
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrogels based on cellulose and chitin: fabrication, properties, and applications
Xiaoping
Shen
ab,
Julia L.
Shamshina
c,
Paula
Berton
ad,
Gabriela
Gurau
cd and
Robin D.
Rogers
*ad
aDepartment of Chemistry, The University of Alabama, Tuscaloosa, AL 35487, USA
bKey Laboratory of Bio-based Material Science and Technology (Ministry of Education), Northeast Forestry University, 26 Hexing Road, Harbin 150040, China
c525 Solutions, Inc., 720 2nd Street, Tuscaloosa, AL 35401, USA
dDepartment of Chemistry, McGill University, 801 Sherbrooke St. West, Montreal, QC H3A 0B8, Canada. E-mail: robin.rogers@mcgill.ca
First published on 16th November 2015
Abstract
This review is focused on the fabrication, properties, and applications of hydrogels prepared from two of the most abundant biopolymers on earth, cellulose and chitin. The review emphasizes the latest developments in hydrogel preparation (including solvent systems, cross-linker types, and preparation methods, which determine the “greenness” of the process) using these biocompatible and biodegradable biopolymers. The preparation of both physical (without covalent cross-links) and chemical (with covalent cross-links) hydrogels via dissolution/gelation is discussed. Additionally, formation of injectable thermoset and/or pH sensitive hydrogels from aqueous solutions of derivatives (chitosan, methyl cellulose, and hydroxypropylmethyl cellulose) with or without a cross-linker are discussed. This review also compares the design parameters for different applications of various pure and composite hydrogels based on cellulose, chitin, or chitosan, including applications as controlled and targeted drug delivery systems, improved tissue engineering scaffolds, wound dressings, water purification sorbents, and others.
1. Introduction
Hydrogels are physically or chemically cross-linked three-dimensional (3D) hydrophilic polymeric networks capable of absorbing large amounts of water (or biological fluids) and swelling.1 Any (semi-)flexible polymer is able to virtually formulate hydrogels in a variety of physical forms including slabs, membranes, beads, microgels (microspheres), and nanogels (nanoparticles), and once freeze-dried or supercritically dried, hydrogels become cryogels or aerogels, respectively.2 Hydrogels are held together by either physical interactions (chain entanglements, van der Waals forces, hydrogen bonds, crystallite associations,3 and/or ionic interactions4) or chemical cross-links (covalent bonding5).Generally, hydrogels are divided into two categories, according to their natural or synthetic origin: biopolymer-based or synthetic.6 Considering the biocompatibility, biodegradability, and tissue-mimicking consistency of biopolymer-based hydrogels, they have acquired increasing attention. It has been shown that biopolymer-based hydrogels are applied in varied fields such as hygiene (disposable diapers and feminine care products),7 agriculture (water retention,8 and pesticide delivery9), biomedical materials (drug carriers,10 wound dressings,11 and tissue engineering scaffolds12), pollutant adsorbents (heavy metal ions,13 dyes,14,15 and pesticides16), biosensors,17etc. Various natural polymers or their salts such as sodium alginate,18 starch,19 protein,20 gelatin,21 hyaluronate,22 hemicelluloses,23 lignin,24 cellulose,25 chitin,26 and their derivatives27,28 have been used to fabricate biopolymer-based hydrogels.
Among the biopolymers, cellulose and chitin are two of the most abundant on earth, thus having great potential in hydrogel preparation. Cellulose consists of a straight chain of β-(1→4)-linked D-glucose units, and chitin, structurally similar to cellulose, is a long-chain co-polymer of β-(1→4)-linked 2-acetamido-2-deoxy-β-D-glucose units, with acetamide groups in the C-2 position. If the degree of acetylation (%DA) of the biopolymer is lower than 50%, it is no longer called chitin, but chitosan.29 The plentiful hydrophilic functional groups (hydroxyl- and/or amino-) in the backbones of either cellulose or chitin qualify them as promising materials for highly absorbent hydrogel systems.
In this review, we discuss cellulose and chitin hydrogels with emphasis on their fabrication, properties, and applications. Given that chemically modified cellulose and chitin polymers may result in improved processability and/or unique characteristics, hydrogels from several common derivatives of cellulose and chitin are also discussed, including methyl cellulose (MC), hydroxypropylmethyl cellulose (HPMC), sodium carboxymethyl cellulose (CMC), hydroxypropyl cellulose (HPC), hydroxyethyl cellulose (HEC), and chitosan.
2. Dissolution of native cellulose and chitin
Most hydrogels based on native cellulose or chitin are usually prepared through a two-step process involving dissolution followed by cross-linking (i.e., gelation), although culturing specific bacteria can produce hydrogels directly (discussed in the “physical hydrogels” section below). Specific solvent systems are required to dissolve native cellulose and chitin, which possess poor solubility characteristic due to the numerous inter- and intra-molecular hydrogen bonds between polymeric chains.To date, only a few solvents have been used for the dissolution of native cellulose or chitin (Table 1). These include some conventional polar solvent systems such as N-methylmorpholine oxide (NMMO),30 lithium chloride/dimethylacetamide (LiCl/DMAc),31,32 paraformaldehyde/dimethylsulfoxide (PF/DMSO), triethylammonium chloride/dimethylsulfoxide (TEAC/DMSO),33 tetrabutylammonium fluoride/dimethylsulfoxide (TBAF/DMSO),34 lithium chloride/N-methyl-2-pyrolidone (LiCl/NMP),35 and calcium chloride dihydrate/methanol (CaCl2·2H2O/MeOH).26 Although the use of these polar solvents has somewhat alleviated the issues with the biopolymer's intractability, the toxicity or corrosivity of these organic components can inhibit batch production and potential applications of the resulting gels.
| Entry | Solvents | Matrix | Dissolution | Gelation | Ref. | |
|---|---|---|---|---|---|---|
| a Choline bromide (ChBr); choline chloride (ChCl); dimethylacetamide (DMAc); dimethylsulfoxide (DMSO); freeze/thaw (F/T); microcrystalline cellulose (MCC); methanol (MeOH); N-methylmorpholine-N-oxide (NMMO); N-methyl-2-pyrolidone (NMP); paraformaldehyde (PF); room temperature (RT); supercritical-drying (Sc-dry); tetrabutylammonium fluoride (TBAF); triethylammonium chloride (TEAC). b Cast: cast on a glass or ceramic plate followed by coagulation; bead: drop,32 inject,38 extrude,73 or spray/atomize/nebulizer65 into the coagulant, spay-drying,74 or ion exchange;31 mold: pour in a vial or a specific mold followed by curing and/or coagulation; cure: stand at various temperatures (5–60 °C) for a certain time (minutes–weeks). c The “a” and “b” refer to the same dissolution process but different gelation procedures. | ||||||
| S1 | Polar Solvents | NMMO | Wood, cotton pulp | 85 °C | 7–11 wt%, castb | 30 |
| S2ac | LiCl/DMAc | Cellulose pulp | 75–90 °C (time not reported) | 0.5–8 wt%, moldb/cast/beadb | 33, 65, 66 | |
| S2bc | 1–8 wt%, ion exchange | 31 | ||||
| S3 | PF/DMSO | Wood pulp | 120 °C, 1 h | 4 wt%, cast | 33 | |
| S4 | TEAC/DMSO | Wood pulp | 90 °C (time not reported) | |||
| S5 | TBAF/DMSO | MCC | 60 °C, 20 min | 0.5–1 wt%, mold, coagulate | 34 | |
| S6a | LiCl/DMAc | Chitin | 19 °C, 12 h | 1, 1.85 wt%, mold, cureb | 67 | |
| S6b | 0.3–1.5% w/v, mold/bead | 32 | ||||
| S7 | LiCl/NMP | Chitin | RT, 48 h | 0.3–5% w/v, mold/bead | 32, 35 | |
| S8 | CaCl2·2H2O/MeOH | Chitin | 100 °C (time not reported) | 1.96 wt%, coagulate, dialyze/filter | 26 | |
| S9ac | Ionic liquids | [C2mim][OAc] | MCC | 80 °C, 3–4 h | 12.5 wt%, cure | 37, 38 |
| S9bc | 5–7 wt%, bead | |||||
| S10 | [C4mim]Cl | MCC | 130 °C (or microwave), 3.5 h | 4.75 wt%, cast | 39, 40 | |
| S11 | [Amim]Cl | Filter paper | 70 °C, 2 h | 1.5 wt%, mold, cool, coagulate | 41 | |
| S12 | [Amim]Br | Chitin | 100 °C, 48 h | 7 wt%, mold, cool | 42 | |
| S13 | [C2mim][OAc] | Chitin | 90–95 °C, 5 h | 1–3 wt%, mold, cure, coagulate | 43 | |
| S14 | [C4mim][OAc] | Chitin | 100 °C (time not reported) | 4 wt%, mold, cool, coagulate | 44 | |
| S15 | Deep eutectic solvents | ChCl/urea; ChBr/urea; ChCl/thiourea | Chitin | 100 °C, 2 h | 10 wt%, cure | 48 |
| S16 | Alkali aqueous systems | Alkali | MCC, cellulose pulp | −6 °C, 2 h | 3–7 wt%, cure, Sc-drya | 68 |
| S17ac | Alkali/urea | Cellulose pulp, filter paper, tunicate cellulose | −12 to −10 °C, 5–10 min; F/T (F: −5 °C, 5 h), 1 cycle | 4 wt%, cure (55 °C) | 54 | |
| S17bc | 0.5–7 wt%, cast | 64, 69–71 | ||||
| S18ac | Alkali/thiourea | Cotton linter | −5 °C, 2–10 min; F/T (F: −8 °C, 12 h), 1 cycle | 5 wt%, cast | 61 | |
| S18bc | 3–6 wt%, mold, cure | 61, 62, 77 | ||||
| S19 | Alkali | Chitin | F/T (F: −18 °C, 12 h), 1 cycle | 2 wt%, mold, cure (60 °C) | 72 | |
| S20 | Alkali/urea | Chitin | F/T (F: −20 °C, 12–18 h), 1–2 cycles | 2–8 wt%, cure/coagulate | 75, 78 | |
Recent alternatives for cellulose and chitin dissolution and hydrogel production include ionic liquids (ILs), deep eutectic solvents (DESs), and alkali or alkali/(thio)urea aqueous systems developed since the 2000s. Various ILs used for cellulose dissolution consist of an imidazolium, pyridinium, ammonium, or phosphonium cation paired with a strongly basic, hydrogen bond accepting anion (e.g., OAc−, HCOO−, HSCH2COO−, (MeO)HPO2−, (MeO)MePO2−, (MeO)2PO2−, Cl−, or Br−),36 and several of these ILs including 1-ethyl-3-methylimidazolium acetate ([C2mim][OAc]),37,38 1-butyl-3-methylimidazolium chloride ([C4mim]Cl),39,40 and 1-allyl-3-methylimidazolium chloride ([Amim]Cl)41 have been applied in the preparation of cellulose gels. In contrast to the enormous attention captured by cellulose, not much information is yet available on chitin-dissolving ILs; typical ILs in this context are 1-allyl-3-methylimidazolium bromide ([Amim]Br),42 [C2mim][OAc],43 and 1-butyl-3-methylimidazolium acetate ([C4mim][OAc]).44
Deep eutectic solvents (DESs) are fluids obtained by heating two or three components that are capable of self-association through hydrogen bonding. DESs have almost identical physicochemical properties to those of ILs except that they do not entirely consist of ionic species and may be cheaper.45,46 The classic examples are the combinations of choline chloride (mp 302 °C) with urea (mp 133 °C) or thiourea (mp 175 °C), forming a DES with a mp of 12 °C or 69 °C, respectively.47 Although dissolution of both chitin and cellulose in DESs has been verified,46 there is only a single report on self-assembly of chitin in such solvents (producing a soft chitin gel) and no research on cellulose gelation yet to date.48
Another solvent system for cellulose or chitin dissolution and gelation are the alkali/urea (or thiourea) aqueous systems developed in L. Zhang's group.49,50 Typically, the dissolution power of alkali/urea solvent systems is in the order NaOH/thiourea > LiOH/urea > NaOH/urea ≫ KOH/urea aqueous solution.49,51 In particular, the dissolution process in alkali/urea systems is very different from that in other solvents mentioned above, i.e., a low temperature treatment (by precooling the solvent52 or freezing/thawing the mixture50) is needed instead of stirring at room temperature (RT) or high temperatures. This may be because the dissolution of polymers in aqueous systems is an entropy-driven process (i.e., the entropy of the polymers in dissociated state increases significantly compared with that in the crystalline state), whereas in other solvents it is not (e.g., the entropy of the polymer chains after dissolution in IL increases very slightly or even decreases).52,53 Although the mechanism of the dissolution of cellulose or chitin in all these solvents has not been fully determined, the widely accepted opinion is that hydrogen bond acceptors (N–O, Cl−, OAc−, etc.) and/or donors (–NH2 in urea or thiourea) of the solvent break up the intra- and inter-molecular hydrogen bonds in the biopolymer chains upon stirring, heating, or low temperature treatment.36,46,50,54,55
3. Physical hydrogels
Physical hydrogels are cross-linked by physical interactions such as chain entanglements, van der Waals forces, hydrogen bonds, hydrophobic or electronic associations. For cellulose and chitin, physical hydrogels have been prepared from four major biopolymer sources: native cellulose or chitin powders (dissolved in specific solvents presented as above), from nanowhiskers (dispersed in water), from biopolymer derivatives (dissolved in water or acid), and from bacterial cellulose (BC; dissolved in specific solvents presented as above; or biosynthesized directly into gels during bacterial culture).3.1. Hydrogel formation from native cellulose or chitin
Cellulose and chitin polymers in solution behave as random coils, semi-flexible (or semi-rigid) chains, or entangled chains, and the degree of entanglement depends on the polymer concentration.56–60 While polymer solutions of low concentrations are completely isotropic, with increasing polymer concentration, the transition from solution to a liquid crystalline gel takes place followed by gelation into a solid gel that has an anisotropic structure.31,37 The gel structure becomes more and more organized and stable as biopolymer concentration increases due to the higher degree of entanglements and the more hydrogen bonding interactions that exist.37 Further, upon curing (i.e., keeping the solution at various temperatures between 5–60 °C for minutes or weeks)61–63 or coagulation (i.e., immersion in certain anti-solvents such as water,38 ethanol,32 methanol,26 H2SO4/Na2SO4,64etc.) in various fashions (e.g., beading, molding, or casting;30,31,34,65–74Table 1), the physicochemical interactions between the polymer chains increases and the stability of the hydrogel enhances.Usually, physical associations are reversible, leading to a thermo-reversible and “green” (without cross-linkers) sol–gel transition process.75,76 However, if (a) the reversible gel is coagulated in an anti-solvent, or (b) the NaOH/urea (or thiourea) solvent denatures at high curing temperatures (>60 °C, resulting in a yellow color) via reaction and thermal decomposition of the solvent molecules,54,77 the solvents are washed out (in “a”) or destroyed (in “b”), resulting in formation of so-called irreversible gels. Such irreversible physical gels, when vacuum- or supercritically-dried, exhibit lower degrees of crystallinity (reduced by 9–22%) than native cellulose or chitin.32,43,78 In addition, cellulose I changes to cellulose II and β-chitin changes to α-chitin, respectively, after dissolution and gelation.26,79
On the other hand, when coagulative regeneration is used, the transmittance can be tuned through control of the degree of phase separation in the sample using a certain amount of coagulant (e.g., water).34 Incorporation of acetone into water as a coagulation solution increases the transparency of the hydrogels, which may be related with a “specific polymer structure” formed resulting from the balance between swelling of the gel by water and shrinking by acetone.33 A novel method, deionization of the cellulose/LiCl/DMAc solution with ion exchange resins, has been reported for preparation of gel beads that appear colorless and transparent without a fibrous texture compared with the “water-coagulated gel”, probably because the molecular association (crystallite formation) precedes the phase separation during ion exchange.31
In addition, the mechanical strength of physical hydrogels can be improved by employing a pre-gelation process before performing coagulation as shown in the example of cellulose hydrogel beads from a NaOH/thiourea solution. In this example, hydrated cellulose membranes were packed densely with many nanospheres (200–510 nm according to the pre-gelation temperature).61 This could be attributed to the thiourea inclusion complex aggregates (47–160 nm) in which the cellulose chains associated with NaOH hydrates as guests were encaged by NaOH and thiourea.62,80
3.2. Hydrogel formation from cellulose or chitin nanowhiskers
The main process for the isolation of cellulose nanowhiskers (CNWs) and chitin nanowhiskers (ChNWs) from cellulose and chitin fibers, respectively, is based on acid hydrolysis (usually sulfuric acid for CNWs and hydrochloric acid for ChNWs) of the polymer(s).81,82 The nanoscale size (3–50 nm in width, 0.1–2 μm in length)83,84 and electrostatic repulsions (negatively charged sulfate groups on CNWs, positively charged amino groups on ChNWs) promote a perfectly uniform dispersion of the whiskers in water. As with native cellulose and chitin, the dispersed whiskers are also able to concentrate and self-organize into nematic or solid gels above a certain concentration.85,86Production of stable solid nanowhisker-based gels was achieved through further treatment of the nanowhisker aqueous suspensions, using solvent exchange,87 ultrasonication,88,89 dialysis (to concentrate),90 and, most often, adjustment of pH91 (Table 2). In ultrasonication-assisted assembly, the sonication bath provides energy with a similar order of magnitude as that of hydrogen bonds (4–50 kJ mol−1),89 allowing for the rearrangement and formation of a 3D-percolated network through hydrogen bonding. The solvent exchange method with a miscible anti-solvent (routinely acetone) results in chain rearrangement and formation of robust macroscopic gels from the nanowhisker suspension.87 Yet another method is pH adjustment; for instance, when the pH value of the ChNW suspension in acetic acid was adjusted to 10–11, the suspension immediately turned into a freestanding hydrogel even at a concentration as low as 0.5 wt% due to the elimination of repulsive interactions between the ChNW elements under basic conditions.91
Additionally, cellulose and chitin nanowhiskers are used to reinforce some polymeric matrices, such as cellulose,92 chitosan,93 poly(vinyl acetate),94 and poly(methyl vinyl ether-co-maleic acid) and poly(ethylene glycol)95,96 due to the high Young's modulus (ca. 140 GPa) of the nanowhiskers.97,98 The nanowhisker density (i.e., content) is generally within 25%, which leads to increased thermal stability, mechanical strength, and overall Young's modulus, but decreased water uptake of the polymeric gels with the increase of whisker content.93–96 Functionalization of nanowhiskers with carboxyl or amine groups prior to their incorporation into the polymer networks results in mechanically and hydrophilically adaptive, pH-responsive nanocomposite gels.99
Once formed, the transparency of nanowhisker-based gels is determined theoretically by the thickness of the gels or the density of the whiskers since a single nanowhisker has a diameter less than one-tenth of the wavelength of visible light and thus does not scatter light.90,100 For example, thin chitin nanowhisker-based gel films are transparent (transmittance = 90.2% at 600 nm);90 and when used as reinforcement, both cellulose and chitin nanowhiskers can retain the transparency of the polymer matrix (e.g., chitosan,93 and poly(acrylic acid)100) to different extents depending on the whisker density.
3.3. Hydrogel formation from cellulose or chitin derivatives
The most widely used biocompatible derivatives of cellulose and chitin are hydrophobically modified cellulose (MC and HPMC) and chitosan (deacetylated chitin), respectively. These derivatives are much easier to fabricate into hydrogels because they are water- or acid-soluble. MC and HPMC are water soluble due to the fact that hydrogen bond formation is prevented by the introduction of pendant groups; and chitosan can be dissolved in dilute acid aqueous solutions (e.g., dilute acetic acid). From aqueous solutions of these derivatives, unique hydrogels such as thermoset and pH-sensitive hydrogels can be formed either with or without the addition of a physical cross-linker that can introduce hydrophobic or electrostatic associations.101–103As an example of such gels, an elastic and thermo-reversible MC gel was formed when heating the MC aqueous solution above a critical temperature, which seemed dependent on MC concentration (63 °C at 0.30–2.5 wt%,104 42.5 °C at ca. 4.7 wt%,101 32 °C at 7.0 wt%, and 27 °C at 9.0 wt%105). An accepted mechanism of this gelation employs the concept of solvent reorganization on heating of a MC solution, where the solvated cage-like MC structure (originally formed through hydrogen bonds of water along MC chains) gets destroyed and thereby hydrophobic regions of MC are exposed, leading to the formation of hydrophobic aggregates.104 The mechanism of formation of HPMC gels is thought to be similar, although solutions have slightly higher gelation temperatures (e.g., 70 °C at 2 wt%) than MC solutions, indicating that the hydroxypropyl substituents inhibit the gelation.106–108 These thermoset hydrogels have much potential as injectable drug or cell carriers in vivo.
Chitosan hydrogels can be obtained by coagulation in alkaline media (such as NaOH109 and NaOH/Na2CO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 110 aqueous systems) to relieve the electronic repulsion of –NH3+ groups between polymeric chains. To increase the strength of chitosan hydrogels, physical ionic cross-linkers (Table 3), sodium citrate (SC)102 and tripolyphosphate (TPP),103,111–114 were employed, which introduced electrostatic interaction between the anion (–COO− or P3O105−) and cation (–NH3+) into the system and led to the formation of pH-sensitive chitosan hydrogels. In such pH-sensitive chitosan hydrogels, the cross-linking density was controlled by adjusting the pH value of the ionic cross-linker solution, e.g., chitosan could be completely ionically cross-linked by P3O105− ions in acidic TPP solution (pH 3), whereas it was slightly ionically cross-linked in the original TPP solution (pH 8.6).103
110 aqueous systems) to relieve the electronic repulsion of –NH3+ groups between polymeric chains. To increase the strength of chitosan hydrogels, physical ionic cross-linkers (Table 3), sodium citrate (SC)102 and tripolyphosphate (TPP),103,111–114 were employed, which introduced electrostatic interaction between the anion (–COO− or P3O105−) and cation (–NH3+) into the system and led to the formation of pH-sensitive chitosan hydrogels. In such pH-sensitive chitosan hydrogels, the cross-linking density was controlled by adjusting the pH value of the ionic cross-linker solution, e.g., chitosan could be completely ionically cross-linked by P3O105− ions in acidic TPP solution (pH 3), whereas it was slightly ionically cross-linked in the original TPP solution (pH 8.6).103
| Type | Cross-linkers | Structure | Solvent | Gelation | Ref. |
|---|---|---|---|---|---|
| a Acetic acid (HOAc); room temperature (RT). b Shape: gelate the chitosan solution in a certain fashion (beading, casting, or molding). c Soak: soak the gel in the cross-linker solution. d Dropping the chitosan solution of high concentrations (e.g., 2% w/v) into the cross-linker solution producing beads with larger diameters, while dropping the cross-linker solution into the chitosan solution of low concentrations (e.g., 0.1% w/v) producing micro or nanogels. | |||||
| Electrostatic interaction | Sodium citrate (SC) |
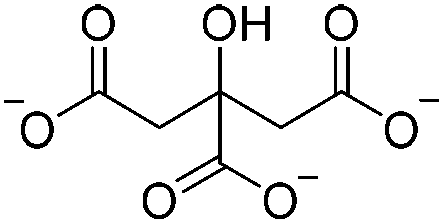
|
4% (w/v) HOAc | 4% w/v, shapeb, soakc, cure (RT, 1 h) | 102 |
| Electrostatic interaction | Tripolyphosphate (TPP) |
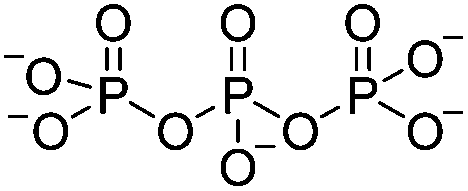
|
1% (w/v or v/v) or 5% (v/v) HOAc; ultrapure H2O | 0.1–3.3% w/v, beadd, cure (RT, 12 h) | 103, 111–114 |
| Hydrophobic interaction | β-Glycerophosphate (β-GP) |
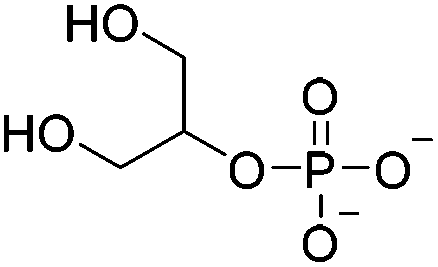
|
0.75% (v/v) HOAc; 0.1 M HCl | 1.5–3% w/v, thermogelate | 115–117 |
Another hydrophobic cross-linker, β-glycerophosphate (β-GP), was used for formation of thermoset chitosan hydrogels that featured a gelation temperature in the vicinity of 37 °C within the pH range of 6.8–7.2 (buffered by the dosage of β-GP).115–117 The addition of β-GP increased pH and ionic strength of the chitosan solution, establishing a favorable environment to form a gel structure by both the screening of electrostatic repulsion and the enhancement of polymer–polymer hydrophobic interactions.118 The protonation of chitosan, meanwhile, decreased strongly in the neutral chitosan/β-GP solution, especially at higher temperatures, thus leading to a minor contribution of ionic cross-linking.118
3.4. Hydrogel formation from bacterial cellulose (BC)
The properties of bacterial cellulose (BC) are quite different from those of plant cellulose, especially its high purity, ultrafine network structure, high hydrophilicity, and moldability during hydrogel formation (culturing without any cross-linker).119 The species of bacteria capable of producing cellulose extracellularly is generally called Acetobacter xylinum (acetic acid bacteria). The culture is carried out normally in static conditions at ca. 28–30 °C in the culture medium containing saccharides or natural saps and juices.120 A white BC gel pellicle generates on the surface, and its thickness increases steadily with time, reaching over 25 mm in four weeks.120 Such BC gels help the aerobic bacterial cells hold their position close to the oxygen-rich surface and protect themselves from water loss, ultraviolet lights, enemies, heavy metal ions, etc.121The as-biosynthesized BC gels show a slightly higher water content and ability to bind water molecules than plant cellulose gels; only 10% of the water molecules in the BC gel behave like free bulk water while the majority are more or less bound to cellulose.122 BC gels can be easily molded into desired shapes and sizes during synthesis, e.g., tubular BC gels with proper fibril orientation were created by culturing bacteria in oxygen-permeable silicone tubes with inner diameter less than 8 mm, which hold promise for use as microvessels in medical and pharmaceutical applications.123 BC gels could also be molded into a shape with dimensions similar to cartilage tissues (e.g., meniscus) as a potential implant124 or a scaffold for chondrocyte proliferation.125
4. Chemical hydrogels
4.1. Chemical cross-linkers
To guarantee the stable structure and effective swelling of cellulose- or chitin-based hydrogels, a covalently bound 3D hydrophilic network is usually required and achieved through the use of chemical cross-linkers during gelation, i.e., small bifunctional or multifunctional molecules such as 1,2,3,4-butanetetracarboxylic dianhydride (BTCA),126,127 succinic anhydride (SA),128,129 citric acid (CA),130,131 epichlorohydrin (ECH),25,132 ethylene glycol diglycidyl ether (EGDE),133–135 and divinyl sulfone (DVS)136–138 (Table 4). Chemical cross-linkers form covalent bonds that link one polymer chain to another. According to the mechanism of cross-linking reactions, chemical cross-linkers for cellulose and chitin can be classified into two types: (a) esterifying agents including carboxylic acids and carboxylic anhydrides; and (b) etherifying agents including organochlorine, epoxide, and vinyl compounds. The first type of cross-linkers results in formation of –COOR bonds and probably a few peptide bonds (–CONH–) in chitin gels, while the second type of cross-linkers results in formation of R–O–R bonds and probably some secondary amine bonds (R–NH–R) for chitin.| Type | Cross-linkers | Structure | Solvent | Gelation | Ref. |
|---|---|---|---|---|---|
| a Dimethylsulfoxide (DMSO); N-methyl-2-pyrolidone (NMP); tetrabutylammonium fluoride (TBAF). b Cellulose derivatives (carboxymethyl cellulose (CMC), hydroxyethylcellulose (HEC), or hydroxypropyl cellulose (HPC)) or chitosan were used. c Nanoparticles with diameters 270–370 nm were obtained and not macrogels, due to the low concentration of the biopolymer in the solution. d Chitosan beads were prepared from the acid chitosan solution through injection into an alkaline coagulant. e Soak: soak the gel in the cross-linker solution. f HPC nanogels were obtained from the 0.1 wt% emulsion with dodecyltrimethylammonium bromide (DTAB) as a surfactant above the critical micelle concentration (cmc) of the surfactant.137 | |||||
| Esterification | 1,2,3,4-Butanetetracarboxylic dianhydride (BTCA) |
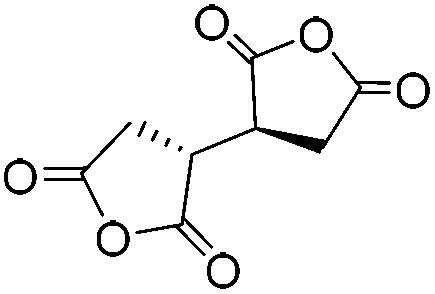
|
LiCl/NMP | 0.9–1.0 wt%, DMAP, cure (RT, 24 h), coagulate | 126, 127 |
| Succinic anhydride (SA) |
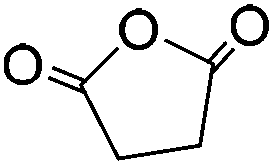
|
LiCl/NMP; TBAF/DMSO | 0.5–1.0 wt%, DMAP, cure (RT, 24 h), coagulate | 128, 129 | |
| Citric acid (CA) |
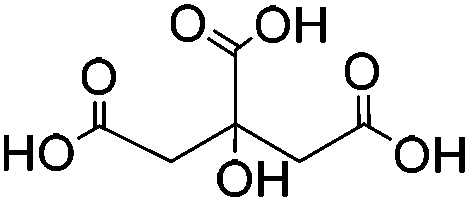
|
Waterb | 2 wt%, mold, heat (80 °C, 24 h); 0.01–0.1 wt%c, carbodiimide, cure (RT, 24 h) | 130, 131 | |
| Etherification | Epichlorohydrin (ECH) |
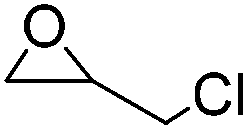
|
NaOH/urea | 1–4 wt%, mold, heat (50–60 °C, 1–20 h) | 25, 132 |
| Ethylene glycol diglycidyl ether (EGDE) |
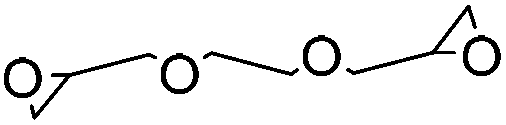
|
Waterb | 3 wt%, NaOH, mold, heat (60 °C, 24 h); beadd (2 wt%), NaOH, soake, cure (50–70 °C, 3–6 h) | 133–135 | |
| Divinyl sulfone (DVS) |
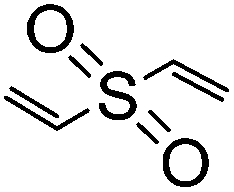
|
Waterb | 6–9 wt%, KOH or NaOH, mold, cure (RT or 47 °C, 24 h); shape (as emulsion)f, NaOH, cure (55 °C, 1 h) | 136–138 | |
Most esterification-type cross-linking reactions of biopolymers with carboxylic anhydrides require effective nucleophilic catalysis, and typically 4-dimethylaminopyridine (DMAP) has been used; examples include using BTCA and SA as cross-linkers.126–129 In these reactions, DMAP initially reacts with an acyl group of the anhydride, forming a positive acylpyridinium intermediate and a negative carboxyl counterion. The polysaccharide molecules are deprotonated by the latter, and then attack the acyl group in the former to form an ester (Fig. 1a).139
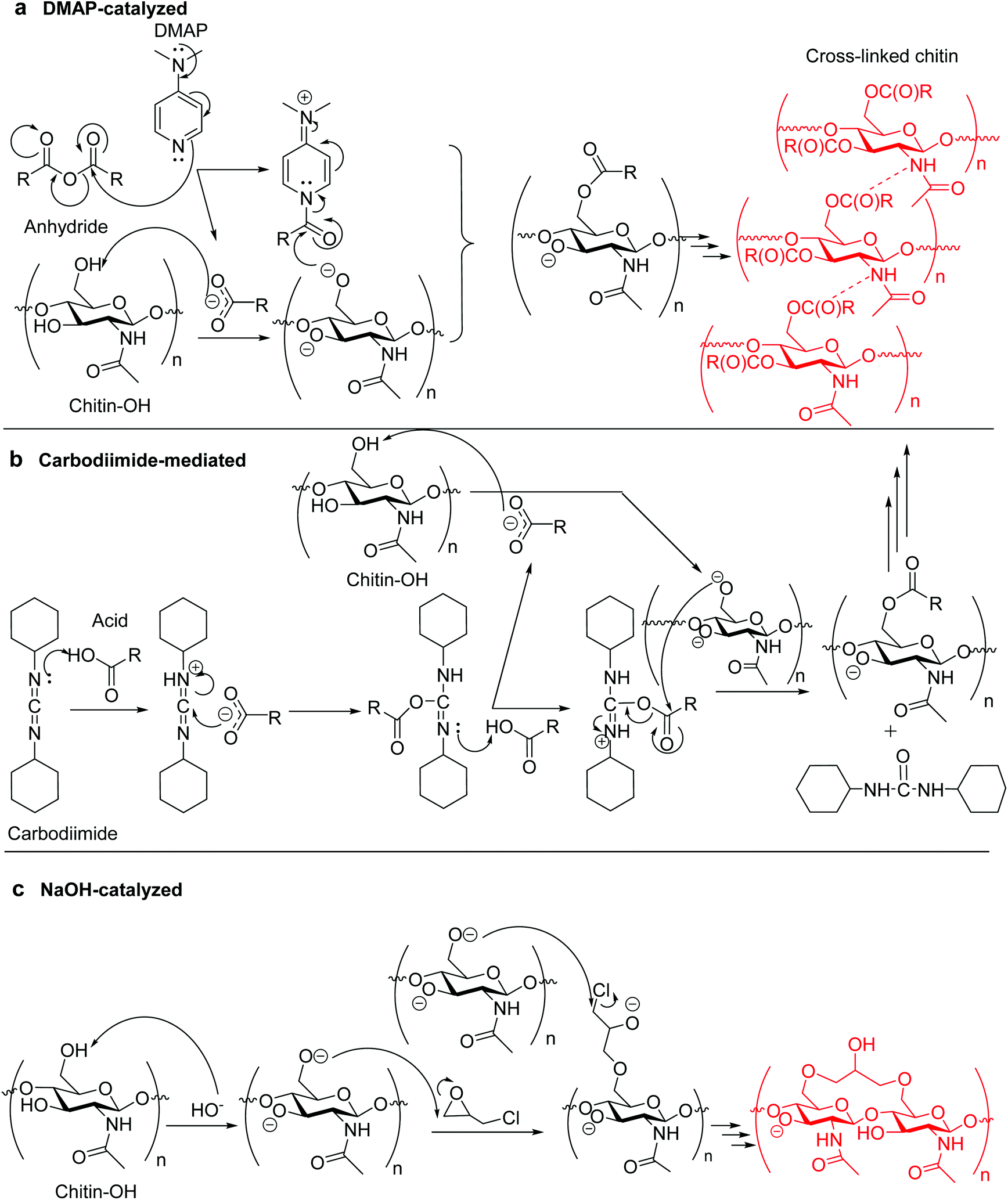 | ||
| Fig. 1 Possible chitin cross-linking mechanisms of (a) DMAP-catalyzed cross-linking using anhydride;139 (b) carbodiimide-mediated cross-linking (N,N′-dicyclohexylcarbodiimide is used as carbodiimide), and (c) alkali-catalyzed cross-linking. | ||
There is an alternative for the condensation route between carboxylic acid (e.g., CA) and biopolymer by using water-soluble carbodiimide (e.g., 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide methiodide, and N,N′-dicyclohexylcarbodiimide) as a mediation agent.131 The mechanism potentially involves formation of a labile acyl-intermediate, capable of reacting with the biopolymer and changing itself into a nontoxic urea derivative (Fig. 1b). When changing the carboxylic acid into CMC, the cross-linking of a CMC/biopolymer composite hydrogel was achievable.140 For etherifying cross-linkers, i.e., organochlorine, epoxide, and vinyl compounds (e.g., ECH, EGDE, and DVS25,132–138), etherification generally involves reactions in aqueous alkaline conditions (alkali-catalysis) for the deprotonation of hydroxyl groups on the biopolymer, making them highly nucleophilic and reactive with the cross-linker (Fig. 1c).
Some chemical cross-linkers are only applicable to deacetylated chitin or chitosan that has amine (–NH2) groups, including glutaraldehyde (GA),28,141,142 malondialdehyde (MDA),143 hexamethylene-1,6-di-(aminocarboxysulfonate) (HDS),93 and genipin (GNP).74,144 One cross-linker molecule reacts with two amine groups in two chitosan units: GA and MDA cross-link to produce two Schiff bases (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–), HDS results in peptide bonds (–CONH–), and GNP produces both a peptide bond and a tertiary amine (Table 5).
N–), HDS results in peptide bonds (–CONH–), and GNP produces both a peptide bond and a tertiary amine (Table 5).
| Type | Cross-linkers | Structure | Solvent | Gelation | Ref. |
|---|---|---|---|---|---|
| a Acetic acid (HOAc); room temperature (RT). b NaCl (2%) can be added to improve the solution properties (viscosity, transparency, etc.). c Chitosan beads were prepared from the chitosan solution through dropping or spraying. d Soak: soak the gel in the cross-linker solution. e Malondialdehyde (MDA) was formed through the hydrolysis of 1,1,3,3-tetramethoxypropane (TMP). | |||||
| –CN– |
Glutaraldehyde (GA) |

|
0.05 M HCl; 5% (units not reported) HOAcb | Beadc (1.5 wt%), soakd, heat (40 °C, 72 h); 1.5 wt%, cure (RT, 24 h) | 28, 141, 142 |
| –C–N– | Malondialdehyde (MDA)e |
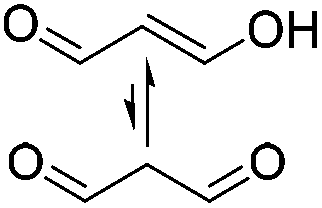
|
1 M HOAc/NaOAc | 3.8% w/v, cure (40 °C, 4 h), reduce (NaBH3CN, 4 days) | 143 |
| –CONH– | Hexamethylene-1,6-di-(aminocarboxysulfonate) (HDS) |
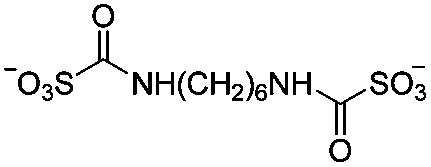
|
5 wt% HOAc | 4.55 wt%, cure (60 °C, 48 h) | 93 |
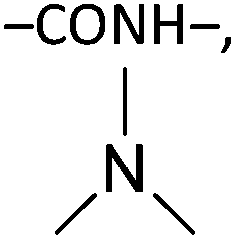
|
Genipin (GNP) |
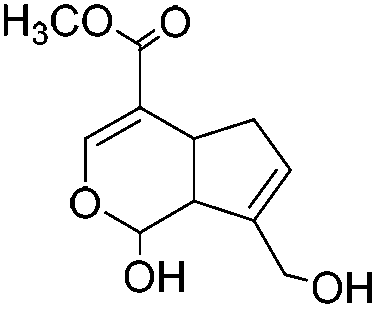
|
0.5 wt% or 2% (units not reported) HOAc | Beadc (1.5 wt%), soak, cure (RT, 3–16 h) | 74, 144 |
Cross-linking of chitosan gels can be performed either during or after the shaping (molding, casting, or beading) of the gel without the use of catalysts. The Schiff base can be further reduced using the weak reductant, sodium cyanoborohydride (NaBH3CN), for the formation of stable secondary amines.143 Out of all cross-linkers, genipin is naturally obtained from gardenia fruit extract and is the most “green” (10000 times less cytotoxic than GA145), which endows the resulting gel with better biocompatibility and slower biodegradation rate.74
If supercritical drying is applied as a drying technique, aerogels with pore sizes in the nanoscale (most frequently 2–50 nm (ref. 43)) are obtained since hydrogels shrink substantially during immersion in organic solvents (methanol, ethanol, or acetone) followed by supercritical drying.68,149 The use of organic solvents to replace the water prior to supercritical drying is generally necessary, as without this step significant foaming (40–160 μm pores) during CO2 processing is observed.67,150 However, there is a report stating that the shrinkage of BC gels after immersing in ethanol and supercritical drying was low, and these authors were able to obtain bigger pore diameters up to 100 μm.151
There is little research on the swelling of physical gels due to their weak structure or low swelling, so majority of the literature is related to swelling of chemical hydrogels. There are several factors that influence the water absorbency of hydrogels. First the drying procedure affects the water absorbency capacity of hydrogels. Supercritically- or freeze-dried hydrogels retain the microporosity of the gel systems and thus significantly increase the swelling properties compared with air-dried, oven-dried, or vacuum dried hydrogels, whose capillary retention decreases due to the recrystallization of the biopolymers in the gel systems. The former drying methods can produce dry gels with a swelling ratio up to 60 g g−1,25 while the latter usually lead to low swelling ratios below 3 g g−1.144 Our research shows that the molecular weight (MW) of the biopolymer plays an important role in the water uptake capacity of the resulting aerogel (i.e. supercritically dried hydrogel), e.g., gels from chitin and CRM of higher MWs isolated with IL possessed higher absorbency than corresponding gels from commercial biopolymers of lower MWs.146 Also, with an increase of polymer concentration, the swelling ratio of the hydrogel decreases significantly as a result of the decrease of pore sizes.132
The degree of cross-linking also affects swelling properties of hydrogels somewhat, although not significantly.25,144 The introduction of electronic repulsion forces into the gel strongly increases the swelling capacity, even using common drying (e.g., vacuum drying). It was reported that quaternized cellulose nanofibril nanopaper could swell in water and become a hydrogel with a maximal water absorbency up to 750 g g−1.154 Superabsorbent hydrogels with a swelling ratio up to 300–400 g g−1 were prepared by using SA or BTCA as the cross-linker because of the presence of grafted cross-linkers that still had carboxyl groups.126–129 Finally, parameters of the aqueous medium, such as ionic strength,128 pH,127,131 and temperature,25 may influence the water uptake of hydrogels (as discussed below).
4.2. Irradiative cross-linking
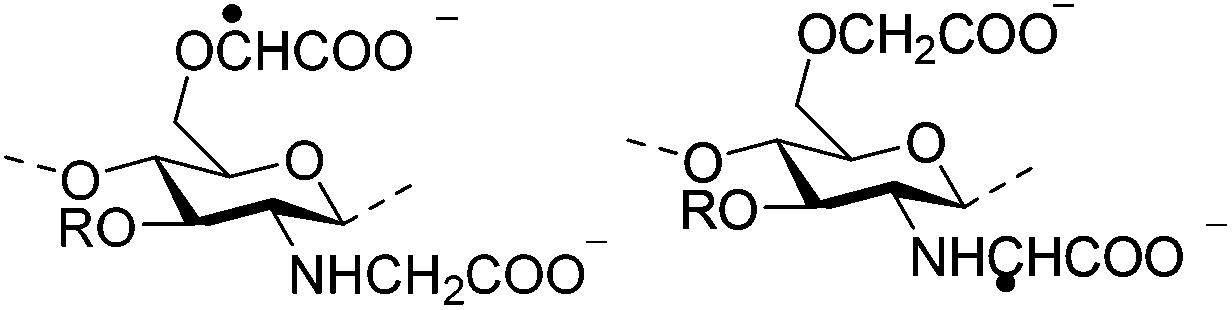
Irradiation is a useful method for the formation of covalent bonding between polymer chains. This method is advantageous because of the high purity of the hydrogel product without use of toxic cross-linkers, thus enlarging the applications in food and pharmaceutical industries. However, only a small fraction (17–30%) of gel aggregates (lumps) could be obtained by γ-ray irradiation at a dose of 20 kGy from 20 wt% biopolymer solutions (such as cellulose/IL/water, CMC and carboxymethyl chitosan (CMCts) aqueous solutions), with the assistance of generated hydroxyl radicals.155–157Electron beam (EB) irradiation in vacuum seems to be able to increase the gel yield, e.g., after EB irradiation, the gel fraction reached up to 55% at 20 kGy, and increased with the irradiation dose.158,159 Further, instead of low yield and a liquid product, Petrov et al. obtained opaque spongy materials via UV irradiation from moderately frozen semi-dilute (3 wt%) aqueous polymer (e.g., HPMC, HEC, and MC) solutions with (4-benzoylbenzyl)trimethylammonium chloride (BBTMAC) as a photoinitiator.160 It was suggested that after freezing, the photoinitiator and water molecules connected to the polymer through hydrogen bonds could form a non-frozen liquid microphase in which the polymer concentration was very high, resulting in a sufficient number of chains in close enough vicinity to bind with each other during irradiation.160 Additionally, a “radical cross-linker” (usually N,N′-methylenebisacrylamide (MBAAm)) could be added to the solution before irradiation to enhance the gelation efficiency.161 It has been reported that macroradicals emerge preferentially in weakened 1 and 4 positions of cellulose as a result of the fracture of C–H bonds upon irradiation,162 while macroradicals of the derivatives are created in the side chains during radical cross-linking as shown in Table 6.157,163
| Biopolymer | Macroradical | |
|---|---|---|
| a R = H or corresponding substituent. | ||
| Cellulose |
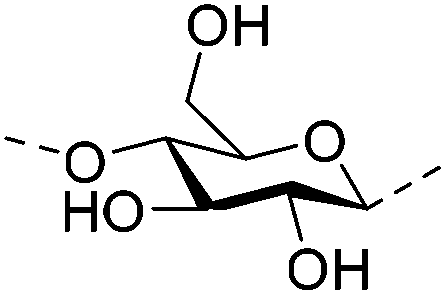
|
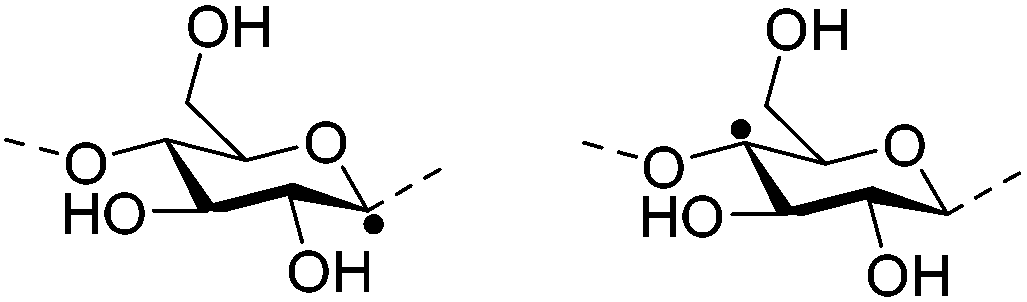
|
| Chitin |
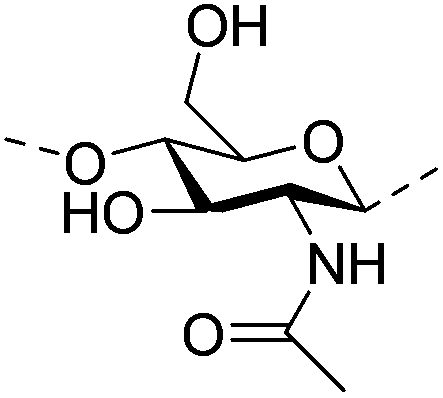
|
|
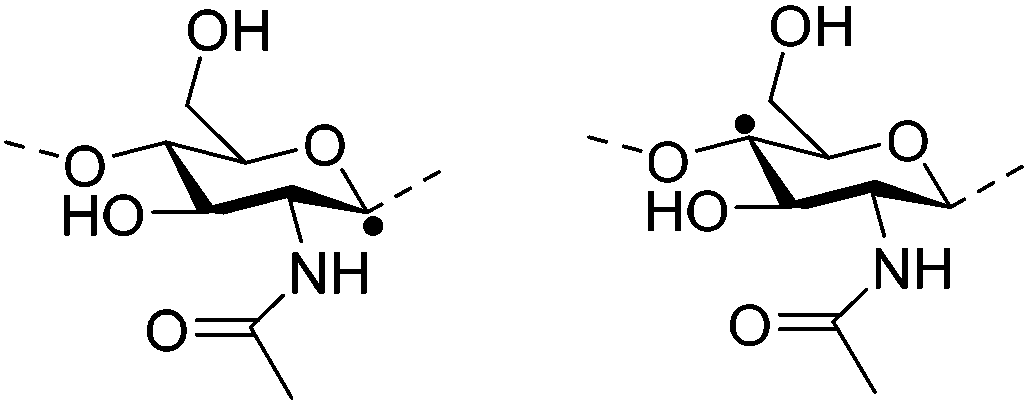
| Methylcellulose (MC) |
|
|
| Hydroxyethyl cellulose (HEC) |
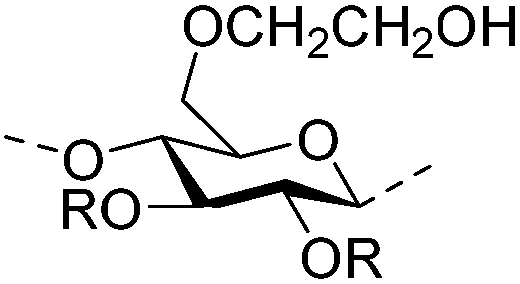
|
|
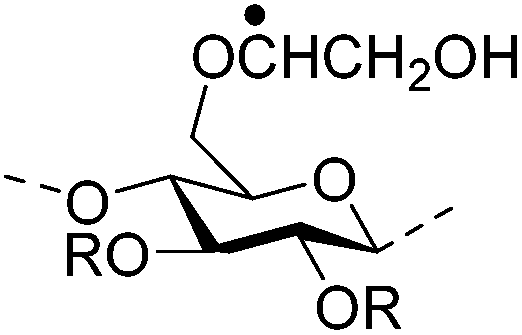
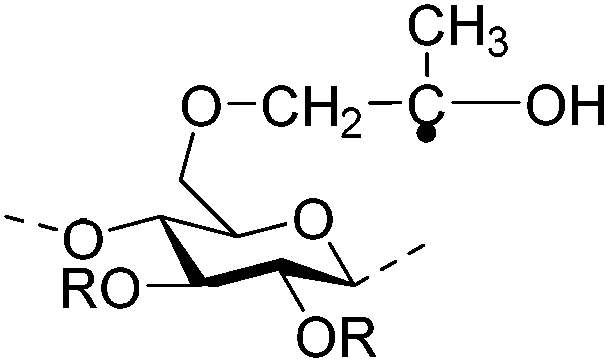
| Hydroxypropylmethyl cellulose (HPMC) |
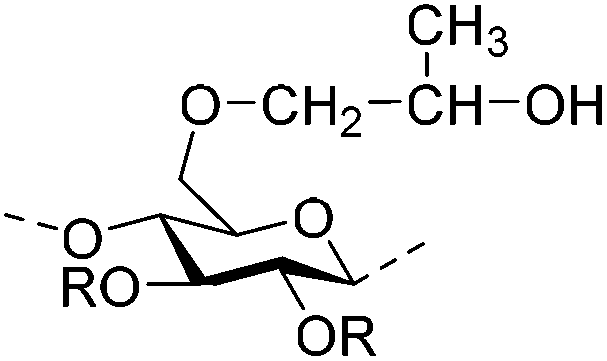
|
|
| Carboxymethyl cellulose (CMC) |
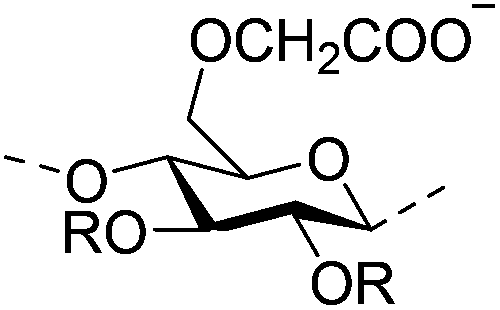
|

|
| Carboxymethyl chitin (CMCh) |
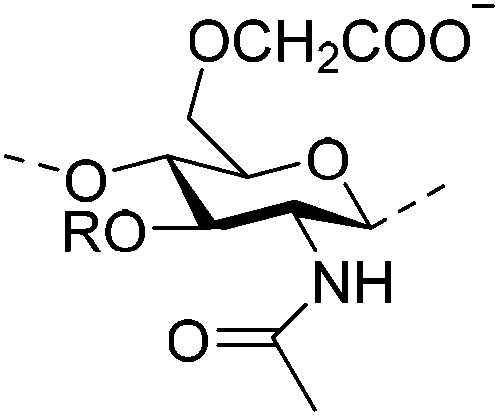
|
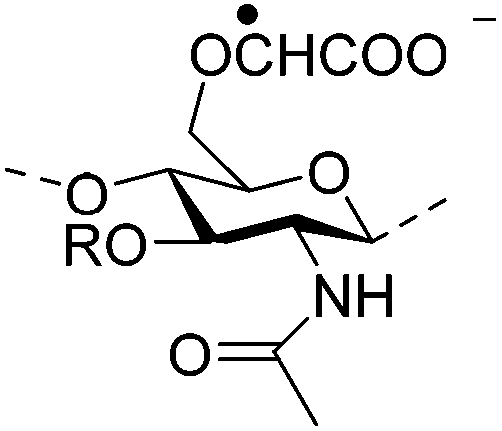
|
| Carboxymethyl chitosan (CMCts) |
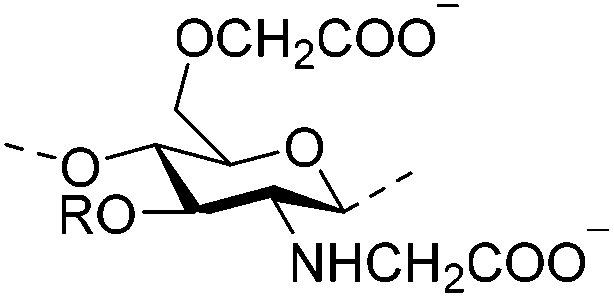
|
|
Degradation of the polymer chains competes with cross-linking during irradiation, especially at high-energy doses and low polymer concentrations (10 wt%), resulting in the destruction of network structure and decrease of tensile strength.159 Degradation could be decreased by (a) using EB irradiation instead of γ-irradiation because of the much higher radical number created in the system under EB irradiation, or by (b) irradiation in an oxygen-free atmosphere to avoid the generation of oxides and peroxides.164
5. Composite hydrogels
There are few concrete and operational applications of pure hydrogels to date, such as injectable thermoset gels (mentioned above), membrane separation,30 and encapsulation of active species,39 although much fundamental research on their preparation has been developed. In fact, most of the pure hydrogels may not fit one specific purpose due to the lack of some feature, structure, or property. By mixing with another polymer or inorganics, novel structural materials with advantages of both components can be obtained, which tremendously enhances the appeal of the resulting composite hydrogels in various areas. In this review, we will concentrate on two-component composite hydrogels made from cellulose, chitin, or chitosan as a matrix for various applications.5.1. Natural polymer-based hybrid hydrogels
The preparation processes of composite hydrogels are based on those of pure hydrogels, including dissolution (or dispersion) and cross-linking. Compared to making pure hydrogels, the only extra step during formation of natural polymer-based hybrid hydrogels is the mixing of two biopolymer solutions (usually using the same solvent) before shaping into a gel with or without a cross-linker (identical to those mentioned above). The second biopolymer can also be incorporated by impregnation after obtaining the cellulose or chitin scaffold.165 When blending with biopolymers such as CMC, sodium alginate and pectin, which have carboxyl groups on the polymer chains, the hydrogel system can be cross-linked particularly with divalent or trivalent ions, e.g., Ca2+ and Al3+, owing to their chelate formation with the carboxyl groups (Fig. 2).166,167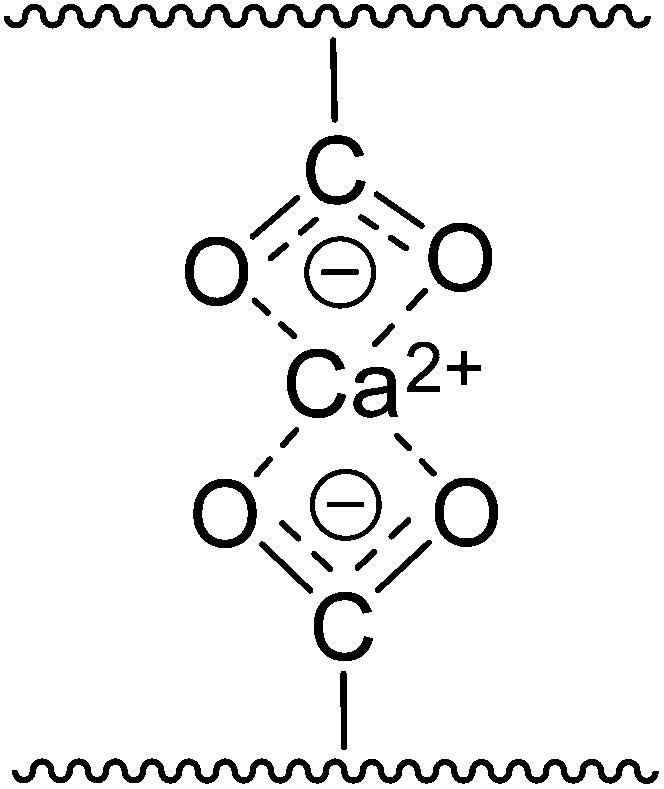 | ||
| Fig. 2 Divalent metal cross-linking through chelate formation with carboxyl groups. | ||
5.2. Biopolymer/synthetic polymer hydrogels
When mixing with synthetic polymers (e.g., polyethylene glycol (PEG) and polyvinyl alcohol (PVA)), the preparation processes are identical to those of natural polymer-based hybrid hydrogels.168–171 However, for the grafting, the monomers of the synthetic polymer are often homopolymerized in the cellulose or chitin solution with the assistance of an initiator upon heating,172,173 irradiation (microwave,174,175 Electron-Beam,176,177 UV irradiation178) or both.179 In some cases, grafting can take place before polymerization, e.g., acrylic acid (AA) was able to link first to the biopolymer via esterification, acting as the active grafting sites on the chains for polymerization.180There are a variety of initiators reported in the literature including potassium persulfate (PPS),181 ammonium persulfate (APS),182 ceric ammonium sulfate (CAS),183 cerium ammonium nitrate (CAN),184 benzoyl peroxide (BPO),173 2,2′-azobisisobutyronitrile (AIBN),172 trihexyltetradecylphosphonium persulfate (TETDPPS),185 and the photoinitiator 2,2-diethoxyacetophenone (DEAP).178 As an example, sulfate ion radicals (SO4−˙) are produced from PPS upon thermal dissociation, which further react with water molecules in the solution to generate hydroxyl radicals (OH˙). Both radicals react with monomers and cellulose and produce active sites (CC˙ of monomers, and –O˙ on side chains of cellulose) for polymerization and grafting to further produce a branched composite network. Meanwhile, the radical cross-linker, MBAAm, can be applied to chemically cross-link the network.
5.3. Biopolymer/inorganic hydrogels
To introduce inorganics into the hydrogel network, several approaches have been adopted: (a) simply mixing the target inorganics with the biopolymer solution followed by shaping into a gel;186–188 (b) transition of the inorganic precursor into target inorganics in the biopolymer solution (or suspension) along with (or followed by) gelation;189–191 (c) in situ transition of the precursor in the hydrated gel or dry scaffold;192–199 or (d) using BC as a template for the ordered deposition of target inorganics during fermentation.200 The precursors for various inorganics and the corresponding transition processes are listed in Table 7.| Organics | Precursor | Transition | Ref. |
|---|---|---|---|
|
a Phosphorylation of cellulose could first be performed to enhance the transition.202
b Reduction catalysts for Ag: NaBH4,206 ethylene glycol (EG),207 polyethylene glycol (PEG),219 ascorbic acid,208 or sodium citrate.187,209
c Reduction catalysts for ZnO: ammonium hydroxide,197 triethanolamine (TEA),198 or NaOH.188,191
d H/C catalysts for SiO2: ammonia,210 heteropoly acids,189 acetic acid,194 or HCl.190
e H/C catalysts for TiO2 and iron oxides: ammonia,193 NaOH,199,212,215–218 or NaOH/KNO3214 upon heating. (Dilute HCl could be used to prevent hydrolysis of the precursors.199)
|
|||
| Hydroxyapatite (HA) | H3PO4/Ca(OH)2; H3PO4/CaCl2;a Ca(NO3)2/(NH4)2HPO4; Ca(NO3)2/NH4H2PO4; CaCl2/Na2HPO4 | Precipitation reaction | 196, 201–205 |
| Silver nanoparticle (Ag) | AgNO3 | Hydrothermal, or catalyticb reduction | 187, 192, 206–209 |
| Zinc oxide (ZnO) | Zn(CH2COO)2·2H2O; Zn(NO)2 | Catalyticc reduction | 188, 191, 197, 198 |
| Silica (SiO2) | Tetraethoxysilane (TEOS) | Hydrolysis and condensation (H/C)d | 189, 190, 194, 210 |
| Na2SiO3 | Reaction in ethanol/water at pH 10–10.5 | 211 | |
| Titanium dioxide (TiO2) | Tetrabutyl titanate (Ti(OBu)4) | Hydrolysis and condensation (H/C)e | 212 |
| Iron oxides | |||
| Fe3O4 | FeCl3·6H2O/FeCl2·4H2O; FeCl3·6H2O/FeSO2·7H2O | Hydrolysis and condensation (H/C)e | 193, 199, 200, 213 |
| CoFe2O4 | FeSO4/CoCl2; FeCl3/CoCl2 | 214, 215 | |
| Fe2O3 | FeCl2; FeCl3 | 216–218 | |
6. Applications
6.1. Drug delivery
| Release type | Hydrogel | Cross-linker | Release mediumb | Ref. |
|---|---|---|---|---|
| a Carboxymethyl cellulose (CMC); cellulose nanowhisker (CNW); epichlorohydrin (ECH); genipin (GNP); N,N′-methylenebisacrylamide (MBAAm); polyacrylic acid (PAAc); polyacrylamide (PAAm); polyethylene glycol (PEG); poly(N-isopropylacrylamide) (PNIPAAm); poly(N-vinylcaprolactam) (PNVCL); sodium acetate–acetic acid (SA/AA) buffer solution; simulated body fluid (SBF); phosphate buffer solution (PBS); simulated colonic fluid (SCF); simulated intestinal fluid (SIF). b Release medium: the most commonly used release medium for each, or the medium in which the gel shows maximal swelling. c Glutaraldehyde (GA) is not a common cross-linker for the cellulose matrix. | ||||
| Controlled release by composition | Cellulose/CMC | ECH | PBS (7.4) | 220 |
| Cellulose/lignin | ECH | 19/1 water/ethanol | 221 | |
| Cellulose/CNW | N/A | SBF (7.4) | 92 | |
| Thermosensitive | Cellulose/PNIPAAm | MBAAm | Water | 223 |
| Cellulose/PNVCL | MBAAm | — | 185 | |
| Chitosan/PNIPAAm | MBAAm and GAc | Water | 224 | |
| Chitosan/PNVCL | N/A | — (Dual responsive) | 225 | |
| pH-Sensitive | Chitosan/alginate | Ca2+ and/or TPP | SIF (6.8) and SCF (7.4) | 227, 228 |
| Chitosan/pectin | Ca2+ and SO42− | PBS (7.4) containing pectinase | 229, 230 | |
| Cellulose/PAAm, or PAAc | MBAAm; N/A | PBS (7.4); SIF (6.8) | 175, 176 | |
| Injectable | Chitosan/CMC | N/A | — | 231 |
| Chitosan/collagen | N/A | Subcutaneous injection | 232 | |
| Chitosan/PEG | GNP; N/A | PBS (7.4) | 233 | |
| Magnetic | Cellulose/Fe3O4 | GAc | PBS (7.4); SA/AA (5.0) | 193 |
Drug release from hydrogels can also be controlled by using gel structures that can change reversibly in response to environmental stimuli. Temperature-sensitive hydrogels are probably the most commonly studied class of environmentally-sensitive polymer systems in drug delivery research.222 The common characteristic of temperature-sensitive polymer networks is the presence of hydrophobic groups, such as methyl, ethyl, propyl groups, etc. As temperature increases, inter-polymer chain associations through hydrophobic interactions among hydrophobic segments strengthen, resulting in the shrinking of the hydrogel. When grafting N-isopropylacrylamide (NIPAAm) or N-vinylcaprolactam (NVCL) on cellulose or chitosan, negatively thermosensitive drug release hydrogel systems are obtained.185,223–225 These hydrogels show a decreased swelling ratio with increasing temperature and thereby can be applied as “on–off” release devices, with “on” (swelling) at low temperature and “off” (shrinking) at high temperature.
While pure chitosan hydrogels (cross-linked or not) usually show a maximal swelling at low pH,102,103,111,112 multilayer chitosan/alginate composite hydrogel beads cross-linked with Ca2+ and/or TPP,227 and single-layer chitosan/alginate beads dually cross-linked with Ca2+ and SO42−,228 exhibit a low drug release in simulated gastric fluid (SGF) but sustained release in simulated intestinal and colonic fluids (SIF and SCF), thus achieving intestine or colon-specific delivery. Chitosan/pectin hydrogels are also capable of colon-specific delivery because of their mucoadhesion characteristics and enzyme-dependent degradation.229,230 In addition, polyacrylic acid (PAAc)- and polyacrylamide (PAAm)-grafted cellulose hydrogels show maximal swelling near neutral pH because of the generation of –COO− groups via the deprotonation of –COOH in PAAc and the hydrolysis of –CONH2 in PAAm, respectively.175,176
Thermoset hydrogel systems, such as some pure derivative hydrogels (mentioned above), chitosan/CMC,231 chitosan/collagen,232 and chitosan/PEG,233 can be injected into the body as a liquid, and form a gel in situ where the body temperature is above their lower critical solution temperature (LCST), offering the potential to serve as targeted drug carriers without the need for invasive surgeries. Moreover, such hydrogels usually significantly prolong the release time up to a few weeks due to the efficient encapsulation of drugs. In addition, magnetic-induced transference is potentially useful for applications in drug targeting delivery, e.g., magnetite/cellulose microspheres loaded with drugs or fluorogen are hypothesized to be able to deliver to and localize in specific locations by using an external magnetic field.193
6.2. Tissue engineering
Tissue engineering is a most recent application of hydrogels, in which they are used as scaffolds to mimic many roles of extracellular matrixes and to engineer new tissues, i.e., these scaffolds provide space and nutrients for new tissue formation, and potentially control the structure and function of the engineered tissue in situ or in vitro.234 Currently, hydrogel scaffolds are at or near clinical uses in engineering many tissues in the body including cartilage, bone, muscle, skin, fat, artery, ligament, tendon, liver, bladder, and neurons.12Hydrogel scaffolds in tissue engineering must meet a number of design criteria to function appropriately and promote new tissue formation, including processability, biodegradability (through hydrolysis167 or enzymatic cleavage165,205,235–237), biocompatibility (i.e., excellent cell viability/proliferation,167,201,238,239 without inflammatory response240), bioactivity (i.e., biomineralization),238,241 cell adhesion ability,165,204,205,240etc. Hybridization of the matrix (cellulose or chitin) with a second component improves one or some of these properties of the scaffolds, e.g., hydroxyapatite (HA) and silica enhance mechanical strength, calcification, as well as accelerate biodegradation,237,238,242,243 while collagen and gelatin increase strength and cell attachment.165,236,244
The optimum pore sizes of scaffolds in tissue engineering have been shown to be 5 μm for neovascularization,245 20–125 μm for regeneration of adult mammalian skin,246 and 100–350 μm for regeneration of bone.247 However, it was later suggested that the microarchitecture plays a role in tissue regeneration, e.g., bone can be regenerated in freeze-dried scaffolds with medium pore sizes as low as 16 μm (>90% porosity and concave spherical shape of the pores) via hematoma osteoinduction instead of osteoconduction in big pores.248
A selection of porous pure and composite scaffolds (dried) based on cellulose, chitin, or chitosan along with their physical properties are given in Table 9. Also, the mechanical properties of human cortical and cancellous bone are listed for comparison. While factors such as drying methods, biopolymer concentration, and cross-linking degree (mentioned above) have impacts on the pore sizes of hydrogels, pore sizes of the scaffolds can also be enhanced either by adding a “porogen” (e.g., polymethylmethacrylate (PMMA)201 and NaCl165) or by adding 1% surfactant to the biopolymer solution.68
| Matrix | The 2nd component | Cross-linker | Porosity (%) | Pore size (μm) | Compressive modulus (MPa) | Tensile strength (MPa) | Young's modulus (MPa)b | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Glutaraldehyde (GA); N-acetyl-D-(+)-glucosamine (GlcNAc); hydroxyapatite (HA). b The large difference between the modulus of the same kind of scaffold may be related to the different materials and preparation methods used. c Cellulose was first esterified with glycine. | ||||||||
| Cellulose | N/A | N/A; Kymene | 71–99 | 0.01–10; 47.5; 100 | 0.20–0.24 | — | 8–9; 200–300 | 68, 70, 151, 252, 253 |
| Collagen | Carbodiimide techniquec | — | 15 | — | 84.6; 275 | 8.8 × 102; 9.5 × 103 | 239, 254 | |
| Pectin | CaCl2 | 88 | 10–250 | — | — | 3.987 | 167 | |
| HA | N/A | 85–90 | 250–450 | — | — | — | 201 | |
| Chitin | N/A | N/A | 82–97 | 2–50 × 10−3; 40–160 | — | 3.0–13.6 | 60–386; 9.32; 3.6 | 43, 67, 78, 89, 255 |
| Collagen | UV-irradiation | 63–78 | 260–330 | — | — | — | 165 | |
| Gelatin | N/A; GlcNAc | — | — | — | 5–44 | — | 241, 256 | |
| Silica | N/A | — | — | — | — | — | 238 | |
| HA | N/A | 69–78 | 200–400 | 0.27 | — | 0.32 | 204, 205, 237, 242 | |
| Chitosan | N/A | N/A | — | 1–250 | — | 5 × 10−3 | — | 148, 257 |
| Collagen | GA | 85 | 37–55; 100–200 | — | 0.31 | — | 235, 240 | |
| Gelatin | GA | 90–95 | 100 | 0.26 | 0.45–1.15 | — | 236, 258 | |
| Alginate | CaCl2 | 92 | 100–300 | 0.46 (C) | — | 8.16 | 244, 259 | |
| Silica | N/A | — | — | — | 0.96 | 55.53 | 243 | |
| HA | N/A | 95 | 20–60 | — | — | 7.8–13.5 | 196, 251, 260 | |
| Cancellous bone | — | 4–12 | — | 100–500 | 261, 262 | |||
| Cortical bone | — | 130–180 | 50–151 | (1.2–1.8) × 104 | 263, 264 | |||
It can be seen that the dry porous scaffolds exhibit at least one order of magnitude lower mechanical strength compared to cancellous bone and orders of magnitude lower than cortical bone. In addition, gel swelling (which will occur during cell culture) usually results in a decrease in the mechanical strength.249 Therefore, hydrogel scaffolds are being widely used in the area of nonload bearing tissue engineering. It has been reported that chitin/HA, chitosan/alginate, and chitosan/silica can be placed into bone defects in a minimally invasive manner to promote bone regeneration.237,243,244 The scaffolds have also been applied in other tissues, e.g., the chitosan/gelatin scaffold prepared using a novel fabrication process by combining rapid prototyping, microreplication, and freeze-drying techniques possessed well-organized hepatic architectures.236
Some dense scaffolds (generally air-dried) match cancellous bone properties and are capable of internal fixation of bone fractures, e.g., a chitosan/HA nonporous composite showed a higher bending strength and modulus of 86 MPa and 3.4 GPa, respectively.250 It is noteworthy, that Li et al. found an injectable thermoset chitosan/HA/Na2CO3 solution, which gelated within 9 min at 37 °C, and led to angiogenesis in vivo when loaded with cells.251
6.3. Wound dressing
An ideal wound gel dressing should allow gaseous exchange, maintain the proper moisture level and constant temperature of the wound bed, remove excess exudates, protect the wound against bacteria and contamination, accelerate healing, and alleviate pain. It should also be non-toxic, non-allergenic, non-adherent, and easily removed without trauma. Currently, various forms (such as fibers, membranes, and sponges) of wound dressing products based on BC, chitin, chitosan, and their derivatives are commercially available,11,265 of which deacetylated chitin or chitosan is a hemostat, and possesses a natural antimicrobial property due to the polycationic nature.266 Some wound dressing gels and their outstanding features are listed in Table 10.| Matrix | The 2nd component | Cross-linker | Unique properties | Ref. |
|---|---|---|---|---|
| a Bacterial cellulose (BC); glutaraldehyde (GA); genipin (GNP); lactic acid (LA); nanoparticles (NPs); polyacrylic acid (PAAc); water vapor (WV); water vapor permeability (WVP); water vapor transmission rate (WVTR). b The gel wound dressing has already been commercialized. c T: tensile strength (MPa); E: Young's modulus (MPa). | ||||
| Cellulose | N/A | N/A | High ability to bind water (BC gels)b | 122, 265 |
| Ag NPs | N/A | Antibacterial | 206, 207 | |
| ZnO NPs | N/A | 197 | ||
| Alginate | Ca2+ | Good tear resistance (0.75–3, Tc; porous) | 268 | |
| Chitin | N/A | N/A | WVTR close to that of intact skin; high water uptakeb | 269, 270 |
| PAAc | N/A | Controlled water uptake and cell attachment | 180 | |
| Ag NPs | N/A | Antibacterial, blood-clotting, weak attachment | 187, 209 | |
| ZnO NPs | N/A | Antibacterial; 0.02–0.05 (T; porous) | 188 | |
| Chitosan | N/A | N/A | Hemostatic, antimicrobial, WV and O2 permeable, non-irritant (chitosan-LA), 59.87 (T; nonporous)b | 109, 110, 272, 273 |
| Gelatin | GA | Improved healing effect; antimicrobial | 258, 274 | |
| Hyaluronan | N/A | Lower WVP and adhesion; antimicrobial | 275 | |
| Ag NPs | GNP | Antimicrobial; 26–29 (T; porous–porogen), 583–795 (Ec) | 219, 276 | |
| ZnO NPs | N/A | Antimicrobial; 0.15 (T; porous) | 277 | |
Since cellulose itself has no antimicrobial activity to prevent wound infection, ZnO or Ag nanoparticles (NPs) can be impregnated into the cellulose gel system to achieve antimicrobial ability.197,206,207 The mechanisms of the bactericidal effects of ZnO and Ag NPs are different: it is assumed that water molecules can react with incorporated ZnO particles, leading to the generation of reactive oxy-radicals or hydroxyl-radicals that may cause oxidative injury inside bacterial cells,197 while Ag NPs may penetrate inside the bacteria or attach to the surface of the bacteria disturbing permeability and respiration functions.267 Higher concentrations of ZnO or Ag NPs are required for eukaryotes to achieve comparable toxic effects than for bacterial cells, leading to biocompatibility of the materials for human cells.187,188 Without the antibacterial property, the BC/alginate sponge can be conceived as a temporary wound dressing material due to its appropriate cell attachment and good tear resistance properties.268
Wet chitin hydrogel membranes show similar water vapor transmission rate (WVTR) to that of intact skin, as well as water uptake ability and flexibility, thus are potentially capable of both transpiring substantial amounts of excess exudate and maintaining occlusivity and wound moisture.269,270 In addition, chitin wound dressings generally show preferred medium or weak adhesion with cells, which is caused by either the globular morphology of proliferative cells or the smaller amount of active free amino groups.187,271 Incorporating hydrophilic PAAc into the gel system offers two advantages: controlled water sorption and cell attachment of the dressing at the wound site, which is important in the wound healing process.180 As with cellulose dressings, the antibacterial ability of chitin dressings can be improved by hybridization with ZnO or Ag NPs.187,188,209
In addition to the hemostatic, antimicrobial, and permeable (for both water vapor (WV) and O2) properties, chitosan dressings are non-irritating and do not cause allergic reactions, especially the ones prepared using lactic acid (LA) as the solvent instead of acetic acid.110,272 Incorporation of antibiotics in the wound dressings has been developed for further inhibition of wound infection.109,273 Hybridization with gelatin can improve the healing effect because of the strong bioactivity of gelatin whose main components are collagen, a few protein amylases, and certain organic substances,274 while hyaluronan helps weaken the water vapor permeability (WVP) and cell adhesion, which are desirable characteristics for wound dressings.275
The strength of wound dressings is related to the drying method and water content, which is consistent with the tissue scaffolds. In addition, nonporous or porogen-induced porous dressings that are obtained by air-drying are two orders of magnitude stronger than the porous ones obtained by freeze-drying.188,219,268,272,276,277 Almost all hybrid composite gels meet the strength standard for wound dressings, since a low strength of 0.1 MPa is adequate for a wound dressing material.277
6.4. Water purification
Many liquid- and solid-phase extraction techniques have been used for the removal of toxic pollutants from water such as chemical precipitation, flocculation, flotation, coagulation, membrane filtration, ion exchange, adsorption, and electrochemical treatment.278 Hydrogels have attracted special attention for water purification through adsorption, due to their high absorbency, porous structure, rich functional groups, and relatively low crystallinity. Incorporation of different components into the gel system endows the resulting hybrid hydrogels with abilities to remove various aquatic pollutants such as metal ions (transition or radioactive), dyes (cationic or anionic), and other ions (nitrogenous or phosphorous), etc. (Table 11).| Pollutant | Hydrogel (shapeb; mass ratio) | Cross-linker |
q
ec (mmol g−1) |
q
md (mmol g−1) |
pHe | Ref. | |
|---|---|---|---|---|---|---|---|
| a Carbon nanotube (CNT); epichlorohydrin (ECH); ethylene glycol diglycidyl ether (EGDE); glutaraldehyde (GA); polyacrylic acid (PAAc); polyacrylamide (PAAm); polyvinyl alcohol (PVA); tripolyphosphate (TPP); anionic dyes: acid red 37 (AR 37), acid blue 25 (AB 25), congo red (CR), erythrosine B (EB), and remazol black B (RB); positive dyes: gentian violet (GV), and neutral red (NR; protonatable); dibenzothiophene sulfone (DBTS). b Bead (B); bulk (Bu); membrane (M); powder (P; obtained by milling the dry gels). c Adsorption capacity at the pollutant concentration of 1 mmol L−1. d Maximal monolayer adsorption capacity calculated by the Langmuir adsorption model. e The most suitable pH condition to achieve the most effective adsorption. f Depending on whether cross-linked or not. g The decrease of biopolymer concentration contributes to a much higher sorption. h Formaldehyde pre-treatment could enhance the adsorption. i Ion-imprinting hydrogel (IIH). j The lower adsorption ability of chitosan/PVA IIH may be due to the much lower uranium concentration tested in this article than in others (0.34 vs. 1.7 mmol L−1). k A core–shell structure could be generated by a sodium dodecyl sulfate (SDS; as an anionic surfactant) gelation process.285 l Adsorption capacity at the DBTS concentration of 4 mmol L−1. m Adsorption was performed in an acetonitrile solution. | |||||||
| Metal ions | Hg2+, Cu2+, Pb2+ | Chitin (Mb) | N/A | Hg2+: 1.8; Cu2+: 1.8; Pb2+: 1.1 | — | 5 | 73, 279 |
| Cellulose (M) | Hg2+: 0.9; Cu2+: 0.7; Pb2+: 0.5 | — | |||||
| Chitin/cellulose (M; 3:1) |
Hg2+: 1.7; Cu2+: 1.5; Pb2+: 1.3 | Hg2+: 3.85; Cu2+: 2.00; Pb2+: 2.44 | |||||
| Cu2+ | Cellulose (Bb) | N/A | 0.10 | 0.37 | 6 | 284 | |
| Cellulose/collagen (B; 1:3) |
0.14 | 1.06 | |||||
| Cellulose/chitosan (B; 1:1) |
EGDE; N/A | 0.44–0.65f | 0.84 | 6 | 280 | ||
| Chitosan (B) | TPP, GA, EGDE, or N/A | 0.31–1.0f; 1.29g; 2.11h | 0.31–1.0f; 1.90g; 2.58h | 4 or 4.5 | 111, 134, 283 | ||
| Chitosan/alginate (B; not reported) | N/A | 1.0 | 1.06 | 283 | |||
| Chitosan/PVA (B; 4:3) |
N/A | — | 0.75 | 6 | 300 | ||
| Hg2+ | Chitosan (B) | EGDE | 0.91 | 0.91 | 4 | 293 | |
| Chitosan/PAAm (B; not reported) | 1.61 | 1.61 | |||||
| UO22+ | Chitosan (B) | TPP | 0.67 | 1.0 | 5 | 112 | |
| Chitosan/PVA IIHi (Bub; 3:1) |
EGDE | — | 0.55j | 297 | |||
| Cd2+ | Chitin/Fe3O4 (B; ca. 7:2) |
N/A | 0.933 | — | >7 | 199 | |
| Pb2+, Mn2+, Cr3+ | BC/Fe3O4 (B; ca. 5:4) |
N/A | Pb2+: 0.25; Mn2+: 0.22; Cr3+: 0.21 | — | ca. 7 | 200 | |
| As3+/As5+ | Cellulose/Fe3O4·NH3 (M; ca. 3:7) |
N/A | — | 1.2 | 3 | 299 | |
| Chitosan/TiO2 (B; ca. 7:3) |
N/A | — | 0.047 | 7 | 289 | ||
| Dyes | EB, NR, GV, RB | Chitin/SiO2 (B; 1:14) |
N/A | — | EB: 0.15; NR: 1.06; GV: 0.14; RB: 0.0062 | EB: 5; NR: 6; GV: 8; RB: 4 | 288 |
| AR 37, AB 25 | Chitosan (B) | EGDE; N/A | — | AR: 0.11 or 0.24f; AB: 0.34 or 0.63f | AR: <6; AB: <4 | 135 | |
| CR | Chitosan (B)k | N/A | 0.30 | 0.30 | 5 | 281 | |
| Chitosan/CNT (B; 100:1)k |
ECH; N/A | 0.53; 0.65 | 0.53; 0.65 | 5 | 285–287 | ||
| Chitosan/PVA/Fe3O4 (B; 1:1:1) |
N/A | 0.67 | 0.67 | 6 | 213 | ||
| Others | NO3− | Chitosan (B) | N/A | 0.65 | 1.45 | 5 | 282 |
| NH4+ | Chitosan/PAAc (Pb; ca. 1:7) |
N/A | ca. 0.4 | 7.8 | 6–7 | 290 | |
| H2PO4− | Cu(II)-loaded Chitosan | GA | 0.97 | 0.97 | 5 | 294 | |
| Ag+ | Chitosan IIHi (B) | ECH | — | 1.2 | 5 | 296 | |
| DBTS | Chitosan IIHi (Bu) | GA | 0.09l | — | —m | 295 | |
| Phenols | Tyrosinase-loaded chitosan (M) | N/A | — | — | 7 | 298 | |
It is known that dissolved pollutant ions or molecules can easily penetrate into cellulose or chitin hydrogels and establish bonds with the amine (–NH2) and/or hydroxyl (–OH) groups at appropriate pH values generally through three different kinds of interactions: (a) complexation (or chelation) between the lone pair electrons of N and/or O and the metal ions;73,199,279,280 (b) crystallization of the metal ions with the complexed metals as nucleation sites;73,278 or (c) electrostatic attraction (or ion exchange) between the protonated amino groups and various anions.281,282 In addition, cross-linking has been found to decrease the adsorption capacity of the gel (although the strength or stability increases) primarily due to the decrease of functional groups, and a decrease in swelling ability of the gel also decreases sorption.111,134,283
The addition of a second component to the biopolymer can increase the adsorption capacity of the final material. For example, the incorporation of collagen is believed to endow cellulose gels with much better adsorption ability because of the active amino groups of collagen.284 Zhu et al. used BC beads as adsorbents without removing entrapped dead bacteria cells and found that the adsorption capacity was improved because of the multiple functional groups (such as carboxyl groups, phosphoryl groups, hydroxyl groups, phosphate groups, amino groups, and amide groups) existing in the proteins, polysaccharides, and lipids of the cells, resulting in formation of coordination complexes between N, P, O, or S and metal ions.200 Also, 0.01 wt% (relative to the chitosan matrix) carbon nanotubes (CNTs) impregnation was useful for enhancing the adsorption capacity of the composite beads due to the large specific surface area and layered hexagonal arrays of carbon atoms in CNTs.285–287 In SiO2 composite hydrogels, the SiO2 skeleton has negative charge density (Si–O−) above pH 3, which can interact with positively charged pollutants.288 When TiO2 is added to chitosan, the resulting beads have a net positive charge (TiIV–OH2+) at pH < pHpzc (point of zero charge (pzc)), which can attract arsenic oxyanions (As(III): AsO(OH)2−, AsO2(OH)2−, AsO33−; As(V): AsO2(OH)2−, AsO3(OH)2−, AsO43−).289 For removal of NH4+ cations, a hydrogel system grafted with polyacrylic acid (PAAc) is well worth considering due to the ionic interaction between the positively charged NH4+ and negative adsorption sites (–COO−) of the adsorbent.290
Research on selective removal of pollutants from contaminated water using cellulose or chitin hydrogels is scanty. Some research has focused on the adsorption of Cu2+, and there is literature suggesting higher adsorption selectivity of adsorbents for Cu2+ over Cd2+.291 However, in most cases, neither cellulose nor chitin (or chitosan) hydrogels show selectivity for a given pollutant. In order to achieve selective adsorption, two approaches are generally followed: introducing a specific functional group in the system or utilizing ion (or molecular) imprinting techniques. Yan et al. modified the surface of chitosan hydrogel beads with chloroacetic acid to introduce carboxyl groups, producing a product with a higher coordination affinity for Cu2+ over Mg2+ and Pb2+,292 while chitosan beads surface grafted with PAAm showed selectivity in adsorbing Hg2+ over Pb2+ due to the action of the amide groups.293 Further, after Cu2+ extraction, the same chitosan beads can be used for further phosphate (H2PO4−) adsorption from aqueous solutions, where the Cu2+ ions on the surface of the beads play a very important role in phosphate adsorption at pH 5.294 (At pH < 3, most of the Cu2+ ions are released; while at higher pH, OH− groups strongly competed with H2PO4− for the chitosan–Cu2+ active sites.)
The ion (or molecular) imprinting technique results in ionic (or molecular) recognition cavities (conformational memory) inside the polymeric network using the ion or molecule (generally identical to the one to be selectively adsorbed) as an imprint template during the cross-linking of polymer chains and before elution.295–297 Although scarcely explored, a third strategy is to incorporate enzymes into the biopolymer matrix to selectively modify the target adsorbate before sorption. Phenols were selectively oxidized by the enzyme tyrosinase (immobilized within chitosan gels) into reactive o-quinones, which reacted with nucleophilic amine functional groups present in the chitosan matrix.298
Adding a second component to the material can also be used to improve the properties and/or functionalities of the resulting composite hydrogels. For example, when incorporating magnetite or other iron oxides into a gel system, the resulting adsorbent can be easily attracted out of the effluent, using a magnetic field.199,213 Nata et al. prepared an aminated Fe3O4/cellulose composite which not only could be easily collected but also demonstrated a very high adsorption ability towards metal ions because of the protonation of the introduced NH2 on Fe3O4 at low pH.299 To enhance the sorbent's mechanical strength or chemical stability, crucial in batch or column water remediation, fillers of high modulus such as SiO2,288 and CNTs285–287 can be incorporated; or the biopolymer can be cross-linked with other polymers, such as the self-assembly chitosan/PVA,213,300 or chitosan/alginate composites.283
Recycling of the gels and reusing them is of extreme importance for the sustainability of the process. There are several eluents proposed to desorb the ions from gels, including SC,200 ethylene diamine tetraacetic acid (EDTA),300 HNO3, AlCl3, CaCl2, NaCl,213,290etc., which are chelating or cation exchange agents. In addition, since most of the gels have better adsorption capacity for pollutants when the pH is within 5–7, acid (HCl)199 or alkali (NaOH)281,282,294 solutions can be used to strip the ions from the adsorbents. Using these methods, the desorption efficiency can reach 99%, and the gels can be reused at least 4 cycles with sometimes even higher sorption efficacy than the first time.199,213,290
6.5. Other applications
Hydrogels from cellulose, chitin, or chitosan are utilized widely in many other diverse fields. One of their major uses is as the supporting material for functional additives, including (a) membranes containing electrolyte molecules used in a capacitor or battery (e.g., cellulose/chitin containing ILs and H2SO4,301,302 and chitosan/PVA containing NH4NO3 and ethylene carbonate (EC)303); (b) porous aerogels containing inorganic catalysts (e.g., Ag,192,304 TiO2,305,306 and SiO2307) for heterogeneous catalysis; and (c) hydrogels containing fluorescent quantum dots (QDs) potentially used in fluoroimmunoassays and biological labeling.186,308 Indeed, chitosan aerogels are potential base catalysts themselves due to the amino groups.91,309
In addition to their role in catalysis, cellulose/SiO2 aerogels are able to act as excellent thermal insulators while being flexible and translucent, and having high mechanical stability and processability.210 Furthermore, pure inorganic nanomaterials, such as TiO2,310 SiO2,210 and Fe2O3217 with porous networks that are useful in photocatalysis, biosensor, bioseparation, respectively, can be obtained by calcining the corresponding composite hydrogels in various forms as sacrificial templates.
There are unique polyelectrolyte hydrogels consisting of polyanionic polymers (e.g., CMC, alginate, and PAAc) and/or polycationic polymers (e.g., chitosan, PAAm, and polyvinylamine (PVAm)311), which can be used as electronic elements. Smitha et al. identified the chitosan/PAAc composite as an ideal proton exchange membrane in the direct methanol fuel cell, as it exhibited high proton conductivity, low methanol permeability, and adequate thermal and mechanical stability.312 CMC/chitosan and cellulose/alginate hybrid hydrogels are potential electroactive sensors or actuators in electronic devices because they swell or shrink differentially on two electrode sides (anode and cathode) as a consequence of mobile ion transport in an electric field, causing bending towards one electrode.313,314 To facilitate this, a plasticizer (e.g., glycerol) can be added into the system to weaken the intra- and inter-molecular hydrogen bonding in the matrix and thereby improve the flexibility of the product.314
7. Conclusions and perspectives
Because of their renewable, biocompatible, and biodegradable characteristics, hydrogels made from cellulose, chitin, and some of their derivatives are attracting and will continue to draw both academic and industrial attention. Considering their other advantages, such as excellent processability, high absorbency, porosity, bioactivity, abundant active groups, etc., it is not surprising that these hydrogels have already found extensive applications in many areas such as drug delivery, tissue engineering, wound dressing, water purification, catalysis, electrical elements, and more. Much fundamental research has been conducted on the preparation of these hydrogels, including the development of solvent systems for native cellulose or chitin, hydrogel formation techniques, physical or chemical cross-linkers, drying methods, etc., however, recent studies have mainly focused on potential applications of the hydrogels in water purification and pharmaceutical and medical systems, and less attention has been paid to other areas such as their use in the electronic and optical fields.For future studies, more attention should be focused on (1) “green” (safe solvents, none or nontoxic cross-linkers), and/or low-energy processing for hydrogel systems; (2) injectable hydrogels forming safely within the body without the need of surgery for targeting drug release or tissue engineering; (3) pH or enzymatic triggered drug release at targeted sites; (4) hydrogel degradation in a controlled manner in tissue engineering or sustained drug release; (5) development of hydrogel functionalization as an economical way to improve efficacy, selectivity, or recycling during water purification; (6) development of new applications of hydrogel systems (e.g., polyelectrolyte complex hydrogels as electrical elements); (7) development of cellulose and chitin nanowhiskers to prepare novel hydrogels with unique characteristics such as high mechanical strength and photonic properties; and (8) development of novel biopolymer isolation methods (e.g., IL isolation) to produce cellulose and chitin with high molecular weights and thus unique hydrogels. Undoubtedly, hydrogels based on cellulose, chitin, and their derivatives still offer abundant promising opportunities in various industries, although significant challenges will need to be overcome before commercialization, and thus fundamental research into the nature of these systems should also continue.
Acknowledgements
The authors would like to thank 525 Solutions, Inc., the DOE SBIR Office of Science (DE-SC0010152), and the China Scholarship Council (no. 201306600007) for financial support.Notes and references
- N. A. Peppas, P. Bures, W. Leobandung and H. Ichikawa, Eur. J. Pharm. Biopharm., 2000, 50, 27–46 CrossRef CAS PubMed.
- N. Job, A. Thery, R. Pirard, J. Marien, L. Kocon, J. N. Rouzaud, F. Beguin and J. P. Pirard, Carbon, 2005, 43, 2481–2494 CrossRef CAS.
- S. Van Vlierberghe, P. Dubruel and E. Schacht, Biomacromolecules, 2011, 12, 1387–1408 CrossRef CAS PubMed.
- X. Z. Shu and K. J. Zhu, Int. J. Pharm., 2002, 233, 217–225 CrossRef CAS PubMed.
- W. E. Hennink and C. F. van Nostrum, Adv. Drug Delivery Rev., 2002, 54, 13–36 CrossRef CAS PubMed.
- G. D. Mogosanu and A. M. Grumezescu, Int. J. Pharm., 2014, 463, 127–136 CrossRef CAS PubMed.
- M. J. Zohuriaan-Mehr, A. Pourjavadi, H. Salimi and M. Kurdtabar, Polym. Adv. Technol., 2009, 20, 655–671 CrossRef CAS.
- V. Arbona, D. J. Iglesias, J. Jacas, E. Primo-Millo, M. Talon and A. Gomez-Cadenas, Plant Soil, 2005, 270, 73–82 CrossRef CAS.
- A. R. Kulkarni, K. S. Soppimath, T. M. Aminabhavi, A. M. Dave and M. H. Mehta, J. Controlled Release, 2000, 63, 97–105 CrossRef CAS PubMed.
- Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2012, 64, 49–60 CrossRef.
- R. Jayakumar, M. Prabaharan, P. T. S. Kumar, S. V. Nair and H. Tamura, Biotechnol. Adv., 2011, 29, 322–337 CrossRef CAS PubMed.
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1879 CrossRef CAS PubMed.
- R. S. Juang and R. C. Shiau, J. Membr. Sci., 2000, 165, 159–167 CrossRef CAS.
- G. X. Li, Y. M. Du, Y. Z. Tao, H. B. Deng, X. G. Luo and J. H. Yang, Carbohydr. Polym., 2010, 82, 706–713 CrossRef CAS.
- A. T. Paulino, M. R. Guilherme, A. V. Reis, G. M. Campese, E. C. Muniz and J. Nozaki, J. Colloid Interface Sci., 2006, 301, 55–62 CrossRef CAS PubMed.
- C. Dwivedi, A. Gupta, A. Chaudhary and C. K. Nandi, RSC Adv., 2014, 4, 39830–39838 RSC.
- B. Adhikari and S. Majumdar, Prog. Polym. Sci., 2004, 29, 699–766 CrossRef CAS.
- A. W. Chan, R. A. Whitney and R. J. Neufeld, Biomacromolecules, 2009, 10, 609–616 CrossRef CAS PubMed.
- C. Elvira, J. F. Mano, J. San Roman and R. L. Reis, Biomaterials, 2002, 23, 1955–1966 CrossRef CAS PubMed.
- J. Guo, Y. Zhang and X. Q. Yang, Food Hydrocolloids, 2012, 26, 277–285 CrossRef CAS.
- H. W. Kang, Y. Tabata and Y. Ikada, Biomaterials, 1999, 20, 1339–1344 CrossRef CAS PubMed.
- T. Sawada, K. Tsukada, K. Hasegawa, Y. Ohashi, Y. Udagawa and V. Gomel, Hum. Reprod., 2001, 16, 353–356 CrossRef CAS PubMed.
- X. W. Peng, L. X. Zhong, J. L. Ren and R. C. Sun, J. Agric. Food Chem., 2012, 60, 3909–3916 CrossRef CAS PubMed.
- X. M. Li and X. J. Pan, J. Biobased Mater. Bioenergy, 2010, 4, 289–297 CrossRef CAS.
- J. Zhou, C. Chang, R. Zhang and L. Zhang, Macromol. Biosci., 2007, 7, 804–809 CrossRef CAS PubMed.
- H. Tamura, H. Nagahama and S. Tokura, Cellulose, 2006, 13, 357–364 CrossRef CAS.
- H. R. Nie, M. Z. Liu, F. L. Zhan and M. Y. Guo, Carbohydr. Polym., 2004, 58, 185–189 CrossRef CAS.
- F. M. Goycoolea, A. Heras, I. Aranaz, G. Galed, M. E. Fernandez-Valle and W. Arguelles-Monal, Macromol. Biosci., 2003, 3, 612–619 CrossRef CAS.
- T. Kiang, H. Wen, H. W. Lim and K. W. Leong, Biomaterials, 2004, 25, 5293–5301 CrossRef CAS PubMed.
- Y. P. Zhang, H. Shao, C. X. Wu and X. C. Hu, Macromol. Biosci., 2001, 1, 141–148 CrossRef CAS.
- D. Ishii, D. Tatsumi, T. Matsumoto, K. Murata, H. Hayashi and H. Yoshitani, Macromol. Biosci., 2006, 6, 293–300 CrossRef CAS PubMed.
- E. Yilmaz and M. Bengisu, Carbohydr. Polym., 2003, 54, 479–488 CrossRef CAS.
- H. Saito, A. Sakurai, M. Sakakibara and H. Saga, J. Appl. Polym. Sci., 2003, 90, 3020–3025 CrossRef CAS.
- A. Ostlund, D. Lundberg, L. Nordstierna, K. Holmberg and M. Nyden, Biomacromolecules, 2009, 10, 2401–2407 CrossRef PubMed.
- D. Bouyer, L. Vachoud, Y. Chakrabandhu and C. Pochat-Bohatier, Chem. Eng. J., 2010, 157, 605–619 CrossRef CAS.
- H. Wang, G. Gurau and R. D. Rogers, Chem. Soc. Rev., 2012, 41, 1519–1537 RSC.
- H. Z. Song, Y. H. Niu, Z. G. Wang and J. Zhang, Biomacromolecules, 2011, 12, 1087–1096 CrossRef CAS PubMed.
- M. H. Kim, S. An, K. Won, H. J. Kim and S. H. Lee, J. Mol. Catal. B: Enzym., 2012, 75, 68–72 CrossRef CAS.
- M. B. Turner, S. K. Spear, J. D. Holbrey and R. D. Rogers, Biomacromolecules, 2004, 5, 1379–1384 CrossRef CAS PubMed.
- M. L. Deng, Q. Zhou, A. K. Du, J. van Kasteren and Y. Z. Wang, Mater. Lett., 2009, 63, 1851–1854 CrossRef CAS.
- L. Li, Z. B. Lin, X. Yang, Z. Z. Wan and S. X. Cui, Chin. Sci. Bull., 2009, 54, 1622–1625 CrossRef CAS.
- K. Prasad, M. Murakami, Y. Kaneko, A. Takada, Y. Nakamura and J. Kadokawa, Int. J. Biol. Macromol., 2009, 45, 221–225 CrossRef CAS PubMed.
- S. S. Silva, A. R. C. Duarte, A. P. Carvalho, J. F. Mano and R. L. Reis, Acta Biomater., 2011, 7, 1166–1172 CrossRef CAS PubMed.
- Y. Wu, T. Sasaki, S. Irie and K. Sakurai, Polymer, 2008, 49, 2321–2327 CrossRef CAS.
- P. D. de Maria and Z. Maugeri, Curr. Opin. Chem. Biol., 2011, 15, 220–225 CrossRef PubMed.
- M. Sharma, C. Mukesh, D. Mondal and K. Prasad, RSC Adv., 2013, 3, 18149–18155 RSC.
- A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 70–71 RSC.
- C. Mukesh, D. Mondal, M. Sharma and K. Prasad, Carbohydr. Polym., 2014, 103, 466–471 CrossRef CAS PubMed.
- J. Cai and L. Zhang, Macromol. Biosci., 2005, 5, 539–548 CrossRef CAS PubMed.
- X. W. Hu, Y. M. Du, Y. F. Tang, Q. Wang, T. Feng, J. H. Yang and J. F. Kennedy, Carbohydr. Polym., 2007, 70, 451–458 CrossRef CAS.
- L. N. Zhang, D. Ruan and S. J. Gao, J. Polym. Sci., Polym. Phys. Ed., 2002, 40, 1521–1529 CrossRef CAS.
- H. S. Qi, C. Y. Chang and L. N. Zhang, Cellulose, 2008, 15, 779–787 CrossRef CAS.
- A. S. Gross, A. T. Bell and J. W. Chu, Phys. Chem. Chem. Phys., 2012, 14, 8425–8430 RSC.
- J. Cai and L. Zhang, Biomacromolecules, 2006, 7, 183–189 CrossRef CAS PubMed.
- L. Petrus, D. G. Gray and J. N. Bemiller, Carbohydr. Res., 1995, 268, 319–323 CrossRef CAS.
- J. P. Zhou, L. N. Zhang and J. Cai, J. Polym. Sci., Polym. Phys. Ed., 2004, 42, 347–353 CrossRef CAS.
- Q. L. Kuang, J. C. Zhao, Y. H. Niu, J. Zhang and Z. G. Wang, J. Phys. Chem. B, 2008, 112, 10234–10240 CrossRef CAS PubMed.
- G. X. Li, Y. M. Du, Y. Z. Tao, Y. T. Liu, S. Li, X. W. Hu and J. H. Yang, Carbohydr. Polym., 2010, 80, 970–976 CrossRef CAS.
- C. L. McCormick, P. A. Callais and B. H. Hutchinson, Macromolecules, 1985, 18, 2394–2401 CrossRef CAS.
- M. Terbojevich, C. Carraro, A. Cosani and E. Marsano, Carbohydr. Res., 1988, 180, 73–86 CrossRef CAS.
- S. M. Liang, L. N. Zhang, Y. F. Li and J. Xu, Macromol. Chem. Phys., 2007, 208, 594–602 CrossRef CAS.
- L. H. Weng, L. N. Zhang, D. Ruan, L. H. Shi and J. Xu, Langmuir, 2004, 20, 2086–2093 CrossRef CAS PubMed.
- C. C. Chen, D. G. Li, H. Yano and K. Abe, Cellulose, 2014, 21, 3339–3346 CrossRef CAS.
- Y. Mao, J. P. Zhou, J. Cai and L. N. Zhang, J. Membr. Sci., 2006, 279, 246–255 CrossRef CAS.
- W. DeOliveira and W. G. Glasser, J. Appl. Polym. Sci., 1996, 60, 63–73 CrossRef CAS.
- Z. G. Wang, S. L. Liu, Y. Matsumoto and S. Kuga, Cellulose, 2012, 19, 393–399 CrossRef CAS.
- C. Tsioptsias and C. Panayiotou, J. Supercrit. Fluids, 2008, 47, 302–308 CrossRef CAS.
- R. Gavillon and T. Budtova, Biomacromolecules, 2008, 9, 269–277 CrossRef CAS PubMed.
- J. Cai, L. X. Wang and L. N. Zhang, Cellulose, 2007, 14, 205–215 CrossRef CAS.
- J. Cai, S. Kimura, M. Wada, S. Kuga and L. Zhang, ChemSusChem, 2008, 1, 149–154 CrossRef CAS PubMed.
- L. N. Zhang, D. Ruan and J. P. Zhou, Ind. Eng. Chem. Res., 2001, 40, 5923–5928 CrossRef CAS.
- C. Chen, D. Li, H. Yano and K. Abe, Cellulose, 2014, 21, 3339–3346 CrossRef CAS.
- D. Zhou, L. Zhang and S. L. Guo, Water Res., 2005, 39, 3755–3762 CrossRef CAS PubMed.
- F. L. Mi, Y. C. Tan, H. F. Liang and H. W. Sung, Biomaterials, 2002, 23, 181–191 CrossRef CAS PubMed.
- X. W. Hu, Y. F. Tang, Q. Wang, Y. Li, J. H. Yang, Y. M. Du and J. F. Kennedy, Carbohydr. Polym., 2011, 83, 1128–1133 CrossRef CAS.
- S. P. Rwei, M. S. Lyu, P. S. Wu, C. H. Tseng and H. W. Huang, Cellulose, 2009, 16, 9–17 CrossRef CAS.
- A. Lue and L. Zhang, J. Phys. Chem. B, 2008, 112, 4488–4495 CrossRef CAS PubMed.
- B. B. Ding, J. Cai, J. C. Huang, L. N. Zhang, Y. Chen, X. W. Shi, Y. M. Du and S. Kuga, J. Mater. Chem., 2012, 22, 5801–5809 RSC.
- J. P. Zhou, L. Zhang, J. Cai and H. Shu, J. Membr. Sci., 2002, 210, 77–90 CrossRef CAS.
- A. Lue, L. Zhang and D. Ruan, Macromol. Chem. Phys., 2007, 208, 2359–2366 CrossRef CAS.
- J. F. Revol, H. Bradford, J. Giasson, R. H. Marchessault and D. G. Gray, Int. J. Biol. Macromol., 1992, 14, 170–172 CrossRef CAS PubMed.
- J. Li, J. F. Revol, E. Naranjo and R. H. Marchessault, Int. J. Biol. Macromol., 1996, 18, 177–187 CrossRef CAS PubMed.
- Y. Habibi, L. A. Lucia and O. J. Rojas, Chem. Rev., 2010, 110, 3479–3500 CrossRef CAS PubMed.
- J. B. Zeng, Y. S. He, S. L. Li and Y. Z. Wang, Biomacromolecules, 2012, 13, 1–11 CrossRef CAS PubMed.
- J. Araki and S. Kuga, Langmuir, 2001, 17, 4493–4496 CrossRef CAS.
- J. F. Revol and R. H. Marchessault, Int. J. Biol. Macromol., 1993, 15, 329–335 CrossRef CAS PubMed.
- J. R. Capadona, K. Shanmuganathan, S. Trittschuh, S. Seidel, S. J. Rowan and C. Weder, Biomacromolecules, 2009, 10, 712–716 CrossRef CAS PubMed.
- L. Heath and W. Thielemans, Green Chem., 2010, 12, 1448–1453 RSC.
- L. Heath, L. F. Zhu and W. Thielemans, ChemSusChem, 2013, 6, 537–544 CrossRef CAS PubMed.
- Y. Lu, Q. F. Sun, X. L. She, Y. Z. Xia, Y. X. Liu, J. Li and D. J. Yang, Carbohydr. Polym., 2013, 98, 1497–1504 CrossRef CAS PubMed.
- Y. Tsutsumi, H. Koga, Z. D. Qi, T. Saito and A. Isogai, Biomacromolecules, 2014, 15, 4314–4319 CrossRef CAS PubMed.
- Y. X. Wang and L. Y. Chen, Carbohydr. Polym., 2011, 83, 1937–1946 CrossRef CAS.
- J. Araki, Y. Yamanaka and K. Ohkawa, Polym. J., 2012, 44, 713–717 CrossRef CAS.
- N. L. G. de Rodriguez, W. Thielemans and A. Dufresne, Cellulose, 2006, 13, 261–270 CrossRef.
- L. Goetz, A. Mathew, K. Oksman, P. Gatenholm and A. J. Ragauskas, Carbohydr. Polym., 2009, 75, 85–89 CrossRef CAS.
- S. S. Nair, J. Y. Zhu, Y. L. Deng and A. J. Ragauskas, ACS Sustainable Chem. Eng., 2014, 2, 772–780 CrossRef CAS.
- T. Kohnke, A. Lin, T. Elder, H. Theliander and A. J. Ragauskas, Green Chem., 2012, 14, 1864–1869 RSC.
- A. Sturcova, G. R. Davies and S. J. Eichhorn, Biomacromolecules, 2005, 6, 1055–1061 CrossRef CAS PubMed.
- A. E. Way, L. Hsu, K. Shanmuganathan, C. Weder and S. J. Rowan, ACS Macro Lett., 2012, 1, 1001–1006 CrossRef CAS.
- M. I. Shams, S. Ifuku, M. Nogi, T. Oku and H. Yano, Appl. Phys. A: Mater. Sci. Process, 2011, 102, 325–331 CrossRef CAS.
- L. Li, P. M. Thangamathesvaran, C. Y. Yue, K. C. Tam, X. Hu and Y. C. Lam, Langmuir, 2001, 17, 8062–8068 CrossRef CAS.
- X. Z. Shu, K. J. Zhu and W. H. Song, Int. J. Pharm., 2001, 212, 19–28 CrossRef CAS PubMed.
- S. T. Lee, F. L. Mi, Y. J. Shen and S. S. Shyu, Polymer, 2001, 42, 1879–1892 CrossRef CAS.
- L. Li, H. Shan, C. Y. Yue, Y. C. Lam, K. C. Tam and X. Hu, Langmuir, 2002, 18, 7291–7298 CrossRef CAS.
- D. Gupta, C. H. Tator and M. S. Shoichet, Biomaterials, 2006, 27, 2370–2379 CrossRef CAS PubMed.
- A. Haque and E. R. Morris, Carbohydr. Polym., 1993, 22, 161–173 CrossRef CAS.
- A. Haque, R. K. Richardson, E. R. Morris, M. J. Gidley and D. C. Caswell, Carbohydr. Polym., 1993, 22, 175–186 CrossRef CAS.
- C. Sammon, G. Bajwa, P. Timmins and C. D. Melia, Polymer, 2006, 47, 577–584 CrossRef CAS.
- S. Aoyagi, H. Onishi and Y. Machida, Int. J. Pharm., 2007, 330, 138–145 CrossRef CAS PubMed.
- F. L. Mi, S. S. Shyu, Y. B. Wu, S. T. Lee, J. Y. Shyong and R. N. Huang, Biomaterials, 2001, 22, 165–173 CrossRef CAS PubMed.
- W. S. W. Ngah and S. Fatinathan, J. Environ. Manage., 2010, 91, 958–969 CrossRef CAS PubMed.
- M. K. Sureshkumar, D. Das, M. B. Mallia and P. C. Gupta, J. Hazard. Mater., 2010, 184, 65–72 CrossRef CAS PubMed.
- N. Csaba, M. Koping-Hoggard and M. J. Alonso, Int. J. Pharm., 2009, 382, 205–214 CrossRef CAS PubMed.
- B. Hu, C. L. Pan, Y. Sun, Z. Y. Hou, H. Ye, B. Hu and X. X. Zeng, J. Agric. Food Chem., 2008, 56, 7451–7458 CrossRef CAS PubMed.
- S. Kim, S. K. Nishimoto, J. D. Bumgardner, W. O. Haggard, M. W. Gaber and Y. Z. Yang, Biomaterials, 2010, 31, 4157–4166 CrossRef CAS PubMed.
- A. Chenite, C. Chaput, D. Wang, C. Combes, M. D. Buschmann, C. D. Hoemann, J. C. Leroux, B. L. Atkinson, F. Binette and A. Selmani, Biomaterials, 2000, 21, 2155–2161 CrossRef CAS PubMed.
- S. M. Richardson, N. Hughes, J. A. Hunt, A. J. Freemont and J. A. Hoyland, Biomaterials, 2008, 29, 85–93 CrossRef CAS PubMed.
- J. Y. Cho, M. C. Heuzey, A. Begin and P. J. Carreau, Biomacromolecules, 2005, 6, 3267–3275 CrossRef CAS PubMed.
- D. Klemm, D. Schumann, U. Udhardt and S. Marsch, Prog. Polym. Sci., 2001, 26, 1561–1603 CrossRef CAS.
- M. Iguchi, S. Yamanaka and A. Budhiono, J. Mater. Sci., 2000, 35, 261–270 CrossRef CAS.
- P. DeWulf, K. Joris and E. J. Vandamme, J. Chem. Technol. Biotechnol., 1996, 67, 376–380 CrossRef CAS.
- K. Gelin, A. Bodin, P. Gatenholm, A. Mihranyan, K. Edwards and M. Stromme, Polymer, 2007, 48, 7623–7631 CrossRef CAS.
- A. Putra, A. Kakugo, H. Furukawa, J. P. Gong and Y. Osada, Polymer, 2008, 49, 1885–1891 CrossRef CAS.
- A. Bodin, S. Concaro, M. Brittberg and P. Gatenholm, J. Tissue Eng. Regener. Med., 2007, 1, 406–408 CrossRef CAS PubMed.
- A. Svensson, E. Nicklasson, T. Harrah, B. Panilaitis, D. L. Kaplan, M. Brittberg and P. Gatenholm, Biomaterials, 2005, 26, 419–431 CrossRef CAS PubMed.
- H. Kono and S. Fujita, Carbohydr. Polym., 2012, 87, 2582–2588 CrossRef CAS.
- H. Kono and M. Zakimi, J. Appl. Polym. Sci., 2013, 128, 572–581 CrossRef CAS.
- T. Yoshimura, K. Matsuo and R. Fujioka, J. Appl. Polym. Sci., 2006, 99, 3251–3256 CrossRef CAS.
- T. Yoshimura, I. Uchikoshi, Y. Yoshiura and R. Fujioka, Carbohydr. Polym., 2005, 61, 322–326 CrossRef CAS.
- C. Demitri, R. Del Sole, F. Scalera, A. Sannino, G. Vasapollo, A. Maffezzoli, L. Ambrosio and L. Nicolais, J. Appl. Polym. Sci., 2008, 110, 2453–2460 CrossRef CAS.
- M. Bodnar, J. F. Hartmann and J. Borbely, Biomacromolecules, 2005, 6, 2521–2527 CrossRef CAS PubMed.
- C. Y. Chang, S. Chen and L. N. Zhang, J. Mater. Chem., 2011, 21, 3865–3871 RSC.
- R. Rodriguez, C. Alvarez-Lorenzo and A. Concheiro, J. Controlled Release, 2003, 86, 253–265 CrossRef CAS PubMed.
- N. Li and R. B. Bai, Ind. Eng. Chem. Res., 2005, 44, 6692–6700 CrossRef CAS.
- A. Kamari, W. N. Wan Saime and L. Lai Ken, J. Environ. Sci., 2009, 21, 296–302 CrossRef.
- B. G. Kabra, S. H. Gehrke and R. J. Spontak, Macromolecules, 1998, 31, 2166–2173 CrossRef CAS.
- X. H. Lu, Z. B. Hu and J. Gao, Macromolecules, 2000, 33, 8698–8702 CrossRef CAS.
- U. Anbergen and W. Oppermann, Polymer, 1990, 31, 1854–1858 CrossRef CAS.
- A. Sakakura, K. Kawajiri, T. Ohkubo, Y. Kosugi and K. Ishihara, J. Am. Chem. Soc., 2007, 129, 14775–14779 CrossRef CAS PubMed.
- A. Sannino, M. Madaghiele, M. G. Lionetto, T. Schettino and A. Maffezzoli, J. Appl. Polym. Sci., 2006, 102, 1524–1530 CrossRef CAS.
- S. H. Pangburn, P. V. Trescony and J. Heller, Biomaterials, 1982, 3, 105–108 CrossRef CAS PubMed.
- S. R. Jameela and A. Jayakrishnan, Biomaterials, 1995, 16, 769–775 CrossRef CAS PubMed.
- D. Capitani, A. A. De Angelis, V. Crescenzi, G. Masci and A. L. Segre, Carbohydr. Polym., 2001, 45, 245–252 CrossRef CAS.
- Y. Yuan, B. M. Chesnutt, G. Utturkar, W. O. Haggard, Y. Yang, J. L. Ong and J. D. Bumgardner, Carbohydr. Polym., 2007, 68, 561–567 CrossRef CAS.
- H. W. Sung, R. N. Huang, L. L. H. Huang and C. C. Tsai, J. Biomater. Sci., Polym. Ed., 1999, 10, 63–78 CrossRef CAS PubMed.
- R. D. Rogers , not yet reported.
- S. G. Hirsch and R. J. Spontak, Polymer, 2002, 43, 123–129 CrossRef CAS.
- S. V. Madihally and H. W. T. Matthew, Biomaterials, 1999, 20, 1133–1142 CrossRef CAS PubMed.
- J. Innerlohinger, H. K. Weber and G. Kraft, Macromol. Symp., 2006, 244, 126–135 CrossRef CAS.
- C. Tsioptsias, A. Stefopoulos, I. Kokkinomalis, L. Papadopoulou and C. Panayiotou, Green Chem., 2008, 10, 965–971 RSC.
- F. Liebner, E. Haimer, M. Wendland, M. A. Neouze, K. Schlufter, P. Miethe, T. Heinze, A. Potthast and T. Rosenau, Macromol. Biosci., 2010, 10, 349–352 CrossRef CAS PubMed.
- K. E. Crompton, J. D. Goud, R. V. Bellamkonda, T. R. Gengenbach, D. I. Finkelstein, M. K. Horne and J. S. Forsythe, Biomaterials, 2007, 28, 441–449 CrossRef CAS PubMed.
- F. Lionetto, A. Sannino and A. Maffezzoli, Polymer, 2005, 46, 1796–1803 CrossRef CAS.
- A. H. Pei, N. Butchosa, L. A. Berglund and Q. Zhou, Soft Matter, 2013, 9, 2047–2055 RSC.
- A. Kimura, N. Nagasawa and M. Taguchi, Radiat. Phys. Chem., 2014, 103, 216–221 CrossRef CAS.
- K. Hara, A. Iida, K. Yano and T. Nishida, Colloids Surf., B, 2004, 38, 227–230 CAS.
- M. Wang, L. Xu, M. L. Zhai, J. Peng, J. Q. Li and G. S. Wei, Carbohydr. Polym., 2008, 74, 498–503 CrossRef CAS.
- L. Zhao, H. Mitomo and F. Yoshii, J. Bioact. Compat. Polym., 2008, 23, 319–333 CrossRef CAS.
- L. Zhao, H. Mitomo, N. Nagasawa, F. Yoshii and T. Kume, Carbohydr. Polym., 2003, 51, 169–175 CrossRef CAS.
- P. Petrov, E. Petrova, R. Stamenova, C. B. Tsvetanov and G. Riess, Polymer, 2006, 47, 6481–6484 CrossRef CAS.
- E. Velickova, E. Winkelhausen, S. Kuzmanova, M. Cvetkovska and C. Tsvetanov, React. Funct. Polym., 2009, 69, 688–693 CrossRef CAS.
- B. G. Yershov and A. S. Klimentov, Polym. Sci. USSR, 1977, 19, 936–941 CrossRef.
- R. A. Wach, H. Mitomo and F. Yoshii, J. Radioanal. Nucl. Chem., 2004, 261, 113–118 CrossRef CAS.
- R. A. Wach, H. Mitomo, F. Yoshii and T. Kume, J. Appl. Polym. Sci., 2001, 81, 3030–3037 CrossRef CAS.
- S. B. Lee, Y. H. Kim, M. S. Chong and Y. M. Lee, Biomaterials, 2004, 25, 2309–2317 CrossRef CAS PubMed.
- G. Yang, L. Zhang, T. Peng and W. Zhong, J. Membr. Sci., 2000, 175, 53–60 CrossRef CAS.
- N. Ninan, M. Muthiah, I. K. Park, A. Elain, S. Thomas and Y. Grohens, Carbohydr. Polym., 2013, 98, 877–885 CrossRef CAS PubMed.
- S. M. Liang, J. J. Wu, H. F. Tian, L. N. Zhang and J. Xu, ChemSusChem, 2008, 1, 558–563 CrossRef CAS PubMed.
- A. Seves, G. Testa, A. M. Bonfatti, E. D. Paglia, E. Selli and B. Marcandalli, Macromol. Mater. Eng., 2001, 286, 524–528 CrossRef CAS.
- Y. Nishio and R. S. Manley, Macromolecules, 1988, 21, 1270–1277 CrossRef.
- Y. M. Lee, S. H. Kim and S. J. Kim, Polymer, 1996, 37, 5897–5905 CrossRef CAS.
- S. L. Williamson, R. S. Armentrout, R. S. Porter and C. L. McCormick, Macromolecules, 1998, 31, 8134–8141 CrossRef CAS.
- G. S. Chauhan, L. Guleria and R. Sharma, Cellulose, 2005, 12, 97–110 CrossRef CAS.
- M. Pandey and M. C. I. M. Amin, Int. J. Polym. Mater. Polym., 2013, 62, 402–405 CrossRef CAS.
- M. Pandey, M. C. I. M. Amin, N. Ahmad and M. M. Abeer, Int. J. Polym. Sci., 2013, 2013, 1–10 CrossRef.
- M. C. I. M. Amin, N. Ahmad, N. Halib and I. Ahmad, Carbohydr. Polym., 2012, 88, 465–473 CrossRef.
- H. M. Said, S. G. A. Abd Alla and A. W. M. El-Naggar, React. Funct. Polym., 2004, 61, 397–404 CrossRef CAS.
- J. A. Kelly, A. M. Shukaliak, C. C. Y. Cheung, K. E. Shopsowitz, W. Y. Hamad and M. J. MacLachlan, Angew. Chem., Int. Ed., 2013, 52, 8912–8916 CrossRef CAS PubMed.
- T. T. Hanh, H. T. Huy and N. Q. Hien, Radiat. Phys. Chem., 2015, 106, 235–241 CrossRef CAS.
- S. Tanodekaew, M. Prasitsilp, S. Swasdison, B. Thavornyutikarn, T. Pothsree and R. Pateepasen, Biomaterials, 2004, 25, 1453–1460 CrossRef CAS PubMed.
- L. B. R. Castro, K. V. Soares, A. F. Naves, A. M. Carmona-Ribeiro and D. F. S. Petri, Ind. Eng. Chem. Res., 2004, 43, 7774–7779 CrossRef CAS.
- T. G. Liu, L. W. Qian, B. Li, J. Li, K. K. Zhu, H. B. Deng, X. H. Yang and X. Wang, Carbohydr. Polym., 2013, 94, 261–271 CrossRef CAS PubMed.
- M. M. Ibrahim, A. Koschella, G. Kadry and T. Heinze, Carbohydr. Polym., 2013, 95, 414–420 CrossRef CAS PubMed.
- L. J. Mao, Y. J. Hu, Y. S. Piao, X. D. Chen, W. S. Xian and D. X. Piao, Curr. Appl. Phys., 2005, 5, 426–428 CrossRef.
- R. Sanna, E. Fortunati, V. Alzari, D. Nuvoli, A. Terenzi, M. F. Casula, J. M. Kenny and A. Mariani, Cellulose, 2013, 20, 2393–2402 CrossRef CAS.
- C. Y. Chang, J. Peng, L. N. Zhang and D. W. Pang, J. Mater. Chem., 2009, 19, 7771–7776 RSC.
- P. T. S. Kumar, S. Abhilash, K. Manzoor, S. V. Nair, H. Tamura and R. Jayakumar, Carbohydr. Polym., 2010, 80, 761–767 CrossRef CAS.
- P. T. S. Kumar, V. K. Lakshmanan, M. Raj, R. Biswas, T. Hiroshi, S. V. Nair and R. Jayakumar, Pharm. Res., 2013, 30, 523–537 CrossRef PubMed.
- S. Sequeira, D. V. Evtuguin, I. Portugal and A. P. Esculcas, Mater. Sci. Eng., C, 2007, 27, 172–179 CrossRef CAS.
- B. Alonso and E. Belamie, Angew. Chem., Int. Ed., 2010, 49, 8201–8204 CrossRef CAS PubMed.
- M. Bagheri and S. Rabieh, Cellulose, 2013, 20, 699–705 CrossRef CAS.
- J. J. Wu, N. Zhao, X. L. Zhang and J. Xu, Cellulose, 2012, 19, 1239–1249 CrossRef CAS.
- X. G. Luo, S. L. Liu, J. P. Zhou and L. N. Zhang, J. Mater. Chem., 2009, 19, 3538–3545 RSC.
- A. Ashori, S. Sheykhnazari, T. Tabarsa, A. Shakeri and M. Golalipour, Carbohydr. Polym., 2012, 90, 413–418 CrossRef CAS PubMed.
- S. A. Hutchens, R. S. Benson, B. R. Evans, H. M. O'Neill and C. J. Rawn, Biomaterials, 2006, 27, 4661–4670 CrossRef CAS PubMed.
- Y. C. Kuo and C. Y. Lin, Biotechnol. Bioeng., 2006, 95, 132–144 CrossRef CAS PubMed.
- C. Katepetch, R. Rujiravanit and H. Tamura, Cellulose, 2013, 20, 1275–1292 CrossRef CAS.
- A. John, H. U. Ko, D. G. Kim and J. Kim, Cellulose, 2011, 18, 675–680 CrossRef CAS.
- H. Tang, W. J. Zhou, A. Lu and L. N. Zhang, J. Mater. Sci., 2014, 49, 123–133 CrossRef CAS.
- H. X. Zhu, S. R. Jia, T. Wan, Y. Y. Jia, H. J. Yang, J. Li, L. Yan and C. Zhong, Carbohydr. Polym., 2011, 86, 1558–1564 CrossRef CAS.
- S. Zadegan, M. Hosainalipour, H. R. Rezaie, H. Ghassai and M. A. Shokrgozar, Mater. Sci. Eng., C, 2011, 31, 954–961 CrossRef CAS.
- Y. Z. Wan, L. Hong, S. R. Jia, Y. Huang, Y. Zhu, Y. L. Wang and H. J. Jiang, Compos. Sci. Technol., 2006, 66, 1825–1832 CrossRef CAS.
- C. J. Grande, F. G. Torres, C. M. Gomez and M. C. Bano, Acta Biomater., 2009, 5, 1605–1615 CrossRef CAS PubMed.
- P. T. S. Kumar, S. Srinivasan, V. K. Lakshmanan, H. Tamura, S. V. Nair and R. Jayakumar, Carbohydr. Polym., 2011, 85, 584–591 CrossRef.
- P. T. S. Kumar, S. Srinivasan, V. K. Lakshmanan, H. Tamura, S. V. Nair and R. Jayakumar, Int. J. Biol. Macromol., 2011, 49, 20–31 CrossRef CAS PubMed.
- T. Maneerung, S. Tokura and R. Rujiravanit, Carbohydr. Polym., 2008, 72, 43–51 CrossRef CAS.
- S. M. Li, N. Jia, M. G. Ma, Z. Zhang, Q. H. Liu and R. C. Sun, Carbohydr. Polym., 2011, 86, 441–447 CrossRef CAS.
- S. M. Li, N. Jia, J. F. Zhu, M. G. Ma, F. Xu, B. Wang and R. C. Sun, Carbohydr. Polym., 2011, 83, 422–429 CrossRef CAS.
- K. Madhumathi, P. T. S. Kumar, S. Abhilash, V. Sreeja, H. Tamura, K. Manzoor, S. V. Nair and R. Jayakumar, J. Mater. Sci. Mater. Med., 2010, 21, 807–813 CrossRef CAS PubMed.
- J. Cai, S. L. Liu, J. Feng, S. Kimura, M. Wada, S. Kuga and L. N. Zhang, Angew. Chem., Int. Ed., 2012, 51, 2076–2079 CrossRef CAS PubMed.
- W. Ogasawara, W. Shenton, S. A. Davis and S. Mann, Chem. Mater., 2000, 12, 2835–2837 CrossRef CAS.
- J. Zeng, S. L. Liu, J. Cai and L. Zhang, J. Phys. Chem. C, 2010, 114, 7806–7811 CAS.
- H. Y. Zhu, Y. Q. Fu, R. Jiang, J. Yao, L. Xiao and G. M. Zeng, Bioresour. Technol., 2012, 105, 24–30 CrossRef CAS PubMed.
- R. T. Olsson, M. A. S. A. Samir, G. Salazar-Alvarez, L. Belova, V. Strom, L. A. Berglund, O. Ikkala, J. Nogues and U. W. Gedde, Nat. Nanotechnol., 2010, 5, 584–588 CrossRef CAS PubMed.
- S. L. Liu, Q. F. Yan, D. D. Tao, T. F. Yu and X. Y. Liu, Carbohydr. Polym., 2012, 89, 551–557 CrossRef CAS PubMed.
- S. L. Liu, J. P. Zhou and L. N. Zhang, Cellulose, 2011, 18, 663–673 CrossRef CAS.
- S. L. Liu, L. N. Zhang, J. P. Zhou, J. F. Xiang, J. T. Sun and J. G. Guan, Chem. Mater., 2008, 20, 3623–3628 CrossRef CAS.
- S. L. Liu, D. D. Tao and L. N. Zhang, Powder Technol., 2012, 217, 502–509 CrossRef CAS.
- K. Vimala, Y. M. Mohan, K. S. Sivudu, K. Varaprasad, S. Ravindra, N. N. Reddy, Y. Padma, B. Sreedhar and K. MohanaRaju, Colloids Surf., B, 2010, 76, 248–258 CrossRef CAS PubMed.
- C. Y. Chang, B. Duan, J. Cai and L. N. Zhang, Eur. Polym. J., 2010, 46, 92–100 CrossRef CAS.
- D. Ciolacu, A. M. Oprea, N. Anghel, G. Cazacu and M. Cazacu, Mater. Sci. Eng., C, 2012, 32, 452–463 CrossRef CAS.
- L. E. Bromberg and E. S. Ron, Adv. Drug Delivery Rev., 1998, 31, 197–221 CrossRef CAS.
- J. Wang, X. S. Zhou and H. N. Xiao, Carbohydr. Polym., 2013, 94, 749–754 CrossRef CAS PubMed.
- C. Alvarez-Lorenzo, A. Concheiro, A. S. Dubovik, N. V. Grinberg, T. V. Burova and V. Y. Grinberg, J. Controlled Release, 2005, 102, 629–641 CrossRef CAS PubMed.
- J. Á. Montes, A. Ortega and G. Burillo, J. Radioanal. Nucl. Chem., 2015, 303, 2143–2150 CAS.
- N. Bhattarai, J. Gunn and M. Q. Zhang, Adv. Drug Delivery Rev., 2010, 62, 83–99 CrossRef CAS PubMed.
- A. K. Anal and W. F. Stevens, Int. J. Pharm., 2005, 290, 45–54 CrossRef CAS PubMed.
- Y. M. Xu, C. Y. Zhan, L. H. Fan, L. Wang and H. Zheng, Int. J. Pharm., 2007, 336, 329–337 CrossRef CAS PubMed.
- F. Bigucci, B. Luppi, T. Cerchiara, M. Sorrenti, G. Bettinetti, L. Rodriguez and V. Zecchi, Eur. J. Pharm. Sci., 2008, 35, 435–441 CrossRef CAS PubMed.
- K. L. B. Chang and J. Lin, Carbohydr. Polym., 2000, 43, 163–169 CrossRef CAS.
- H. Q. Chen and M. W. Fan, J. Bioact. Compat. Polym., 2008, 23, 38–48 CrossRef CAS.
- L. L. Y. Chiu and M. Radisic, J. Controlled Release, 2011, 155, 376–385 CrossRef CAS PubMed.
- N. Bhattarai, H. R. Ramay, J. Gunn, F. A. Matsen and M. Q. Zhang, J. Controlled Release, 2005, 103, 609–624 CrossRef CAS PubMed.
- J. J. Marler, J. Upton, R. Langer and J. P. Vacanti, Adv. Drug Delivery Rev., 1998, 33, 165–182 CrossRef CAS.
- L. Ma, C. Y. Gao, Z. W. Mao, J. Zhou, J. C. Shen, X. Q. Hu and C. M. Han, Biomaterials, 2003, 24, 4833–4841 CrossRef CAS PubMed.
- J. K. He, D. C. Li, Y. X. Liu, B. Yao, H. X. Zhan, Q. Lian, B. H. Lu and Y. Lv, Acta Biomater., 2009, 5, 453–461 CrossRef CAS PubMed.
- Z. G. Ge, S. Baguenard, L. Y. Lim, A. Wee and E. Khor, Biomaterials, 2004, 25, 1049–1058 CrossRef CAS PubMed.
- K. Madhumathi, P. T. S. Kumar, K. C. Kavya, T. Furuike, H. Tamura, S. V. Nair and R. Jayakumar, Int. J. Biol. Macromol., 2009, 45, 289–292 CrossRef CAS PubMed.
- S. Saska, L. N. Teixeira, P. T. de Oliveira, A. M. M. Gaspar, S. J. L. Ribeiro, Y. Messaddeq and R. Marchetto, J. Mater. Chem., 2012, 22, 22102–22112 RSC.
- C. H. Zhu, D. D. Fan, Z. Z. Duan, W. J. Xue, L. A. Shang, F. L. Chen and Y. E. Luo, J. Biomed. Mater. Res., Part A, 2009, 89, 829–840 CrossRef PubMed.
- H. Nagahama, V. V. D. Rani, K. T. Shalumon, R. Jayakumar, S. V. Nair, S. Koiwa, T. Furuike and H. Tamura, Int. J. Biol. Macromol., 2009, 44, 333–337 CrossRef CAS PubMed.
- C. Chang, N. Peng, M. He, Y. Teramoto, Y. Nishio and L. Zhang, Carbohydr. Polym., 2013, 91, 7–13 CrossRef CAS PubMed.
- E. J. Lee, D. S. Shin, H. E. Kim, H. W. Kim, Y. H. Koh and J. H. Jang, Biomaterials, 2009, 30, 743–750 CrossRef CAS PubMed.
- Z. S. Li, H. R. Ramay, K. D. Hauch, D. M. Xiao and M. Q. Zhang, Biomaterials, 2005, 26, 3919–3928 CrossRef CAS PubMed.
- J. H. Brauker, V. E. Carrbrendel, L. A. Martinson, J. Crudele, W. D. Johnston and R. C. Johnson, J. Biomed. Mater. Res., 1995, 29, 1517–1524 CrossRef CAS PubMed.
- I. V. Yannas, E. Lee, D. P. Orgill, E. M. Skrabut and G. F. Murphy, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 933–937 CrossRef CAS.
- B. P. Robinson, J. O. Hollinger, E. H. Szachowicz and J. Brekke, Otolaryng Head Neck, 1995, 112, 707–713 CrossRef CAS.
- K. Whang, K. E. Healy, D. R. Elenz, E. K. Nam, D. C. Tsai, C. H. Thomas, G. W. Nuber, F. H. Glorieux, R. Travers and S. M. Sprague, Tissue Eng., 1999, 5, 35–51 CrossRef CAS PubMed.
- K. S. Anseth, C. N. Bowman and L. BrannonPeppas, Biomaterials, 1996, 17, 1647–1657 CrossRef CAS PubMed.
- Q. L. Hu, B. Q. Li, M. Wang and J. C. Shen, Biomaterials, 2004, 25, 779–785 CrossRef CAS PubMed.
- F. F. Li, Y. Liu, Y. Ding and Q. F. Xie, Soft Matter, 2014, 10, 2292–2303 RSC.
- H. L. Cai, S. Sharma, W. Y. Liu, W. Mu, W. Liu, X. D. Zhang and Y. L. Deng, Biomacromolecules, 2014, 15, 2540–2547 CrossRef CAS PubMed.
- M. Paakko, J. Vapaavuori, R. Silvennoinen, H. Kosonen, M. Ankerfors, T. Lindstrom, L. A. Berglund and O. Ikkala, Soft Matter, 2008, 4, 2492–2499 RSC.
- Z. J. Cai and G. Yang, J. Appl. Polym. Sci., 2011, 120, 2938–2944 CrossRef CAS.
- C. Tsioptsias, C. Michailof, G. Stauropoulos and C. Panayiotou, Carbohydr. Polym., 2009, 76, 535–540 CrossRef CAS.
- H. Nagahama, T. Kashiki, N. Nwe, R. Jayakumar, T. Furuike and H. Tamura, Carbohydr. Polym., 2008, 73, 456–463 CrossRef CAS.
- P. J. VandeVord, H. W. T. Matthew, S. P. DeSilva, L. Mayton, B. Wu and P. H. Wooley, J. Biomed. Mater. Res., 2002, 59, 585–590 CrossRef CAS PubMed.
- J. S. Mao, L. G. Zhao, Y. J. Yin and K. D. Yao, Biomaterials, 2003, 24, 1067–1074 CrossRef CAS PubMed.
- Z. S. Li and M. Q. Zhang, J. Biomed. Mater. Res., Part A, 2005, 75, 485–493 CrossRef PubMed.
- L. J. Kong, Y. Gao, G. Y. Lu, Y. D. Gong, N. M. Zhao and X. F. Zhang, Eur. Polym. J., 2006, 42, 3171–3179 CrossRef CAS.
- E. B. W. Giesen, M. Ding, M. Dalstra and T. M. G. J. van Eijden, J. Biomech., 2001, 34, 799–803 CrossRef CAS PubMed.
- Y. N. Yeni and D. P. Fyhrie, J. Biomech., 2001, 34, 1649–1654 CrossRef CAS PubMed.
- P. Zioupos and J. D. Currey, Bone, 1998, 22, 57–66 CrossRef CAS PubMed.
- K. Rezwan, Q. Z. Chen, J. J. Blaker and A. R. Boccaccini, Biomaterials, 2006, 27, 3413–3431 CrossRef CAS PubMed.
- R. Jonas and L. F. Farah, Polym. Degrad. Stab., 1998, 59, 101–106 CrossRef CAS.
- S. McCarthy, K. Gregory and J. Morgan, US Patent, Application No. US20050147656 A1, 2005 Search PubMed.
- V. K. Sharma, R. A. Yngard and Y. Lin, Adv. Colloid Interface Sci., 2009, 145, 83–96 CrossRef CAS PubMed.
- N. Chiaoprakobkij, N. Sanchavanakit, K. Subbalekha, P. Pavasant and M. Phisalaphong, Carbohydr. Polym., 2011, 85, 548–553 CrossRef CAS.
- N. L. B. M. Yusof, A. Wee, L. Y. Lim and E. Khor, J. Biomed. Mater. Res., Part A, 2003, 66, 224–232 CrossRef PubMed.
- N. L. B. M. Yusof, L. Y. Lim and E. Khor, J. Biomed. Mater. Res., 2001, 54, 59–68 CrossRef CAS PubMed.
- M. Prasitsilp, R. Jenwithisuk, K. Kongsuwan, N. Damrongchai and P. Watts, J. Mater. Sci. Mater. Med., 2000, 11, 773–778 CrossRef CAS PubMed.
- T. A. Khan, K. K. Peh and H. S. Ch'ng, J. Pharm. Pharm. Sci., 2000, 3, 303–311 CAS.
- F. L. Mi, Y. B. Wu, S. S. Shyu, J. Y. Schoung, Y. B. Huang, Y. H. Tsai and J. Y. Hao, J. Biomed. Mater. Res., 2002, 59, 438–449 CrossRef CAS PubMed.
- C. M. Deng, L. Z. He, M. Zhao, D. Yang and Y. Liu, Carbohydr. Polym., 2007, 69, 583–589 CrossRef CAS.
- H. T. Xu, L. Ma, H. F. Shi, C. Y. Gao and C. M. Han, Polym. Adv. Technol., 2007, 18, 869–875 CrossRef CAS.
- B. S. Liu and T. B. Huang, Macromol. Biosci., 2008, 8, 932–941 CrossRef CAS PubMed.
- P. T. S. Kumar, V. K. Lakshmanan, T. V. Anilkumar, C. Ramya, P. Reshmi, A. G. Unnikrishnan, S. V. Nair and R. Jayakumar, ACS Appl. Mater. Interfaces, 2012, 4, 2618–2629 Search PubMed.
- F. L. Fu and Q. Wang, J. Environ. Manage., 2011, 92, 407–418 CrossRef CAS PubMed.
- H. Tang, C. Y. Chang and L. N. Zhang, Chem. Eng. J., 2011, 173, 689–697 CrossRef CAS.
- N. Li and R. B. Bai, Sep. Purif. Technol., 2005, 42, 237–247 CrossRef CAS.
- S. Chatterjee, T. Chatterjee and S. H. Woo, Bioresour. Technol., 2010, 101, 3853–3858 CrossRef CAS PubMed.
- S. Chatterjee and S. H. Woo, J. Hazard. Mater., 2009, 164, 1012–1018 CrossRef CAS PubMed.
- W. S. W. Ngah and S. Fatinathan, Chem. Eng. J., 2008, 143, 62–72 CrossRef CAS.
- J. L. Wang, L. G. Wei, Y. C. Ma, K. L. Li, M. H. Li, Y. C. Yu, L. Wang and H. H. Qiu, Carbohydr. Polym., 2013, 98, 736–743 CrossRef CAS PubMed.
- S. Chatterjee, T. Chatterjee, S. R. Lim and S. H. Woo, Bioresour. Technol., 2011, 102, 4402–4409 CrossRef CAS PubMed.
- S. Chatterjee, M. W. Lee and S. H. Woo, Bioresour. Technol., 2010, 101, 1800–1806 CrossRef CAS PubMed.
- S. Chatterjee, M. W. Lee and S. H. Woo, Carbon, 2009, 47, 2933–2936 CrossRef CAS.
- G. J. Copello, A. M. Mebert, M. Raineri, M. P. Pesenti and L. E. Diaz, J. Hazard. Mater., 2011, 186, 932–939 CrossRef CAS PubMed.
- S. M. Miller and J. B. Zimmerman, Water Res., 2010, 44, 5722–5729 CrossRef CAS PubMed.
- Y. Zheng and A. Q. Wang, J. Hazard. Mater., 2009, 171, 671–677 CrossRef CAS PubMed.
- D. Mohan, C. U. Pittman and P. H. Steele, J. Colloid Interface Sci., 2006, 297, 489–504 CrossRef CAS PubMed.
- H. Yan, J. Dai, Z. Yang, H. Yang and R. S. Cheng, Chem. Eng. J., 2011, 174, 586–594 CrossRef CAS.
- N. Li, R. B. Bai and C. K. Liu, Langmuir, 2005, 21, 11780–11787 CrossRef CAS PubMed.
- J. Dai, H. Yang, H. Yan, Y. G. Shangguan, Q. A. Zheng and R. S. Cheng, Chem. Eng. J., 2011, 166, 970–977 CrossRef CAS.
- J. Aburto and S. Le Borgne, Macromolecules, 2004, 37, 2938–2943 CrossRef CAS.
- X. Song, C. Li, R. Xu and K. Wang, Ind. Eng. Chem. Res., 2012, 51, 11261–11265 CrossRef CAS.
- Y. H. Liu, X. H. Cao, R. Hua, Y. Q. Wang, Y. T. Liu, C. Pang and Y. Wang, Hydrometallurgy, 2010, 104, 150–155 CrossRef CAS.
- W. Q. Sun and G. F. Payne, Biotechnol. Bioeng., 1996, 51, 79–86 CrossRef CAS PubMed.
- I. F. Nata, M. Sureshkumar and C. K. Lee, RSC Adv., 2011, 1, 625–631 RSC.
- W. S. W. Ngah, A. Kamari and Y. Koay, Int. J. Biol. Macromol., 2004, 34, 155–161 CrossRef PubMed.
- S. Yamazaki, A. Takegawa, Y. Kaneko, J. Kadokawa, M. Yamagata and M. Ishikawa, J. Electrochem. Soc., 2010, 157, A203–A208 CrossRef CAS.
- S. Yamazaki, A. Takegawa, Y. Kaneko, J. Kadokawa, M. Yamagata and M. Ishikawa, Electrochem. Commun., 2009, 11, 68–70 CrossRef CAS.
- M. F. Z. Kadir, S. R. Majid and A. K. Arof, Electrochim. Acta, 2010, 55, 1475–1482 CrossRef CAS.
- A. Murugadoss and A. Chattopadhyay, Nanotechnology, 2008, 19 Search PubMed.
- M. Kemell, V. Pore, M. Ritala, M. Leskela and M. Linden, J. Am. Chem. Soc., 2005, 127, 14178–14179 CrossRef CAS PubMed.
- A. El Kadib, K. Molvinger, C. Guimon, F. Quignard and D. Brunel, Chem. Mater., 2008, 20, 2198–2204 CrossRef CAS.
- K. Molvinger, F. Quignard, D. Brunel, M. Boissiere and J. M. Devoisselle, Chem. Mater., 2004, 16, 3367–3372 CrossRef CAS.
- Q. L. Nie, W. B. Tan and Y. Zhang, Nanotechnology, 2006, 17, 140–144 CrossRef CAS.
- R. Valentin, K. Molvinger, F. Quignard and D. Brunel, New J. Chem., 2003, 27, 1690–1692 RSC.
- D. Y. Zhang and L. M. Qi, Chem. Commun., 2005, 2735–2737 RSC.
- X. H. Feng and R. Pelton, Macromolecules, 2007, 40, 1624–1630 CrossRef CAS.
- B. Smitha, S. Sridhar and A. A. Khan, Macromolecules, 2004, 37, 2233–2239 CrossRef CAS.
- J. Shang, Z. Z. Shao and X. Chen, Biomacromolecules, 2008, 9, 1208–1213 CrossRef CAS PubMed.
- J. Kim, N. G. Wang, Y. Chen, S. K. Lee and G. Y. Yun, Cellulose, 2007, 14, 217–223 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |