymer gelator),100 for a gel formed by supramolecular self-assembly with a LMWG,101 and in the preparation of materials where a porous carbon structure is first formed and backfilled with an ionic liquid.102 The term ionogel has also been used to describe a material generated from an ionic liquid with silicon alkoxide (triethoxysilane) functional groups attached to the imidazolium cation, so that the cation of the ionic liquid is involved in the network formation. The anion is associated with the surface and often an additional silica precursor is added to enhance rigidity.103 This has led to different interpretations of the term ‘ionogel’ depending on the research community.
The term ion gel has been used to describe ionic liquid incorporated through the self-assembly of a triblock copolymer in a room temperature ionic liquid104 or prepared by in situ radical polymerization of vinyl monomers in an ionic liquid105 to describe the dispersion of colloidal particles in an ionic liquid leading to the formation of a gel such as the dispersion of silica nanoparticles made in ionic liquid.106 The related term ionic gel was used to describe a quasi-solid state ionic gel for use in a dye sensitised solar cell (DSSC).107 The term ‘supported nanoliquid’ was coined by Deng and co-workers in a paper which described a material with a sol–gel silica and an ionic liquid.108 Confined ionic liquids and ionic liquids in confined geometries are described in a number of different communications.80 Such confined ionic liquids have shown enhanced charge transport,85 bulk liquid-like mobility77,83,109 and displayed unusual spectroscopic behaviour in FT-IR and FT-Raman.110 In addition to these commonly adopted naming systems, there have been several studies that have adopted rare or one-off names. The term ion jelly was used to describe the formation of ion gels using gelatine as the solid (continuous phase of the gel),111,112 and the term bucky gels113 is used to describe gels made by the inclusion of carbon nanotubes into ionic liquids.114 Other examples include zwitterionic liquid compounds (anion and the cation on the same molecule) have been of specific interest in battery applications and polymer gel electrolytes from zwitterionic liquids have been synthesised.115 Gel formation with DES was shown by the use of DES to produce a hierarchical porous carbon monolith through the polycondensation of formaldehyde DES gel and the transformation of this into a porous carbon material 600 m2 g−1 a by carbonization.116 Similar techniques were used to form carbon and carbon nanotube composites where the DES was removed after initial templating, therefore the final material was not a gel but the formation of the gel lead to the templating of the porous microstructure.117 These ZIL gels and DES gels will continue to provide further examples of interesting materials for many applications. Similarly alkali metal ionic liquids (AMILs) have been reported as gels with a polymer as the continuous phase.118 Molten salt gel examples are found in the stabilization/solidification of radioactive molten salts.119 Further examples, one off names and acronyms are discussed and treated individually as they arise.
Since the liquid component of the gels in this case are all ionic liquids or ionic liquid variants, a good way to differentiate between ionic liquid gels is by the nature of the solid component. Using the gel classification by nature of the gelator we find examples of each type of ionic liquid gel. The three gel systems are evident in the literature for the formation of ionic liquid gels: macromolecular/polymer, colloidal, and molecular (low molecular weight gelators (LMWG)).
Macromolecular/polymer ionic liquid gels
These Ionic liquid gels can be formed in a number of ways. Gels have been prepared by the in situ polymerization of vinyl monomers in an ionic liquid,105,120,121 and by dissolution and casting to form flexible membranes.122 In one example a gel was formed by in situ polymerisation of poly(ethyleneglycol) diacrylate with an ionic liquid, and the resulting material was referred to as a gelled ionic medium.123 Photoreversible polymer ionic liquid gels124 and thermoreversible125 polymer ionic liquid gels have been reported. The addition of actives into a polymer ionic liquid gel showed the ability to make magnetic gel materials termed MagIGs.126 These gels were prepared from poly(N-isopropylacrylamide) (as the continuous phase), Trihexyl(tetradecyl)phosphonium Dicyanamide (IL), and dispersed iron oxide nanoparticles. Gels can also be formed from natural polymers (biomacromolecules). Kadokowa and co-workers synthesised a gel of chitin with an ionic liquid, (1-allyl-3-methylimidazolium bromide)127 and chitin/cellulose128 where chitin/cellulose composite gels and films were made by dissolving chitin and cellulose in two ionic liquids, 1-allyl-3-methylimidazolium bromide and 1-butyl-3-methylimidazolium chloride ([BMIM][Cl]). The mixture was heated and left to gel, which took 4 days. Gels have also been prepared using a combination of chitin and agarose as the solid phase.129 Ionic liquid gels have also been synthesised based on gelatin (ion jelly).111,112,130Colloidal ionic liquid gels
In the initial ionic liquid gels made by the sol–gel method the ionic liquids were used as a template and the ionic liquid was removed after the material was formed.131,132 In the same vein ionic liquids and DESs have been used successfully as templates in the preparation of mesoporous solids where the IL was removed after the material was prepared.133–135 As the IL was removed to reveal the intact porous structure, these materials are no longer considered to be in the gel state. The nature and amount of an ionic liquid contained in a silica ionogel has a direct influence on the structure of the solid continuous phase.136,137 Tin dioxide ionogels have also been formed using a sol–gel approach138 The formation of gels from ionic liquids and preformed silica particles gives rise to nanocomposite gel electrolytes,106,139,140 and similarly TiO2 spheres were used to prepare self assembled gels with ionic liquid,141 from a mixed system of emulsified [BMIM] bis(trifluoromethane)sulfonimide ([BMIM][NTF2]) and TiCl4 in an emulsion using methanol as the solvent and formic acid as the catalyst. Nanocomposite quasi-solid electrolyte containing ionic liquids from hybrid organic inorganic sol–gel precursors known as ormosils (organically modified silica) have also been prepared.142,143 The inorganic oligomer poly(dimethysiloxane) (PDMS) was synthesized with triethoxysilylethyl terminated end groups. This sol–gel precursor was then used to form gels with 80% 1-ethyl-3-methylimidazolium [NTf2] ([EMIM][NTf2])144 Another example of modification of a sol–gel alkoxide precursor involves the bonding of the cation of the IL to the silicon alkoxide forming a hybrid sol–gel precursor, (the anion is associated) the sol–gel precursor is mixed with TMOS and the sol–gel reaction is initiated. In this case there is no free ionic liquid acting as the dispersed phase and the only liquid present comes as a by-product of the sol–gel reactions (ethanol and methanol).103The ordering of molten salts by the addition of carbon nanotubes has also been employed to form a gel,113,114 and modified graphene oxide ionic liquid gels have been reported.145 The sol–gel synthesis of ionic liquid gels have been shown to have an influence on the particle size colour and structure of silica made by the sol–gel process.146 Gel materials for catalysis will be discussed in section 3.
Molecular/supramolecular low molecular weight gelators (LMWG)
Low molecular weight gelators (LMWG) are most frequently shown to have the ability to gel organic solvents and water creating organogels and hydrogels by self-assembly and physical interactions. The solvent used is volatile and loss of the solvent always results in a loss of structure in such soft systems. Replacing an organic solvent with an ionic liquid creates soft ionic liquid gels with longevity as the low vapour pressure of the ionic liquid stabilises the gel, and extends the possible uses of these LMWG gels. Kimizuka and Nakashima noticed a gelation occurring when they used an ether containing ionic liquid to dissolve amide-group-enriched glycolipids.101 Again gels were formed with a cholesterol based structure with a 2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl group as a gelator. The formation of a gel by this method requires the LMWG to be soluble in the liquid before the reorganisation and gelation occurs, however in this case the gelator was not soluble in the IL used (N,N′-dialkyl-substituted imidazolium ions and N-alkyl pyridinium) and required a co-solvent (acetone), gelation occurred on evaporation of the acetone from the mixture.147 Ionic liquid gels as electrolytes will be discussed further in section 4. Quasi-solid-state electrolytes made with LMWGs, were made to combine the benefits of liquid electrolytes and solid electrolytes, and impart enhanced mechanical stability.148 LMWG ionic liquid gel made from a molecular gelator9 for 1-alkyl-3-methylimidazolium iodide (alkyl: C3–C9), were reported which showed temperature stability up to 85 °C.149 Hanabusa and co-workers150 also tested twenty-four kinds of ionic liquids with twelve kinds of LMWGs, and found two excellent gelators, both based on aspartame. Both molecules can gel ionic liquids at a minimum concentration of lower than 1 wt%. The ionic conductivities of the ionic liquid gels were very similar to those of the pure ionic liquids. In another example 12-hydroxystearic acid has been observed to act as a LMWG to form a gel with 1-hexyl-3-methylimidazolium [NTf2] ([HMIM][NTf2]).151 Tan and co-workers have used bis(4-octanoylaminophenyl) ether, bis(4-octanoylaminophenyl) methane and 2,4-bis(octanureido) toluene to gel ionic liquid, and they found that the electrochemical properties of the ionic liquid gels were similar to those of the pure ionic liquid.152 Grätzel and co-workers presented quasi-solid-state DSSC using the LMWG 1,3:2,4-di-O-benzylidene-D-sorbitol (DBS)153 to gel ILs as an electrolyte for a DSSC.154–156 LMWG of biological origin (amino acids) have also been shown to form gels with ionic liquids.157 Metallogelators, LMWGs containing a metal, are a subset of LMWGs.158 Organometallic LMWG have been prepared from pyridine bridged bis(benzimidazolylidene)–palladium complexes. These carbene complexes have been shown to be efficient low-molecular-mass metallogelators for organic solvents and ionic liquids (such as imidazolium, pyridinium, pyrazolidinium, piperidinium, and ammonium salts) at concentrations as low as 0.5 mg mL−1.159Ionic liquids as gelators
There are examples of ionic liquids that have been reported as capable of acting as molecular gelators for organic solvents (organogelators). Organic salts have charge-assisted hydrogen bonds which are stronger than average hydrogen bonds in normal organic compounds160 and this plays an important role in self-assembly, gel formation, strength, morphology, and longevity. D'Anna, Noto and co-workers investigated the ability of some diimidazolium salts as LMWG for organic solvents.161 The ability of an ionic liquid to gel an organic solvent seems not to be determined by the cation structure, (alteration of the shape and size was investigated). However it was found that the overall properties and structural features of the obtained gel phases seem to be ruled by the anion properties. The gelling ability significantly improved on moving from mono to dianions. This is in agreement with what was found by Hanabusa and co-workers.150 In their study changing the cation had no effect on the ability of the gelator to form gels when the anion remained the same. Cross-linked polymeric nanogels (CLPNs) have been reported, prepared by one-step cross-linking copolymerization of ionic liquids featuring geminal dicationic monomers and cross-linkers for the preparation of methanol organogels.162 The copolymerisation and cross-linking of tetrabutylphosphonium styrenesulfonate and tributyl-hexyl phosphonium 3-sulfopropylacrylate, have been shown to form thermoresponsive poly(ionic liquid) hydrogels.163 Addition of poly(N-isopropylacrylamide) (PNIPAAM) into the gel enhanced and swelling and shrinking properties of these polymeric ionic liquid hydrogels.164 In these cases as the ionic liquid is the solid phase, they are not ionic liquid gels as the liquid phase is reported as water (hydrogel) or an organic solvent (organogel). Gels constructed from poly(ionic liquids) as the solid (continuous phase) and ionic liquid as the liquid phase have been shown to have applications as electrolytes.165 As ionic liquids can self-organise into bulk liquids, surfactants, liquid crystals or semi ordered/solids,89,166 and we have seen that some ILs have the ability to themselves behave as gelators for solvents, perhaps one ionic liquid could self assemble with another different ionic liquid to form domains where one ionic liquid acts as the liquid phase and the other as the solid continuous phase.3. Ionic liquid gel catalysts for chemical transformations
Catalytic materials consisting of active catalysts entrapped in ionic liquid gels are discussed in the following sections. Catalysts which comprise ionic liquids layered on solid matrices, or ions covalently bonded to a solid are not covered. These systems, often referred to as SILCs, SILPS and SCILs, have been reviewed elsewhere.167–172 Also not covered in this section are the use of poly(ionic liquids) as catalysts. This has been also been reviewed elsewhere.173 Soft gels used in electrocatalysis will only be briefly mentioned, and will be discussed further in section 4.The entrapment of an active catalyst inside a solid oxide matrix by sol–gel methods was pioneered by Blum and Avnir174 in the 1990s.175 The extension of the method to ionic liquid gels followed the introduction of ionic liquid based methods for the formation of silica gels in 2000.131 In the subsequent sections the catalysts entrapped have been classified as nanoparticle (supported metal) catalysts, organometallic catalysts, functionalised ionic liquids or biocatalysts.
Nanoparticles entrapped in ionic liquid gels
Following the synthesis of catalysts by the sol–gel entrapment of Pd nanoparticles in silica assisted by supercritical fluids,176,177 in 2003 P. C. Marr and co-workers combined the facile synthesis of metal nanoparticles in ionic liquids, with the sol–gel method, to prepare catalytic gels.132 [Pd(OAc)2] (OAc = acetate) and PPh3 were heated in [BMIM][NTf2] to form a suspension of nanoparticles.178 These were subsequently entrapped in a stiff gel upon addition of TEOS, and formic acid (as the gelation catalyst). The aged and washed silica contained highly dispersed Pd nanoparticles of a fairly uniform size (approximately 2 nm diameter) at 0.37 wt% loading. The catalytic activity was demonstrated for the Heck coupling of iodobenzene and butyl acrylate and the selective hydrogenation of cinnamaldehyde to hydro cinnamaldehyde (Scheme 1). | ||
| Scheme 1 Heck–Mizoroki coupling and hydrogenation catalysed by Pd nanoparticle doped IL gels. | ||
The synthesis of Rh nanoparticles on silica by an analogous method was published by Marr and Marr in 2009 and the catalytic activity demonstrated for styrene hydrogenation (Scheme 2).77
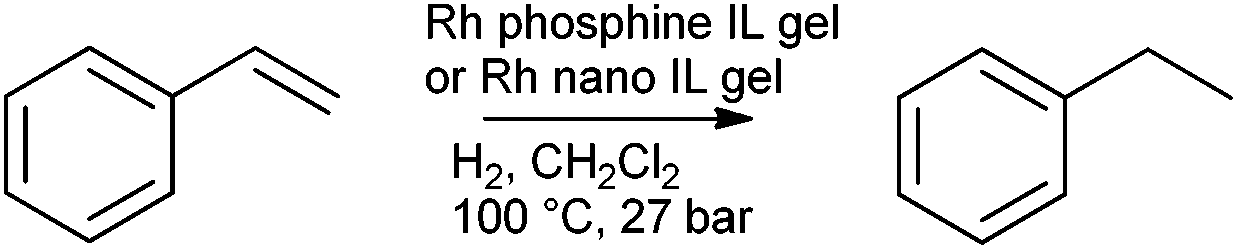 | ||
| Scheme 2 Styrene hydrogenation catalysed by rhodium doped IL gels. | ||
A silica gel doped with Rh nanoparticles was prepared by entrapment of a Rh colloid, prepared by the thermal decomposition of a Rh(I) complex under hydrogen in [BMIM][NTf2], the material had a high surface area (563 m2 g) and was an active hydrogenation catalyst, however lower activities and poorer recycling were observed than that obtained for catalysts prepared by entrapping Rh(I) complexes in analogous ionic liquid gels.77 Building on their extensive expertise on the synthesis of metal nanoparticles in ionic liquids, in 2007 Santos, Dupont and co-workers published sol–gel methods for the preparation of silica doped with 0.1 wt% Rh(0) nanoparticles by ionic liquid assisted sol–gel methods.179
Preformed nanoparticles were added in to mixtures for the preparation of [BMIM][BF4] silica gels from TEOS. Acid (HF or HCl) and base (NH4OH) catalysed routes were compared. The resultant gels were used as catalysts for neat 1-decene and cyclohexene hydrogenation. Acid catalysed synthesis was found to yield better catalysts. Catalysts could be recycled at least ten times without significant loss of activity and selectivity. The activity and stability of the catalyst was attributed to a higher quantity of co-entrapped ionic liquid. Later Pt nanoparticles were entrapped for benzene and aliphatic alkene hydrogenation (Scheme 3).180
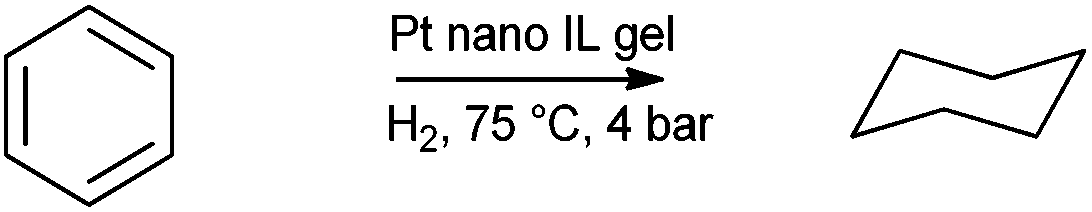 | ||
| Scheme 3 Benzene hydrogenation catalysed by Pt nanoparticle doped IL gels. | ||
Recently Ru nanoparticles in silica ion gels have been applied to the low temperature Water Gas Shift reaction (WGS) (Scheme 4).181
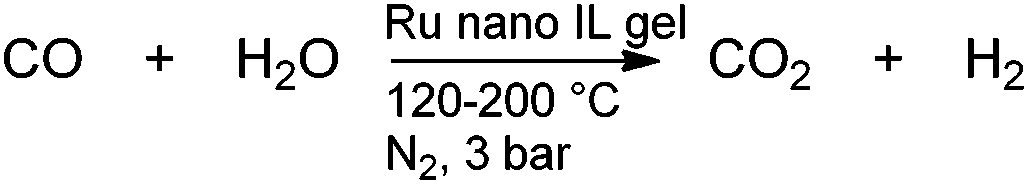 | ||
| Scheme 4 Water Gas Shift catalysed by Ru nanoparticles in IL. | ||
The gel preparation method was similar to that employed by Santos, Dupont and co-workers. Ru nanoparticles were prepared by reaction of [Ru(NO)(NO3)3] with NaBH4 in [BMIM][BF4]. Again the materials prepared in acid were found to outperform those prepared using base. This is a rare example of a gaseous phase application of a gel catalyst. The catalyst, prepared by sol–gel in the presence of HCl, was found to outperform an analogous calcined mesocellular foam silica catalyst. CO conversion of 37.8% was achieved at 140 °C. The catalyst exhibited high stability; after 25 h 36.2% CO conversion was achieved.
Han and co-workers have prepared supported metal catalysts with hierarchical pores by employing a templating procedure.182 An ionic liquid–water–CaCl2/MgCl2 gel was formed using 1-octyl-3-methylimidazolium chloride ([OMIM][Cl]), and used as a template for the formation of silica by sol–gel from TEOS. The chloride gel could be removed, and metal precursor ([HAuCl4] for gold catalysts and [RuCl3·3H2O] for Ru catalysts) reduced in one step using a solution of NaBH4 in water. The formation of small nanoparticles of narrow size distribution was observed. The unique structure of the CaCl2 gel was believed to assist the formation of small metal nanoparticles. Metal doping of 1–8 wt% was achieved. Catalytic activity was demonstrated for oxidative esterification (Au), oxidation of benzyl alcohol to benzaldehyde (Pd) (Scheme 5) and hydrogenation of benzene (Ru). High activities, selectivities and stabilities were observed.
 | ||
| Scheme 5 Oxidative esterification catalysed by Au nanoparticles in an IL gel with hierarchical pores. | ||
The use of ionic liquid–water gels to template the formation of a silica supported metal nanoparticle catalysts created materials with unique structures that would be difficult to achieve by other methods. The presence of hierarchical pores maximises mass transfer, helping to create an active catalyst, whilst providing a stable system with low leaching. The entire process, including metal entrapment and silica growth, was performed without intermediate separation; making the catalyst synthesis operationally simple.
Safavi and co-workers183 demonstrated the application of nanoparticle doped ionic liquid gels to electrocatalysis. Ionic liquid (octylpyridinum [PF6] or imidazolium-based phosphinite) and Pd nanoparticle doped gels were coated on a carbon ionic liquid electrode surface by a sol–gel method from TEOS catalysed by HCl. Good electrochemical and electrocatalytic performances were demonstrated. Advantages of this composite coating, compared to conventional silica gel layers, included increased stability and crack resistance, and good mass transport and conductivity.
The success of these methods can be attributed to the unique environment provided by ionic liquid gel materials. The ability of ionic liquids to enable the facile synthesis of metal nanoparticles is well known,173 and can be attributed to the stabilisation of the nanoparticles within the ionic liquid phase. The creation of an ionic liquid phase inside a porous solid not only stabilises the nanoparticles, but also creates a heterogeneous catalyst that is simple to separate from the product mixture.
Metal complexes entrapped in ionic liquid gels
In 2005 catalysts prepared by entrapment of metal complexes within ionic liquid–silica gels were introduced by Deng and co-workers,108 and Marr, Marr and co-workers.184 These methods were inspired by the catalyst entrapment methods developed by Blum and Avnir in the 1990's.174,185Both groups employed acid catalysed gelation methods, and observed the reliable formation of stiff gels containing the metal catalyst precursor. Deng and co-workers reported ionic liquid gel catalysts doped with [RuCl2(H)(PPh3)2], [RhCl(PPh3)3], [PdCl2(PPh3)2] and [CoCl2(PPh3)3] for the carbonylation of amines and nitrobenzene (Scheme 6).186
 | ||
| Scheme 6 Carbonylation of amines and nitrobenzene catalysed by metal phosphine doped IL gels. | ||
A range of ionic liquids were investigated including 1-decyl-3-methylimidazolium [BF4] ([DMIM][BF4]), [EMIM][BF4] and [BMIM][BF4]. Use of the DMIM cation enabled the ionic liquid to be retained in the gel upon recycling. Deng and co-workers termed this class of catalyst “nanoliquid” catalysts,108 as the reaction is performed by a dissolved catalyst within the filled pores of a gel. Raman spectra revealed that the confined ionic liquid was affected by the unusual environment (pores). The gel doped with 0.11 wt% Rh and 35 wt% [DMIM][BF4] was found to be most active, giving up to 93% conversion and >98% selectivity, and turnover frequency of over 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1. Activity was enhanced relative to homogeneous operation. Upon recycling some loss of activity was observed. An analogous SILP system gave very poor results.186 Marr, Marr and co-workers developed a recyclable hydrogenation catalyst by entrapping [RhCl(PPh3)3] in an ionic liquid silica gel.184 The complex was entrapped within silica by employing a formic acid promoted gelation of TEOS in [BMIM][NTf2]. The catalyst was found to hydrogenate styrene in dichloromethane or toluene, with leach rates lower than the limit of detection. Facile recycling could be achieved without the need for intermediate treatment of the gel catalyst, this set these methods apart from previous gel catalyst systems. Initially the synthesis, catalytic activity and recycling were reported. In a later report77 the catalyst was characterised by a range of solid state techniques, including solid state NMR and XPS. This revealed the presence of ionic liquid before and after catalytic activity. The catalytic activity was compared to the parent organometallic catalyst precursor. The average performance (by turnover frequency) was found to be greater in the gel than the initial rate of the homogeneous catalyst. The effects of changing parameters such as particle size were investigated, and the catalyst was shown to be active also for the challenging renewable substrate levoglucosenone.
000 h−1. Activity was enhanced relative to homogeneous operation. Upon recycling some loss of activity was observed. An analogous SILP system gave very poor results.186 Marr, Marr and co-workers developed a recyclable hydrogenation catalyst by entrapping [RhCl(PPh3)3] in an ionic liquid silica gel.184 The complex was entrapped within silica by employing a formic acid promoted gelation of TEOS in [BMIM][NTf2]. The catalyst was found to hydrogenate styrene in dichloromethane or toluene, with leach rates lower than the limit of detection. Facile recycling could be achieved without the need for intermediate treatment of the gel catalyst, this set these methods apart from previous gel catalyst systems. Initially the synthesis, catalytic activity and recycling were reported. In a later report77 the catalyst was characterised by a range of solid state techniques, including solid state NMR and XPS. This revealed the presence of ionic liquid before and after catalytic activity. The catalytic activity was compared to the parent organometallic catalyst precursor. The average performance (by turnover frequency) was found to be greater in the gel than the initial rate of the homogeneous catalyst. The effects of changing parameters such as particle size were investigated, and the catalyst was shown to be active also for the challenging renewable substrate levoglucosenone.
The unprecedented performance of this class of gel catalyst relative to homogeneous and traditional heterogeneous catalysts may be attributed to the creation of a unique environment inside the gel for catalytic activity. This is analogous to the action of enzymes, within which the active site operates ultra-efficiently due to specific intermolecular interactions in wet soft matter. The addition of ionic liquid to the traditional entrapped system will provide intermolecular forces akin to those provided by the enzyme polypeptide, and the confinement of the ionic liquid within the pores of the matrix provides local ordering effects. For reactions in which ionic liquid solvents provide a particular benefit (such as the carbonylation of aniline186) incorporation of the ionic liquid within a gel enables liquid like reactivity, whilst employing a much smaller amount of ionic liquid. Furthermore the confinement can have the effect of creating a concentrated ionic liquid reaction environment, leading to enhancement of reactivity beyond that observed in the liquid phase.108,186 These benefits are in addition to the advantages in separation gleaned from heterogenising the catalyst.
The hydroformylation study of Hamza and Blum is worthy of mention,187 (although it does not apply the same methodology), as an example of the related tethering procedure for preparing catalytic gels. A recyclable gel catalyst for the hydroformylation of alkenes (styrene and derivatives) was prepared from tetramethyl orthosilicate (TMOS), 1-butyl-3-[3-(trimethoxysilyl)propyl]imidazolium chloride, [Rh(COD)Cl]2 (where COD is 1,5-cyclooctadiene), and sodium 3-(diphenylphosphanyl)benzenesulfonate dehydrate. High selectivity (>95%) towards branched products was observed (Scheme 7).
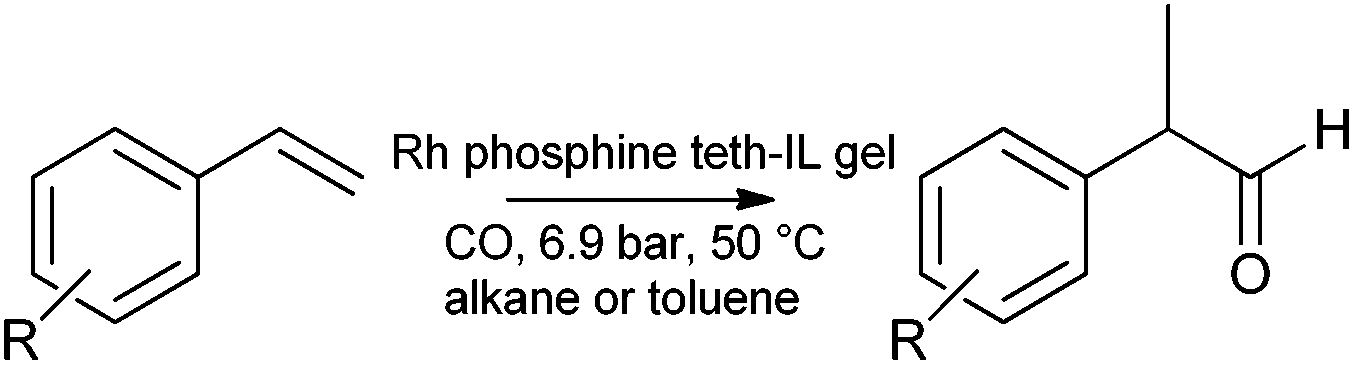 | ||
| Scheme 7 Hydroformylation of vinyl arenes by an ionic liquid tethered gel doped with rhodium and phosphine. | ||
The catalyst could be recycled without leaching for at least 4 runs. The formation of a recyclable catalyst was found to depend on the inclusion of both the silica and ionic components. This investigation highlighted the importance of the ionic component for stabilising and retaining the active metal dopant. Omission of the ionic component led to an unstable system.
Although tethering ions reliably produces a virtually leach-free system, there are major disadvantages over the lifetime of the catalyst. Tethering renders the catalyst more expensive and wasteful to prepare, with many more synthetic steps, and the spent catalyst is much harder to recycle. A major advantage of self-assembled catalytic gels is that the components can be removed and recycled after use. Many ionic liquid gels are stable in one solvent system (such as toluene), but unstable in more polar solvents, leading to facile extraction of the ionic liquid phase for reuse.
In 2009 Pd(OAc)2 doped ionic liquid gels were reported by Vioux, Lavastre and co-workers.188 The Heck–Mizoroki coupling of iodobenzene and ethyl acrylate; and 4-vinylpyridine and 4-iodo-N,N-dibutylaniline (in toluene containing NEt3) was demonstrated, catalysed by Pd [BMIM][Br] ionogels in the presence and absence of PPh3. For the phosphine promoted system an active catalyst could be prepared with one tenth of the concentration of Pd. The catalyst was shown to be heterogeneous by hot filtration, and exhibited comparable activity to analogous homogeneous systems.
Following on from an ion tethering method detailed by Pagliaro and co-workers189 a gel catalyst for the oxidation of alcohols was prepared by Tong and co-workers.190 The CuCl2/TEMPO catalytic system was dependent on tethering TEMPO to an imidazolium cation, but the bulk material was a [BMIM][PF6] silica gel prepared from TEOS with the addition of HCl. The oxidation of alcohols (most successfully benzyl alcohols) was affected in n-octane under 1 Atm. of O2. The gel catalyst was reused 5 times, upon which the conversion gradually reduced, but the high selectivity to the carbonyl product was unaffected.
Entrapped functional ionic liquid catalysts
Early in the evolution of ionic liquid gel catalysts Deng and co-workers191 introduced the concept of entrapped functional ionic liquid catalysts. This premise depends upon the retention of the ionic liquid within the matrix as the ionic liquid acts as the catalyst directly. As such this is the purest form of ionic liquid gel catalyst as it requires only two components, solid matrix and ionic liquid.Carboxylic acid functionalized imidazolium and pyridinium chloride, bromide, PF6− and BF4− ionic liquids were entrapped by a sol–gel method from TEOS catalyzed by HCl. These Brønsted acidic ionic liquid (BAIL) gel catalysts were used to deoximate aliphatic and aromatic oximes in acetone and water (Scheme 8).
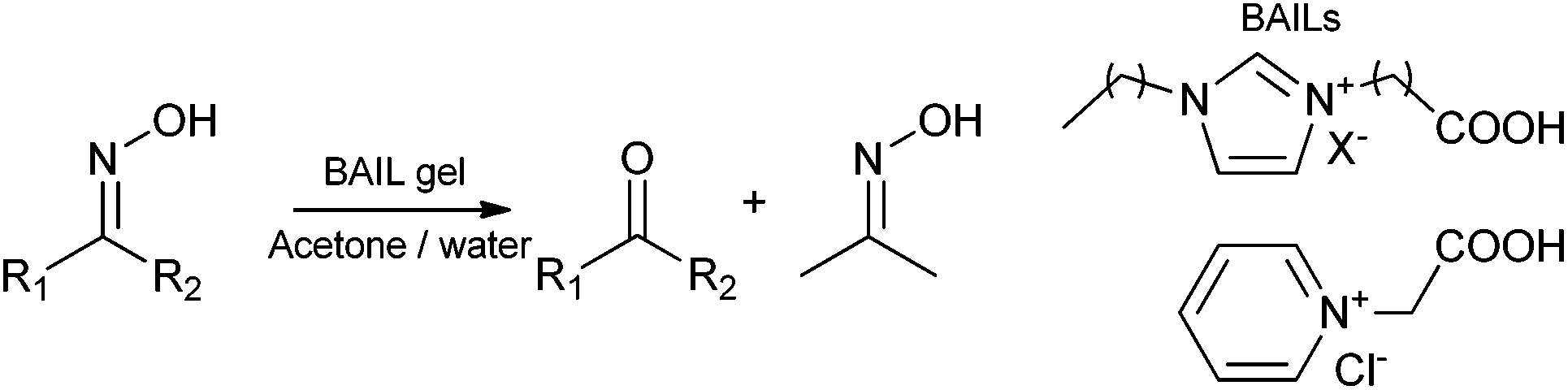 | ||
| Scheme 8 Deoximation of oximes catalysed by BAIL gels. | ||
In the following decade there followed reports of tethering acidic ionic groups onto silica to form catalytic gels, and acidic ionic liquids absorbed on the surface of silica; but little progress on the synthesis of recyclable ionic liquid gel catalysts.192–196
It is good practice in green chemistry to attempt to remove reagents that may be superfluous in a synthesis or process, as every material used has an associated legacy of waste and energy consumption. Therefore it was logical to develop the synthesis of a gel catalyst that dispensed with an additional gelation catalyst, such as HCl in the Deng and co-workers syntheses. To this end Marr, Marr and co-workers197 attempted the gelation of tens of BAILs in the absence of an additional promoter, and found that triethylammonium propanesulfone [NTf2] ([TEAPS][NTf2]), treated with TEOS formed stiff reproducible gels, that could be applied to liquid phase catalytic transformations in toluene. The excellent recyclability of the catalyst was demonstrated for the dehydration of rac-1-phenyl ethanol (Scheme 9), and the gel-phase activity was proven by hot filtration test. The excellent retention of the ionic liquid was contrasted to the high leaching observed for an analogous SILP system. Solid state NMR revealed that the sol–gel synthesized ionic liquid gel contained a large proportion of silanol groups, these are able to hydrogen bond to the ionic liquid and assist the retention of the BAIL.
 | ||
| Scheme 9 Dehydration of rac-1-phenyl ethanol catalysed by a BAIL gel. | ||
These systems benefit from being two component catalysts. Their assembly is straightforward and low energy, and subsequent recycling of the ionic liquid involves only extraction using a polar solvent, leaving behind only silica. They have the potential therefore to be some of the “greenest” catalysts available. Panzer and co-workers198 showed that, in some cases, recovery of a hydrophobic ionic liquid from a silica matrix is achievable by submerging the ion gel in water. This is thought to be because the water competes for the silica surface, pushing the ionic liquid out.
Entrapped biocatalysts
Ionic liquids have been shown to be excellent media for certain classes of enzyme.199–201This can be attributed to advantageous protection of the protein structure by the ionic liquid. This was swiftly verified, as merely coating of an isolated enzyme in an ionic liquid can improve the performance of the biocatalyst in organic solvents.202
The entrapment of isolated enzymes within silica by sol–gel methods has been long established as a method of increasing the stability and making the biocatalyst easier to handle.203 The combination of ionic liquid coating and silica entrapment is therefore a logical progression for the immobilisation of enzymes. Li and co-workers204 entrapped horseradish peroxidase (HRP) in [BMIM][BF4] doped silica by treating a TEOS solution with small quantities of an HCl solution. The enzyme was entrapped whilst in phosphate buffer, (pH 6.86). Performance was compared with a gel prepared in the absence of ionic liquid. Compared to the traditional silica matrix, the ionic liquid gel afforded superior activity (specific activity was 30 times higher), and excellent thermal stability.
Koo and co-workers205 demonstrated the stabilizing effect of an ionic liquid doped gel on Candida rugosa lipases. A range of ILs were co-entrapped with the enzyme and tested in esterifications, the best results were observed with [OMIM][NTf2]. The difference in performance was even more marked, as the conventional sol–gel entrapment virtually killed the activity of the enzyme. Later the same year the group published a modified procedure, employing a mixture of a more hydrophobic ionic liquid and a less hydrophobic one, HCl was still used as the gelation catalyst.206
Highly recyclable enzyme doped gel materials were prepared by Ribeiro and co-workers.207 Naringinase doped gels were prepared containing combinations of different ionic liquids and glycerol as co-dopants. TMOS and HCl were used to generate silica. Co-entrapment of glycerol was found to be beneficial. The stability of the entrapped enzyme depended on the cation used, with hydrophobic cations such as [OMIM]+ conferring the best recyclability. Hydrophobic anions such as [BF4]−, [PF6]− and [NTf2]− were also necessary to obtain the best performance. Protic ionic liquids (Scheme 10) formed by the combination of monoethanol amine and aliphatic carboxylic acids of various chain lengths were used to stabilize lipase entrapped in silica by Soares and co-workers.208 The group also observed a vast improvement in enzyme performance compared to entrapment without ionic liquid. More hydrophobic ionic liquids were again found to be superior.
 | ||
| Scheme 10 Protic ionic liquids co-entrapped with lipases. | ||
The potential of ionic liquid gel catalysts
The potential of ionic liquid gels has been demonstrated across many fields of catalysis. Ionic liquid gels provide facile nanoparticle entrapment, enhanced homogeneous catalyst performance, contamination free Brønsted acidity, and protective environments for biocatalysis. Clear advantages are apparent when using an ionic liquid gel rather than a liquid phase, heterogeneous or SILP catalyst. Heterogenising catalysts in this way not only improves separation, but also conserves liquid like reactivity; and in some cases confinement enhances liquid reactivity. Ionic liquid gel methods have led to catalysts with unique structure due to templating effects. The absence of volatile solvents in the material synthesis avoids damage to the matrix during catalyst preparation. These advantages combine to provide catalysts with remarkably low leach rates. The range of ionic liquids and catalysts available is constantly expanding, and therefore catalytic ionic liquid gels will continually evolve, providing innovative solutions to the challenges emerging in chemical synthesis.4. Applications of ionic liquid gels in energy
Ionic liquids have a great potential in energy applications and have been the subject of several highlights and perspectives.71,97,209,210Applications include use as capacitors211/supercapacitor materials212 and in electrochemistry.213,214 The production and storage of energy will continue to gain importance over the coming decades, and ionic liquid gels are anticipated to find an increasing number of applications in this field. In this section some examples of ionic liquid gel materials classified by the nature of the gelator, are given, including macromolecular (polymeric), colloidal and molecular LMWG as electrolytes. Specific examples are cited of ionic liquid gel materials in applications such as DSSCs, low energy displays, electrocatalysis, capacitors/supercapacitors, and fuel cells. The drive for energy production to be sustainable and clean is a significant priority. At the heart of a new energy landscape will be versatile materials manufactured in a green way.
Ionic liquid gels as electrolytes
Polymer electrolytes, have changed the face of modern batteries.215–221 The use of ionic liquids as electrolytes and/or hosts for other electroactive species has enormous potential that has yet to be harnessed.71 Ionic liquid gels are providing interesting and versatile electrolyte materials from soft solid to solid, and have shown both enhancement and retardation of conductivity.Macromolecular/polymer ionic liquid gels
Ionic liquid gels made with polymers as the solid phase have been the longest studied IL gel systems applied as electrolytes. In 1996 rubbery gel electrolytes were reported made with N,N,N,N-tetraethylammonium BF4 ([TEA][BF4]), polyvinylidene fluoride (PVdF) homopolymer, and propylene carbonate (PC) as the solvent possessing room temperature conductivity of 6.3 mS cm−1. These measurements were made with the PC remaining in the gel.222 In a later report120 the solvent (PC) was removed before the conductivity was measured, in this case the solid continuous phase of these polymer ionic liquid gels was provided by a poly(vinylidene fluoride)-hexafluoropropylene copolymer [PVdF(HFP)]. Two ionic liquids were tested [EMIM][OTf] (where OTf is the triflate anion) and [EMIM][BF4]. Ratios of polymer to ionic liquid 2:1 [EMIM][OTf]–PVdF(HFP) and 2:1 [EMIM][BF4]–PVdF(HFP) gels were prepared having conductivites of 5.6 and 5.8 mS cm−1, respectively, absence of solvent didn't make an appreciable difference to the conductivity.
Another early example of an ionic liquid polymer gel electrolyte was reported by Nakagawa and co-workers.123 The gel was formed by in situ polymerisation of poly(ethyleneglycol) diacrylate with a mixture of [EMIM][BF4] and LiBF4. The gelled ionic medium was termed GLiEMIBF4. (The G prefix denoting the Gel state). The ionic conductivity of the gelled electrolyte, was less than the binary mixture but stability was excellent and 50 charge/discharge cycles were achieved.
Highly flexible elastic non volatile electrolytes were prepared using poly(vinylidene fluoride-co-hexafluoropropylene) (PVdF-HFP). These polymer electrolyte materials were prepared by mixing a conducting salt (Li-TFSA), the polymer PVdF-HFP and the ionic liquid. Many ionic liquids were screened including [EMIM][NTf2], [EMPyrr][NTf2] (where EMPyrr is 1-ethyl-1-methylpyrrolidinium), [MPPyrr][NTf2] (where MPPyrr is 1-methyl-1-propylpyrrolidinium), and [DMMA][NTf2] (where DMMA is N,N-diethyl-N-methyl-N-(2 methoxyethyl)ammonium).223 Of note was the need for solvent, in this case acetone was used to homogenise the mixture. The mixture was then left overnight to remove the acetone. The resulting gel disks in the glass vial were evaporated in vacuum. Conductivities of the gel electrolytes at room temperature were about 1–2 mS cm−1. Xiao-Guang Sun and co-workers224 showed the copolymerization of polyethylene glycol methyl ether methacrylate with polyethylene glycol dimethacrylate in the presence of the room temperature ionic liquid, methylpropylpyrrolidinium [NTf2] ([MPPY][NTf2]), this produced a polymer ionic liquid gel. The monomers and the ionic liquid were mixed and polymerisation was initiated with AIBN. The mixture was cast and left to polymerise at 70 °C. The conductivity was shown to increase with increasing concentration of ionic liquid. Panzer and co-workers225 reported an ion gel made by the UV-initiated cross-linking of poly(ethylene glycol) diacrylate (PEGDA) in the presence of [EMIM][NTf2].
Ohno and co-workers115 introduced an example of a polymer gel electrolytes prepared from a ZIL. These polymer gel electrolytes were obtained by the dissolution of the ZIL and the polymer (poly(vinylidene fluoride-co-hexafluoropropylene) in 4-methyl-2-pentanone, and films containing films with zwitterionic liquid of up to 80 wt% were formed on evaporation of the solvent. When the zwitterionic liquid content was 66 wt%, the ionic conductivity of the polymer gel electrolytes was around 0.01 mS cm−1 at 50 °C, at room temperature.115 In collaboration Ohno, MacFarlane and co-workers118 introduced examples of AMIL gel electrolytes where AMIL gels were made by the direct mixing of the AMIL with poly(3-sulfopropyl acrylate) lithium salt or poly(2-acrylamido-2-methylpropanesulfonic acid) lithium salt to form polymer gels. At low polymer concentrations, these gels displayed ionic conductivity of 0.1 to 1 mS cm−1 at room temperature. Gel electrolytes from natural macromolecules have also been reported using gelatine as the continuous phase. A conducting material based on ion jelly (gelatine ionic liquid polymeric gel) was investigated112 and the ionic liquids 1-ethyl-3 methylimidazolium ethylsulfate, ([EMIM][C2SO4]), [EMIM][OAc] and trimethyl-ethanolammonium (choline) acetate, ([Ch][OAc]), and [EMIM][C2SO4] investigated. The gels were made by heating gelatine and water to 50 °C before the addition of ionic liquid, glycerol, formaldehyde and a cross-linking agent. The ionic conductivities were measured of various polymer electrolytes over the temperature range from 25 to 105 °C. The highest room temperature (T = 30 °C) conductivity of the electrolyte system, was reported as 0.118 mS cm−1, for the gelatine gel based on [EMIM][OAc].130 Electrolytes consisting of a poly(ionic liquid) as the solid phase and an ionic liquid as the dispersed phase have been made by various methods and shown to be electrolyte materials. The stabilisation of the IL within the poly(ionic liquid) is thought to be enhanced as the charges can easily interact with each other. A recent review on poly(ionic liquids) outlines the three major ways that poly(ionic liquid) ionic liquid gels are prepared,226 and a review of polymer based ionic liquid gels for electrochemistry also covering poly(ionic liquid) ionic liquid electrolytes has been published.227 Two common methods are mentioned for illustrative purposes. The first method is cross-linking and polymerisation: a co-polymer 1-[(2-methacryloyloxy)ethyl]-3-ethylimidazolium [NTf2] was cross-linked with methacrylate or styrene based cross-linker in the presence of ionic liquid by in situ free radical polymerisation as reported by Ohno and Nakajima.228 In a simpler casting approach the poly(ionic liquid) was dissolved in an ionic liquid and cast into a flexible material.229
Colloidal ionic liquid gels
The use of colloidal particles as gelling agents in ionic liquids has been employed to build devices for energy applications. For example silica particles have been studied and modelled,139 and the use of this type of colloidal ionic liquid gel as an electrolyte has been shown.106,230–232 In this case the conductivity is comparable, or slightly lower, than that of the pure ionic liquids.106 Titania spheres141 and carbon nanotubes113,114 have also been used to gel ionic liquids and produce electrolyte materials.140,141 Ionic liquid gels formed by the sol–gel process using functional ionic liquids have been found to have potential as electrolytes.233,234Hybrid inorganic organic sol–gel precursors (ormosils) based on a di-ureasil precursor have been successfully used for the synthesis of ionic liquid gels.235–237 A protic ionic liquid N-ethylimidazolium trifluoromethanesulfonate ([EIM][OTf]) has been incorporated into a poly(oxyethyelene) (POE)/siloxane hybrid host matrix belonging to the di-ureasil family. These gels were shown to be thermally stable up to 200 °C and showed a conductivity of 3.7 mS cm−1 at 160 °C.238 Vioux and co-workers report a polymer nanocomposite ionogel using a poly(methyl methacrylate) (PMMA) bearing trimethoxysilane groups which was mixed with TEOS and ionic liquid, this material was flexible and at some compositions the conductivity was very close to the ionic liquid.239
Molecular/supramolecular/LMWG ionic liquid gels
The use of low molecular weight gelators (LMWG) for the formation of ionic liquid gel electrolytes was reported by Hanabusa and co-workers.149 Ionic liquid gels were successfully reported for a range of 1-alkyl-3-methylimidazolium iodides (propyl [PMIM][I], [HMIM][I], and nonyl [NMIM][I]) using a LMWG gelator.9 In 2005 the same group reported Ionic liquid gels as electrolytes made from the LMWGs cyclo(L-β-3,7-dimethyloctylasparaginyl-L-phenylalanyl) and cyclo(L-β-2-ethylhexylasparaginyl-L-phenylalanyl).150 These gelators prepared by the esterification of L-asparaginyl-L-phenylalanine methyl ester (aspartame) with 3,7-dimethyl-1-octanol were shown to be capable of gelling the following cations, imidazolium, pyridinium, pyrazolidinium, piperidinium, morpholinium, and ammonium, and gelation occurred irrespective of whether the anion used was [BF4]− or [NTf2]−. These gels were shown to have ionic conductivities very similar to those of the pure ionic liquids, however the highest conductivity was seen for [BMIM][NTf2] and [BMIM][BF4]. Cyclo(L-β-3,7-dimethyloctylasparaginyl-L-phenylalanyl) formed gels at a concentration of less than 1 wt% of the gelator, and it was noted that the strength of the gel material increased with increasing concentration of the solid component. Tan, Dong, Wang and Yang152 used the LMWGs bis(4-octanoylaminophenyl) ether, bis(4-octanoylaminophenyl) methane and 2,4-bis(octanureido) toluene to gel ionic liquid, and they found that the electrochemical properties of the ionic liquid gels were slightly inferior to those of the parent ionic liquid. Šantić and co-workers240 also showed that high conductivity was not guaranteed in gel materials. They found that a concentration effect was in operation in the gelation of [BMIM][BF4] with (S,S)-bis(leucinol)oxalamide as the LMWG. There were differences seen in the conductivity depending on the amount of gelator used, with an increasing concentration leading to a drop in conductivity; this is explained by a loss of liquid like mobility on increasing the solid component of the gel.Ionic liquid gels in fuel cells
Fuel cells can convert a wide variety of fuels to electricity. The conversion of chemical energy into electrical energy is achieved by exploiting many different designs. One common component needed for all fuel cells is an electrolyte. Ionic liquids have been studied as electrolytes for fuel cells. The development of ionic liquid electrolytes for fuel cells has been discussed in detail elsewhere.241 In particular protic ionic liquids are of specific interest for fuel cell membranes such as proton exchange membranes.242,243 Sol–gel processing has always shown potential for the synthesis of membranes in fuel cells, and ionic liquid gel materials have been applied as proton exchange membranes for H2/O2 fuel cells.244 An ionic liquid gel made by the sol–gel method, (described as a glass membrane) was shown to have high conductivity. The protic ionic liquid diethylmethylammonium [OTf], [dema][OTf] was used as the liquid phase and the anhydrous proton conductor. The [dema][OTf]/SiO2 hybrid glass membranes were prepared via a sol–gel process. The stability and ionic conductivity of the glass membrane were investigated. The [dema][OTf]/SiO2 hybrid glass monoliths exhibited high anhydrous ionic conductivities that exceeded 10 mS cm−1 at 120–220 °C.245 An example of a biofuel cell in which a bucky gel (referred to in this paper as CNTs/IL gel) was used as the bioanode for a glucose/O2 biofuel cell.246 These devices were made by modifying carbon nanotubes by covalently attaching ferrocenecarboxaldehyde. The modified carbon tubes were then added to the ionic liquid [BMIM][PF6], glucose oxidase was then added, and the mixture was ground together to form a bucky gel.Solar cells: ionic liquid gels as electrolytes for DSSCs
A DSSC is an elegant example of a solar cell.247 In a DSSC a photosensitive dye is adsorbed onto the surface of TiO2. The TiO2-dye is surrounded by an electrolyte. Light is absorbed by the dye causing excitation of electrons from the ground state to the excited state. The excited electrons are injected into the conduction band of the TiO2 leaving the dye oxidised. In this state the dye is unable to produce another excitation. The electrons formed from the dye excitation are transferred to the conduction band of the TiO2 and flow through the TiO2 to the anode. The electrolyte helps to transfer electrons from the cathode to the dye molecules to replace the lost electron, allowing the whole process to continue.One of the major limitations of the conventional DSSC is the use of a liquid electrolyte. The use of a gel electrolyte provides a more stable structure, and the use of an ionic liquid gel offers further stability, as the exceptional low volatility of the ionic liquid is combined with the supporting structure afforded by the continuous (solid) phase of the gel. Thus ionic liquids gels could significantly advance the efficiency, longevity and user-friendliness of this class of solar cell. Polymer ionic liquid gels,248 colloidal ionic liquid gels using colloidal silica nanoparticles,249 and sol–gel nanocomposite quasi-solid electrolytes containing ionic liquid from hybrid silica organic materials143 have all been applied to DSSCs. Hanabusa used LMWG N-benzyloxycarbonyl-L-valyl-L-valine n-octadecylamide9 to prepare ionic liquid gels as gel electrolytes for DSSCs149 Other examples from Grätzel and co-workers154 are based on 1,3:2,4-di-O-benzylidene-D-sorbitol (DBS)153 derivatives as low molecular weight organic gelators. Grätzel and co-workers observed that the structure of the LMWG had an influence on the structure and the solar conversion efficiency of the DSSC.155 In this case the LMWGs used were didodecanoylamides of α,ω-alkylidenediamines250 and the IL was 1,2-methyl-3-propylimidazolium iodide ([PMMIM][I]). LMWGs containing odd-numbers of CH2s between the two amide carbonyl groups in the molecules of the gelators were shown to be different to those with an even-number. The gel constructed containing five CH2 groups showed a photoelectric conversion efficiency of 7.53%,155 and further improvements in photoelectric conversion efficiency (9.61%) was seen using a similar LMWG system.156
Capacitors/supercapacitors/ultracapacitors based on ionic liquid gel electrolytes
Energy storage is essential and will be of increasing importance as energy sources become more diverse and renewable. Capacitors are used to store energy electrostatically in an electric field. The simplest form consists of two conducting plates separated by an insulator. When charged with opposite charges, an electric field exists between the two plates. Supercapacitors and ultracapacitors combine the high energy density of conventional batteries with the high power density of conventional capacitors. Ionic liquid gels have again proven useful. Wen Lu and co-workers251 described the preparation and testing of three different materials. In method A) a polymer IL gel with the polymer (polyvinylidene fluoride-co-hexafluoropropylene (PVdF-HFP)), was mixed with N-methyl-2-pyrrolidone (NMP). Gelation occurred on heating and the gel was left under vacuum to remove the solvent. Method B) differed from Method A by the inclusion of a small amount of zeolite to the mixture. In method C) a commercial Teflon membrane was soaked in the IL. The ionic liquids used were [EMIM][BF4] and [EMIM][NTf2]. Ionic conductivity of all ionic liquid gel polymer electrolytes (ILGPEs) tested was lower than the pure ionic liquids. Addition of the zeolite to the [EMIM][BF4] polymer ionic liquid gel led to a drop in conductivity, but made no difference with the equivalent material made with [EMIM][NTf2].Flexible supercapacitors were made by coating a carbon nanotube impregnated office paper with an ionic liquid gel made by mixing the ionic liquid with preformed silica spheres (approx 14 nm diameter). These solid-state supercapacitors gave a performance for specific capacitance of 134.6 F g−1, maximum power 164 kW kg−1 and energy density 41 Wh kg−1, and over 4000 charge/discharge cycles.252 Panzer and Visentin225 prepared UV-initiated cross-linked poly(ethylene glycol) diacrylate (PEGDA) ionic liquid gel. These gels were tested as capacitors. In this study gels were prepared using different ratios of solid and liquid phase. When conductivity was measured at room temperature a decrease in conductivity with increasing concentration solid phase was observed. The same group reported a soft silica based ionogel made from TMOS and high [EMIM][NTf2] ionic liquid loading. They noted that these materials had ionic conductivity and double layer capacitance values close to those of the neat ionic liquid.253
Zaumseil and co-workers103 report the use of an ionic liquid with silicon alkoxide (triethoxysilane) functional groups attached to the imidazolium cation (3-methyl-1-(3-(triethoxysilyl)propyl)-imidazolium [NTf2] ([(EtO)3SiPMIM][NTf2]) The cation of the ionic liquid is involved in the network formation via the sol–gel process. The cationic sol–gel precursor was mixed with TMOS and the sol–gel process was catalysed with formic acid. A viscous, spin coatable sprayable sticky material was formed with a capacitance of between 10.4 and 18 μF cm−2 depending on the concentration of the added TMOS. They concluded that more rigid the structure, the lower the measurement, this is in agreement with Panzer and Visentin.225
Another example of a sol–gel material for capacitors was reported by Negre and co-workers,254 electrical double layer capacitors using sol–gel derived ionic liquid gels were synthesised as solvent free supercapacitors with a high IL content (70%). An increase of both the operating voltage window and the electrolyte ionic conductivity was seen. This gel was made by mixing [EMIM][NTf2] with a mixture of TEOS and TMOS silica precursors, formic acid was used as the catalyst for the sol–gel reaction. In another study supercapacitor cells were assembled layer by layer using a casting approach. First gold platinum current collectors were coated with an activated carbon based active material film. This carbon layer was formed from a mixture of 95 wt% of PICACTIF SC, (a standard high specific surface area activated carbon), and 5 wt% of PTFE (60 wt% PTFE in water). A few droplets of silane-IL sol were deposited onto the electrodes, which were sandwiched together. The TMOS/TEOS/IL/acid mixture gelled to complete the supercapacitor cell. It has recently been demonstrated that it is possible to gel a silica precursor with an acidic ionic liquid without the need for formic or other additional acid197 this should lead to a further enhancement and the greener processing of solvent free materials for capacitors.
Ionic liquid gel materials for displays
Ionic liquid gels are finding use in the development of low energy displays and electrochromic windows for energy efficient buildings. Electrochromic devices change colour on the application of an applied electrical field. As such they can be used to regulate the internal temperatures of buildings, leading to reduced energy consumption. The ability of a gel to combine active materials, with an electrolyte within the same material has many uses. As detailed in the catalysis section, nanoparticles and molecular species can show enhanced function within gels. Such synergy would be of great benefit within display and electrochromic materials, as these comprise complex components that must act cooperatively. Conjugated polymers (CPs)255 have many uses. One of the many applications of CPs include their use in electrochromic256,257 devices. Electrochromism is seen in some conjugated polymers. CPs are also much studied as active materials for many electronic devices such as organic light emitting diodes, solar cells and actuators.Poly(3,4-ethylenedioxythiophene) (PEDOT) has aroused particular interest as an electrochromic material, it switches between deep-blue and pale-blue hues.258–261 Electrochromic devices based on PEDOT as the cathodic colouring electrode, and polyaniline (PANI) or Prussian blue as the counter electrode creates a self-supporting transparent polymer ionic liquid gel have been reported by Deepa et al.262 Moon et al.263 have prepared ion gels composed of polystyrene-block-poly(methyl methacrylate)-block-polystyrene and [EMIM][NTf2] capable of electrochromism by adding the electrochromic (EC) redox molecule, methyl viologen. Ferrocene was also added to the EC gel as an anodic species. Fundamentally the use of photoluminescent probes can tell us a great deal about how the material is formed. Analyses of a light emitting CP, poly(9,9-dioctylfluorine) (PFO) in an ionic liquid silica gel by photoluminescence and FTIR showed that the conjugated polymer self-organised inside the pores to form an ordered phase through a confinement effect.81 The self organisation of the ionic liquid within the pores influenced the secondary ordering of the conjugated polymer within the pores leading to the formation of the organised β-phase of the PFO. In a different study phenylene–ethynylene monomer and copolymer with pendant imidazolium groups were synthesized to be soluble in [BMIM][NTf2]. The ionogels were made using a mixture of TMOS and methyltrimethoxysilane (MTMOS) in the presence of formic acid as the catalyst.264 In other studies lanthanides were incorporated as the luminescent entity.265 The ionogel was formed using [HMIM][NTf2]and then removed using acetonitrile and then refilled with a lanthanide containing ionic liquid ([HMIM] tetrakis(naphthoyltrifluoroacetonato)europate(III)). In a later study a series of lanthanide ionic liquids were studied [HMIM][Ln(tta)4], where tta is 2-thenoyltrifluoroacetonate and Ln = Nd, Sm, Eu, Ho, Er, Yb, and [choline]3[Tb(dpa)3], where dpa = pyridine-2,6-dicarboxylate (dipicolinate).266 Understanding how to include and control light emitting/harvesting materials and electroactive materials within ionic liquid gels will unlock the great potential of these materials in photovoltaics, low energy displays, sensors and actuators.
Ionic liquid gels in electrocatalysis
An electrocatalyst is a catalyst that increases the rate of an electrochemical reaction at the surface of an electrode. The catalyst can be homogeneous or heterogeneous, chemical or biological. Electrocatalysts are an important part of some fuel cell designs. Materials made for electrocatalysis could also inform the design of catalysts for chemical transformations in the bulk and vice versa. Ionic liquid gels have also been successfully applied to electrocatalysis. This is again supported by the ability to include active molecules within an electrolyte material. Colloidal gels based on carbon nanotubes113 and are being used to build catalyst materials in the presence of other actives. A multi-walled carbon nanotube ionic liquid bucky gel was prepared with the addition of two additives, cobalt porphyrin and Prussian blue nanoparticles. This mixed material was shown to be active for the 4e-reduction of O2, and could have potential for use as non-platinum cathode materials for fuel cells.267 Of note is the fact that both catalysts work independently in the same material. First cobalt porphyrin catalyses the 2e-reduction of O2 into H2O2, and the Prussian blue nanoparticles are responsible for the 2e-reduction of the H2O2 produced. A similar ionic liquid gel modified glass carbon electrode (termed MWCNTs-RTIL/GCE), was formed by coating an electrode in gel. This material was shown to be active for the electrocatalytic oxidation of venlafaxine (1-2-(dimethylamino)-1-(4-methoxyphenyl)ethylcyclohexanol), a pharmaceutical compound used in the treatment of depression.268 These gel materials were made by grinding carbon nanotubes with an ionic liquid, which was reported to form a gel substance stable enough to be used as an electrode coating.Some interesting bio-electrocatalytic applications of ionic liquid gels have been reported. Direct electrochemistry and electrocatalysis of myoglobin was shown on a material based on silica-coated gold nanorods/room temperature ionic liquid/silica sol–gel composite films.269 Zeng and co-workers270 described biocatalyst entrapment for electrocatalysis, showing the direct electrochemistry of the enzyme, horseradish peroxidase, in an ion jelly biomacromolecular gelatine based ionic liquid gel. The gel which was made from [OMIM] [PF6] gelatine hydrogel was found to stabilise the enzyme and the structure of the enzyme was found to remain intact. The gel supported enzyme electrode showed good thermal stability. A bioactive hemoglobin was immobilized in a chitosan based ionic liquid [BMIM][BF4] gel.271 In this system hemoglobin was reported to retain its bioactivity in the ionic liquid gel electrode for the electrochemical reduction of O2. The conductivity of the IL was also thought to contribute to the electron transfer. These materials could have potential for biosensors and in biocatalysis. The inclusion of catalytically active components (of all types) in ionic liquid gels will continue to gain influence in electrocatalysis and the electrochemical enhancement of photocatalysis.
Choosing an ionic liquid gel for a given application
Choice of solid phase and choice of ionic liquid allow a material to be tailored specifically for a given application. Polymer/macromolecular solid phases can be tailored to work at a specific temperature range. The implicit processability of polymer materials means that they have the ability to be cast into films, rolled up, spun into fibres, coated onto an electrode surface, or indeed be coated with electrodes. Polymers with functional pendant groups can impart further functionality. Polymer/macromolecular ionic liquid gels can be tailored from soft to rigid by changing the ratio of the solid phase to the liquid, and indeed the liquid nature of the gel can also be controlled by the extent of polymer crosslinking.Colloidal gels can range from soft to glassy and be made from many substances including carbon nanotubes and silica, and thus can be used to coat substrates. Further stability and rigidity can be attained by sol–gel methods. Sol–gel offers a range of metal oxide materials available for ionic liquid gel formation and these solid phases can be utilised when stability and robustness of the gel at elevated temperatures are required. Sol–gel processing can produce thin films, monoliths, powder and coated substrates.
In DSSC devices the liquid nature of the electrolyte is one of the material limitations. The benefits of using molecular ionic liquid gels in DSSCs comes from the ability of the gel to retain the electrolyte. We see evidence that the more liquid like the gel (low solid phase concentration), the closer the conductivity is to that of the parent ionic liquid; and so where ion mobility is of paramount importance, molecular gels are specifically interesting. Molecular gels tend to be softer and the concentration of the solid phase of this type of gel can be very low <1 wt%. This has been seen to have specific benefit in the production of DSSCs where ion mobility and fast ion transport is crucial to the overall efficiency of the cell operation; however, molecular gels can have some thermal and mechanical stability issues, which will need to be overcome for some applications.
Once the type of gel (liquid like, flexible, glassy, ceramic, thermally stable) needed for an application is identified, further complexity can be introduced. Solid phases capable of conductivity or photoluminescence can be produced for polymer, colloidal or molecular gels. Ionic liquid gels have been shown to successfully contain entrapped functional materials such as catalysts (homogeneous and metal nanoparticle), functional polymer and colloidal entities, enzymes and cells. Add to this the ability to dial in functionality into the ionic liquid component of the gel (acidity, basicity, conductivity, metal extraction, biocompatibility, therapeutic pharmaceutical, cell and enzyme support), and a further level of complexity can be utilised. Traditional processing such as casting and coating, and mass printing techniques can be employed to produce gel based electronics; ink jet and gel jet printers are suitable for liquid like gels, and 3D printing is suitable for others.
5. Conclusions
Ionic liquid gels offer an abundance of materials for study and application. The combination of the extensive variety of gel methods, with the myriad of ionic liquids, provides an endless variety of materials for green and sustainable applications. Confinement of ILs within a gel can lead to novel behaviour and open new applications. Highlights of work so far include the confinement of task specific/functional ionic liquids, and the incorporation of active species such as enzymes, metal nanoparticles, molecular species, homogeneous catalysts, and polymers. This buildable system provides the ability to tailor-make a functional material for any of a wide range of applications. Real progress has been made in the application of ionic liquid gels to the fields of catalysis and energy materials, and the vast potential is becoming apparent. In addition to the advantages over liquid systems, such as ease of handling and stability, the gel state provides several advantages over solid systems. In comparison with traditional functional solid synthesis gel preparation is low cost, low energy and straightfoward. Recycling of ionic liquid gel materials is a simple process, and the ionic liquids can be removed from the gel and reused. The continuous phase whether silica, titania, carbon, molecular or macromolecular can be easily disassociated, reclaimed, and reused. From monoliths to thin films the diversity of these materials will continue to offer fascinating insights into ionic liquid behaviour and structure, together with dedicated solutions for catalysis, and sustainable energy.References
- IUPAC gold Book, PAC, 2007, vol. 79, p. 1801. (Definitions of terms relating to the structure and processing of sols, gels, networks, and inorganic–organic hybrid materials (IUPAC Recommendations 2007)) Search PubMed.
- T. Graham, Philos. Trans., 1861, 151, 183–224 CrossRef.
- T. Graham, Proc. R. Soc., 1864, 13, 335–341 CrossRef.
- K. Kajiwara, Gels Handbook, Academic Press, 2000 Search PubMed.
- Y. Osada and A. Khokhlov, Polymer Gels and Networks, CRC Press, 2001 Search PubMed.
- K. Almdal, J. Dyre, S. Hvidt and O. Kramer, Polym. Gels Networks, 1993, 1, 5–17 CrossRef CAS.
- J. R. Stokes and W. J. Frith, Soft Matter, 2008, 4, 1133–1140 RSC.
- P. J. Flory, Faraday Discuss. Chem. Soc., 1974, 57, 7–18 RSC.
- K. Hanabusa, J. Tange, Y. Taguchi, T. Koyama and H. J. Shirai, J. Chem. Soc., Chem. Commun., 1993, 390–392 RSC.
- F. Placin, J. P. Desvergne, C. Belin, T. Buffeteau, B. Desbat, L. Ducasse and J. Lasségues, Langmuir, 2003, 19, 4563–4572 CrossRef CAS.
- N. M. Sangeetha, S. Bhat, G. Raffy, C. Belin, A. Loppinet-Serani, C. Aymonier, P. Terech, U. Maitra, J. P. Desvergne and A. Del Guerzo, Chem. Mater., 2009, 21, 3424–3432 CrossRef CAS.
- K. J. C. van Bommel, A. Friggeri and S. Shinkai, Angew. Chem., Int. Ed., 2003, 42, 980–999 CrossRef CAS PubMed.
- K. Sugiyasu, S. I. Tamaru, M. Takeuchi, D. Berthier, I. Huc, R. Oda and S. Shinkai, Chem. Commun., 2002, 1212–1213 RSC.
- S. J. Craythorne, C. L. Pollock, A. J. Blake, M. Nieuwenhuyzen, A. C. Marr and P. C. Marr, New J. Chem., 2009, 33, 479–483 RSC.
- J. W. Steed, Chem. Commun., 2011, 47, 1379–1383 RSC.
- P. Dastidar, Chem. Soc. Rev., 2008, 37, 2699–2715 RSC.
- N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC.
- M. O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. L. Steed, Chem. Rev., 2010, 110, 1960–2004 CrossRef CAS PubMed.
- D. K. Smith, in Organic Nanostructures, ed. J. L. Atwood and J. W. Steed, Wiley VCH, Weinheim, 2008, pp. 111–154 Search PubMed.
- F. Fages, Top. Curr. Chem., 2005, 256, 1–275 CrossRef.
- S. S. Babu, V. K. Preveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS PubMed.
- K. Hanabusa and M. Suzuki, Polym. J., 2014, 46, 776–782 CrossRef CAS.
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3160 CrossRef CAS PubMed.
- R. G. Weiss and P. Terech, Molecular Gels: Materials with Self-Assembled Fibrillar Networks, Springer, 2006 Search PubMed.
- Topics in current chemistry, in Low Molecular Mass Gelators, ed. F. Fages, Springer-Verlag, Berlin, Heidelberg, 2005 Search PubMed.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1217 CrossRef CAS PubMed.
- J. Liu, P. He, J. Yan, X. Fang, J. Peng, K. Liu and Y. Fang, Adv. Mater., 2008, 20, 2508–2511 CrossRef CAS.
- N. Javid, S. Roy, M. Zelzer, Z. Yang, J. Sefcik and R. V. Ulijn, Biomacromolecules, 2013, 14, 4368–4376 CrossRef CAS PubMed.
- K. Liu, P. He and Y. Fang, Sci. China: Chem., 2011, 54(4), 575–586 CrossRef CAS.
- J. Jagur-Godzinski, Polym. Adv. Technol., 2010, 21, 27–47 Search PubMed.
- J. Elisseeff, Nat. Mater., 2008, 7, 271–273 CrossRef CAS PubMed.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- C. Peak, J. J. Wilker and G. Schmidt, Colloid Polym. Sci., 2013, 291, 2031–2047 CAS.
- Y. Osada and A. Matsuda, Nature, 2002, 376, 219 CrossRef PubMed.
- Y. Osada, H. Okuzaki and H. Hori, Nature, 1992, 355, 242–244 CrossRef CAS.
- C. L. Pollock, K. J. Fox, S. D. Lacroix, J. McDonagh, P. C. Marr, A. M. Nethercott, A. Pennycook, S. Qian, L. Robinson, G. C. Saunders and A. C. Marr, Dalton Trans., 2012, 41, 13423–13428 RSC.
- M. Prokopowicz, Drug Delivery, 2007, 3, 129–138 CrossRef PubMed.
- C. Shende, A. Kabir, E. Townsend and A. Malik, Anal. Chem., 2003, 75, 3518–3530 CrossRef CAS PubMed.
- P. Lesot, S. Chapuis, J. P. Bayle, J. Rault, E. Lafontaine, A. Campero and P. Judeinstein, J. Mater. Chem., 1998, 8, 147–151 RSC.
- D. Stauffer and A. Aharony, Introduction to Percolation Theory, CRC Press, 1994 Search PubMed.
- L. L. Hench and J. K. West, Chem. Rev., 1990, 90, 33–72 CrossRef CAS.
- P. Pfeifer and D. Avnir, J. Chem. Phys., 1983, 79, 3558 CrossRef CAS.
- Theory of the Stability of Lyophobic Colloids - The Interaction of Soil Particles Having an Electrical Double Layer, ed. E. J. W. Verwey, J. T. G. Overbeek and K. Van Ness, Elsevier, 1948 Search PubMed.
- J. R. Stokes and W. J. Frith, Soft Matter, 2008, 4, 1133–1140 RSC.
- C. J. Brinker and G. W. Scherer, Sol–Gel Science: The Physics and Chemistry of Sol–Gel Processing, Academic Press, Boston, 1990 Search PubMed.
- R. D. Rogers and K. Seddon, Science, 2003, 302, 792–793 CrossRef PubMed.
- R. A. Sheldon, R. M. Lau, M. J. Sorgedrager, F. Van Rantwijk and K. R. Seddon, Green Chem., 2002, 4, 147–151 RSC.
- F. Van Rantwijk and R. A. Sheldon, Chem. Rev., 2007, 107, 2757–2785 CrossRef CAS PubMed.
- B. F. Gilmore, G. P. Andrews, G. Borberly, M. J. Earle, M. A. Gilea, S. P. Gorman, A. F. Lowry, M. McLaughlin and K. R. Seddon, New J. Chem., 2013, 37, 873–876 RSC.
- J. H. Davis, K. J. Forrester and T. Merrigan, Tetrahedron Lett., 1998, 39, 8955–8958 CrossRef CAS.
- E. B. Carter, S. L. Culver, P. A. Fox, R. D. Goode, I. Ntai, M. D. Tickell, R. K. Traylor, N. W. Hoffman and J. H. Davis, Chem. Commun., 2004, 630–631 RSC.
- H. D. Williams, Y. Sahbaz, L. Ford, T. Nguyen, P. J. Scammells and C. J. H. Porter, Chem. Commun., 2014, 50, 1688–1690 RSC.
- A. Romero, A. Santos, J. Tojo and A. Rodriguez, J. Hazard. Mater., 2008, 151, 268–273 CrossRef CAS PubMed.
- E. D. Bates, R. D. Mayton, I. Ntai and J. H. Davis Jr., J. Am. Chem. Soc., 2002, 124, 926–927 CrossRef CAS PubMed.
- M. Ramdin, T. W. de Loos and T. J. H. Vlugt, Ind. Eng. Chem. Res., 2012, 51, 8149–8177 CrossRef CAS.
- M. Smiglak, J. M. Pringle, X. Lu, L. Han, S. Zhang, H. Gao, D. R. MacFarlane and R. D. Rogers, Chem. Commun., 2014, 50, 9228–9250 RSC.
- A. E. Visser, R. P. Swatloski, W. M. Reichert, R. Mayton, S. Sheff, A. Wierzbicki, J. H. Davis Jr. and R. D. Rogers, Chem. Commun., 2001, 135–136 RSC.
- T. Vander Hoogerstraete, S. Wellens, K. Verachtert and K. Binnemans, Green Chem., 2013, 15, 919–927 RSC.
- A. P. Abbott, G. Frisch, J. Hartley and K. S. Ryder, Green Chem., 2011, 13, 471–481 RSC.
- P. Nockemann, B. Thijs, S. Pittois, J. Thoen, C. Glorieux, K. Van Hecke, L. Van, L. Meervelt, B. Kirchner and K. Binnemans, J. Phys. Chem. B, 2006, 110, 20978–20992 CrossRef CAS PubMed.
- A. E. Visser, R. P. Swatloski, W. M. Reichert, R. Mayton, S. Sheff, A. Wierzbicki, J. H. Davis Jr. and R. D. Rogers, Environ. Sci. Technol., 2002, 36, 2523–2529 CrossRef CAS PubMed.
- T. P. T. Pham, C.-W. Cho and Y.-S. Yun, Water Res., 2010, 44, 352–372 CrossRef CAS PubMed.
- N. Gathergood, M. T. Garcia and P. J. Scammells, Green Chem., 2004, 6, 166–175 RSC.
- M. T. Garcia, N. Gathergood and P. J. Scammells, Green Chem., 2005, 7, 9–14 RSC.
- N. Gathergood, P. J. Scammells and M. T. Garcia, Green Chem., 2006, 8, 156–160 RSC.
- H. Ohno and K. Fukumoto, Acc. Chem. Res., 2007, 40(11), 1122–1129 CrossRef CAS PubMed.
- N. Ferlin, M. Courty, S. Gatard, M. Spulak, B. Quilty, I. Beadham, M. Ghavre, A. Haiß, K. Kümmerer, N. Gathergood and S. Bouquillon, Tetrahedron, 2013, 69, 6150–6161 CrossRef CAS.
- T. Welton, Chem. Rev., 1999, 99, 2071–2084 CrossRef CAS PubMed.
- P. Wasserscheid and W. Keim, Angew. Chem., Int. Ed., 2000, 39(21), 3772–3789 CrossRef CAS.
- C. Hardacre and V. Parvulescu, Catalysis in ionic liquids, RSC catalysis series, Royal society of chemistry, 2014 Search PubMed.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621–629 CrossRef CAS PubMed.
- M. C. Buzzero, R. G. Evans and R. G. Compton, ChemPhysChem, 2004, 5, 1106–1120 CrossRef PubMed.
- H. Q. N. Gunaratne, P. Nockemann and K. R. Seddon, Chem. Commun., 2015, 51, 4455–4457 RSC.
- M. Earle and K. R. Seddon, Pure Appl. Chem., 2000, 72, 1391–1398 CrossRef CAS.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- A. Mahpuzah, M. P. Atkins, A. Hassan, J. D. Holbrey, Y. Kuah, P. Nokemann, A. A. Oliferenko, N. V. Plechkova, S. Rafeen, A. A. Rahman, R. Ramli, S. M. Shariff, K. R. Seddon, G. Srinivasan and Y. Zou, Dalton Trans., 2015, 44, 8617–8624 RSC.
- S. J. Craythorne, K. Anderson, F. Lorenzini, C. McCausland, E. F. Smith, P. Licence, A. C. Marr and P. C. Marr, Chem. – Eur. J., 2009, 15, 7094–7100 CrossRef CAS PubMed.
- C. Iacob, J. R. Sangoro, W. K. Kipnusu, R. Valiullin, J. Kärger and F. Kremer, Soft Matter, 2012, 8, 289–293 RSC.
- R. Atkin and G. G. Warr, J. Phys. Chem. C, 2007, 111, 5162–5168 CAS.
- S. Perkin, Phys. Chem. Chem. Phys., 2012, 14, 5052–5062 RSC.
- R. C. Evans and P. C. Marr, Chem. Commun., 2012, 48, 3742–3744 RSC.
- Y. Zhou, J. H. Schattka and M. Antonietti, Nano Lett., 2004, 4, 477–481 CrossRef CAS.
- J. Le Bideau, P. Gaveau, S. Bellayer, M. A. Néouze and A. Vioux, Phys. Chem. Chem. Phys., 2007, 9, 5419–5422 RSC.
- G. Xie, J. Luo, D. Guo and S. Liu, Appl. Phys. Lett., 2010, 96, 043112–043123 CrossRef.
- C. Iacob, J. R. Sangoro, W. K. Kipnusu, R. Valiullin, J. Karger and F. Kremer, Soft Matter, 2012, 8, 289–293 RSC.
- C. Li, X. Guo, Y. He, Z. Jiang, Y. Wang, S. Chen, H. Fu, Y. Zou, S. Dai, G. Wu and H. Xu, RSC Adv., 2013, 3, 9618–9621 RSC.
- M. A. Firestone, J. A. Dzielawa, P. Zapol, L. A. Curtiss, S. Seifert and M. L. Dietz, Langmuir, 2002, 18, 7258–7260 CrossRef CAS.
- C. Tourne-Peteilh, B. Cosne, M. In, D. Brevet, J.-M. Devoisselle and A. Vioux, Langmuir, 2014, 30, 1229–1238 CrossRef CAS PubMed.
- R. Hayes, G. G. Warr and R. Atkin, Chem. Rev., 2015, 115, 6357–6426 CrossRef CAS PubMed.
- G. Ori, F. Villemot, L. Viau, A. Vioux and B. Coasne, Mol. Phys., 2014, 112, 1350–1361 CrossRef CAS.
- R. Göbel, R. J. White, M. M. Titirici and A. Taubert, Phys. Chem. Chem. Phys., 2012, 14, 5992–5997 RSC.
- M. Neouze, J. Le Bideau, P. Gaveau, S. Bellayer and A. Vioux, Chem. Mater., 2006, 18(17), 3931–3936 CrossRef CAS.
- Molten Salts and Ionic Liquids: Never the Twain?, ed. M. G. Escard and K. R. Seddon, John Wiley and sons, 2010 Search PubMed.
- Molten Salts Chemistry and Technology, ed. M. G. Escard and G. M. Haarberg, John Wiley & Sons, 2014 Search PubMed.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- D. Carriazo, M. C. Serrano, M. C. Gutierrez, M. L. Ferrer and F. del Monte, Chem. Soc. Rev., 2012, 41, 4996–5014 RSC.
- H. Ohno, Electrochemical Aspects of Ionic Liquids, John Wiley & Sons, 2011 Search PubMed.
- J. Le Bideau, L. Viau and A. Vioux, Chem. Soc. Rev., 2011, 40, 907–925 RSC.
- M.-A. Néouse, J. Le Bideau, F. Leroux and A. Vioux, Chem. Commun., 2005, 1082–1084 RSC.
- A. Kavanagh, R. Byrne, D. Diamond and K. J. Fraser, Membranes, 2012, 2, 16–39 CrossRef CAS PubMed.
- N. Kimizuka and T. Nakashima, Langmuir, 2001, 17, 6759–6761 CrossRef CAS.
- R. Göbel, R. J. White, M. M. Titirici and A. Taubert, Phys. Chem. Chem. Phys., 2012, 14, 5992 RSC.
- S. Thiemann, S. J. Sachnov, M. Gruber, F. Gannott, S. Spieker, P. Wasserscheid and J. Zaumseil, J. Mater. Chem. C, 2014, 2, 2423–2430 RSC.
- H. Yiyong, P. G. Boswell, P. Buhlmann and T. P. Lodge, J. Phys. Chem. B, 2007, 111, 4645–4652 CrossRef PubMed.
- M. A. B. H. Susan, T. Kaneko, A. Noda and M. Watanabe, J. Am. Chem. Soc., 2005, 127(13), 4976–4983 CrossRef CAS PubMed.
- K. Ueno, K. Hata, T. Katakabe, M. Kondoh and M. Watanabe, J. Phys. Chem. B, 2008, 112, 9013–9019 CrossRef CAS PubMed.
- W. Kubo, S. Kambe, S. Nakade, T. Kitamura, K. Hanabusa, Y. Wada and S. Yanagida, J. Phys. Chem. B, 2003, 107, 4374–4381 CrossRef CAS.
- F. Shi, Q. Zhang, D. Li and Y. Deng, Chem. – Eur. J., 2005, 11, 5279–5288 CrossRef CAS PubMed.
- R. Goebel, P. Hesemann, J. Weber, E. Moeller, A. Friedrich, S. Beuermann and A. Taubert, Phys. Chem. Chem. Phys., 2009, 11, 3653–3662 RSC.
- F. Shi and Y. Deng, Spectrochim. Acta, Part A, 2005, 62, 239–244 CrossRef PubMed.
- N. M. T. Lourenço, A. V. M. Nunes, C. M. M. Duarte and P. Vidinha, Ionic Liquids Gelation with Polymeric Materials: The Ion Jelly Approach in Applications of Ionic Liquids in Science and Technology, ed. S. Handy, InTech, 2011 Search PubMed.
- P. Vidinha, N. M. T. Lourenco, C. Pinheiro, A. R. Bras, T. Carvalho, T. Santos-Silva, A. Mukhopadhyay, M. J. Romao, J. Parola, M. Dionisio, J. M. S. Cabral, C. A. M. Afonso and S. Barreiros, Chem. Commun., 2008, 5842–5844 RSC.
- T. Fukushima, A. Kosaka, Y. Ishimura, T. Yamamoto, T. Takigawa, N. Ishii and T. Aida, Science, 2003, 300, 2072–2074 CrossRef CAS PubMed.
- M. Tunckol, J. Durand and P. Serp, Carbon, 2012, 50, 4303–4334 CrossRef CAS.
- H. Ohno, M. Yoshizawa and W. Ogihara, Electrochim. Acta, 2003, 2079–2083 CrossRef CAS.
- D. Carriazo, M. C. Gutierrez, M. L. Ferrer and F. del Monte, Chem. Mater., 2010, 22, 6146–6152 CrossRef CAS.
- M. C. Gutierrez, F. Rubio and F. del Monte, Chem. Mater., 2010, 22, 2711–2719 CrossRef CAS.
- W. Ogihara, J. Sun, M. Forsyth, D. R. MacFarlane, M. Yoshizawa and H. Ohno, Electrochim. Acta, 2004, 1797–1801 CrossRef CAS.
- H. S. Park, I. T. Kim, H. Y. Kim, S. K. Ryu and J. H. Kim, Environ. Sci. Technol., 2007, 15, 345–351 Search PubMed.
- J. Fuller, A. C. Breda and R. T. Carlin, J. Electrochem. Soc., 1997, 144(4), L67–L70 CrossRef CAS.
- A. Noda and M. Watanabe, Electrochim. Acta, 2000, 45, 1265–1270 CrossRef CAS.
- R. T. Carlin and J. Fuller, Chem. Commun., 1997, 1345–1346 RSC.
- H. Nakagawa, S. Izuchi, K. Kuwana, T. Nukuda and Y. Aihara, J. Electrochem. Soc., 2003, 150, 6A695–6A700 CrossRef.
- T. Ueki, Y. Nakamura, R. Usui, Y. Kitazawa, S. So, T. P. Lodge and M. Watanabe, Angew. Chem., Int. Ed., 2015, 127, 3061–3065 CrossRef.
- Y. Zhang, X. Fan, Z. Shen and Q. Zhou, Macromolecules, 2015, 48, 4927–4935 CrossRef CAS.
- B. Ziółkowski, K. Bleek, B. Twamley, K. J. Fraser, R. Byrne, D. Diamond and A. Taubert, Eur. J. Inorg. Chem., 2012, 32, 5245–5251 CrossRef.
- K. Prasad, M. Murakami, Y. Kaneko, A. Takada, Y. Nakamura and J. Kadokawa, Int. J. Biol. Macromol., 2009, 45, 221–225 CrossRef CAS PubMed.
- A. Takegawa, M. Murakami, Y. Kaneko and J. Kadokawa, Carbohydr. Polym., 2010, 79, 85–90 CrossRef CAS.
- T. Singh, T. J. Trivedi and A. Kumar, Green Chem., 2010, 12, 1029–1035 RSC.
- R. Leones, F. Sentanin, L. C. Rodrigues, R. A. S. Ferreira, I. M. Marrucho, J. M. S. S. Esperança, A. Pawlicka, L. D. Carlos and M. Manuela Silva, Opt. Mater., 2012, 35, 187–195 CrossRef CAS.
- S. Dai, Y. H. Ju, H. J. Gao, J. S. Lin, S. J. Pennycook and C. E. Barnes, Chem. Commun., 2000, 243–244 RSC.
- K. Anderson, S. C. Fernandez, C. Hardacre and P. C. Marr, Inorg. Chem. Commun., 2004, 7, 73–76 CrossRef CAS.
- C. J. Adams, A. E. Bradley and K. R. Seddon, Aust. J. Chem., 2001, 54, 679–681 CrossRef CAS.
- E. R. Cooper, C. D. Andrews, P. S. Wheatley, P. B. Webb, P. Wormald and R. E. Morris, Nature, 2004, 430, 1012–1016 CrossRef CAS PubMed.
- E. R. Parnham, E. A. Drylie, P. S. Wheatley, A. M. Z. Slawin and R. E. Morris, Angew. Chem., Int. Ed., 2006, 45, 4962–4966 CrossRef CAS PubMed.
- L. Viau, M.-A. Neouze, C. Biolley, S. Volland, D. Brevet, P. Gaveau, P. Dieudonne, A. Galarneau and A. Vioux, Chem. Mater., 2012, 24, 3128–3134 CrossRef CAS.
- B. Coasne, L. Viau and A. Vioux, J. Phys. Chem. Lett., 2011, 2, 1150–1154 CrossRef CAS PubMed.
- S. Bellayer, L. Viau, Z. Tebby, T. Toupance, J. Le Bideau and A. Vioux, Dalton Trans., 2009, 1307–1313 RSC.
- K. Ueno and M. Watanabe, Silica Colloidal Suspensions in Ionic Liquids: Colloidal Stability and Fabrication of Ion Gels on the basis of Colloidal Self-Assembly in Ionic Liquids in From Knowledge to Application Ionic Liquids: From Knowledge to Application, ed. N. V. Plechkova, R. D. Rogers and K. R. Seddon, ACS Symposium Series, 2009, vol. 1030 Search PubMed.
- S. Shimano, H. Zhou and I. Honma, Chem. Mater., 2007, 19, 5216–5221 CrossRef CAS.
- W. U.-H. Lee, T. Kudo and I. Honma, Chem. Commun., 2009, 3068–3070 RSC.
- E. Stathatos, P. Lianos, U. Lavrencic-Stangar and B. Orel, Adv. Mater., 2002, 14, 354–357 CrossRef CAS.
- E. Stathatos, P. Lianos, S. M. Zakeeruddin, P. Liska and M. Grätzel, Chem. Mater., 2003, 15, 1825–1829 CrossRef CAS.
- A. I. Horowitz and M. J. Panzer, Angew. Chem., Int. Ed., 2014, 53, 9780–9783 CrossRef CAS PubMed.
- S. Srinivasan, W. H. Shin, J. W. Choi and A. Coskun, J. Mater. Chem. A, 2013, 1, 43–48 CAS.
- K. Z. Donato, R. K. Donato, M. Lavorgna, L. Ambrosio, L. Matejka, R. S. Mauler and H. S. Schrekker, J. Sol–Gel Sci. Technol., 2015, 76, 414–427 CrossRef CAS.
- A. Ikeda, K. Sonoda, M. Ayabe, S. Tamaru, T. Nakashima, N. Kimizuka and S. Shinkai, Chem. Lett., 2001, 30, 1154–1155 CrossRef.
- L. Tao, Z. Huo, X. Pan, C. Zhang and S. Dai, Prog. Chem., 2013, 25, 990–998 CAS.
- W. Kubo, S. Kambe, S. Nakade, T. Kitamura, K. Hanabusa, Y. Wada and S. Yanagida, J. Phys. Chem. B, 2003, 107, 4374–4381 CrossRef CAS.
- K. Hanabusa, H. Fukui, M. Suzuki and H. Shirai, Langmuir, 2005, 21, 10383–10390 CrossRef CAS PubMed.
- B. A. Voss, J. E. Bara, D. L. Gin and R. D. Noble, Chem. Mater., 2009, 21, 3027–3029 CrossRef CAS.
- L. Tan, X. Dong, H. Wang and Y. Yang, Electrochem. Commun., 2009, 11, 933–936 CrossRef CAS.
- S. Yamasaki and H. Tsutsumi, Bull. Chem. Soc. Jpn., 1994, 67, 906–911 CrossRef CAS.
- N. Mohmeyer, P. Wang, H.-W. Schmidt, S. M. Zakeeruddin and M. Grätzel, J. Mater. Chem., 2004, 14, 1905–1909 RSC.
- L. Tao, Z. Huo, Y. Ding, L. Wang, J. Zhu, C. Zhang, X. Pan, M. K. Nazeeruddin, S. Dai and M. Grätzel, J. Mater. Chem. A, 2014, 2, 9251–9259 Search PubMed.
- L. Tao, Z. Huo, Yo Ding, Y. Li, S. Dai, L. Wang, J. Zhu, X. Pan, B. Zhang, J. Yao, M. K. Nazeeruddin and M. Grätzel, J. Mater. Chem. A, 2015, 3, 2344–2352 CAS.
- L. Tao, Z. Huo, X. Pan, C. Zhang and S. Dai, Prog. Chem., 2013, 25, 990–998 CAS.
- F. Fages, Angew. Chem., Int. Ed., 2006, 45, 1680–1682 CrossRef CAS PubMed.
- T. Tu, X. Bao, W. Assenmacher, H. Peterlik, J. Daniels and K. H. Dötz, Chem. – Eur. J., 2009, 15, 1853–1861 CrossRef CAS PubMed.
- J. Majumder, J. Deb, M. R. Das, S. S. Jana and P. Dastidar, Chem. Commun., 2014, 50, 1671–1674 RSC.
- F. D'Anna, P. Vitale, F. Ferrante, S. Marullo and R. Noto, ChemPlusChem, 2013, 78, 331–342 CrossRef.
- Y. Xiong, J. Liu, Y. Wang, H. Wang and R. Wang, Angew. Chem., Int. Ed., 2012, 51, 9114–9118 CrossRef CAS PubMed.
- B. Ziolkowski and D. Diamond, Chem. Commun., 2013, 49, 10308–10310 RSC.
- S. Gallagher, L. Florea, K. J. Fraser and D. Diamond, Int. J. Mol. Sci., 2014, 15, 5337–5349 CrossRef PubMed.
- G. B. Appetecchi, G.-T. Kim, M. Montanino, M. Carewska, R. Marcilla, D. Mecerreyes and I. De Meatza, J. Power Sources, 2010, 195, 3668–3675 CrossRef CAS.
- T. L. Greaves and C. J. Drummond, Chem. Soc. Rev., 2008, 37, 1709–1726 RSC.
- C. P. Mehnert, Chem. – Eur. J., 2005, 11, 50–56 CrossRef PubMed.
- A. Riisager, R. Fehrmann, M. Haumann and P. Wasserscheid, Eur. J. Inorg. Chem., 2006, 695–706 CrossRef CAS.
- P. Wasserscheid, J. Ind. Eng. Chem., 2007, 13, 325–338 CAS.
- H. Olivier-Bourbigou, L. Magna and D. Morvan, Appl. Catal., A, 2010, 373, 1–56 CrossRef CAS.
- C. Van Doorslaer, J. Wahlen, P. Mertens, K. Binnemans and D. De Vos, Dalton Trans., 2010, 39, 8377–8390 RSC.
- A. M. Kluwer, C. Simons, Q. Knijnenburg, J. I. van der Vlugt, B. de Bruinc and J. N. H. Reek, Dalton Trans., 2013, 42, 3609–3616 RSC.
- P. Zhang, T. Wu and B. Han, Adv. Mater., 2014, 26, 6810–6827 CrossRef CAS PubMed.
- J. Blum, D. Avnir and H. Schumann, CHEMTECH, 1999, 29, 32–38 CAS.
- A. C. Marr and P. C. Marr, Dalton Trans., 2011, 40, 20–26 RSC.
- K. S. Morley, P. C. Marr, P. B. Webb, A. R. Berry, F. J. Allison, G. Moldovan, P. D. Brown and S. M. Howdle, J. Mater. Chem., 2002, 12, 1898–1905 RSC.
- K. S. Morley, P. Licence, P. C. Marr, J. R. Hyde, P. D. Brown, R. Mokaya, Y. D. Xia and S. M. Howdle, J. Mater. Chem., 2004, 14, 1212–1217 RSC.
- N. A. Hamill, C. Hardacre and S. E. J. McMath, Green Chem., 2002, 4, 139–142 RSC.
- M. A. Gelesky, S. S. X. Chiaro, F. A. Pavan, J. H. Z. dos Santos and J. Dupont, Dalton Trans., 2007, 5549–5553 RSC.
- L. Foppa, J. Dupont and C. W. Scheeren, RSC Adv., 2014, 4, 16583–16588 RSC.
- Y. Ma, B. Liu, Y. Zhang and J. Li, Int. J. Hydrogen Energy, 2015, 40, 9147–9154 CrossRef CAS.
- X. Kang, J. Zhang, W. Shang, T. Wu, P. Zhang, B. Han, Z. Wu, G. Mo and X. Xing, J. Am. Chem. Soc., 2014, 136, 3768–3771 CrossRef CAS PubMed.
- A. Safavi and A. R. Banazadeh, Electroanalysis, 2011, 23, 1536–1542 CrossRef CAS.
- S. J. Craythorne, A. R. Crozier, F. Lorenzini, A. C. Marr and P. C. Marr, J. Organomet. Chem., 2005, 690, 3518–3521 CrossRef CAS.
- H. Sertchook, D. Avnir, J. Blum, F. Joó, A. Kathó, H. Schumann, R. Weimann and S. Wernick, J. Mol. Catal. A: Chem., 1996, 108, 153 CrossRef CAS.
- F. Shi, Q. Zhang, Y. Gu and Y. Deng, Adv. Synth. Catal., 2005, 347, 225–230 CrossRef CAS.
- K. Hamza and J. Blum, Eur. J. Org. Chem., 2007, 4706–4710 CrossRef CAS.
- S. Volland, M. Gruit, T. Régnier, L. Viau, O. Lavastre and A. Vioux, New J. Chem., 2009, 33, 2015–2021 RSC.
- R. Ciriminna, P. Hesemann, J. J. E. Moreau, M. Carraro, S. Campestrini and M. Pagliaro, Chem. – Eur. J., 2006, 12, 5220–5224 CrossRef CAS PubMed.
- L. Liu, J. Ma, J. Xia, L. Li, C. Li, X. Zhang, J. Gong and Z. Tong, Catal. Commun., 2011, 12, 323–326 CrossRef CAS.
- D. Li, F. Shi, S. Guo and Y. Deng, Tetrahedron Lett., 2004, 45, 265–268 CrossRef CAS.
- J. Miao, H. Wan and G. Guan, Catal. Commun., 2011, 12, 353–356 CrossRef CAS.
- M. Erbeldinger, A. J. Mesiano and A. J. Russel, Biotechnol. Prog., 2000, 16, 1129–1131 CrossRef CAS PubMed.
- J. Miao, H. Wan, Y. Shao, G. Guan and B. Xu, J. Mol. Catal. A: Chem., 2011, 348, 77–82 CrossRef CAS.
- W. Ma, W. Wang, Z. Liang, S. Hu, R. Shen and C. Wu, Kinet. Catal., 2014, 55, 665–670 CrossRef CAS.
- K. Funabiki, T. Komeda, Y. Sakaida, Y. Kubota and M. Matsui, J. Mol. Catal. A: Chem., 2013, 367, 116–120 CrossRef CAS.
- Y.-M. Wang, V. Ulrich, G. F. Donnelly, F. Lorenzini, A. C. Marr and P. C. Marr, ACS Sustainable Chem. Eng., 2015, 3, 792–796 CrossRef CAS.
- A. I. Horowitz, Y. Wang and M. J. Panzer, Green Chem., 2013, 15, 3414–3420 RSC.
- R. M. Lau, F. van Rantwijk, K. R. Seddon and R. A. Sheldon, Org. Lett., 2000, 2, 4189–4191 CrossRef CAS.
- P. Lozano, T. De Diego, J. P. Guegan, M. Vaultier and J. L. Iborra, Biotechnol. Bioeng., 2001, 75, 563–569 CrossRef CAS PubMed.
- K. W. Kim, B. Song, M. Y. Choi and M. J. Kim, Org. Lett., 2001, 3, 1507–1509 CrossRef CAS PubMed.
- J. K. Lee and M. J. Kim, J. Org. Chem., 2002, 67, 6845–6847 CrossRef CAS PubMed.
- S. Braun, S. Rappoport, R. Zusman, D. Avnir and M. Ottolenghi, Mater. Lett., 1990, 10, 1–5 CrossRef CAS.
- Y. Liu, M. Wang, J. Li, Z. Li, P. He, H. Liu and J. Li, Chem. Commun., 2005, 1778–1780 RSC.
- S. H. Lee, T. T. N. Doan, S. H. Ha and Y.-M. Koo, J. Mol. Catal. B: Enzym., 2007, 45, 57–61 CrossRef CAS.
- S. H. Lee, T. T. N. Doan, S. H. Ha, W.-J. Chang and Y.-M. Koo, J. Mol. Catal. B: Enzym., 2007, 47, 129–134 CrossRef CAS.
- H. Vila-Real, A. J. Alfaia, J. N. Rosa, P. M. P. Gois, M. E. Rosa, A. R. T. Calado and M. H. Ribeiro, Activity and stability studies, J. Biotechnol., 2011, 152, 147–158 CrossRef CAS PubMed.
- R. L. de Souza, E. L. P. de Faria, R. T. Figueiredo, L. dos S. Freitas, M. Iglesias, S. Mattedic, G. M. Zanin, O. A. A. dos Santos, J. A. P. Coutinho, Á. S. Lima and C. M. F. Soares, Enzyme Microb. Technol., 2013, 52, 141–150 CrossRef CAS PubMed.
- D. R. MacFarlane, N. Tachikawa, M. Forsyth, J. M. Pringle, P. C. Howlett, G. D. Elliott, J. H. Davis, M. Watanabe, P. Simon and C. A. Angell, Energy Environ. Sci., 2014, 7, 232–250 CAS.
- D. W. Bruce, D. O'Hare and R. I. Walton, Inorganic Materials Series, John Wiley & Sons, 2011 Search PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- A. Brandt, S. Pohlmann, A. Varzi, A. Balducci and S. Passerini, MRS Bull., 2013, 38, 554–559 CrossRef CAS.
- C. Buzzeo, R. G. Evans and R. G. Compton, ChemPhysChem, 2004, 5, 1106–1120 CrossRef PubMed.
- A. A. J. Torriero, Electrochemistry in Ionic Liquids: Volume 1: Fundamentals, Springer, 2015 Search PubMed.
- P. V. Wright, Electrochim. Acta, 1998, 43, 1137–1143 CrossRef CAS.
- M. B. Armand, J. M. Chabagno, M. Duclot, P. Vashisha, J. N. Mundy and G. K. Shenoy, Fast Ion Transport in Solids, North Holland, 1979 Search PubMed.
- F. M. Gray, Solid Polymer Electrolytes-Fundamentals and Technological Applications, VCH, New York, 1991 Search PubMed.
- J. R. MacCallum and C. A. Vincent, Polymer Electrolyte Reviews-1, Elsevier, London, 1987 Search PubMed.
- J. R. MacCallum and C. A. Vincent, Polymer Electrolyte Reviews-2, Elsevier, London, 1989 Search PubMed.
- D. F. Shriver and P. G. Bruce, in Solid State Electrochemistry, ed. P. G. Bruce, Cambridge Univ. Press, Cambridge, 1995 Search PubMed.
- P. G. Bruce and F. M. Gray, in Solid State Electrochemistry, ed. P. G. Bruce, Cambridge Univ. Press, Cambridge, 1995 Search PubMed.
- M. Ishikawa, M. Morita and Y. Matsuda, The Electrochemical Society Meeting Abstracts, San Antonio, TX, 1996, vol. 96–2 Search PubMed.
- A. Hofmann, M. Schulz and T. Hanemann, Electrochim. Acta, 2013, 89, 823–831 CrossRef CAS.
- A. Liao, X. Sun and S. Dai, Electrochim. Acta, 2013, 87, 889–894 CrossRef.
- A. F. Visentin and M. J. Panzer, ACS Appl. Mater. Interfaces, 2012, 4, 2836–2839 CAS.
- A. S. Shaplov, R. Marcilla and D. Mecerreyes, Electrochim. Acta, 2015, 175, 18–34 CrossRef CAS.
- M. Salsamendi, L. Rubatat and D. Mecerreyes, in Electrochemistry in Ionic Liquids: Volume 1: Fundamentals, ed. Angel A. J. Torriero, Springer, 2015 Search PubMed.
- H. Nakajima and H. Ohno, Polymer, 2005, 46, 11499–11504 CrossRef CAS.
- S. Jeremias, M. Kunze, S. Passerini and M. Schönhoff, J. Phys. Chem. B, 2013, 117, 10596–10602 CrossRef CAS PubMed.
- J. Gao, R. S. Ndong, M. B. Shiflett and N. J. Wagner, ACS Nano, 2015, 9, 3243–3252 CrossRef CAS PubMed.
- K. Ueno, S. Imaizumi, K. Hata and M. Watanabe, Langmuir, 2009, 25, 825–831 CrossRef CAS PubMed.
- K. Ueno and M. Watanabe, Langmuir, 2011, 27, 9105–9115 CrossRef CAS PubMed.
- T. Mizumo, T. Watanabe and H. Ohno, Polym. J., 2008, 40, 1099–1104 CrossRef CAS.
- S. A. M. Noora, P. M. Bayley, M. Forsyth and D. R. MacFarlane, Electrochim. Acta, 2013, 91, 219–226 CrossRef.
- M. Armand, C. Poinsignon, J.-Y. Sanchez and V. de Zea Bermudez, U.S. Patent, 5283310, 1994 Search PubMed.
- V. de Zea Bermudez, C. Poinsignon and M. B. Armand, J. Mater. Chem., 1997, 7, 1677–1692 RSC.
- V. de Zea Bermudez, D. Baril, J.-Y. Sanchez, M. Armand and C. Poinsignon, in Proc. SPIE, ed. A. Hugot-Le Goff, C.-G. Granqvist and C. M. Lampert, Bellingham, Washington, 1992, vol. 1728, pp. 180–182 Search PubMed.
- M. A. Cardoso, M. Fernandes, L. C. Rodrigues, R. Leones, M. M. Silva and V. de Zea Bermudez, Electrochim. Acta, 2014, 147, 288–293 CrossRef CAS.
- F. Gayet, L. Viau, F. Leroux, S. Monge, J.-J. Robin and A. Vioux, J. Mater. Chem., 2010, 20, 9456–9462 RSC.
- A. Maršavelski, V. Smrečki, R. Vianello, M. Žinić, A. Mogus-Milankovič and A. Šantić, Chem. – Eur. J., 2015, 21, 12121–12128 CrossRef PubMed.
- M. Díaz, A. Ortiz and I. Ortiz, J. Membr. Sci., 2014, 469, 379–396 CrossRef.
- T. Yasuda and M. Watanabe, MRS Bull., 2013, 38, 560–566 CrossRef CAS.
- M. A. B. H. Susan, A. Noda, S. Mitsushima and M. Watanabe, Chem. Commun., 2003, 938–939 RSC.
- G. Lakshminarayana and M. Nogami, Electrochim. Acta, 2010, 55, 1160–1168 CrossRef CAS.
- H. Li, F. Jiang, Z. Di and J. Gu, Electrochim. Acta, 2012, 59, 86–90 CrossRef CAS.
- L. Sun, X. Zhang, W. Wang and J. Chen, Anal. Methods, 2015, 7, 5060–5066 RSC.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737 CrossRef.
- P. Wang, S. M. Zakeeruddin, I. Exnar and M. Grätzel, Chem. Commun., 2002, 2972–2973 RSC.
- P. Wang, S. M. Zakeeruddin, P. Comte, I. Exnar and M. Grätzel, J. Am. Chem. Soc., 2003, 125, 1166–1167 CrossRef CAS PubMed.
- K. Tomioka, T. Sumiyoshi, S. Narui, Y. Nagaoka, A. Iida, Y. Miwa, T. Taga, M. Nakano and T. Handa, J. Am. Chem. Soc., 2001, 123, 11817–11818 CrossRef CAS PubMed.
- W. Lu, K. Henry, C. Turchi and J. Pellegrino, J. Electrochem. Soc., 2008, 55, A361–A367 CrossRef.
- Y. J. Kang, H. Chung, C. Han and W. Kim, Nanotechnology, 2012, 23, 289501–289509 CrossRef.
- A. I. Horowitz and M. J. Panzer, J. Mater. Chem., 2012, 22, 16534–16539 RSC.
- L. Negre, B. Daffos, P. L. Taberna and P. Simon, J. Electrochem. Soc., 2015, 162(5), A5037–A5040 CrossRef CAS.
- H. Shirakawa, E. J. Louis, A. G. MacDiarmid, C. K. Chiang and A. J. Heeger, J. Chem. Soc., Chem. Commun., 1977, 578–580 RSC.
- S. K. Deb, Appl. Opt., 1969, 8(S1), 192–195 CrossRef PubMed.
- S. K. Deb, Philos. Mag., 1973, 27, 801 CrossRef CAS.
- L. B. Groenendaal, F. Jonas, D. Freitag, H. Pielartzik and J. R. Reynolds, Adv. Mater., 2000, 12, 481–494 CrossRef CAS.
- G. Li and P. G. Pickup, Phys. Chem. Chem. Phys., 2000, 2, 1255–1260 RSC.
- S. L. Cho and S. B. Lee, Acc. Chem. Res., 2008, 41, 699–701 CrossRef CAS PubMed.
- S. Bhandari, M. Deepa, A. K. Srivastava, C. Lal and R. Kant, Macromol. Rapid Commun., 2008, 29, 1959–1964 CrossRef CAS.
- M. Deepa, A. Awadhia and S. Bhandari, Phys. Chem. Chem. Phys., 2009, 11, 5674–5685 RSC.
- H. C. Moon, T. P. Lodge and C. D. Frisbie, Chem. Mater., 2015, 27, 1420–1425 CrossRef CAS.
- N. Cheminet, T. Jarrosson, J.-P. Lere-Porte, F. Serein-Spirau, L. Cury, J. Moreau, L. Viau and A. Vioux, J. Mater. Chem., 2011, 21, 13588 RSC.
- K. Lunstroot, K. Driesen, P. Nockemann, C. Gorller-Walrand, K. Binnemans, S. Bellayer, J. Le Bideau and A. Vioux, Chem. Mater., 2006, 18, 5711–5715 CrossRef CAS.
- K. Lunstroot, K. Driesen, P. Nockemann, K. Van Hecke, L. Van Meervelt, C. Gorller-Walrand, K. Binnemans, S. Bellayer, L. Viau, J. Le Bideau and A. Vioux, Dalton Trans., 2009, 298–306 RSC.
- P. Yu, J. Yan, H. Zhao, L. Su, J. Zhang and L. Mao, J. Phys. Chem. C, 2008, 112, 2177–2182 CAS.
- L. Ding, L. Li, W. You, Z.-N. Gao and T.-L. Yang, Croat. Chem. Acta, 2015, 88, 81–87 CrossRef CAS.
- W.-L. Zhu, Y. Zhou and J. R. Zhang, Talanta, 2009, 80, 224–230 CrossRef CAS PubMed.
- R. Yan, F. Q. Zhao, J. W. Li, F. Xiao, S. S. Fan and B. Z. Zeng, Electrochim. Acta, 2007, 52, 7425–7431 CrossRef CAS.
- X. B. Lu, J. Q. Hu, X. Yao, Z. P. Wang and J. H. Li, Biomacromolecules, 2006, 7, 975–980 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |