
An efficient and recyclable tetraoxo-coordinated zinc catalyst for the cycloaddition of epoxides with carbon dioxide at atmospheric pressure†
Ran
Ma
ab,
Liang-Nian
He
*ab and
Yue-Biao
Zhou
ab
aState Key Laboratory and Institute of Elemento-Organic Chemistry, Nankai University, China
bCollaborative Innovation center of Chemical Science and Engineering, Nankai University, Tianjin 300071, China. E-mail: heln@nankai.edu.cn; Fax: (+86) 22-23503878; Tel: (+86) 22-23503878
First published on 8th September 2015
Abstract
An efficient and recyclable catalytic system of a Zn(O)4-coordinated complex e.g. 2-hydroxypyridine N-oxide zinc(II), Zn(OPO)2 and tetrabutylammonium iodide (TBAI) was developed for the cyclic carbonate synthesis from epoxides and CO2 without the use of any organic solvents. This easily prepared, low-toxic Zn(OPO)2 may have a strong Lewis acidity for the activation of epoxides and exhibited high activity (TOF up to 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1) even at a CO2 pressure as low as 1 bar with a broad substrate scope. Furthermore, the catalyst can be easily recovered and reused five times without a significant loss of its catalytic activity.
000 h−1) even at a CO2 pressure as low as 1 bar with a broad substrate scope. Furthermore, the catalyst can be easily recovered and reused five times without a significant loss of its catalytic activity.
Introduction
CO2 is one of the greenhouse gases and has been attracting much attention as an inexpensive, nontoxic, nonflammable, renewable and abundant C1 building block for organic synthesis.1 Many procedures have been developed towards cost-effective chemical fixation of CO2 by forming C–N, C–O, C–C and C–H bonds to convert CO2 into value-added chemicals, polymers, and fuels.2–5 However, the thermodynamic stability and kinetic inertness of CO2 represent a challenge for its conversion under mild conditions. In order to overcome such barriers, a reactant with a relatively high free energy is commonly employed. In this aspect, the addition of CO2 to epoxides is a 100% atom-economical reaction that can generate either cyclic carbonates or polycarbonates and represents one of the most promising ways for CO2 fixation. Synthesis of ethylene and propylene carbonates from CO2 has been developed for over 50 years and cyclic carbonates are widely used as aprotic high-boiling point polar solvents with low odour and toxicity, electrolytes for lithium-ion batteries, starting materials of polymeric materials, and intermediates in the synthesis of fine chemical intermediates.3,6 Numerous catalysts for the efficient synthesis of cyclic carbonates from epoxides and CO2 have been developed including metal oxides,7 porphyrin,8 salen metal compounds,9–11 alkali metal salts,12,13 transition-metal complexes,14 quaternary ammonium15,16 or quaternary phosphonium salts,17,18 ionic liquids,19 organocatalysts20,21 and so on. Among these catalysts, the fixation of CO2 by transition-metal catalysts especially metallosalen complexes has seen significant progress. Metallosalen complexes with a salen-N2O2 ligand are privileged catalysts22 and have been widely used in fundamental organic transformations, such as oxidations, asymmetric ring-opening reactions and addition of trimethylsilyl cyanide to aldehydes and ketones.23–26 Al,11,27 Zn,10,28 Cr29 and Co30,31 have been extensively explored to efficiently catalyze the fixation of CO2 with epoxides to produce polycarbonates and cyclic carbonates.32 The activated epoxide by the electrophilic metallosalen catalyst is able to facilely undergo ring-opening at the less substituted C–O bond upon nucleophilic attack by Br− or I−, to form the alkoxide anion further affording the corresponding cyclic carbonate after reaction with CO2 (Scheme 1).
 | ||
| Scheme 1 Possible mechanism for catalytic formation of cyclic carbonate. | ||
Mashima et al. reported the μ-oxo-tetranuclear zinc cluster Zn4(OCOCF3)6O and TBAI as a dual catalyst system for the synthesis of carbonate from CO2 (1 atm) at room temperature. The epoxide was activated by the Lewis acidity of Zn metal and CO2 could be activated by a second zinc metal.33 Recently, Kleij et al. have developed a trinuclear bifunctional catalyst, derived from a tetraoxo bis-Zn(salphen) metalloligand, for the cycloaddition of epoxides and CO2.10 The central Zn center surrounded by a tetradentate O4-coordinated metallohost would afford significant stabilization and represents a Lewis acidic center for epoxide's activation; meanwhile, an iodide anion dissociated from the outer salen N2O2–Zn could act as an active nucleophile for the ring-opening of the epoxide.10 In this context, we envisaged that a single Zn(O)4 coordinated complex with an appropriate ligand might also display higher activity in the cycloaddition reaction of CO2 with epoxides in comparison with a classic salen-N2O2–Zn catalyst.
The coordination chemistry with pyridine N-oxide as a ligand and its substitution derivatives have been thoroughly investigated.34–36 The 2-hydroxy derivative of pyridine N-oxide (a, Scheme 2) is thought to exist mainly as the tautomer 1-hydroxy-2-pyridone (HOPO) (b, Scheme 1).37 Metal complexes with the deprotonated ligand (OPO) have been reported such as Cu(OPO)2, Fe(OPO)3, Co(OPO)2, and Co(OPO)3. The Zn(OPO)2 complex (otherwise known as ZPH: di(2-hydroxypyridine-N-oxidato)zinc) with a Zn(O)4 coordination mode was found to have insulinomimetic activity.38,39 HOPO can be regarded as a cyclic hydroxamic acid and the deprotonated ligand OPO may coordinate with Zn through an oxyanion and an oxo group. A substantial amount of delocalization in bonding between OPO and Zn may account for stabilizing Zn(OPO)2.40 In this work, this low-toxic, stable Zn(OPO)2 was used as a high-efficiency catalyst with TBAI for the synthesis of cyclic carbonates with the maximum TOF of 22000 h−1 (Scheme 3). Cyclic carbonates were obtained under mild reaction conditions for example, low CO2 pressure (0.1–1 MPa). The developed binary catalyst system was easily recovered and can be reused at least five times without significant loss in activity. Furthermore, solvent-free conditions also render this catalytic system sustainable and economical.
 | ||
| Scheme 2 1-Hydroxy-2-pyridone (HOPO) as a ligand in this study. | ||
 | ||
| Scheme 3 Cycloaddition of epoxides with CO2 catalyzed by Zn(OPO)2/TBAI. | ||
Results and discussion
The synthesis and characterization of the Zn(OPO)2 complex are well described in the literature,37,38 using one-pot precipitate–metallation that involves 2-hydroxypyridine-N-oxide, ZnSO4 and Ba(OH)2 in water (ESI†). The results show that the activity of the binary system originates from a synergistic effect between Zn(OPO)2 and TBAI, as no or low yield was obtained when using only Zn(OPO)2 or TBAI (Table 1, entries 1–3). Notably, Zn(OPO)2 showed higher activity than ZnSO4, ZnI2 or salen-Zn, suggesting that the Zn site with the OPO ligand may possess a stronger Lewis acidity (entries 4–7). To make a comparison with the reported catalysts, we summarized the representative metal catalysts in Table S1.† As shown in Table S1,† the catalytic activity of Zn(OPO)2 was even comparable with the most active metal catalysts. Bifunctional triporphyrin-Zn and triporphyrin-Mg catalysts were reported to have decent catalytic activities in the cycloaddition reaction of CO2 with 1,2-epoxyhexane with remarkable TOFs (turnover frequencies) up to 40000 h−1 and 46000 h−1, respectively.41a The TOF using the Al-complex based on an amino triphenolate ligand and PPN-Br [PPN = bis-(triphenylphosphine)iminium] as a co-catalyst could reach 36000 h−1 with 1 MPa CO2 pressure.41b ZnBr2/hexabutylguanidinium bromide exhibited an activity [TOFs 8670 h−1] for the coupling reaction of CO2 (3 MPa) with PO at 120 °C.41c The TOF using Mg-porphyrin as a catalyst was 19000 h−1 for the reaction of 1,2-epoxyhexane with CO2.42 We found that the TOF of Zn(OPO)2 can reach 22000 h−1 at 3 MPa CO2 pressure (entry 8).
|
|
||||
|---|---|---|---|---|
| Entry | Catal (mol%) | Co-catal (mol%) | t (h) | Yieldb (%) |
| a Reaction conditions: 50 mL stainless-steel autoclave equipped with an inner glass tube, styrene oxide (5 mmol, 0.6015 g), CO2 1 MPa (initial), 80 °C. b Determined by 1H NMR using 1,1,2,2-tetrachloroethane as the internal standard. c N,N-Phenylene-1,2-bis-salicylideneimine (salen Zn). d Propylene oxide (150.0 mmol, 8.712 g), 120 °C, 3.0 MPa initial CO2 pressure. | ||||
| 1 | — | TBAI (0.5) | 12 | 34 |
| 2 | Zn(OPO)2 (0.15) | — | 12 | — |
| 3 | Zn(OPO)2 (0.15) | TBAI (0.5) | 12 | 97 |
| 4 | ZnSO4 (0.15) | TBAI (0.5) | 12 | 74 |
| 5 | ZnI2 (0.15) | TBAI (0.5) | 6 | 44 |
| 6 | Zn(OPO)2 (0.15) | TBAI (0.5) | 6 | 97 |
| 7c | Salen-Zn (0.15) | TBAI (0.5) | 6 | 89 |
| 8d | Zn(OPO)2 (0.0025) | TBAI (0.9) | 1 | 55 |

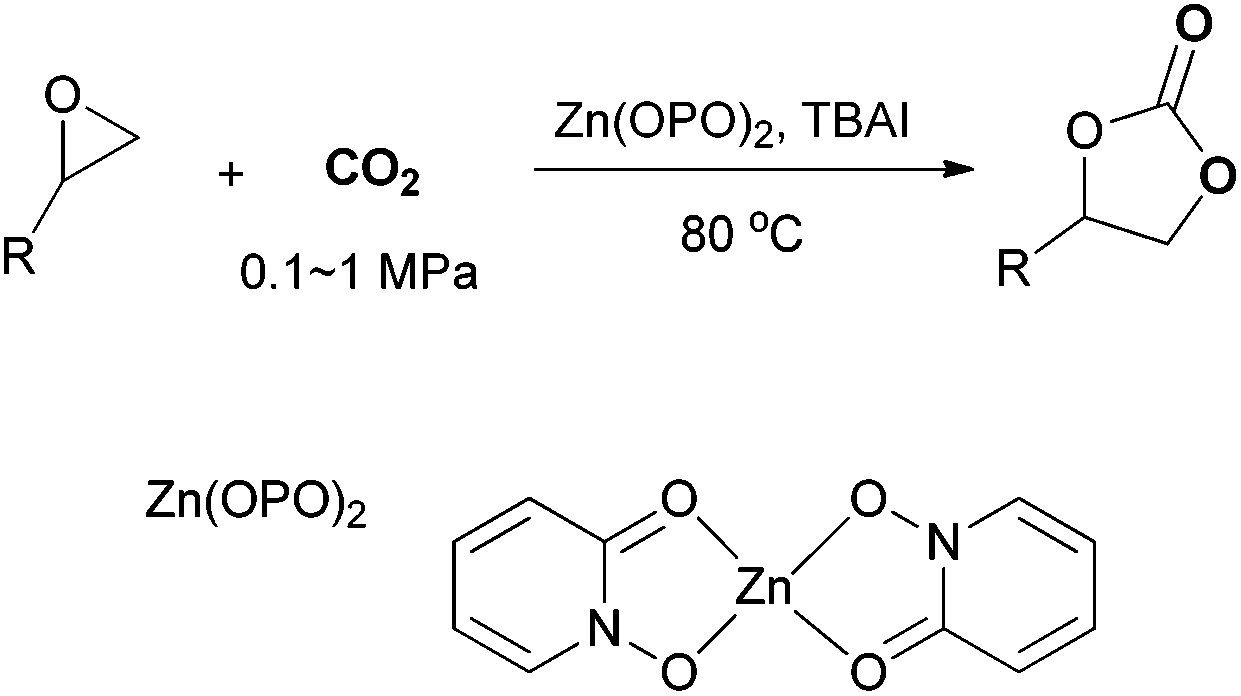
To further investigate the wider applicability of the newly developed binary-catalyst system Zn(OPO)2/TBAI, a series of functionalized terminal and internal epoxide substrates were examined. As listed in Table 2, a variety of terminal epoxides could be smoothly converted to the corresponding cyclic carbonates with high yield and excellent selectivity (entries 1–4). Interestingly, 1,2-epoxyhexane also performed at 1 bar CO2 with relatively high catalyst loading and took a long time to afford 4-butyl-1,3-dioxolan-2-one in quantitative yield (entry 5). Aliphatic ethers showed similar reactivity (entries 6 and 7). The internal epoxides, such as isobutylene oxide and cyclohexene oxide exhibited lower activity even under harsh conditions compared with their terminal counterparts (entries 8 and 9), this being ascribed to the steric effect.43 Furthermore, cyclic carbonate from cyclohexene oxide and CO2 is exclusively the cis isomer, as confirmed by 1H NMR and 13C NMR (ESI†).31
| Entry | Catalyst (mol%) | CO2 (MPa) | Product | Yieldb (%) | |
|---|---|---|---|---|---|
| Zn(OPO)2 | TBAI | ||||
| a Reaction conditions: epoxide (5 mmol), 80 °C, 6 h. b Determined by 1H NMR using 1,1,2,2-tetrachloroethane as the internal standard. c 20 mL Schlenk tube, 1 bar, CO2 (1.5 L balloon), 16 h. d 120 °C, 16 h. e 100 °C, 12 h. | |||||
| 1 | 0.15 | 0.5 | 1 |

|
99 |
| 2 | 0.15 | 0.5 | 1 |
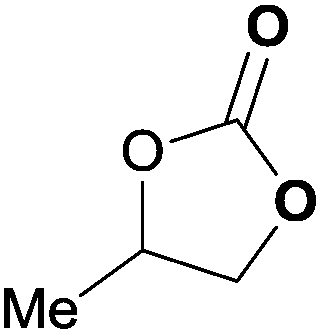
|
99 |
| 3 | 0.15 | 0.5 | 1 |

|
97 |
| 4 | 0.15 | 0.5 | 1 |
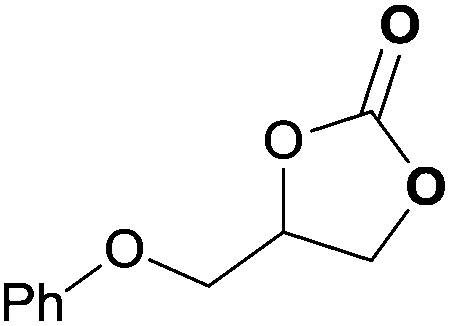
|
99 |
| 5c | 1.5 | 1.5 | 0.1 |

|
99 |
| 6c | 1.5 | 1.5 | 0.1 |
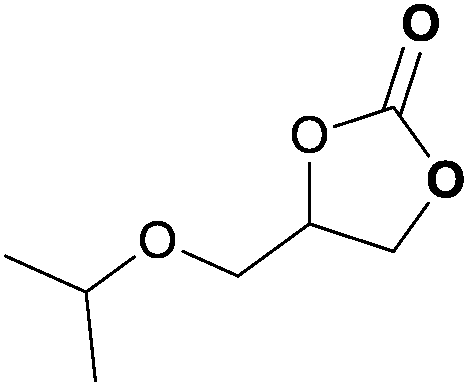
|
99 |
| 7c | 1.5 | 1.5 | 0.1 |
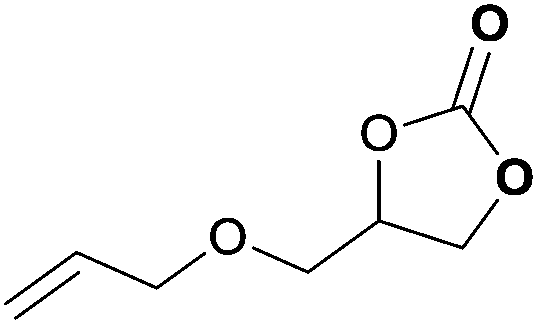
|
96 |
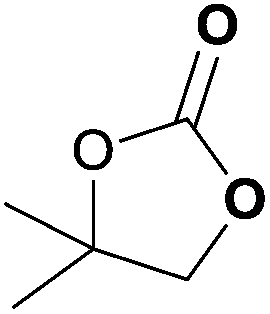
| 8d | 1 | 1 | 2 |
|
50 |
| 9e | 1 | 1 | 5 |

|
34 |
The reusability of the Zn(OPO)2/TBAI system was examined by using 1,2-epoxyhexane as the substrate under the same conditions. After precipitation, the catalysts were recovered by centrifugation and the product 4-butyl-1,3-dioxolan-2-one was extracted with diethyl ether, the catalysts were dried in vacuo (see the Experimental section for details) and directly used for the next run without replenishment of Zn(OPO)2/TBAI. As shown in Fig. 1, the developed binary catalyst system could be reused for five successive runs without any significant loss in its catalytic activity.
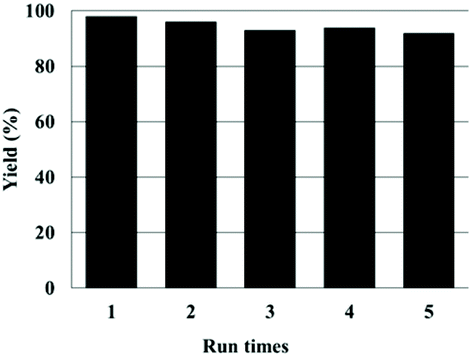 | ||
| Fig. 1 Reusability of Zn(OPO)2/TBAI. Reaction conditions: 1,2-epoxyhexane (5 mmol), Zn(OPO)2 1.5 mol%, TBAI 1.5 mol%, CO2 balloon, 80 °C, 16 h. | ||
The comparative FT-IR analysis of the fresh catalyst Zn(OPO)2 and the recovered Zn(OPO)2/TBAI after the 5th cycle was also performed as shown in Fig. 2. The v(NO) and v(CO) frequencies of the fresh Zn(OPO)2 and the recovered catalyst Zn(OPO)2/TBAI overlapped completely (v(NO) = 1185 cm−1, v(CO) = 1624 cm−1) (Fig. 2avs.2b), thus indicating the good stability of the Zn(OPO)2. Notably, an absorption peak at 1471 cm−1 was assigned as the infrared absorption of TBAI.
 | ||
| Fig. 2 FT-IR spectra of Zn(OPO)2 (a), the recovered Zn(OPO)2/TBAI after 5 cycles (b). | ||
Moreover, the reaction could also be monitored by the use of in situ FT-IR spectroscopy (Fig. 3). Notably, the absorption peak of the carbonyl group at 1782 cm−1 increased gradually as the reaction was progressing, thus providing a convenient way to gain insight into the reaction. The weakening of the signal of 1,2-epoxyhexane at 847 cm−1 is accompanied by the increase in new peaks at 1384, 1168, 1060 and 773 cm−1 of 4-butyl-1,3-dioxolan-2-one. No formation of polycarbonate at 1750 cm−1 suggests that the depolymerization mechanism could be ruled out.
 | ||
| Fig. 3 In situ FT-IR spectra of the synthesis of 4-butyl-1,3-dioxolan-2-one from 1,2-epoxyhexane under solvent-free conditions by using Zn(OPO)2 (2.5 mol%) and TBAI (2.5 mol%) as catalysts. The up arrows indicate peaks resulting from 4-butyl-1,3-dioxolan-2-one at 1782, 1384, 1169, 1060 and 773 cm−1, whereas the down arrow indicates the peak referring to 1,2-epoxyhexane at 847 cm−1. | ||
Accordingly, a possible mechanism for the Zn(OPO)2/TBAI-catalyzed coupling of epoxides with CO2 was proposed and is shown in Scheme 4. The catalytic cycle starts with the coordination of the epoxide to the Lewis acidic Zn center of Zn(OPO)2, followed by the ring-opening by nucleophile attack of the iodine anion to form the metal-coordinated iodo alkoxide. Next, the formed alkoxide species acts in turn as a nucleophile that attacks CO2 to form the metal carbonate species, which undergoes an intramolecular ring-closure reaction to form the cyclic carbonate and regenerate the catalyst.
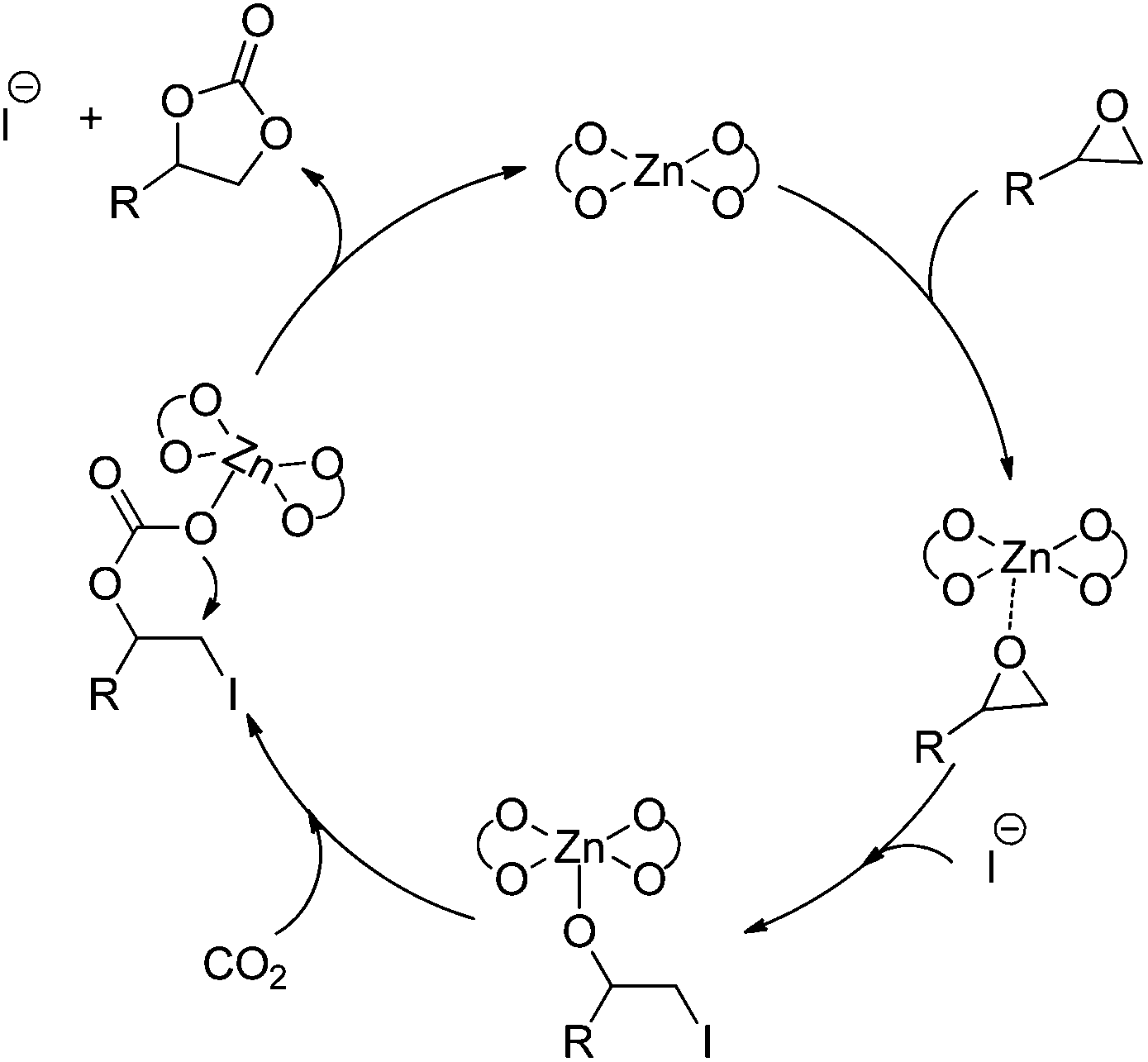 | ||
| Scheme 4 Tentative mechanism for the coupling of epoxides with CO2 catalyzed by Zn(OPO)2/TBAI. | ||
Conclusions
In summary, Zn(O)4-type complexes e.g. Zn(OPO)2 have been developed as exceptionally active catalysts for the synthesis of cyclic carbonates from epoxides and CO2 with the TOF of 22000 h−1 at 120 °C and 3.0 MPa CO2. Various cyclic carbonates were obtained under quite mild conditions (1 bar CO2) in the presence of TBAI as a co-catalyst. This low-toxic Zn(O)4 coordinated complex has the advantage of easy preparation, stability under general reaction conditions and can be easily recovered and reused at least five times without significant loss of its catalytic activity. The results achieved make this protocol an attractive one for potential future industrial applications and it represents a greener pathway for the synthesis of organic carbonates starting from CO2.
Experimental
General information
TBAI and propylene oxide were purchased from Alfa Aesar, other epoxides and 2-hydroxypyridine N-oxide were of analytical grade and obtained from Aladdin. ZnSO4 and Ba(OH)2 were purchased from Tianjin Guangfu Fine Chemical Research Institute. All reagents were used without further purification. Carbon dioxide with a purity of 99.99% was commercially available. The products were characterized by NMR. 1H NMR spectra were recorded on 400 MHz spectrometers using CDCl3 as the solvent referenced to CDCl3 (7.26 ppm). 13C NMR were recorded at 100.6 MHz in CDCl3 (77.16 ppm). Multiplets were assigned as singlet, doublet, triplet, doublet of doublet, multiplet and broad singlet. A gas chromatograph (Shimadzu 2014 chromatograph) is equipped with a RTX-50 capillary column (30 m × 0.25 mm) using a flame ionization detector (FID).General procedure for the cycloaddition reaction of epoxides with CO2 at atmospheric pressure
The cycloaddition reaction of epoxides and CO2 was conducted in a 20 mL Schlenk flask. In a typical reaction, the flask was charged with Zn(OPO)2, TBAI and epoxide successively at room temperature. The flask was capped with a stopper and sealed. Then, the reaction mixture was stirred at 80 °C for the desired time under a CO2 atmosphere (1.5 L balloon). After the reaction was completed, the mixture was cooled to room temperature and diluted with dichloromethane. The conversion of epoxide and yield of cyclic carbonate were determined by 1,1,2,2-tetrachloroethane as the internal standard in CDCl3 by 1H NMR. Then, the residue was obtained by removing the solvent under vacuum and further purified by column chromatography with ethyl acetate–petroleum ether as the eluent to afford the product. The products were further identified by NMR.General procedure for the cycloaddition reaction of epoxides with CO2 at high pressure
The cycloaddition reaction of epoxides and CO2 was conducted in a stainless steel autoclave (50 mL inner volumes). In a typical reaction, the reactor was charged with Zn(OPO)2, TBAI and epoxide successively at room temperature. Then, CO2 was introduced into the reactor and the pressure was adjusted to 1 MPa at 80 °C. The autoclave was heated at this temperature for 6 h. After the reaction was completed, the reactor was cooled to 0 °C in ice-water bath, and then excess of CO2 was carefully vented. The mixture was diluted with dichloromethane. The conversion of epoxide and yield of cyclic carbonate were determined by 1,1,2,2-tetrachloroethane as the internal standard in CDCl3 by 1H NMR. Then, the residue was obtained by removing the solvent under vacuum and further purified by column chromatography with ethyl acetate–petroleum ether as the eluent to afford the product. The products were further identified by NMR.Catalyst recycling studies
After the sample for the 1H NMR analysis was taken, diethyl ether was added into the reactor for precipitation of the catalysts and extraction of the unreacted substrate and carbonate product. The precipitated catalysts were washed with diethyl ether and dried in a vacuum for 1 h. The next catalytic cycle was started with the same amount of epoxide (5 mmol) as described above without replenishment of TBAI.Acknowledgements
We are grateful to the National Natural Sciences Foundation of China, the Specialized Research Fund for the Doctoral Program of Higher Education (20130031110013), the MOE Innovation Team (IRT13022) of China, and the “111” Project of the Ministry of Education of China (project No. B06005) for financial support.References
- T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- I. Omae, Coord. Chem. Rev., 2012, 256, 1384–1405 CrossRef CAS.
- M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514–1539 RSC.
- Y. N. Li, R. Ma, L. N. He and Z.-F. Diao, Catal. Sci. Technol., 2014, 4, 1498–1512 CAS.
- B. Yu and L. N. He, ChemSusChem, 2015, 8, 52–62 CrossRef CAS PubMed.
- T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312–1330 RSC.
- H. Yasuda, L. N. He, T. Sakakura and C. Hu, J. Catal., 2005, 233, 119–122 CrossRef CAS.
- T. Ema, Y. Miyazaki, J. Shimonishi, C. Maeda and J. y. Hasegawa, J. Am. Chem. Soc., 2014, 136, 15270–15279 CrossRef CAS PubMed.
- W. Clegg, R. W. Harrington, M. North and R. Pasquale, Chem. – Eur. J., 2010, 16, 6828–6843 CrossRef CAS PubMed.
- M. V. Escárcega-Bobadilla, M. Martínez Belmonte, E. Martin, E. C. Escudero-Adán and A. W. Kleij, Chem. – Eur. J., 2013, 19, 2641–2648 CrossRef PubMed.
- X. B. Lu, Y. J. Zhang, B. Liang, X. Li and H. Wang, J. Mol. Catal. A: Chem., 2004, 210, 31–34 CrossRef CAS.
- J. Qu, C. Y. Cao, Z. F. Dou, H. Liu, Y. Yu, P. Li and W. G. Song, ChemSusChem, 2012, 5, 652–655 CrossRef CAS PubMed.
- Z. Wu, H. Xie, X. Yu and E. Liu, ChemCatChem, 2013, 5, 1328–1333 CrossRef CAS.
- M. L. Man, K. C. Lam, W. N. Sit, S. M. Ng, Z. Zhou, Z. Lin and C. P. Lau, Chem. – Eur. J., 2006, 12, 1004–1015 CrossRef CAS PubMed.
- A. Monassier, V. D'Elia, M. Cokoja, H. Dong, J. D. A. Pelletier, J.-M. Basset and F. E. Kühn, ChemCatChem, 2013, 5, 1321–1324 CrossRef CAS.
- J. Langanke, L. Greiner and W. Leitner, Green Chem., 2013, 15, 1173–1182 Search PubMed.
- Y. Ren and J. J. Shim, ChemCatChem, 2013, 5, 1344–1349 CrossRef CAS.
- Q. W. Song, L.-N. He, J.-Q. Wang, H. Yasuda and T. Sakakura, Green Chem., 2013, 15, 110–115 RSC.
- Z. Z. Yang, Y. N. Zhao, L. N. He, J. Gao and Z. S. Yin, Green Chem., 2012, 14, 519–527 Search PubMed.
- C. J. Whiteoak, A. Nova, F. Maseras and A. W. Kleij, ChemSusChem, 2012, 5, 2032–2038 CrossRef CAS PubMed.
- M. E. Wilhelm, M. H. Anthofer, M. Cokoja, I. I. E. Markovits, W. A. Herrmann and F. E. Kuhn, ChemSusChem, 2014, 7, 1357–1360 CrossRef CAS PubMed.
- T. P. Yoon and E. N. Jacobsen, Science, 2003, 299, 1691–1693 CrossRef CAS PubMed.
- C. Baleizão and H. Garcia, Chem. Rev., 2006, 106, 3987–4043 CrossRef PubMed.
- N. E. Leadbeater and M. Marco, Chem. Rev., 2002, 102, 3217–3274 CrossRef CAS PubMed.
- E. M. McGarrigle and D. G. Gilheany, Chem. Rev., 2005, 105, 1563–1602 CrossRef CAS PubMed.
- R. M. Haak, S. J. Wezenberg and A. W. Kleij, Chem. Commun., 2010, 46, 2713–2723 RSC.
- X. B. Lu, Y. J. Zhang, K. Jin, L. M. Luo and H. Wang, J. Catal., 2004, 227, 537–541 CrossRef CAS.
- A. Decortes and A. W. Kleij, ChemCatChem, 2011, 3, 831–834 CrossRef CAS.
- D. J. Darensbourg and R. M. Mackiewicz, J. Am. Chem. Soc., 2005, 127, 14026–14038 CrossRef CAS PubMed.
- T. Chang, H. Jing, L. Jin and W. Qiu, J. Mol. Catal. A: Chem., 2007, 264, 241–247 CrossRef CAS.
- C. X. Miao, J. Q. Wang, Y. Wu, Y. Du and L. N. He, ChemSusChem, 2008, 1, 236–241 CrossRef CAS PubMed.
- G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618–6639 CrossRef CAS PubMed.
- Y. Yang, Y. Hayashi, Y. Fujii, T. Nagano, Y. Kita, T. Ohshima, J. Okuda and K. Mashima, Catal. Sci. Technol., 2012, 2, 509–513 CAS.
- N. M. Karayannis, L. L. Pytlewski and C. M. Mikulski, Coord. Chem. Rev., 1973, 11, 93–159 CrossRef CAS.
- A. E. Landers and D. J. Phillips, Inorg. Chim. Acta, 1977, 25, L39–L40 CrossRef CAS.
- A. E. Landers and D. J. Phillips, Inorg. Chim. Acta, 1981, 51, 109–115 CrossRef CAS.
- E. Shaw, J. Am. Chem. Soc., 1949, 71, 67–70 CrossRef CAS PubMed.
- S. Fujimoto, H. Yasui and Y. Yoshikawa, J. Inorg. Biochem., 2013, 121, 10–15 CrossRef CAS PubMed.
- Y. Yoshikawa, E. Ueda, K. Kawabe, H. Miyake, H. Sakurai and Y. Kojima, Chem. Lett., 2000, 29, 874–875 CrossRef.
- R. C. Scarrow, P. E. Riley, K. Abu-Dari, D. L. White and K. N. Raymond, Inorg. Chem., 1985, 24, 954–967 CrossRef CAS.
- (a) C. Maeda, T. Taniguchi, K. Ogawa and T. Ema, Angew. Chem., Int. Ed., 2015, 54, 134 CrossRef CAS PubMed; (b) C. J. Whiteoak, N. Kielland, V. Laserna, E. C. Escudero-Adan, E. Martin and A. W. Kleij, J. Am. Chem. Soc., 2013, 135, 1228 CrossRef CAS PubMed; (c) H. Xie, S. Li and S. Zhang, J. Mol. Catal. A: Chem., 2006, 250, 30–34 CrossRef CAS.
- T. Ema, Y. Miyazaki, J. Shimonishi, C. Maeda and J. y. Hasegawa, J. Am. Chem. Soc., 2014, 136, 15270–15279 CrossRef CAS PubMed.
- C. J. Whiteoak, E. Martin, M. M. Belmonte, J. Benet-Buchholz and A. W. Kleij, Adv. Synth. Catal., 2012, 354, 469–476 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5gc01826a |
| This journal is © The Royal Society of Chemistry 2016 |