
Dual-porous metal organic framework for room temperature CO2 fixation via cyclic carbonate synthesis†
Robin
Babu
a,
Amal Cherian
Kathalikkattil
a,
Roshith
Roshan
a,
Jose
Tharun
a,
Dong-Woo
Kim
b and
Dae-Won
Park
*a
aDivision of Chemical and Biomolecular Engineering, Pusan National University, Busan, 609-735, Korea. E-mail: dwpark@pusan.ac.kr
bDivision of Ulsan Research Center for Green Fine Chemicals, Korea Research Institute of Chemical Technology, Ulsan 681-802, Korea
First published on 15th September 2015
Abstract
A novel approach of employing a dual-porous metal organic framework (MOF) in CO2 fixation at room temperature was demonstrated using a micro–mesoporous MOF, UMCM-1-NH2, in the synthesis of various five-membered cyclic carbonates under solventless conditions. Mesopores allow easy guest diffusion and molecular accessibility, while micropores are predominantly helpful in regulating the catalytic interactions in active centres; thus outperforming the properties of pure microporous or pure mesoporous MOFs in cycloaddition. Structural features, acid–base characteristics and physical properties were studied in detail for carrying out a systematic investigation on the cooperative influences of porosity, functionalization and synergism with quaternary ammonium salts in the cycloaddition reaction of CO2 with propylene oxide, so as to arrive at the underlying mechanism. The catalyst was totally recyclable up to five times without compromising the activity and the extent of heterogeneity was also studied. The effects of various reaction parameters like catalyst–cocatalyst ratio, reaction time and reaction temperature have been investigated.
Introduction
Comprehensive scientific assessments of current climatic behavior and its future have made it clear that the atmospheric CO2 concentration will rise beyond 450 ppm before the mid-21st century as a result of anthropogenic activities. Several conceptual schemes such as integrated carbon capture and storage (CCS) techniques are still under development for mitigating the excessive emission of CO2 from industrial exhausts.1a–e Meanwhile, the abundance of CO2, and its physicochemical properties viz.; non-toxicity and non-flammability, have inspired the scientific community to invent technologies that use CO2 as C1 feed stock. Considering the toxic nature of the currently used C1 sources such as phosgene and CO, technologies that use CO2 could be beneficial due to its advantages like safety in handling, economic viability and environmental friendliness. The cycloaddition of CO2 with epoxides to produce cyclic carbonates is highly desirable because of the efficient resource utilization and 100% atom economicity of the process. Cyclic carbonates, non-toxic liquids with low odor, are excellent in applications such as green and aprotic solvents, electrolytes in lithium-ion batteries, precursors for polymer synthesis including polycarbonates, in pharmaceuticals and agriculture etc., and are expected to expand their applicability with the increasing importance of green and sustainable chemistry in coming decades.2a–gHowever, high operating pressures and temperatures are required for cyclic carbonate production, owing to the thermodynamic stability of CO2, which in turn, limits the exploitation of the process both in terms of energy and economy.2h–j Therefore, highly efficient catalysts that are feasible in terms of energy and reusability are essential for making it a green, sustainable and economic pathway that also offers easy product separation and operation simplification. Numerous homogeneous catalysts like ionic liquids, organic and metal complexes have been reported for cyclic carbonate synthesis.3a–d The recognition of some inherent limitations i.e., non-viability for industrial scale up due to the challenges in separation, purification and recycling, led to flourishing activity that attempts the development of heterogeneous catalysts like metal oxides, zeolites, titanosilicates and ion-exchanged resins.4a,b By docking highly active homogeneous species such as ionic liquids on support materials, even higher activities were obtained, and the heterogeneity and reusability issues were addressed. However, several reports including ours in which ionic liquids were immobilized on supports such as silica, polystyrene and biopolymers were still fraught with inefficiencies in catalytic activities under ambient conditions or suffered from leaching of the active phase into the reaction mixture.5 Fixation under the lowest energy supply is crucial in maintaining a positive balance between CO2 release from power plants and energy expenditure for its mitigation.
Metal organic frameworks (MOFs) are providing new fields of opportunities as active and selective catalysts for environment relevant applications. The assembly of frameworks through a judicious choice of constituent ligands and transition metals provide MOFs’ high internal surface area, ordered pore structure, tuneable porosities and functionalities, putting them at the frontier between zeolites, metal–organic catalysts, and enzymes apparently extending their scope over the heterogeneous catalysis regime.6a–c In 2009, Han et al.7a reported the first MOF-5/n-Bu4Br binary system as a catalyst for the synthesis of cyclic carbonates from CO2 and epoxides. In a short span of time, a series of carboxylate MOFs and their diverse structural and functional counterparts viz., MIXMOFs, IRMOF-3, Cu-BTC, MIL-68, 101 and 125, UIO-66 etc. were reported.7b–h Apart from this, various zeolitic imidazolate frameworks (ZIF-8, ZIF-68 and ZIF-90),8a–c mixed carboxylate-N-donor MOFs (ZnHipBpy, CHB, and CCB), and metal complex-containing MOFs (Ni–salphen and Ni–saldpen complexes) were also reported as efficient in cycloaddition by various groups including ours.9a–e Among these reports, most were microporous or non-porous in nature. High surface area and large pore volumes are desirable for many catalytic applications, since narrow pores or the anchoring of molecular functions may hinder the mass diffusion and transfer of substrates, intermediates or product molecules.
Mesoporous MOFs are regarded as hosts to accommodate bulky molecules, allowing their free reaction or transformation without any diffusional limitations.10a–c With metal organic frameworks, a class of materials with versatility in pore sizes, catalysis is expected to be more efficiently carried out if pores are mesopores. Only a few MOFs of a mesoporous nature have been reported in CO2 cycloaddition to date.11,7f A combination of a meso- and micro-porous nature is expected to provide attractive features which overcome the limitations of purely microporous or purely mesoporous structures and to combine the advantages of each pore size regime (Fig. 1).12a–c Taking into account the lessons learned from the previous reports, it is clear that the co-existence of metal nodes (Lewis/Bronsted acid sites) and basic functional groups such as amine groups into the organic linkers are effective in cycloaddition, where the metal nodes are used to activate the epoxides while the CO2 is mostly activated by basic sites.12d Herein, we report an amine functionalized micro–mesoporous MOF UMCM-1-NH2 employing two different linkers with Zn4O secondary building units (SBUs), and its synergistic enhancement in catalytic activity with a cocatalyst containing strong nucleophilic anions for the room temperature synthesis of cyclic carbonates from epoxide and CO2 (Scheme 1).13 While the first type of linker (1,3,5-tris(4-carboxyphenyl)benzene, BTB) engages in constructing mesoporous architecture, the second linker (2-aminoterephthalic acid, NH2-BDC) affords amine functional groups that are beneficial in enhancing the CO2 binding and subsequent transformation during catalysis.
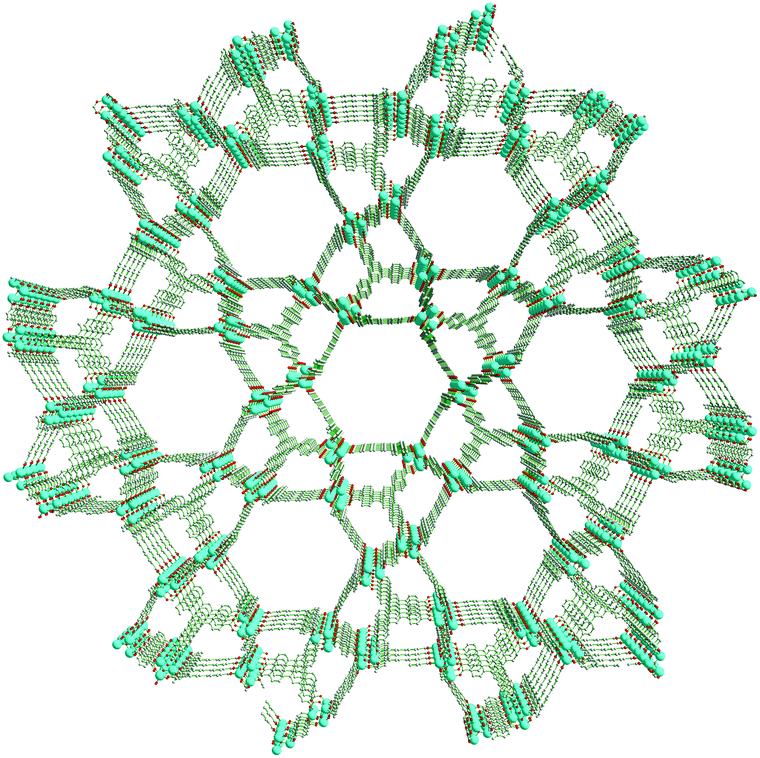 | ||
| Fig. 1 Perspective view of UMCM-1-NH2 along the c-axis depicting the arrangement of 1D hexagonal mesopores and microporous clusters. | ||
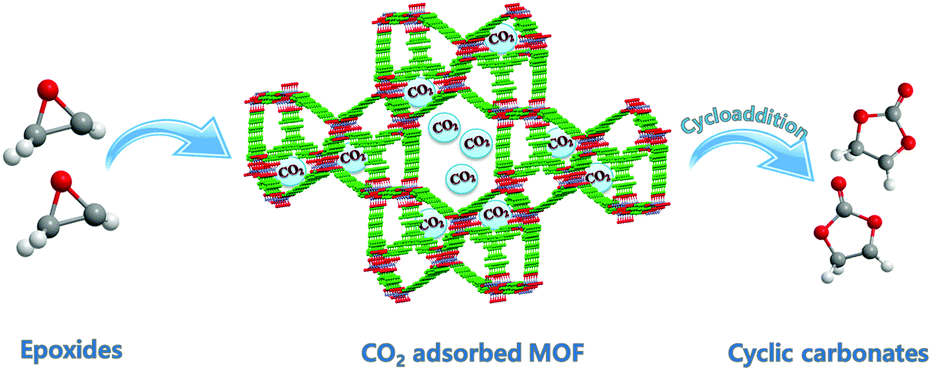 | ||
| Scheme 1 Schematic representation of the synthesis of cyclic carbonate from epoxides and CO2. | ||
Results and discussion
Generally, MOF structures are assembled by the bridging of metal or metal oxide clusters by exo-multidentate ligands that grow regularly to 1D, 2D or 3D structures. Though the factors such as the nature of counter anions, solvent, and synthesis conditions are influential in the self-assembly of MOFs, the final topology and pore dimensions are the reflection of the length and orientation of the donor atoms of the ligands towards metal/metal clusters that adopt a suitable coordination mode. In UMCM-1-NH2 also, Zn4O clusters are present as nodes, and the BTB and NH2-BDC as the bridging ligands are existent; but it is not limited to the concept of SBUs in MOF chemistry. UMCM-1-NH2 has its broad arrangement proceeding in a multi-level, hierarchical assembly as follows (Scheme 2). Nine Zn4O clusters, six NH2-BDC units and five BTB units form three dimensional microporous cages with the pore dimensions 14 × 17 Å. Such microporous cages are arranged further in an edge-sharing fashion such that six cages give rise to a mesoporous space (dimensions 27 × 32 Å).13,14a This mesoporous structure extends as a 1D channel through the whole framework, as we move along the crystallographic c-axis. Thus, the whole structure may be seen as a hexagonally channeled mesoporous material developed from microporous building units (Fig. S1, ESI†). A comparison with the mesoporous silica material MCM-41 (hexagonal mesoporous silica) revealed that MCM-41 is built from edge-sharing [SiO4] units (non-porous units), while UMCM-1-NH2 is built from edge-sharing, microporous, [(Zn4O)9(NH2-BDC)6(BTB)5] units. As per the crystallographic data, in UMCM-1-NH2, 43% of the volume belongs to mesopores, while the remaining 57% are in the microporous range.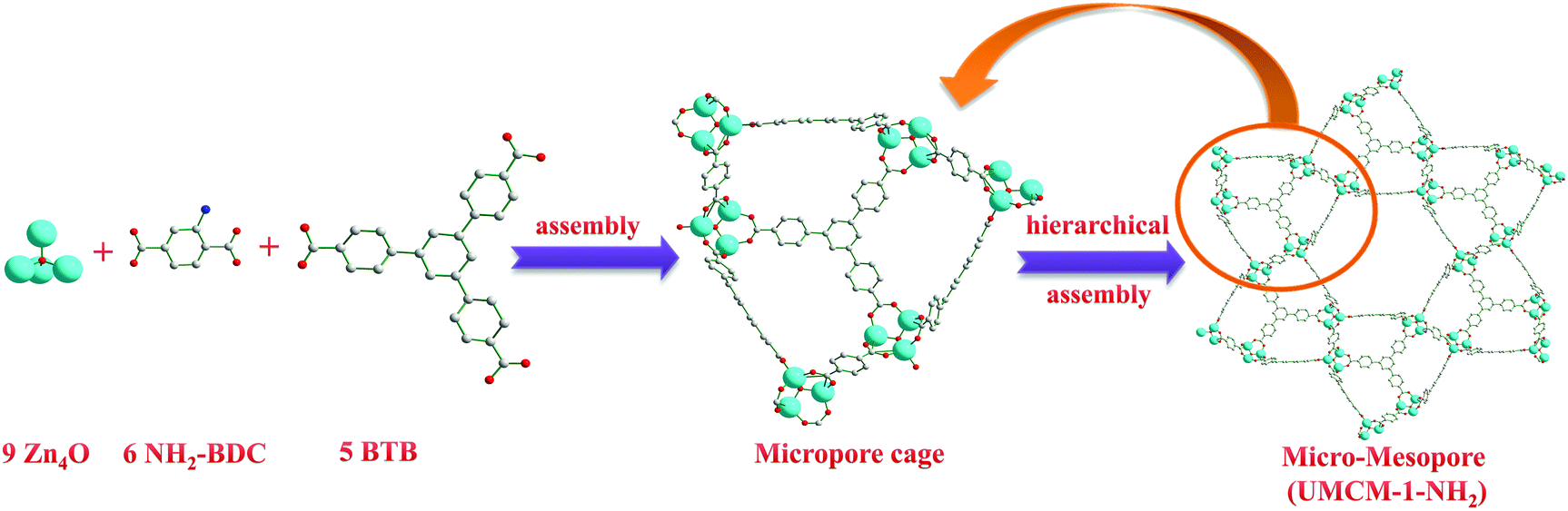 | ||
| Scheme 2 Representation of the hierarchical assembly in UMCM-1-NH2; where the micropores are built from Zn4O clusters and linkers, and subsequent formation of the 1D mesopores from the hexagonal arrangements of the microporous cages. | ||
Thus UMCM-1-NH2 is not purely microporous or mesoporous, but a hybrid micro–mesoporous material with its catalytic active centres most probably being the metal oxide clusters and amino groups. It is noteworthy that the amino group does not participate directly in cementing the framework, and rather it possesses a high order of mobility (along the rotational axis), due to which the single crystal structure of UMCM-1-NH2 has failed to fix the amino groups at a particular orientation. Interestingly, these sorts of hybrid micro–mesoporous MOFs have seldom been explored for their potential in the cycloaddition of epoxides and carbon dioxide, where both types of pores have their own significance. Mesopores provide improved accessibility for the reactants with minimal diffusional limitations, so that in a limited time they can diffuse much more and interact with more catalytic sites. Meanwhile, the micropores provide a favorable platform for the catalysis to proceed in a particular pathway leading to the desired products by virtue of its reduced retention times. Further, it is interesting that the NH2 groups are dynamic, such that they are accessible for substrates that reside in the micropores as well as mesopores.
UMCM-1-NH2 was conceived by the solvothermal treatment of the metal salt (zinc nitrate tertrahydrate), tripodal ligand (1,3,5-tris(4-carboxyphenyl)benzene, BTB) and the amine functional exobidentate ligand (2-aminoterephthalic acid, NH2-BDC) at 85 °C for 3 days. Needle shaped single crystals were obtained upon solvothermal synthesis.14b
In order to verify the phase homogeneity of the bulk solvothermal sample, and its structural integrity, the powder X-ray diffraction (PXRD) pattern of the UMCM-1-NH2 catalyst was compared to the single crystal simulated pattern generated from the CIF file of the original report for UMCM-1-NH2. The XRD reflections in Fig. 2(b) match perfectly with the simulated pattern in Fig. 2(a), confirming that the catalysts correspond to the hexagonal crystal system as reported.13 Solvothermal synthesis allows precise control over the size, morphology (sphere (3D), rod (2D), or wire (1D)), shape distribution, and crystallinity by employing solvent supersaturation and kinetic control.6b
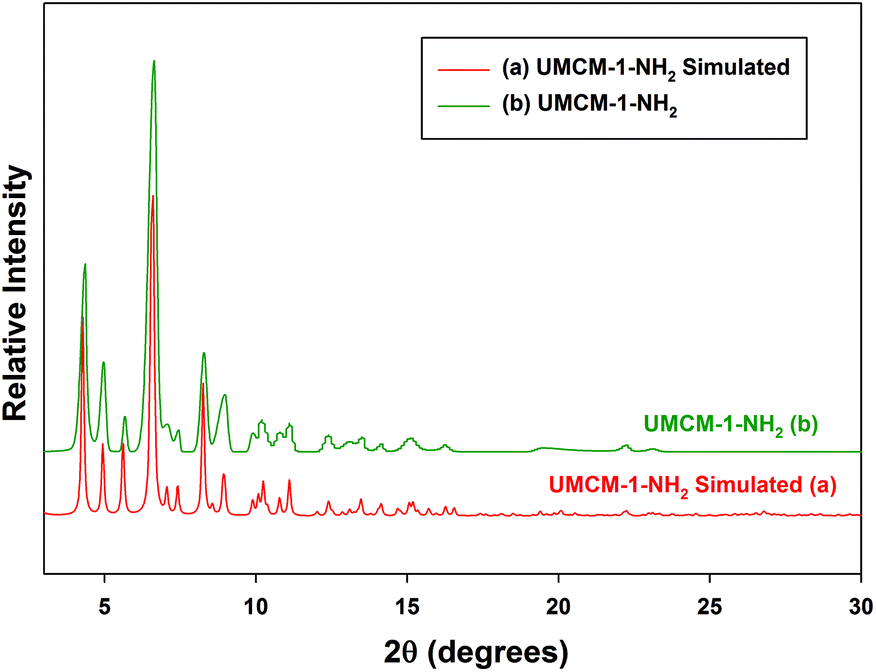 | ||
| Fig. 2 XRD pattern of UMCM-1-NH2 compared to the simulated single crystal pattern. | ||
Further verification of UMCM-1-NH2 for its chemical integrity was analyzed using FT-IR (Fig. 3). The broad peak observed at 3426 cm−1 corresponds to the N–H stretching vibration. The peak at 1600 cm−1 corresponds to the N–H bending vibration of the –NH2 group of UMCM-1-NH2. The absence of a peak at 1650 cm−1 confirms the removal of the DEF molecules from the framework. The peak at 500 cm−1 is related to the stretching vibrations of the Zn–O bonds in the framework. The peak around 2853 to 3000 cm−1 corresponds to C–H stretching vibrations. Similarities in the FTIR peaks with the previous reports also validate the originality of the chemical environments of this MOF.14c,d The presence of the active NH2 group in the surface of the UMCM-1-NH2 group was analyzed using N 1s spectra from the XPS data (Fig. S2, ESI†). The N 1s peak for the amine group appeared at 400 eV.
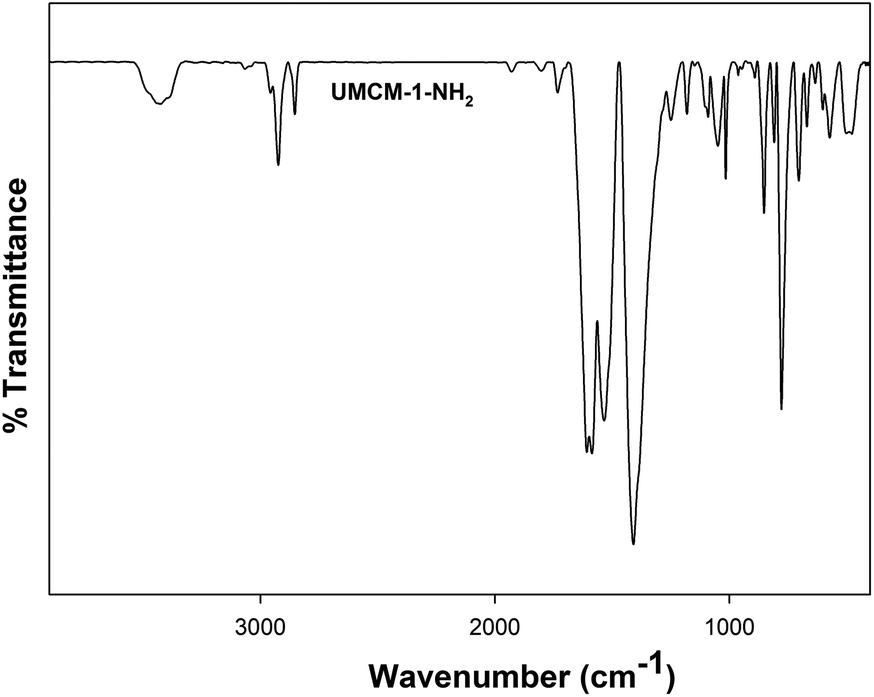 | ||
| Fig. 3 FT-1R spectra of UMCM-1-NH2. | ||
Further, the thermal stability and the nature of the framework construction was evaluated by means of thermogravimetric analysis (TGA) (Fig. 4). TGA was conducted after the complete drying of the sample under vacuum, so that the degradation curve does not reflect the loss of solvent molecules. The first major loss was observed within 400–420 °C for UMCM-1-NH2 indicating that the catalyst is thermally stable up to approx. 400 °C.13 N2 adsorption experiments have been carried out at 77 K on the thermally activated UMCM-1-NH2 (Fig. S3, ESI†). The isotherm, which has a typical Type IV behavior, has been analyzed by the BET method yielding a very high surface area of 4200 m2 g−1. The micro- and meso-porous nature of UMCM-1-NH2 that was observed in the crystallographic structure was physically confirmed from the pore size distribution curve (Fig. S4, ESI†). The total pore volume of the UMCM-1-NH2 was approximately estimated as 2.4 cm3 g−1.
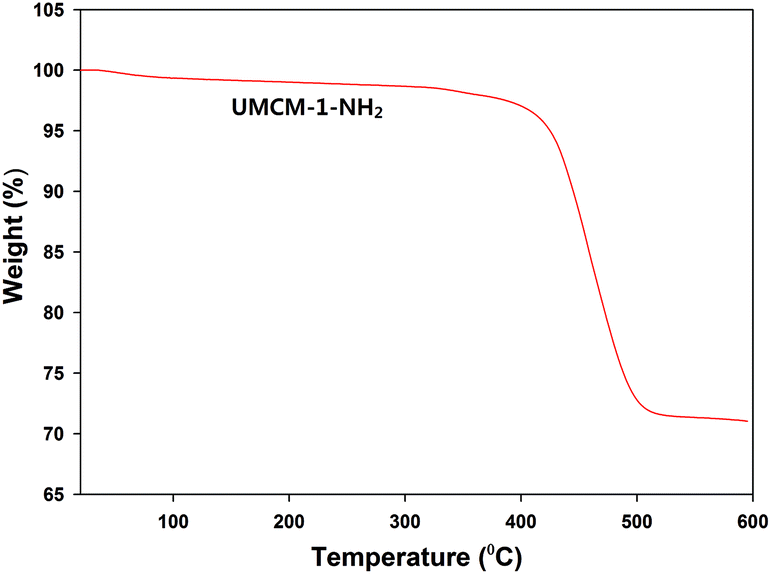 | ||
| Fig. 4 Thermogravimetric analysis (TGA) of UMCM-1-NH2. | ||
From the cumulative insights obtained from various recent reports,7–9 we intend to investigate the catalytic ability of UMCM-1-NH2 at ambient conditions (room temperature) in the presence of the TBAB co-catalyst.15a–c,7a Neither UMCM-1-NH2 nor TBAB separately produced any noticeable PO conversion under an employed reaction condition of RT, 1.2 MPa with 0.64 mol% catalyst in 24 h (Table 1, entries 2 and 3). However, a tremendous rise in the PC yield was observed with the combination of UMCM-1-NH2 and TBAB which served as a binary catalyst system, that manifested a promising result of 90% PO conversion with nearly 100% selectivity (propylene carbonate) under the same reaction conditions (Table 1, entry 7). Since the individual catalyst precursors did not display any catalytic activity with TBAB (Table 1, entries 4 and 5), the above mentioned catalytic activity of the UMCM-1-NH2/TBAB system shall be positively surmised to the crucial role played by the metal organic framework environment, such as its porosity, in modifying the catalytic route. It has been reported that iodides are a better nucleophile for eventuating the synergism with Lewis/Bronsted acid catalysts in CO2-epoxide cycloaddition reaction.16a,b However, it was observed that the UMCM-1-NH2 rendered lower PO conversion with iodide containing co-catalysts (TBAI) than its bromide analogue (TBAB), in contrast to the literature reports. This shall be attributed most probably to the easier access offered to the bromide ions (which are of smaller size in comparison to the iodide ion) inside the meso- and micropores of UMCM-1-NH2 which again is direct evidence that the catalysis occurs in the major part inside the micro/mesopores of UMCM-1-NH2 and not on its external surface. Should the catalysis have happened over the surface of the MOF, the UMCM-1-NH2/TBAB must have had lesser catalytic activities than the UMCM-1-NH2/TBAI system, but the reverse was observed. This supplements the rationale that the porous nature of the MOF plays a critical role in devising the catalytic route (Table 1, entry 9). Thus altogether, the reduced activity of iodides compared to bromide indicates that the diffusion of bulky iodide ions into the pore, where the reaction predominantly occurs, is hampered. A similar result was reported by Baiker et al.7b in the case of MIXMOF. We also noticed that UMCM-1-NH2 can act as a standalone catalyst at high temperatures. A 95% conversion of PO occurred using UMCM-1-NH2 at 120 °C and 1.2 MPa pressure over 24 h without the use of any cocatalyst or solvent (Table 1, entry 10). However, the PC selectivity was slightly affected by small amounts of dimers as side products. This could be ascribed to the absence of strong nucleophilic anions for epoxide ring opening, which are crucial in the addition of the ring opened epoxide to CO2 molecules. In the absence of such nucleophilic species in UMCM-1-NH2, the reverse binding is reported to occur, i.e., the catalyst polarized CO2 attacks the epoxide to cause an addition step. Therefore, in the absence of enough polarized CO2 molecules, epoxide may prefer to dimerize with another epoxide rather than undergoing addition with the inert molecule.
| No | Catalysta | Temperature (°C) | Conversionb (%) | Selectivityb (%) | Yieldb (%) |
|---|---|---|---|---|---|
| a Reaction conditions: propylene oxide (PO) = 42.8 mmol (3 ml), 1.2 MPa PCO2, 24 h, 600 rpm, catalyst mol% = 0.64, semi-batch. b Determined by GC. c Catalyst mol% = 0.44. d Catalyst mol% = 0.88. | |||||
| 1 | None | RT | 0 | 0 | 0 |
| 2 | UMCM-1-NH2 | RT | <3 | — | <3 |
| 3 | TBAB | RT | <3 | — | <3 |
| 4 | Zn(NO3)2/TBAB | RT | <5 | — | <5 |
| 5 | Zn(NO3)2 + BTB + NH2-BDC/TBAB | RT | <5 | — | <5 |
| 6c | UMCM-1-NH2/TBAB | RT | 52 | >99 | 52 |
| 7 | UMCM-1-NH2/TBAB | RT | 90 | >99 | 90 |
| 8d | UMCM-1-NH2/TBAB | RT | 78 | >99 | 78 |
| 9 | UMCM-1-NH2/TBAI | RT | 34 | >99 | 34 |
| 10 | UMCM-1-NH2 | 120 | 95 | 95 | 91 |
Having identified the potential of UMCM-1-NH2 to materialize the cyclic carbonate synthesis with the assistance of co-catalysts even at room temperature, we deemed it worthwhile to compare the efficiency of UMCM-1-NH2 to the earlier MOFs reported in CO2-PO cycloaddition. For this purpose, the catalytic abilities of a series of carboxylate spacer MOFs were compared with that of the UMCM-1-NH2 in Table 2. Although different sets of reaction conditions were employed in the literature, we attempted a comparison of the MOF catalysts at the closest reaction conditions reported. While the microporous zinc dicarboxylate framework, widely known as MOF-5, afforded a 56% yield of PC (Table 2, entry 1), the meso–micropore hybrid MOF UMCM-1-NH2 manifested 78% PC yield (Table 2, entry 2) at room temperature, 0.4 MPa CO2 pressure, with 2.5 mol% each of catalyst and TBAB co-catalyst in 4 h. IRMOF-3, an amine functionalized microporous carboxylate spacer MOF, produced a moderate yield of 57% (Table 2, entry 3), while at the same time, UMCM-1-NH2 gave a 90% PC yield under the same reaction conditions (RT, 1.2 MPa, 24 h, 0.64 mol% each of catalyst and co-catalyst) (Table 2, entry 10). From entries 4–8 (Table 2, Hf-Nu-1000, Cr-MIL-100, Ni–saldpen MOF),12a,7f,9e,18a,b it is evident that the UMCM-1-NH2 performed the CO2-PO cycloaddition in the most effective manner. An interesting thing observed from this comparison was that while the activity (conversion) with the micropore containing MOF/TBAB system (MOF-5, IRMOF-3 etc.) was inferior to that of the mesopore possessing MOF/TBAB binary system (Cr-MIL-101, UMCM-1-NH2), better selectivity to the desired product (cyclic carbonates) was manifested by the microporous MOF than the latter. This shall be considered in connection with the theory that the shortened diffusion pass length may lead to the reduced retention times of products in the micropore, which in turn leads to a lower probability of secondary reactions and hence affording enhanced selectivity to the primary products which in this case is the thermodynamic product, cyclic carbonates.17 Thus, the higher activity of UMCM-1-NH2 in comparison to the other MOFs on aspects of both epoxide conversion and selectivity shall be attributed to the presence of both the micro- and meso-pores as depicted in Scheme 2. While the mesopore channels of UMCM-1-NH2 facilitate easier diffusion of reactant molecules, the catalytic interactions might have occurred predominantly in the micropores of UMCM-1-NH2, as indicated by its high selectivity. Thus, in summary, a hybrid micro–mesoporous MOF serves the synergism with halide nucleophiles better than either a purely micro- or meso-porous MOF, which with a broader view indicates that, even with the same Lewis acidic centers, the porosity of the MOF plays a critical role in exploiting the synergistic catalysis effects of co-catalysts.
| No | Catalyst | Catalyst (mol%) | Co-catalyst (mol%) | Reaction conditions | Conversion (%). | Yield (%) | Ref. | |
|---|---|---|---|---|---|---|---|---|
| Pres. (MPa) | Time (h) | |||||||
| Epoxide: Propylene oxide, temperature: RT, cocatalyst: tetrabutylammonium bromide (TBAB), +: this work. | ||||||||
| 1 | MOF-5 | 2.5 | 2.5 | 0.4 | 4 | — | 57 | 7a |
| 2 | UMCM-1-NH2 | 2.5 | 2.5 | 0.4 | 4 | 78 | 78 | + |
| 3 | IRMOF-3 | 0.64 | 0.64 | 1.2 | 24 | 56 | 56 | — |
| 4 | Hf-Nu-1000 | 4 | 10 | 0.1 | 26 | 100 | 100 | 15c |
| 5 | Cr-MIL-101 | 1.2 | 0.62 | 0.8 | 24 | 91 | 82 | 7f |
| 6 | Ni–saldpen-MOF | 0.7 | 2 | 2 | 4 | 28 | — | 9e |
| 7 | MMCF-2 | 0.13 | 7.2 | 0.1 | 48 | — | 95.4 | 18a |
| 8 | MMPF-9 | 0.13 | 7.2 | 0.1 | 48 | — | 87.4 | 18b |
| 9 | UMCM-1 | 0.64 | 0.64 | 1.2 | 24 | 85 | 85 | + |
| 10 | UMCM-1-NH2 | 0.64 | 0.64 | 1.2 | 24 | 90 | 90 | + |
In order to understand whether the amine groups on UMCM-1-NH2 play any significant role in this room temperature synthesis of cyclic carbonates, a UMCM-1/TBAB system was also investigated. As shown in Table 2, entries 9 and 10, the catalytic activity of both systems was not so different, with the UMCM-1-NH2 excelling over the amine devoid UMCM-1 only slightly. This comparable yield of UMCM-1 with UMCM-1-NH2 leads to the conclusion that when it comes to a broader aspect, the pore size of the catalyst becomes as important as the effect of the amine or similar functional group. This suggests that the combined effects of suitable acidity/basicity and thereby micro–mesoporosity play roles in the promotion of the diffusion and the accessibility to the catalytic active sites. It should also be concluded that the Lewis base sites in the metal oxide cluster actively participate when it comes to the reaction occurring in both micropores and mesopores.
Since the catalysis for the cycloaddition of carbon dioxide and epoxide is expected to occur in a concerted pathway involving both acid and base sites, a physical evaluation of the potential acid and base sites in the UMCM-1-NH2 material was conducted using temperature programmed desorption (TPD) viz., NH3- and CO2-TPD respectively (Table 3). The total adsorbed CO2 during the course of analysis was estimated as 53 mmol g−1, which is also equivalent to the number of base sites in the catalyst. It is assumed that the NH2– functional groups of the ligand and the carboxylate O− atoms attached to the metal centers in its Zn4O metal clusters are responsible for CO2 adsorption. Similarly, in the case of NH3-TPD, the acid sites are equivalent to 7 mmol g−1, which corresponds to the metal centers (Lewis acid sites).
| Catalyst | CO2-TPD (total mmol g−1) | NH3-TPD (total mmol g−1) |
|---|---|---|
| UMCM-1-NH2 | 53 | 7 |
Previous reports for the MOF catalyzed cycloaddition of CO2 and epoxides suggest that in the presence of a strong nucleophilic anion, Lewis/Bronsted acid sites can activate the epoxides for ring opening, and certain groups which can polarize the thermodynamically stable CO2 molecule to facilitate CO2 insertion can effectively catalyze the epoxide-CO2 cycloaddition.12d A plausible mechanism based on these observations for the cycloaddition of PO in the presence of UMCM-1-NH2 co-catalyzed by TBAB is illustrated in Scheme 3. In UMCM-1-NH2, the O− atom of PO interacts with Zn Lewis acid sites. Subsequently, the Br− of TBAB attacks the least hindered carbon atom, resulting in the epoxide ring opening. Concomitantly, CO2 is polarized by amine groups of UMCM-1-NH2. This interaction is facilitated further by the possibilities of rotating the –NH2 groups present in the micropores. Experimental evidence of this disorder is available from the single crystal X-ray diffraction of UMCM-1-NH2, reported by Cohen et al.13 Smaller substrates that diffuse into micropores help enhanced interaction with reactant molecules. In the succeeding step, the O− atom of the polarized CO2 molecule attacks the β-carbon of the ring opened epoxide, forming an intermediate (b) with the elimination of Br−. Finally, the ring closure takes place to generate PC and the regenerated UMCM-1-NH2 moves to the next cycle of cycloaddition by coordinating the next epoxide molecule. In the absence of TBAB, the mechanism proceeds via a UMCM-1-NH2-PO interaction that undergoes a ring-opening by the O− atom of the polarized CO2 molecule (polarized by the presence of NH2 groups) at the β-carbon of the epoxide. Finally the ring closure takes place similar to that of the UMCM-1-NH2/TBAB catalyzed reaction as mentioned above (Scheme 4). The high temperature required for this pathway is justified by the higher energy associated with the polarizing molecule.
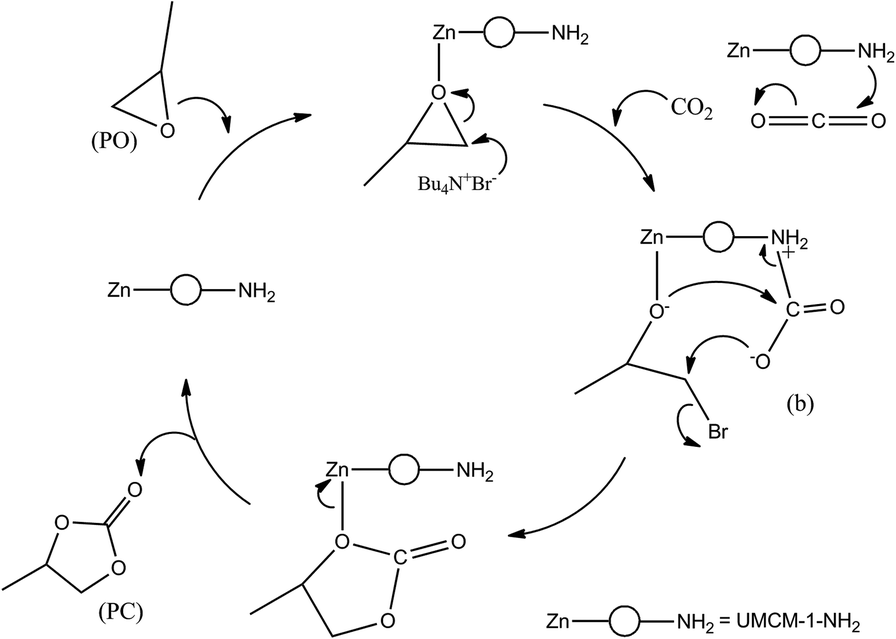 | ||
| Scheme 3 Proposed mechanism for the cycloaddition of PO and CO2 catalyzed by UMCM-1-NH2/TBAB. | ||
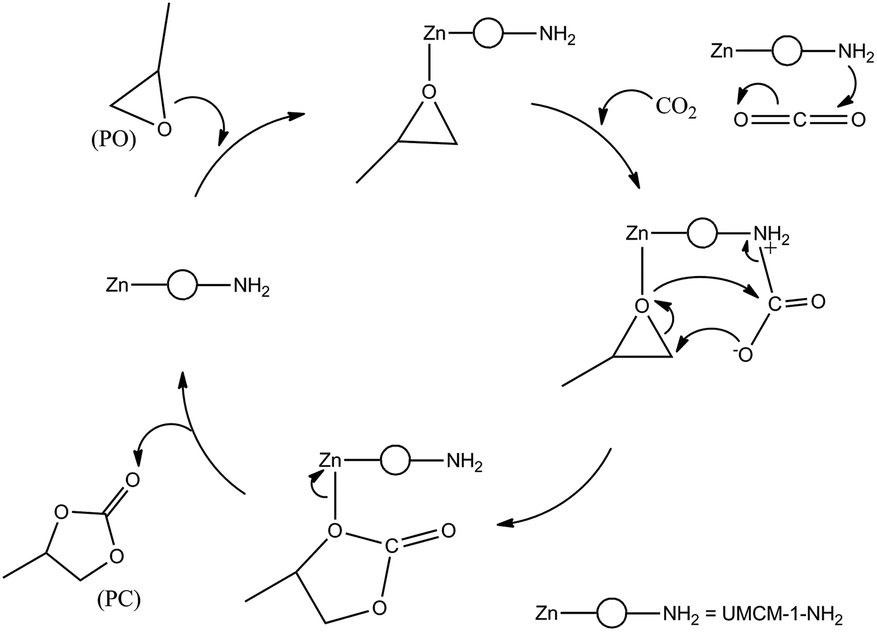 | ||
| Scheme 4 Cycloaddition of PO and CO2 catalyzed by UMCM-1-NH2. | ||
Since a synergism is involved between the catalyst/cocatalyst/substrate in this system, the effects of varying the catalyst concentrations were studied for different ratios of UMCM-1-NH2 in the range of 0.24–0.64 mol% (Table 4). A gradual increment in the PC yield was observed from 0.24–0.64 mol% for UMCM-1-NH2 with a constant cocatalyst concentration of 0.64 mol% (Table 4, entries 1–3). The same trend was observed for further studies that use 0.24–0.64 mol% TBAB with 0.64 mol% UMCM-1-NH2 (Table 4, entries 4 and 5). Thus, 0.64 mol% of UMCM-1-NH2 and TBAB was identified as the optimal catalyst ratio that gives the maximum PC yield with >99% selectivity under room temperature and in 24 h (Table 4, entry 3). Also, a time dependent study on the catalytic activity was carried out at RT using 0.64 mol% UMCM-1-NH2/TBAB (Fig. 5). PC conversion increases gradually with an increasing reaction duration in the range of 0 to 24 h. Prolonged reactions beyond 24 h did not give any noticeable increase in the PC yield, indicating that the optimum reaction time was 24 h with 90% PC yield and >99% PC selectivity.
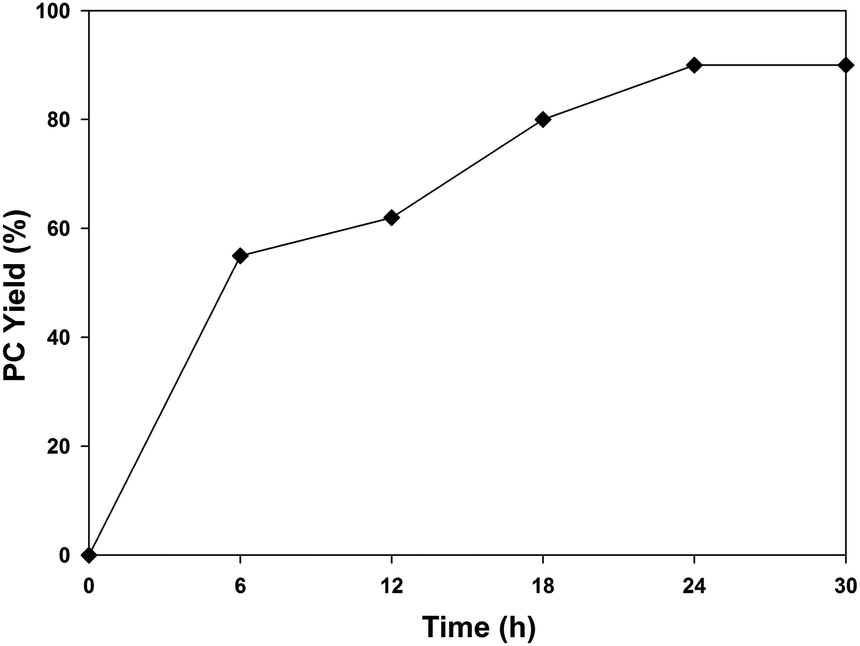 | ||
| Fig. 5 Effect of reaction time on the cycloaddition of propylene oxide and CO2 using 0.64 mol% UMCM-1-NH2 at room temperature and 1.2 MPa PCO2. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TBAB ratio in the yield of propylene carbonatea
:TBAB ratio in the yield of propylene carbonatea
| Entry | UMCM-1-NH2 (mol%) | TBAB (mol%) | Ratio of UMCM-1-NH2/TBAB | Yield (%) |
|---|---|---|---|---|
| a Reaction conditions: PO = 42.8 mmol, 1.2 MPa PCO2, 24 h, RT. | ||||
| 2 | 0.44 | 0.64 | 2:3 |
75 |
| 3 | 0.64 | 0.64 | 1:1 |
90 |
| 4 | 0.64 | 0.44 | 3:2 |
78 |
| 5 | 0.64 | 0.24 | 3:1 |
40 |
Since the catalyst concentration and time are important factors in determining the TON and TOF of the catalysts, the yield of PC at various temperatures at different time intervals was studied. A 92% PC yield was achieved in 10 h at 40 °C and 1.2 MPa. A yield of around 89% PC was obtained in 6 h when the temperature was set to 50 °C. The TON and TOF of the UMCM-1-NH2 were calculated at different temperatures based on the yield per equivalent of metal ions. The TON was calculated for the catalyst based on its formula weight available from the crystallographic information file (CIF) from the Cambridge Structural Data base, based on the assumption that the synthesized sample is purely homogeneous crystals. It was found that when the temperature was increased from RT to 50 °C, the TOF of the UMCM-1-NH2/TBAB mediated catalysis was raised four fold. Since 50 °C is still low compared to most of the previous reports on the synthesis of PC, this multifold increase in TOF is significant (Fig. 6).
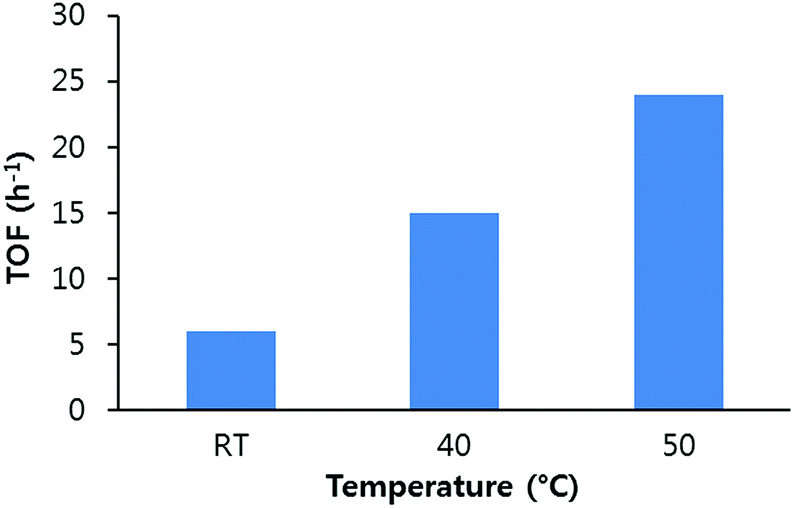 | ||
| Fig. 6 Turnover frequency (TOF) calculated based on the yield of propylene carbonate at different temperatures, 1.2 MPa PCO2 and 0.64 mol% UMCM-1-NH2. | ||
Another crucial criterion to be considered in heterogeneous catalysis is the recyclability of the catalyst. An easier catalyst separation is critical for industrial processes to minimize waste streams and to develop potential catalyst recycling strategies, owing to its economic and environmental aspects.19 We performed catalyst recycling studies for UMCM-1-NH2 at RT, 1.2 MPa pressure (Table 5). UMCM-1-NH2 maintained its activity (90%) and >99% selectivity throughout the first five recycles. The recycled UMCM-1-NH2 catalyst was analyzed by PXRD analysis (Fig. 7). The catalyst maintained its structure throughout the recycling process, which was determined by the retention of the peaks in comparison with the fresh catalyst. FT-IR analysis was also conducted on the recycled UMCM-1-NH2 catalyst. The similarities in the peaks with the fresh catalyst confirmed that the chemical integrity is maintained throughout the recycling process (Fig. 8). We also assessed the heterogeneity and stability of UMCM-1-NH2 in a more accurate way by using an inductively coupled plasma optical emission spectroscopy (ICP-OES) analysis of the supernatant for metal leaching. The ICP-OES showed that less than 1.5% of the zinc ions had leached into the reaction mixture after five recycles.
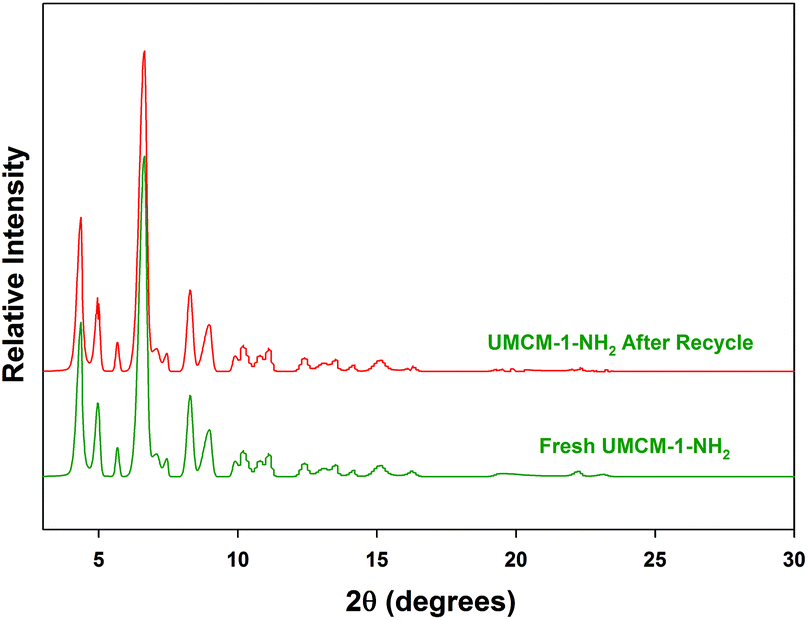 | ||
| Fig. 7 Powder XRD patterns of the fresh and recycled UMCM-1-NH2 catalyst. | ||
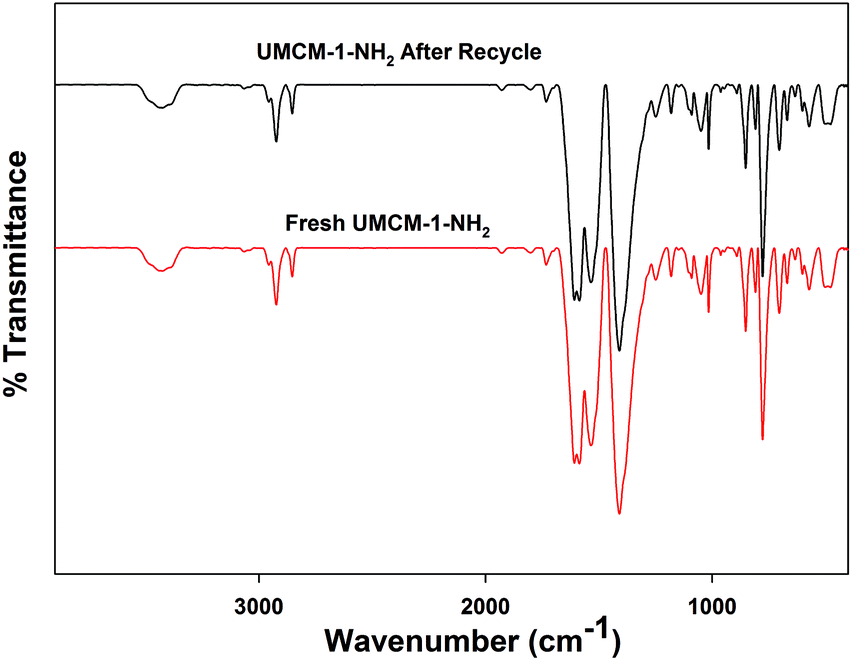 | ||
| Fig. 8 FTIR spectra of the fresh and recycled UMCM-1-NH2. | ||
| No | Recycles | Conversion (%) | Selectivity (%) | Yield (%) |
|---|---|---|---|---|
| Reaction conditions: 42.8 mmol propylene oxide, 0.64 mol% UMCM-1-NH2, room temperature, 1.2 MPa PCO2, 24 h. | ||||
| 1 | Fresh | 90 | >99 | 90 |
| 2 | 1st | 90 | >99 | 90 |
| 3 | 2nd | 90 | >99 | 90 |
| 4 | 3rd | 90 | >99 | 90 |
| 5 | 4th | 90 | >99 | 90 |
| 6 | 5th | 89 | >99 | 89 |
A fundamental and very popular method to estimate the presence of any leached species such as metal ions from the catalyst in the reaction media is the hot filtration test.20 But since the UMCM-1-NH2/TBAB system will always contain halide ions, the hot filtration test with this system would be meaningless. Hence, the hot filtration test was conducted with UMCM-1-NH2 alone but at a high temperature, since, at 120 °C, UMCM-1-NH2 alone has furnished 95% PO conversion (Table 1, entry 10). The cycloaddition was carried out with 0.64 mol% UMCM-1-NH2 at 1.2 MPa pressure and 120 °C until a 55% conversion of PO was achieved (at 12 h) (Fig. 9). The contents were filtered, analyzed by gas chromatography and the remaining filtrate was fed back into the reactor. Sampling was performed every 2 h thereafter. No further substrate conversion in the filtrate occurred after the catalyst removal at the reaction temperature, indicating that there are no homogeneous species catalyzing the reaction in solution.
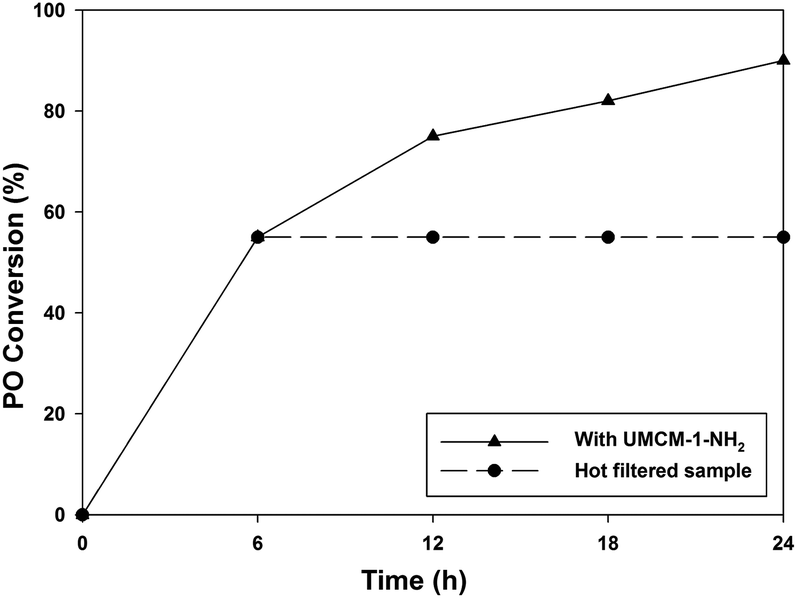 | ||
| Fig. 9 Hot filtration test. Reaction conditions: 120 °C, 0.64 mol% UMCM-1-NH2 and 1.2 MPa PCO2. Hot filtration was done at 55% conversion at 6 h. | ||
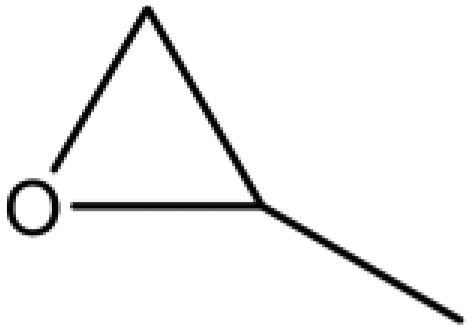
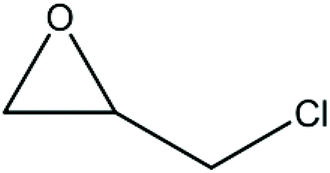
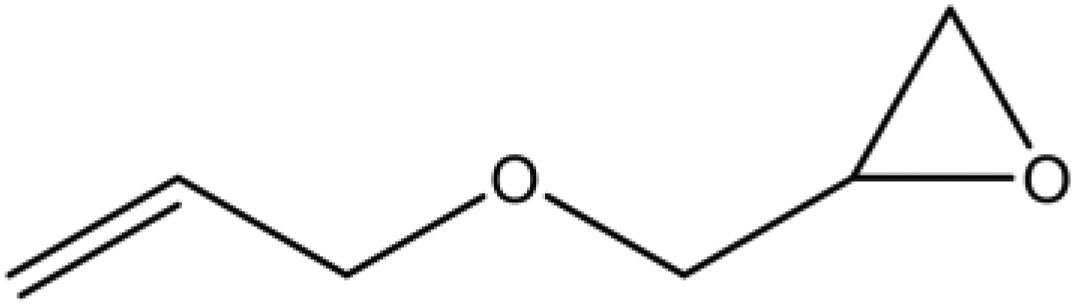
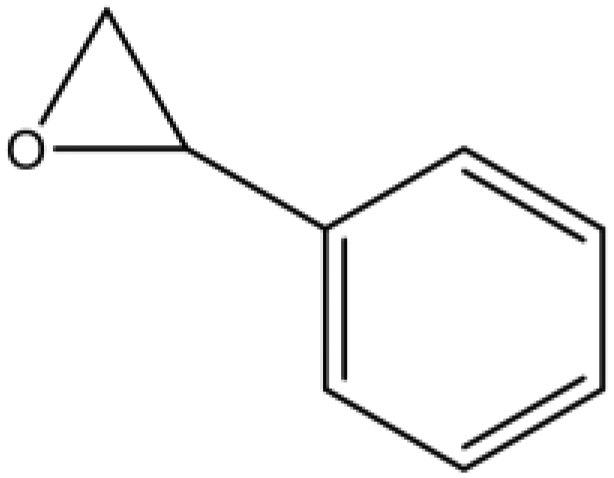
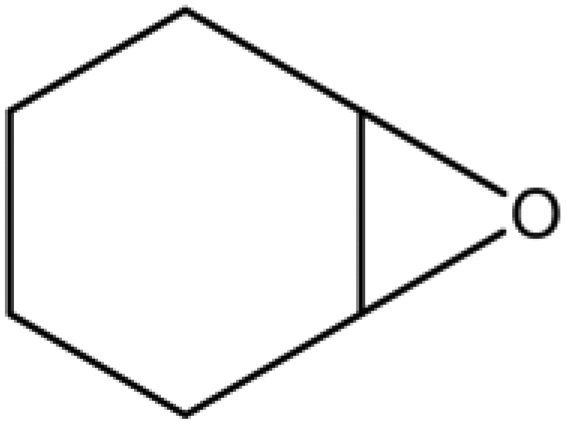
A series of different epoxides were examined for the chemical fixation of CO2 to produce the corresponding cyclic carbonates in the presence of UMCM-1-NH2 under 1.2 MPa pressure, 24 h and room temperature (Table 6). Comparatively lower conversions were observed for the epoxides using styrene oxide, allyl glycidyl ether and cyclohexene oxide. The lower conversion of epichlorohydrin than PO could be explained on the basis of the electron withdrawing effect of the CH2Cl group, which reduces the electron density of the epoxide oxygen system (Table 6, entry 1). The poor reactivity of SO is probably due to the bulky nature of epoxide that hinders the reaction with the CO2 that resides inside micropores, and the low reactivity of its β-carbon center (Table 6, entry 3). Cyclohexene oxide, the internal epoxide, was the least active one among these, probably because of its high steric hindrance (Table 6, entry 4). Therefore, it should be concluded that the bulkiness of the substrate is highly relevant in micro–mesoporous MOFs since the reacting molecules that reside inside the micropores may not be easily vulnerable to the bulky epoxides, even though the epoxide could successfully diffuse into the mesopores. This in turn supplements our rationale that the catalysis mostly occurs inside the micropores of UMCM-1-NH2 rather than the mesopore channels. Thus, micro–mesoporous hybrid materials are more efficient at CO2 cycloaddition with smaller epoxides such as propylene oxide or epichlorohydrin. Still, it is more noteworthy that cyclic carbonates can be obtained at room temperature with a higher selectivity than many of the reported MOFs.
| Entry | Epoxide | Conversion (%) | Selectivity (%) | Yield (%) |
|---|---|---|---|---|
| Reaction conditions: UMCM-1-NH2 = 0.64 mol%, TBAB = 0.64 mol%, epoxide = 42.8 mmol, PCO2 = 1.2 MPa, RT, 24 h, semi-batch. | ||||
| 1 |
|
90 | >99 | 90 |
| 2 |
|
78 | >99 | 78 |
| 3 |
|
55 | >99 | 55 |
| 4 |
|
53 | >99 | 53 |
| 5 |
|
10 | >99 | 10 |
The previous results reported so far about CO2 adsorption in MOFs prove that CO2 adsorption mainly depends on surface area and pore volume.21a UMCM-1-NH2, possessing high surface area and large pore volume, was tested as a potential candidate for CO2 capture applications.21b The excellent catalytic performance of propylene oxide is attributed to the combined effects of suitable acidity/basicity and the micro–mesoporosity. So a better mass transfer between CO2 and the catalytic centre is expected. Catalysis is expected to occur inside the micropores of UMCM-1-NH2, because the bulky epoxides are found to be less reactive (Table 6, entries 4 and 5). Mesopores appear mainly as highways, which improves the mass transport of the reactants and products to/from active sites located inside the micropores. In general, micro–mesoporous materials with a high surface area and proper functionalities22 confer a high catalytic performance towards the synthesis of cyclic carbonates at room temperature with 100% selectivity.
Conclusions
A mixed linker dual-porous amine functionalized 3D MOF, UMCM-1-NH2, constituting Zn4O SBUs with two-fold symmetric (NH2-BDC) and three-fold symmetric (BTB) ligands, was synthesized, characterized and its structural peculiarities were exploited for its catalytic potentials. We proposed a novel approach of synchronizing micro and mesopores in catalysis, so as to overcome the limitations of microporous materials, viz., hindrance to pore accessibility, molecular transport and mass transfer, while harvesting the potential of micropores in enhancing the catalyst-substrate interactions. This enhancement of catalytic efficiencies was demonstrated in the chemical fixation of CO2 by its cycloaddition with epoxides to yield cyclic carbonates. UMCM-1-NH2 showed a TON of 141 at room temperature with a high selectivity for propylene carbonate (>99%) in the cycloaddition of propylene oxide and CO2 using TBAB as cocatalyst. The catalyst was separable by simple filtration, and the heterogeneous nature of the catalyst was investigated using physico-chemical techniques. The catalyst framework did not degrade even after several repetitions of the cycles and, promisingly, no fall in the conversion rate of the cycloaddition occurred. A plausible mechanism was suggested based on the literature and experimental inferences. It was demonstrated that the cautious design and judicious selection of building blocks so as to accommodate apt pores (hybrid micro–mesopores) are equally important as the role of the active sites and functional groups in attaining a viable chemical fixation of CO2. This hybrid nature of UMCM-1-NH2, which offers easy accessibility to the substrates, along with easily modifiable amine groups, provides a handle for post-synthetic functionalization in forming a single component standalone catalytic species, which shall remove the need for any co-catalysts, and thereby serves an inspiration to future catalysis using MOFs.Experimental section
Synthesis of UMCM-1-NH2
The synthesis of purely needle-shaped UMCM-1-NH2 was performed by a solvothermal reaction between Zn(NO3)2·4H2O (≥ 98%, Sigma-Aldrich), 2-aminoterephthalic acid (99%, Sigma-Aldrich) and 1,3,5-tris(4-carboxyphenyl)benzene (97%, Alfa-Aesar) in N,N-diethylformamide (99%, Alfa-Aesar) at 85 °C for 72 h. The optimal molar ratio of H2NH2BDC and H3BTB for the pure phase production was found to be within the range of 3:2 to 1:1. Liquid exchange of the encapsulated solvent DEF was carried out by dipping the material in dichloromethane (≥99.8%, Sigma-Aldrich) for 2 days, followed by pore evacuation under vacuum. Anal. calcd (%) for the sample C44H25NO13Zn4; C: 50.95; H: 2.43; N: 1.35; O: 20.05; Zn: 25.21. Exp. found. (%); C: 51.53; H: 2.851; N: 1.82; O: 20.19 (elemental analysis), Zn: 23.2 (ICP-OES).
Cycloaddition of epoxide and CO2
42.8 mmol propylene oxide and 0.64 mol% of the catalyst were mixed together and put into a 25 mL stainless steel reactor equipped with a magnetic stirrer. The reactor was pressurized with CO2 to the required pressure at room temperature, and stirred at 600 rpm. The reaction vessel was kept connected to a CO2 source throughout the reaction via a one-way check valve to maintain the pressure at the desired level. After reaction completion, the reactor was cooled and excess CO2 was carefully vented off. Toluene (3 mL) was added to the product mixture and filtered. The filtrate was subsequently analyzed by gas chromatography (GC, HP 6890, Agilent Technologies) to determine conversion, selectivity, and yield using toluene as internal standard.Acknowledgements
This study was supported by the National Research Foundation of Korea (GF-HIM 2015-065264).Notes and references
- (a) D. W. Keith, Science, 2009, 325, 1654 CrossRef CAS PubMed; (b) K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724 CrossRef CAS PubMed; (c) M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709 CrossRef CAS PubMed; (d) J. Wang, L. Huang, R. Yang, Z. Zhang, J. Wu, Y. Gao, Q. Wang, D. O. Hare and Z. Zhong, Energy Environ. Sci., 2014, 7, 3478 RSC; (e) P. Markewitz, W. Kuckshinrichs, W. Leitner, J. Linssen, P. Zapp, R. Bongartz, A. Schreibera and T. E. Muller, Energy Environ. Sci., 2012, 5, 7281 RSC.
- (a) M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514 RSC; (b) P. P. Pescarmona and M. Taherimehr, Catal. Sci. Technol., 2012, 2, 2169 RSC; (c) M. North and R. Pasquale, Angew. Chem., Int. Ed., 2009, 48, 2946 CrossRef CAS PubMed; (d) K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724 CrossRef CAS PubMed; (e) S. Klaus, M. W. Lehenmeier, C. E. Anderson and B. Rieger, Coord. Chem. Rev., 2011, 255, 1460 CrossRef CAS; (f) N. Kielland, C. J. Whiteoak and A. W. Kleij, Adv. Synth. Catal., 2013, 355, 2115 CrossRef CAS; (g) T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312 RSC; (h) I. S. Metcalfe, M. North, R. Pasquale and A. Thursfiel, Energy Environ. Sci., 2010, 3, 212 RSC; (i) W. J. Peppel, Ind. Eng. Chem., 1958, 50, 767 CrossRef CAS; (j) D. J. Darensbourg and M. W. Holtcamp, Coord. Chem. Rev., 1996, 153, 155 CrossRef CAS.
- (a) J. Sun, S. I. Fujita and M. Arai, J. Organomet. Chem., 2005, 690, 3490 CrossRef CAS; (b) R. L. Paddock and S. T. Nguyen, J. Am. Chem. Soc., 2001, 123, 11498 CrossRef CAS PubMed; (c) Q. He, J. W. O'Brien, K. A. Kitselman, L. E. Tompkins, G. C. T. Curtisa and F. M. Kerton, Catal. Sci. Technol., 2014, 4, 1513 RSC; (d) J. Wang, J. Wu and N. Tang, Inorg. Chem. Commun., 2007, 10, 1493 CrossRef CAS.
- (a) K. Yamaguchi, K. Ebitani, T. Yoshida, H. Yoshida and K. Kaneda, J. Am. Chem. Soc., 1999, 121, 4526 CrossRef CAS; (b) Y. Du, F. Cai, D. L. Kong and L. N. He, Green Chem., 2005, 7, 518 RSC.
- D. W. Kim, R. Roshan, J. Tharun, A. C. Kathalikkattil and D. W. Park, Korean J. Chem. Eng., 2013, 30, 1973 CrossRef CAS.
- (a) H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673 CrossRef CAS PubMed; (b) N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933 CrossRef CAS PubMed; (c) H.-C. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415 RSC.
- (a) J. Song, Z. Zhang, S. Hu, T. Wu, T. Jiang and B. Han, Green Chem., 2009, 11, 1031 RSC; (b) W. Kleist, F. Jutz, M. Maciejewski and A. Baiker, Eur. J. Inorg. Chem., 2009, 3552 CrossRef CAS; (c) Y. J. Kim and D. W. Park, J. Nanosci. Nanotechnol., 2013, 13, 2307 CrossRef CAS PubMed; (d) E. E. Macias, P. Ratnasamy and M. A. Carreon, Catal. Today, 2012, 198, 215 CrossRef CAS; (e) T. Lescouet, C. Chizallet and D. Farrusseng, ChemCatChem, 2012, 4, 1725 CrossRef CAS; (f) O. V. Zalomaeva, A. M. Chibiryaev, K. A. Kovalenko, O. A. Kholdeeva, B. S. Balzhinimaev and V. P. Fedin, J. Catal., 2013, 298, 179 CrossRef CAS; (g) S. N. Kim, J. Kim, H. Y. Kim, H. Y. Cho and W. S. Ahn, Catal. Today, 2013, 204, 85 CrossRef CAS; (h) J. Kim, S. N. Kim, H. G. Jang, G. Seo and W. S. Ahn, Appl. Catal., A, 2013, 453, 175 CrossRef CAS.
- (a) C. M. Miralda, E. E. Macias, M. Zhu, P. Ratnasamy and M. A. Carreon, ACS Catal., 2012, 2, 180 CrossRef CAS; (b) L. Yang, L. Yu, G. Diao, M. Sun, G. Cheng and S. Chen, J. Mol. Catal. A: Chem., 2014, 392, 278 CrossRef CAS; (c) J. Tharun, G. Mathai, A. C. Kathalikkattil, R. Roshan, Y. S. Won, S. J. Cho, J. S. Chang and D. W. Park, ChemPlusChem, 2015, 80, 715 CrossRef CAS.
- (a) A. C. Kathalikkattil and D. W. Park, J. Nanosci. Nanotechnol., 2013, 13, 2230 CrossRef CAS PubMed; (b) A. C. Kathalikkattil, D. W. Kim, J. Tharun, H. G. Soek, R. Roshan and D. W. Park, Green Chem., 2014, 16, 1607 RSC; (c) A. C. Kathalikkattil, R. Roshan, J. Tharun, H. G. Soek, H. S. Ryu and D. W. Park, ChemCatChem, 2014, 6, 284 CrossRef CAS; (d) Y. Ren, Y. Shi, J. Chen, S. Yang, C. Qi and H. Jiang, RSC Adv., 2013, 3, 2167 RSC; (e) Y. Ren, X. Cheng, S. Yang, C. Qi, H. Jiang and Q. Mao, Dalton Trans., 2013, 42, 9930 RSC.
- (a) W. Xuan, C. Zhu, Y. Liu and Y. Cui, Chem. Soc. Rev., 2012, 41, 1677 RSC; (b) A. Corma, Chem. Rev., 1997, 97, 2373 CrossRef CAS PubMed; (c) N. Linares, A. M. Silvestre-Albero, E. Serrano, J. Silvestre-Albero and J. Garcia-Martinez, Chem. Soc. Rev., 2014, 43, 7681 RSC.
- D. A. Yang, H. Y. Cho, J. Kim, S. T. Yang and W. S. Ahn, Energy Environ. Sci., 2012, 5, 6465 CAS.
- (a) M. H. Beyzavi, R. C. Klet, S. Tussupbayev, J. Borycz, N. A. Vermeulen, C. J. Cramer, J. F. Stoddart, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2014, 136, 15861 CrossRef CAS PubMed; (b) M. H. F. Kox, E. Stavitski, J. C. Groen, J. Pérez-Ramírez, F. Kapteijn and B. M. Weckhuysen, Chem. – Eur. J., 2008, 14, 1718 CrossRef CAS PubMed; (c) J. Roeser, K. Kailasam and A. Thomas, ChemSusChem, 2012, 5, 1793 CrossRef CAS PubMed; (d) M. Zhu and M. A. Carreon, J. Appl. Polym. Sci., 2014, 131, 39738 Search PubMed.
- Z. Wang, K. K. Tanabe and S. M. Cohen, Inorg. Chem., 2009, 48, 296 CrossRef CAS PubMed.
- (a) K. Koh, A. G. Wong-Foy and A. J. Matzger, Angew. Chem., Int. Ed., 2008, 47, 677 CrossRef CAS PubMed; (b) K. K. Tanabe and S. M. Cohen, Angew. Chem., Int. Ed., 2009, 121, 7560 CrossRef; (c) N. Ko and J. Kim, Bull. Korean Chem. Soc., 2011, 32, 2705 CrossRef; (d) J. G. Nguyen, K. K. Tanabe and S. M. Cohen, CrystEngComm, 2010, 12, 2335 RSC.
- (a) W. Cheng, Q. Su, J. Wang, J. Sun and F. T. T. Ng, Catalysts, 2013, 3, 878 CrossRef; (b) W. Clegg, R. W. Harrington, M. North and R. Pasquale, Chem. – Eur. J., 2010, 16, 6828 CrossRef CAS PubMed; (c) J. Martinez, J. A. Castro-Osma, A. Earlam, C. Alonso-Moreno, A. Otero, A. Lara-Sanchez, M. North and A. Rodriguez-Dieguez, Chem. – Eur. J., 2015, 21, 9850 CrossRef CAS PubMed.
- (a) J. Sun, S. Fujita, F. Zhao and M. Arai, Green Chem., 2004, 6, 613 RSC; (b) F. Jutz, J.-D. Grunwaldt and A. Baiker, J. Mol. Catal. A: Chem., 2008, 279, 94 CrossRef CAS.
- I. I. Ivanova and E. E. Knyazeva, Chem. Soc. Rev., 2013, 42, 3671 RSC.
- (a) W. Y. Gao, Y. Chen, Y. Niu, K. Williams, L. Cash, P. J. Perez, L. Wojtas, J. Cai, Y. S. Chen and S. Ma, Angew. Chem., Int. Ed., 2014, 53, 2615 CrossRef CAS PubMed; (b) W. Y. Gao, L. Wojtas and S. Ma, Chem. Commun., 2014, 50, 5316 RSC.
- M. Arai and F. Zhao, Catalysts, 2015, 5, 868 CrossRef CAS.
- R. A. Sheldon, M. Wallau, I. W. C. E. Arends and U. Schuchardt, Acc. Chem. Res., 1998, 31, 485 CrossRef CAS.
- (a) J. Liu, P. K. Thallapally, B. P. McGrail, D. R. Brown and J. Liu, Chem. Soc. Rev., 2012, 41, 2308 RSC; (b) Z. Xiang, S. Leng and D. Cao, J. Phys. Chem. C, 2012, 116, 10573 CrossRef CAS.
- J. Wang, W. Sng, G. Yi and Y. Zhang, Chem. Commun., 2015, 51, 12076 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Arrangement of adjacent 1D hexagonal mesopores in UMCM-1-NH2, XPS spectra, nitrogen adsorption–desorption isotherm, pore size distribution curve, instrumentation. See DOI: 10.1039/c5gc01763g |
| This journal is © The Royal Society of Chemistry 2016 |