DOI:
10.1039/C5GC01741F
(Paper)
Green Chem., 2016,
18, 220-225
Cooperative catalysis of Pt/C and acid resin for the production of 2,5-dimethyltetrahydrofuran from biomass derived 2,5-hexanedione under mild conditions†
Received
28th July 2015
, Accepted 3rd September 2015
First published on 3rd September 2015
Abstract
Conversion of biomass into 2,5-dimethyltetrahydrofuran (DMTHF), an excellent fuel additive, solvent, and industrial intermediate, is an important reaction in green chemistry. In this work, different combinations of supported metal catalysts and solid acids were investigated for the synthesis of DMTHF from biomass derived diketone (2,5-hexanedione, 2,5-HD). The results showed that commercial Pt/C and solid acid Amberlite®IR-120H had an excellent cooperative effect on the reaction. 2,5-HD could be converted into DMTHF with a yield up to 99% under milder conditions. Further experiments indicated that 2,5-hexanediol (2,5-HDO) is a key intermediate in the reaction. The catalyst system could be facilely recovered due to the heterogeneity and reused several times without notable change in the performance.
Introduction
The rapid depletion of fossil resources and growing energy and resource demands have driven the conversion of biomass into useful chemicals to become an important topic in green chemistry.1 Lignocellulose and its derivatives are abundant and potential sustainable sources for bulk chemicals, fuel additives, and materials.1b–e,2 Through rational design of the catalysts or catalyst system, lignocellulose and its derivatives can be converted into value-added chemicals, such as 5-hydroxymethylfurfural (HMF),3 gamma-valerolactone (GVL),4 2,5-dimethylfuran (DMF),5 2,5-bis(hydroxymethyl)furan (BHMF),5a,6 and 2,5-dimethyltetrahydrofuran (DMTHF),5a,c,7 indoles,8etc., which have broad applications in the fields of fuel additives,5a,d,e,9 solvents,4c–e and industrial intermediates.3a,5a,b,6,10
Among the biomass derived chemicals, DMTHF is an important molecule with a wide range of applications. For example, DMTHF is an excellent candidate for gasoline replacement.11 Compared with ethanol, DMTHF has a higher boiling point (90–92 °C) and research octane number (119), and lower miscibility with water.5d The higher energy density of DMTHF (31 MJ L−1) is comparable to gasoline (34.2 MJ L−1) and far superior to ethanol (23 MJ L−1).11,12 DMTHF is even superior to DMF as a gasoline additive. It is similar to DMF in terms of energy density (31.8 MJ L−1), volatility, and solubility in water, but the saturated structure makes DMTHF have good storage and transportation stability and become a better candidate for liquid fuel.13 Besides fuel additives, DMTHF can also be used as reaction solvents,14 chemical intermediates,15 catalyst ligands,16 battery electrolyte,17etc.
Different approaches have been reported to produce DMTHF. Both secondary aliphatic alcohols and diols (2,5-hexanediol) were used as starting molecules to synthesize DMTHF under chemical or enzymatic catalysis.18 However, either complex steps or unfriendly chemicals were used in these reported methods. With the development of green chemistry, the synthesis of DMTHF from renewable biomass has drawn much attention.5a,c,7,13 During the synthesis of DMTHF from biomass, DMF and 2,5-hexanedione (2,5-HD) are commonly used substrates or crucial intermediates,5a–d,7,12,13,19 and 2,5-HD could be derived from biomass.19b It was demonstrated that the DMTHF yield was higher from 2,5-HD than that from DMF, attributed to the better stability of 2,5-HD than DMF, especially under the acidic conditions.7 The reaction rate of 2,5-HD to DMTHF was much higher than that of DMF to DMTHF.12 Direct conversion of DMF to DMTHF also suffered from high reaction temperatures.5c Accordingly, 2,5-HD is a more suitable substrate to produce DMTHF than DMF. However, up to now, studies on the direct conversion of 2,5-HD to DMTHF have been very limited, and either expensive homogeneous Ru or Ir catalysts and corrosive liquid acids or harsh reaction conditions (high temperature and H2 pressure) were adopted.7,12 Therefore, it is still highly desirable to explore efficient, robust, and readily recyclable catalyst systems for the DMTHF production.
Herein, we reported an efficient catalyst system consisting of supported metal catalysts and solid acids for the production of DMTHF from biomass derived 2,5-HD (Scheme 1). Among the various combinations of metal and acid catalysts studied, the combination of commercial Pt/C and Amberlite®IR-120H was demonstrated to be the most efficient, with DMTHF yield up to 99% without using liquid acids and organic solvents, and the reaction condition was milder than those reported. Our catalytic system has some obvious advantages, such as high efficiency and stability, milder reaction conditions, and facile recycling.
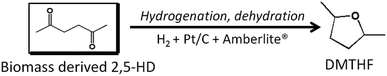 |
| | Scheme 1 Catalyst system for catalytic conversion of 2,5-HD into DMTHF in the presence of H2, Pt/C, and Amberlite®IR-120H. | |
Experimental
Materials
Amberlyst® 15H (denoted as AM-1, wet, ion exchange resin, beads), Amberlite® IR-120H (denoted as AM-2, wet, ion exchange resin), Zeolite Y (hydrogen), and Pd/Al2O3 (palladium, 1 wt% on alumina powder, reduced, 300 m2 g−1) were from Alfa Aesar®. Pt/C (1 wt% on carbon) was provided by Hubei Deli New Material Co., Ltd, China. Skeletal nickel catalysts (slurry in water) and 2,5-dimethyltetrahydrofuran (>98.0%) were from TCI Chemicals. Silicotungstic acid (H4[SiO4(W3O9)4], AR), phosphomolybdic acid (H3Mo12O40P, AR), and tetrahydrofurfuryl alcohol (THFAL, AR) were provided by Sinopharm Chemical Reagent Co. Ltd. 2,5-Hexanedione (>99.0%) and 2-hexanol were obtained from J&K Chemical. 2,5-Hexanediol (>99.0%) was from Acros Organics. Nafion® SAC-13 (denoted as NS, pore diameter > 10 nm, pore volume > 0.6 mL g−1) was purchased from Aldrich Chemistry. H2 was provided by Beijing Analytical Instrument Company.
Catalyst preparation
The solid acid resins, Amberlyst® 15H and Amberlite® IR-120H (denoted as AM-1 and AM-2, respectively) were washed with water and ethanol thoroughly and then dried under 100 °C overnight. Other acid catalysts were used as received. The preparation procedures of metal catalysts, including Pt/C (1 wt%), Ru/C (1 wt%), and Pt/AM-2 (1 wt%) are given in the ESI.† The Co/AM-1 (10 wt%) was prepared by the impregnation and NaBH4 reduction method according to our previous work.20 The acid density of the acids used in this work was characterized according to the reported method, and the data are given in Table S1.†21
Reaction
The hydrogenation reaction was performed in a 10 mL Teflon-lined stainless steel autoclave equipped with a magnetic stirrer. In a typical experiment, 0.5 g 2,5-HD (4.4 mmol), 2 g water and a certain amount of catalysts were introduced into the reactor. After removing the air via a vacuum, hydrogen was charged into the reactor to the desired pressure. Then the reactor was placed in a constant temperature air bath and the stirrer was started. After the reaction, the reactor was cooled in ice-water to quench the reaction and the organic phase was diluted by ethanol. The sample was analyzed by gas chromatograph (GC, HP 4890) equipped with a flame ionization detector (FID), and THFAL was used as the internal standard. Identification of the products and reactants was performed using a GC-MS (SHIMADZU-QP2010) as well as by comparing the retention times to respective standards in GC traces. The yield of DMTHF was expressed in the molar ratio of the added 2,5-HD and DMTHF. In the reusability experiments, the catalysts were separated via centrifugation, washed with water, and then reused for the next run directly. The virgin and used Pt/C catalysts were characterized by transmission electron microscopy (TEM, JEOL JEM-1011).
Results and discussion
Selection of the metal and acid catalysts
We first studied the conditions where only the metal catalyst or acid catalyst was applied as the catalyst (entries 1, 2, Table 1). It can be seen that the DMTHF yield was very low or even no DMTHF was produced when only Pt/C or AM-1 existed. When only Pt/C was used, the product was mainly 2,5-hexanediol (2,5-HDO). But when the metal catalyst and the acid catalyst co-existed, the DMTHF yield significantly improved (entries 3 and 4, Table 1). These results indicated that both the metal catalyst and the acid catalyst were essential and they exhibited a cooperative effect for the reaction. Therefore, we investigated different combinations of metals and acid catalysts on the reaction. The effect of different metal species, including noble metals (Ru, Pt, Pd) and transition metals (Ni, Co), was studied using commercial acid resin AM-1 as the acid catalyst (entries 3–8, Table 1). It was found that the performance of the catalysts was significantly affected by both the metal species and the supports. The noble metal catalysts (Ru/C, Pt/C, Pd/C) generally showed higher conversion and product yield than those of transition metal catalysts (RANEY® Ni, Co/AM-1), and Pt/C (1 wt%) had the highest conversion and product yield (>99% and 93%, respectively), while Pd/C (1 wt%) and Ru/C (1 wt%) had a lower DMTHF yield under the same conditions (entries 3–5, 7, and 8, Table 1). For the Pd catalyst, Pd/C showed better performance than that of Pd/Al2O3, indicating the important role of the support (entries 5 and 6, Table 1). We also attempted to load Pt directly on AM-1 to form a bifunctional Pt/AM-1 (1 wt%) catalyst. The results showed that the conversion was moderate, but no DMTHF was formed (entry 9, Table 1). The reasons for this are complex, and one possible reason is that the H2PtCl6 precursor was not completely reduced under H2 at 150 °C (adopted in this work). Further increasing the reduction temperature may destroy the –SO3H group and the skeletal structures of the acid resin. Therefore, we focused on the simple and efficient combination of metal and acid catalysts in this work. Subsequently, we chose the Pt/C as the metal catalyst to compare the performances of different acid catalysts (entries 10–14, Table 1). The results showed that most of the acid catalysts exhibited good to excellent activity on the reaction, including the acid resins (AM-1, AM-2, NS) (entries 4, 10, and 11, Table 1), silicotungstic acid (SA) (entry 12, Table 1), and molecular sieve (MS) (entry 13, Table 1), among which AM-2 achieved the highest conversion and yield (entry 10, Table 1). It can be seen from Table 1 that although some catalyst combinations had high conversions (for example, entries 3, 13, and 14), some amounts of other products such as 2,5-hexanediol (2,5-HDO), 2-hexanol (2-HO), and some unknown by-products were produced. Based on the above results, the combination of Pt/C + AM-2 was chosen for further studies.
Table 1 Performances of different combinations of metal and acid catalystsa
| Entry |
Catalysts |
Conv./% |
Yield/% |
| DMTHF |
2,5-HDOb |
2-HOc |
Othersd |
|
Typical reaction conditions were as follows until otherwise stated: 2,5-HD, 0.5 g; H2O, 2 mL; metal catalyst, 0.28 mol% of the substrate; acid catalyst, 0.05 g; reaction temperature, 120 °C; H2 pressure, 3.0 MPa; reaction time, 2 h; The metal loading for Pt/C, Ru/C, Pd/C, Pd/Al2O3, and Pt/AM-1 was 1 wt%; AM-1 = Amberlyst®15(H); AM-2 = Amberlite®IR-120(H); NS = Nafion®SAC-13; SA = Silicotungstic acid; MS = Molecular sieve (Zeolite Y); PA = Phosphomolybdic acid.
2,5-HDO, 2,5-hexanediol.
2-HO, 2-hexanol.
Others represented the unknown by-products.
Only Pt/C was used.
Only AM-1 was used.
0.017 g RANEY® Ni (wet) was added.
0.05 g AM-1 supported 0.005 g Co.
|
| 1e |
Pt/C |
>99 |
15 |
80 |
0 |
4 |
| 2 f |
AM-1 |
23 |
0 |
0 |
0 |
23 |
| 3 |
Ru/C + AM-1 |
>99 |
30 |
67 |
2 |
<1 |
| 4 |
Pt/C + AM-1 |
>99 |
93 |
0 |
6 |
<1 |
| 5 |
Pd/C + AM-1 |
43 |
16 |
0 |
0 |
27 |
| 6 |
Pd/Al2O3 + AM-1 |
38 |
10 |
0 |
0 |
28 |
| 7g |
RANEY® Ni + AM-1 |
58 |
23 |
0 |
0 |
35 |
| 8h |
Co/AM-1 |
30 |
8 |
0 |
0 |
22 |
| 9 |
Pt/AM-1 |
32 |
0 |
0 |
0 |
32 |
| 10 |
Pt/C + AM-2 |
>99 |
>99 |
0 |
0 |
<1 |
| 11 |
Pt/C + NS |
>99 |
84 |
0 |
7 |
8 |
| 12 |
Pt/C + SA |
>99 |
95 |
0 |
4 |
<1 |
| 13 |
Pt/C + MS |
>99 |
75 |
0 |
5 |
19 |
| 14 |
Pt/C + PA |
>99 |
41 |
20 |
0 |
38 |
The effect of reaction temperature
The effect of reaction temperature on the conversion of 2,5-HD is shown in Table 2. 2,5-HD could be completely converted within 2 h in the temperature range of 70––120 °C. At a lower temperature (70 °C), a moderate DMTHF yield of 55% was achieved. But when we prolonged the reaction time under 70 °C to 20 h, a 93% yield was obtained. The DMTHF yield could reach up to 95% at 80 °C. When the temperature was increased to 90 °C or to a higher level, the DMTHF yields increased to higher than 99%, indicating that 2,5-HD could nearly be quantitatively converted into DMTHF. The final DMTHF selectivities were higher than 90% in the temperature range of 70–120 °C. These results indicated that the temperature mainly affected the reaction rate, and had no significant influence on the selectivity. Keeping in mind that the energy input should be decreased as far as possible within acceptable product yields and reaction rate, 90 °C is enough for the efficient synthesis of DMTHF from 2,5-HD.
Table 2 Effect of reaction temperature on the conversion of 2,5-HD to DMTHFa
| Entry |
Temp./°C |
Time/h |
Conv./% |
Yield/% |
Sel./% |
|
Reaction conditions: 2,5-HD, 0.5 g; H2O, 2 mL; Pt/C, 0.05 g (0.28 mol% of the substrate); AM-2, 0.05 g; H2 pressure, 3.0 MPa; reaction temperature and time are given in the table.
|
| 1 |
70 |
2 |
>99 |
55 |
55 |
| 2 |
70 |
20 |
>99 |
93 |
93 |
| 3 |
80 |
2 |
>99 |
95 |
95 |
| 4 |
90 |
2 |
>99 |
97 |
97 |
| 5 |
100 |
2 |
>99 |
>99 |
100 |
| 6 |
120 |
2 |
>99 |
>99 |
100 |
The effect of H2 pressure
The effect of H2 pressure on the reaction is shown in Table 3. It can be seen that H2 pressure also played an important role in the reaction. A moderate conversion (51%) with a 28% yield of DMTHF was observed within 2 h even H2 pressure was as low as 1 MPa. When the reaction time was increased to 20 h, a yield of 80% was observed. Increasing the H2 pressure could improve the reaction rate significantly. When the H2 pressure was increased to 3 MPa or a higher level, almost total conversion of 2,5-HD was fulfilled, and the DMTHF yield could reach >99%. The final DMTHF selectivity increased from 80% under 1 MPa to 100% under 3 MPa, indicating that H2 pressure had an effect on both the reaction rate and the selectivity. Therefore, 3 MPa was high enough to convert 2,5-HD into DMTHF.
Table 3 Effect of H2 pressure on the conversion of 2,5-HD to DMTHFa
| Entry |
Pressure/MPa |
Time/h |
Conv./% |
Yield/% |
Sel./% |
|
Reaction conditions: 2,5-HD, 0.5 g; H2O, 2 mL; Pt/C, 0.05 g (0.28 mol% of the substrate); AM-2, 0.05 g; reaction temperature, 90 °C; H2 pressure and reaction time are given in the table.
|
| 1 |
1 |
2 |
51 |
28 |
55 |
| 2 |
1 |
20 |
>99 |
80 |
80 |
| 3 |
2 |
2 |
71 |
49 |
69 |
| 4 |
2 |
20 |
>99 |
90 |
90 |
| 5 |
3 |
2 |
>99 |
>99 |
100 |
| 6 |
4 |
2 |
>99 |
>99 |
100 |
The effect of catalyst ratio
The effect of the ratio of two active species in our catalyst system was also investigated (Table 4). We first fixed the amounts of AM-2 (0.05 g, 10 wt% of the substrate) and varied the dosage of Pt/C (entries 1–3, Table 4). The results showed that even under a very low dosage of Pt/C (0.02 g, corresponding to 0.02 mol% of the substrate), almost total conversion and a DMTHF yield of 86% were achieved (entry 3, Table 4), indicating the high efficiency of the Pt catalyst. Then we fixed the amount of Pt/C at 0.05 g (corresponding to 0.05 mol% of the substrate) to study the effect of the acid catalyst dosage (entries 2, 4, 5, Table 4). A DMTHF yield of 88% was fulfilled even under a low acid catalyst dosage of 2 wt% of the substrate (entry 5, Table 4). When the catalyst ratio was varied by decreasing the amount of the metal catalyst to an extremely low level (0.004 g, 0.022 mol% of the substrate), the conversion of 2,5-HD was not complete (44%) within 2 h (entry 6, Table 4), with a product selectivity of 23%. When the reaction time was prolonged to 20 h, complete conversion of the substrate was fulfilled, and the final DMTHF yield was 80% (entry 7, Table 4). These results indicated that the metal/acid ratio affected both the reaction rate and the selectivity.
Table 4 Influence of the catalyst ratio on the conversion of 2,5-HD to DMTHFa
| |
Catalysts |
|
|
|
| Entry |
m
Pt/C/mAM-2 (g/g) |
Conv./% |
Yield/% |
Sel./% |
|
Typical reaction conditions were as follows until otherwise stated in the table: 2,5-HD, 0.5 g; H2O, 2 mL; Pt/C and AM-2 were added as stated in the table; reaction temperature, 90 °C; H2 pressure, 3.0 MPa; reaction time, 2 h.
The reaction time was prolonged to 20 h.
|
| 1 |
0.10/0.05 |
>99 |
>99 |
100 |
| 2 |
0.05/0.05 |
>99 |
>99 |
100 |
| 3 |
0.02/0.05 |
>99 |
86 |
86 |
| 4 |
0.05/0.03 |
>99 |
96 |
96 |
| 5 |
0.05/0.01 |
>99 |
88 |
88 |
| 6 |
0.004/0.05 |
44 |
10 |
23 |
| 7b |
0.004/0.05 |
>99 |
80 |
80 |
The heterogeneity and reusability of the catalyst system
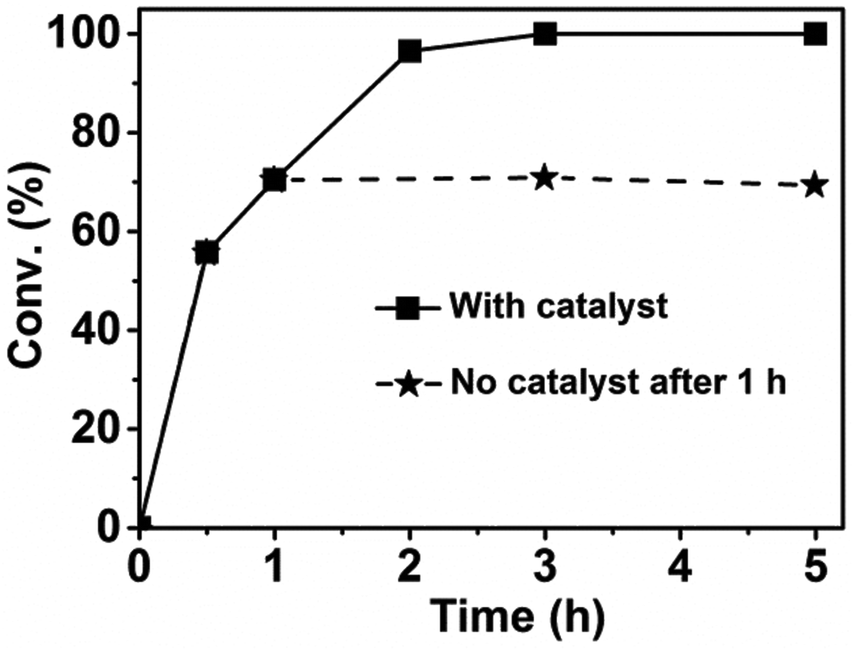
To verify the heterogeneous nature of our dual catalyst system, we removed the catalysts via hot filtration to see whether the reaction could proceed without the solid catalysts. In the experiments, a small amount of catalysts was used to slow down the reaction rate and detect the change of the conversion. It can be seen from Fig. 1 that the conversion of 2,5-HD did not change with the reaction time after the removal of the catalysts. We also detected the yield change of DMTHF and found that no more DMTHF was produced after the removal of the catalysts. These results proved the heterogeneous property of the catalyst system.
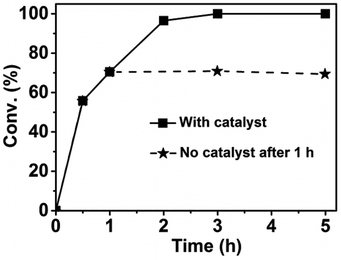 |
| | Fig. 1 Heterogeneous nature of the dual catalyst system. Reaction conditions: 2,5-HD, 0.5 g; H2O, 2 mL; Pt/C, 0.02 g; AM-2, 0.02 g; reaction temperature, 90 °C; H2 pressure, 3.0 MPa. | |
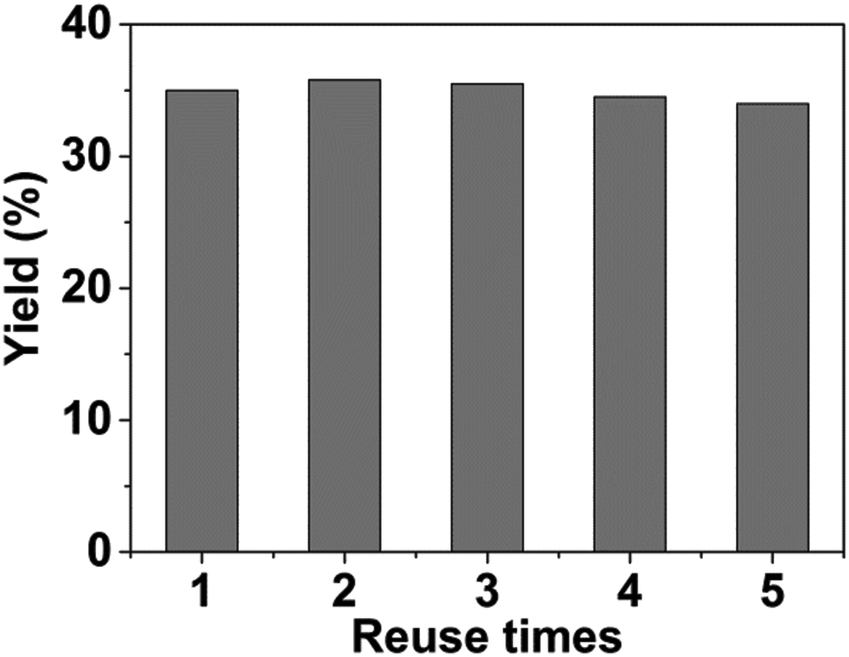
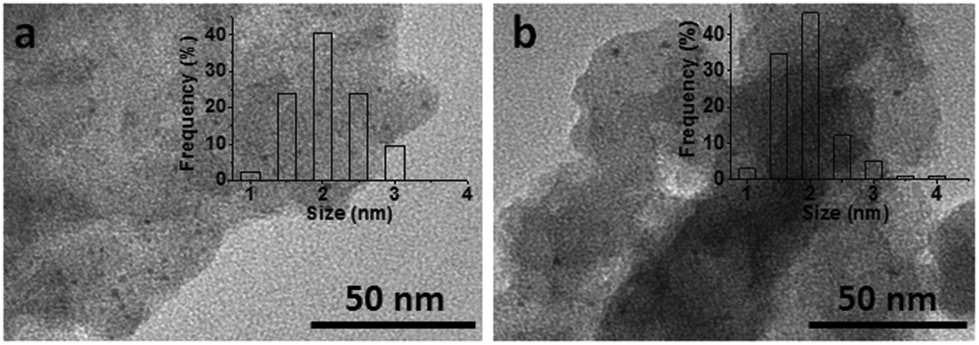
Then we studied the reusability of the catalyst system with product yield controlled at ca. 35% by using a small amount of catalysts (Fig. 2). After the reaction, the catalysts were separated by centrifugation, washed with water, and then directly used for the next run. The results showed that the catalyst system could be used at least 5 times without considerable change in the yields of DMTHF. TEM images of the virgin and the recycled catalysts are shown in Fig. 3. It can be shown from the images that the changes of the catalyst morphologies and size distributions of Pt particles were not considerable, and the average size of Pt particles in the recycled catalyst was similar to that of the virgin catalyst (ca. 2.0 nm), indicating the excellent stability of the Pt/C catalyst in the system.
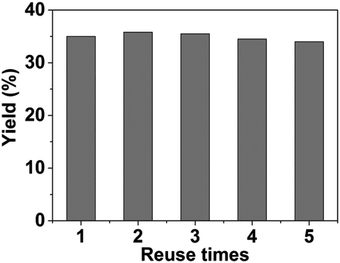 |
| | Fig. 2 Reusability of the dual catalyst system. The product yields were controlled at ca. 35% by using a smaller amount of catalysts. Reaction conditions: 2,5-HD, 0.5 g; H2O, 2 mL; Pt/C, 0.02 g; AM-2, 0.02 g; reaction temperature, 90 °C; H2 pressure, 3.0 MPa; reaction time, 1 h. | |
 |
| | Fig. 3 TEM images of the virgin (a) and the recycled Pt/C catalyst (b) after being reused for 5 times. The insets are the size distributions of Pt particles in the virgin and used catalysts. | |
Mechanism analysis
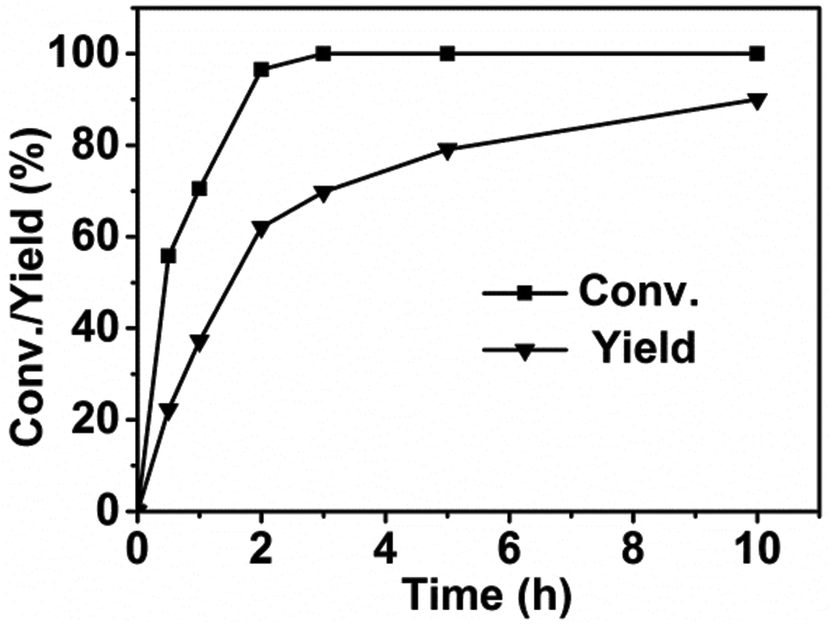
To give an insight into the reaction mechanism, we studied the concentration change of the substrates and products with the reaction time and analysed the intermediates. A small amount of catalysts was used in order to slow down the reaction rate and detect the concentration change of the reactant and product with the reaction time in detail. As can be seen in Fig. 4, more than 50% of the reactant was converted within the first 30 min. Nearly total conversion was finished within 2 h, indicating the conversion of 2,5-HD was a fast step. However, the rate for the formation of DMTHF was much slower. These results indicated that the reactant was converted first to certain intermediates, and the conversion rate of the intermediates was relatively slow. A new chemical identified as 2,5-hexanediol (2,5-HDO) was detected in our GC and GC-MS analysis (Table 1). Thus it was reasonable to deduce that 2,5-HDO was the intermediate for conversion of 2,5-HD to DMTHF. Shirai et al. reported that 2,5-HDO could indeed be converted to DMTHF under acid catalysis via intramolecular dehydration.22 In our experiments, we also used 2,5-HDO as the direct substrate using AM-2 as the acid catalyst under the same conditions as 2,5-HD conversion. It was found that almost quantitative DMTHF was produced, while nearly no DMTHF was produced without the acid catalyst. From these results, we deduced that 2,5-HDO was an intermediate during the conversion of 2,5-HD into DMTHF and the acid catalysts promoted the conversion of 2,5-HDO into DMTHF.
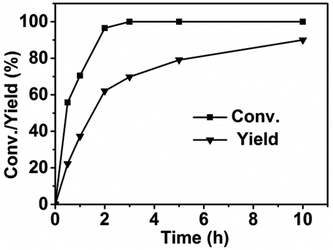 |
| | Fig. 4 Conversion and product yield as a function of reaction time. Reaction conditions: 2,5-HD, 0.5 g; H2O, 2 mL; Pt/C, 0.02 g; AM-2, 0.02 g; reaction temperature, 90 °C; H2 pressure, 3.0 MPa. | |
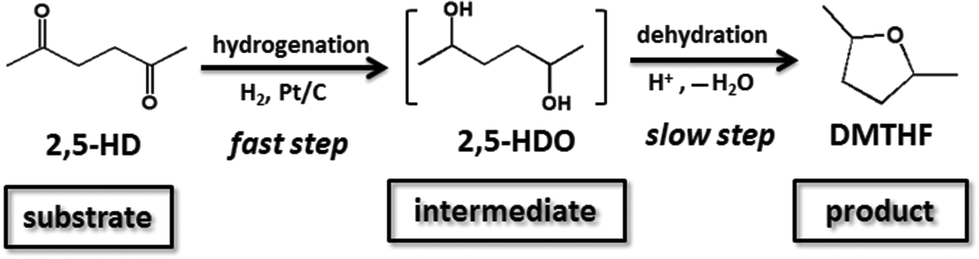
Based on the above experimental results and previous reports,7,18c,22,23 a plausible pathway was proposed (Scheme 2). Firstly, the carbonyl groups in 2,5-HD were hydrogenated to hydroxyl groups under the catalysis of Pt, which was a fast step. Secondly, the formed diol intermediate was converted to cyclic ether structures via intramolecular dehydration under the catalysis of the acid catalyst. The second step proceeded smoothly at a slower rate compared with the first step under the present conditions in this work.
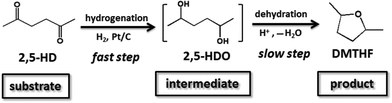 |
| | Scheme 2 Plausible mechanism for the conversion of 2,5-HD to DMTHF under the catalysis of the Pt/C–AM-2 system. | |
Conversion of DMF to DMTHF
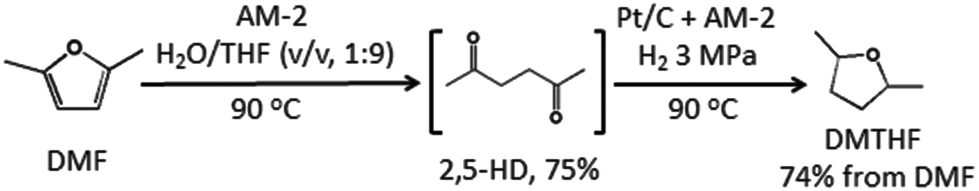
Finally, we attempted to apply the present catalyst system to the conversion of another biomass derived molecule DMF into DMTHF via 2,5-HD (Scheme 3). It has been reported that the CO2/H2O system was efficient for the conversion of DMF to 2,5-HD at 150 °C and CO2 pressure of 4 MPa.19b In our experiments, we found that AM-2 was also efficient for this reaction in a H2O/THF (v![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :v, 1:9) system. A moderate 2,5-HD yield (75%) was obtained, and the reaction conditions were much milder with lower temperature at ambient pressure. Nearly quantitative conversion of the produced 2,5-HD into DMTHF was achieved with a final yield of 74% under the catalysis of Pt/C + AM-2 without the separation of 2,5-HD (Scheme 3). Therefore, the catalyst system for the synthesis of DMTHF from biomass derived 2,5-HD and DMF are greener and more efficient than those via the chemical synthetic route,18 and has potential application for DMTHF production.
:v, 1:9) system. A moderate 2,5-HD yield (75%) was obtained, and the reaction conditions were much milder with lower temperature at ambient pressure. Nearly quantitative conversion of the produced 2,5-HD into DMTHF was achieved with a final yield of 74% under the catalysis of Pt/C + AM-2 without the separation of 2,5-HD (Scheme 3). Therefore, the catalyst system for the synthesis of DMTHF from biomass derived 2,5-HD and DMF are greener and more efficient than those via the chemical synthetic route,18 and has potential application for DMTHF production.
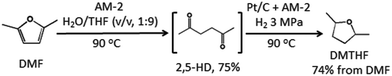 |
| | Scheme 3 Conversion of DMF into DMTHF via 2,5-HD under the catalysis of the Pt/C and AM-2 system. | |
Conclusion
In this work, a simple and efficient catalyst combination consisting of commercial Pt/C and AM-2 was applied for the conversion of biomass derived 2,5-HD to DMTHF. Almost quantitative conversion of 2,5-HD to DMTHF could be fulfilled with high yield up to 99% under 90 °C and 3 MPa H2 within 3 h, which was a milder condition than those reported for this reaction with satisfactory efficiency. The Pt/C + AM-2 catalyst system can be easily recovered and reused at least five times without decreasing the activity and selectivity. In the reaction process, the reactant (2,5-HD) is first converted into the intermediate 2,5-HDO by hydrogenation, which is further dehydrated to the desired product DMTHF. Pt/C and AM-2 promote the reaction cooperatively. We believe that the highly efficient, greener, facilely prepared, and easily recoverable catalytic system has great potential application for the reaction.
Acknowledgements
The authors thank the National Natural Science Foundation of China and the Chinese Academy of Sciences for financial support. This work was supported by the National Natural Science Foundation of China (21003133, 21321063), and the Chinese Academy of Sciences (KJCX2.YW.H30).
Notes and references
-
(a) M. Y. He, Y. H. Sun and B. X. Han, Angew. Chem., Int. Ed., 2013, 52, 9620 CrossRef CAS PubMed;
(b) G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044 CrossRef CAS PubMed;
(c) C. H. Zhou, X. Xia, C. X. Lin, D. S. Tong and J. Beltramini, Chem. Soc. Rev., 2011, 40, 5588 RSC;
(d) M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827 CrossRef CAS PubMed;
(e) J. L. Song, H. L. Fan, J. Ma and B. X. Han, Green Chem., 2013, 15, 2619 RSC.
-
(a) A. Q. Wang and T. Zhang, Acc. Chem. Res., 2013, 46, 1377 CrossRef CAS PubMed;
(b) B. B. Zhang, J. L. Song, G. Y. Yang and B. X. Han, Chem. Sci., 2014, 5, 4656 RSC.
-
(a) J. B. Binder and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 1979 Search PubMed;
(b) H. B. Zhao, J. E. Holladay, H. Brown and Z. C. Zhang, Science, 2007, 316, 1597 CrossRef CAS PubMed.
-
(a) X. L. Du, L. He, S. Zhao, Y. M. Liu, Y. Cao, H. Y. He and K. N. Fan, Angew. Chem., Int. Ed., 2011, 50, 7815 CrossRef CAS PubMed;
(b) Z. Yang, Y. B. Huang, Q. X. Guo and Y. Fu, Chem. Commun., 2013, 49, 5328 RSC;
(c) D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Green Chem., 2013, 15, 584 RSC;
(d) J. L. Song, L. Q. Wu, B. W. Zhou, H. C. Zhou, H. L. Fan, Y. Y. Yang, Q. L. Meng and B. X. Han, Green Chem., 2015, 17, 1626 RSC;
(e) H. C. Zhou, J. L. Song, H. L. Fan, B. B. Zhang, Y. Y. Yang, J. Y. Hu, Q. G. Zhu and B. X. Han, Green Chem., 2014, 16, 3870 RSC.
-
(a) G. Bottari, A. J. Kumalaputri, K. K. Krawczyk, B. L. Feringa, H. J. Heeres and K. Barta, ChemSusChem, 2015, 8, 1323 CrossRef CAS PubMed;
(b) M. Chatterjee, T. Ishizaka and H. Kawanami, Green Chem., 2014, 16, 1543 RSC;
(c) X. Kong, R. X. Zheng, Y. F. Zhu, G. Q. Ding, Y. L. Zhu and Y. W. Li, Green Chem., 2015, 17, 2504 Search PubMed;
(d) B. Saha, C. M. Bohn and M. M. Abu-Omar, ChemSusChem, 2014, 7, 3095 CrossRef CAS PubMed;
(e) Y. Román-Leshkov, C. J. Barrett, Z. Y. Liu and J. A. Dumesic, Nature, 2007, 447, 982 CrossRef PubMed.
-
(a) J. Mitra, X. Zhou and T. Rauchfuss, Green Chem., 2015, 17, 307 RSC;
(b) A. J. Kumalaputri, G. Bottari, P. M. Erne, H. J. Heeres and K. Barta, ChemSusChem, 2014, 7, 2266 CrossRef CAS PubMed.
- R. J. Sullivan, E. Latifi, B. K. M. Chung, D. V. Soldatov and M. Schlaf, ACS Catal., 2014, 4, 4116 CrossRef CAS.
- L. J. Xu, Y. Y. Jiang, Q. Yao, Z. Han, Y. Zhang, Y. Fu, Q. X. Guo and G. W. Huber, Green Chem., 2015, 17, 1281 RSC.
-
(a) Q. N. Xia, Q. Cuan, X. H. Liu, X. Q. Gong, G. Z. Lu and Y. Q. Wang, Angew. Chem., Int. Ed., 2014, 53, 9755 CrossRef CAS PubMed;
(b) M. Mascal, S. Dutta and I. Gandarias, Angew. Chem., Int. Ed., 2014, 53, 1854 CrossRef CAS PubMed;
(c) X. Y. Li, R. Shang, M. C. Fu and Y. Fu, Green Chem., 2015, 17, 2790 RSC.
-
(a) Y. H. Xiu, A. J. Chen, X. R. Liu, C. Chen, J. Z. Chen, L. Guo, R. Zhang and Z. S. Hou, RSC Adv., 2015, 5, 28233 RSC;
(b) W. W. Zhu, H. M. Yang, J. Z. Chen, C. Chen, L. Guo, H. M. Gan, X. G. Zhao and Z. S. Hou, Green Chem., 2014, 16, 1534 RSC.
- H. K. Chakravarty and R. X. Fernandes, J. Phys. Chem. A, 2013, 117, 5028 CrossRef CAS PubMed.
- M. R. Grochowski, W. R. Yang and A. Sen, Chem. – Eur. J., 2012, 18, 12363 Search PubMed.
- W. R. Yang and A. Sen, ChemSusChem, 2010, 3, 597 CrossRef CAS PubMed.
-
(a) T. L. Church, Y. D. Y. L. Getzler and G. W. Coates, J. Am. Chem. Soc., 2006, 128, 10125 CrossRef CAS PubMed;
(b) M. Hansson, P. I. Arvidsson, S. O. N. Lill and P. Ahlberg, J. Chem. Soc., Perkin Trans. 2, 2002, 763 RSC.
-
(a) Á. Molnár, I. Ledneczki, I. Bucsi and M. Bartók, Catal. Lett., 2003, 89, 1 CrossRef;
(b) J. F. Costello, W. N. Draffin and S. P. Paver, Tetrahedron, 2005, 61, 6715 CrossRef CAS;
(c) L. Vilcocq, A. Cabiac, C. Especel, S. Lacombe and D. Duprez, J. Catal., 2014, 320, 16 CrossRef CAS.
-
(a) F. Marchetti, G. Pampaloni and S. Zacchini, Inorg. Chem., 2008, 47, 365 CrossRef CAS PubMed;
(b) R. Krishnan and R. H. Schultz, Organometallics, 2001, 20, 3314 CrossRef CAS;
(c) C. Su, J. Guang and P. G. Williard, J. Org. Chem., 2014, 79, 1032 CrossRef CAS PubMed.
- Y. Sasaki, M. Handa, S. Sekiya, K. Kurashima and K. Usami, J. Power Sources, 2001, 97–98, 561 CrossRef CAS.
-
(a) M.-J. Kim and I. S. Lee, J. Org. Chem., 1993, 58, 6483 CrossRef CAS;
(b) M. L. Mihailović, S. Gojković and S. Konstantinović, Tetrahedron, 1973, 29, 3675 CrossRef;
(c) Á. Molnár, K. Felfoldi and M. Bartók, Tetrahedron, 1981, 37, 2149 CrossRef.
-
(a) W. R. Yang and A. Sen, ChemSusChem, 2011, 4, 349 CrossRef CAS PubMed;
(b) F. Liu, M. Audemar, K. D. O. Vigier, J.-M. Clacens, F. D. Campo and F. Jérôme, ChemSusChem, 2014, 7, 2089 CrossRef CAS PubMed.
- H. C. Zhou, J. L. Song, X. C. Kang, J. Y. Hu, Y. Y. Yang, H. L. Fan, Q. L. Meng and B. X. Han, RSC Adv., 2015, 5, 15267 RSC.
-
(a) Z. H. He, Z. S. Hou, Y. P. Luo, Y. Dilixiati and W. M. J. Eli, Catal. Sci. Technol., 2014, 4, 1092 RSC;
(b) G. Morales, M. Paniagua, J. A. Melero, G. Vicente and C. Ochoa, Ind. Eng. Chem. Res., 2011, 50, 5898 CrossRef CAS;
(c) Q. L. Meng, H. L. Fan, H. Z. Liu, H. C. Zhou, Z. H. He, Z. W. Jiang, T. B. Wu and B. X. Han, ChemCatChem, 2015, 7, 2831 CrossRef CAS;
(d) B. B. Bardin, S. V. Bordawekar, M. Neurock and R. J. Davis, J. Phys. Chem. B, 1998, 102, 10817 CrossRef CAS.
- A. Yamaguchi, N. Hiyoshi, O. Sato and M. Shirai, ACS Catal., 2011, 1, 67 CrossRef CAS.
- M. L. Mihailović, S. Gojković and Ž. Čeković, J. Chem. Soc., Perkin Trans. 1, 1972, 2460 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5gc01741f |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.