
Light driven styrene epoxidation and hydrogen generation using H2O as an oxygen source in a photoelectrosynthesis cell†
P.
Farràs
ab,
C.
Di Giovanni
a,
J. N.
Clifford
a,
P.
Garrido-Barros
a,
E.
Palomares
*ac and
A.
Llobet
*ad
aInstitute of Chemical Research of Catalonia (ICIQ), Barcelona Institute of Science and Technology, Avinguda Països Catalans 16, E-43007 Tarragona, Spain. E-mail: epalomares@iciq.es; allobet@iciq.es
bMolecular Photonics Laboratory, School of Chemistry, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK
cICREA, Avinguda Lluís Companys 28, Barcelona E-08030, Spain
dDepartament de Química, Universitat Autònoma de Barcelona, Cerdanyola del Vallès, E-08193 Barcelona, Spain
First published on 2nd October 2015
Abstract
A dye-sensitized photoelectrosynthesis cell (DSPEC) has been prepared for the oxidation of alkenes to epoxides and evolution of hydrogen using water as an oxygen source and sunlight. A Ru oxidation catalyst, 2,2+, is used in the homogeneous phase in the anodic compartment to oxidize a water-soluble alkene, 4-styrene sulfonic acid (4-HSS), to the corresponding epoxide that in acidic solution is hydrolyzed to the diol (4-(1,2-dihydroxyethyl)-benzenesulfonic acid). Concomitantly, protons are generated that diffuse through a proton exchange membrane to a Pt cathode where they are transformed into hydrogen. Illumination under 1.5 AMG (100 mW cm−2) together with an external bias of 0.3 V vs. NHE after 24 h leads to the generation of 1.28 Coulombs together with the formation of 6.1 μmol of H2 at the cathodic compartment that corresponds to a faradaic efficiency of 92%. In addition 0.7 mM of the 4-HSS substrate has been oxidized at the anodic compartment with a conversion yield of 7%. The rate of hydrogen evolution is limited by the oxidation of the organic substrate, and the TOF for both reactions is measured to be 0.8 ks−1.
Introduction
Epoxides are highly useful intermediates for the production of a variety of important commercial products,1,2 and therefore their synthesis is a subject of substantial academic and industrial interest. Olefin epoxidation is one of the main paths that lead to the production of epoxides both at a laboratory scale and at the industrial scale.3 While molecular oxygen can be used for the epoxidation of ethylene, the majority of alkene epoxidation reactions are carried out using stoichiometric amounts of alkylhydroperoxides or peracids, with the formation of the corresponding alcohol or carboxylic acid respectively as an undesired side product. In addition, from a safety perspective, peracids are relatively hazardous chemicals. In this respect, there is a strong need for the development of new epoxidation methods that make use of safer oxidants and minimize waste products. The employment of hydrogen peroxide is an attractive option both on environmental and economic grounds,4–7 giving just water as a byproduct. Another option is to use water as the source of oxygen for the epoxidation reaction with the formation of hydrogen as a valuable side product as indicated in the equation below,8 which involves the input of 37.35 kcal mol−1 given its uphill thermodynamics that can be provided thermally9,10 or photochemically11 with sunlight. This strategy also benefits from the formation of hydrogen as a byproduct that is also a clean energy vector.12,13 Given the fact that the worldwide production of ethylene oxide represents 20 million tons per year14 and assuming that the reaction is driven by sunlight and with 100% atom economy, it would represent around 0.9 million tons of hydrogen produced in that process or 2500 tons of hydrogen per day. In terms of energy, this represents the production of an impressive 2700 MWh per day just for this reaction. The same conceptual scheme can be applied for the oxidation of aldehydes to acids, the oxidation of organic sulfides to sulfoxides, etc.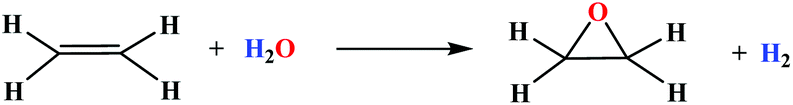 | (1) |
To achieve the reaction shown in eqn (1), a set of reactions catalyzed by transition metal complexes is needed. In the thermal case,9 Ru pincer type complexes have been used for the oxidation of aldehydes to carboxylic acids and the formation of hydrogen with a single catalyst in the homogeneous phase.
A different strategy consists of taking advantage of the intrinsic redox nature of the chemical reactions occurring and separating the single reaction shown in eqn (1) into two half-reactions. This would allow building an electrochemical cell where the oxidation of the alkene will take place at the anode and proton reduction will occur at the cathode, as depicted for styrene in eqn (2) and (3) respectively.
 | (2) |
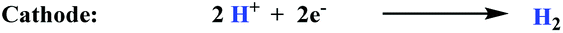 | (3) |
Furthermore, if the reaction is to be driven photochemically, a light harvesting molecule is needed since sunlight interacts with H2O and alkenes mainly at a vibrational level. Thus a series of additional reactions involving light harvesting molecules capable of interacting with suitable catalysts will have to be designed in order to break and form the desired bonds at a reasonable rate.
The oxidation of substrates induced by visible light using water as an oxygen source has previously been described in the literature in the homogeneous phase using [Ru(bpy)3]2+ (bpy is 2,2′-bypyridine) as a light harvesting molecule coupled to a sacrificial electron acceptor (SEA), such as sodium persulfate or Co(III) complexes like [Co(NH3)5Cl]2+.15–19 Other examples include the use of catalyst–photosensitizer dyad systems that can oxidize alcohols, sulfides and alkenes.20–25 On the other hand the reduction of protons has also been achieved with a variety of sacrificial electron donors (SEDs).26,27 In both cases the sacrificial agents react fast and in an irreversible manner so that they favor only the desired reactions. The development of photoelectrosynthesis cells (DSPECs) to carry out the reaction in eqn (1) implies necessarily the absence of sacrificial agents and thus introduces further difficulties in its overall implementation.28–37 Today there is an urgent need to build DSPECs and understand and master all the kinetic and thermodynamic parameters involved in such a cell so that efficient devices can be obtained.
With all this in mind, we have designed a DSPEC using FTO/TiO2-P-bpy-Ru (where FTO is fluorine doped tin oxide, P-bpy-Ru is [RuII(P-bpy)(bpy)2]2+, P-bpy is 2,2′-bipyridine-4,4′-bis(phosphonic acid), bpy is 2,2′-bypyridine) as a photoanode and a Pt mesh as a cathode. The oxidation catalyst will be present in the homogeneous phase since this configuration gives a high degree of flexibility with regard to the selection and use of potential catalysts and reactions. The latter has to comply with two indispensable requirements: (a) the catalyst has to act efficiently with regard to substrate oxidation and (b) the active site of the catalyst needs to be regenerated very fast by the photoanode. Finally, a specific proton exchange membrane based on a combination of Nafion® and poly(3,4-ethylenedioxythiophene) (PEDOT) has been designed so that cationic species in the anodic compartment do not block the proton channel.
Here we report a two compartment DSPEC for H2 generation and epoxidation of alkenes using visible light and water and its complete kinetic and thermodynamic characterization.
Results and discussion
Alkene epoxidation catalyzed by 2,2+ in the homogeneous phase
We have recently reported a dinuclear Ru-aqua 2,2+ complex, whose structure is shown in Fig. 1 and that contains a dinucleating trianionic ligand backbone (pyrazolate-3,5-dicarboxylate, pdc3−) that bridges the two Ru metal centers and a tridentate meridional ligand (2,2′:6′,2′′-terpyridine, trpy) that completes the octahedral geometry around the metal center. Complex 2,2+ has been shown to be a powerful epoxidation catalyst for a variety of alkenes including styrene (TOFi > 2.9 s−1)38 in organic solvents and using PhIO as a sacrificial oxidant. The catalytic mechanism proposed is shown in Fig. 1. In order to introduce the 2,2+ catalyst into a DSPEC it is indispensable that it also works in water and is activated by 1e− outer sphere electron transfer oxidants such as [Ru(bpy)3]3+. For this purpose we have carried out the photochemically induced generation of [Ru(bpy)3]3+ in water using [Co(NH3)5Cl]2+ (CoIII) as a sacrificial electron acceptor in the presence of the water soluble sodium 4-styrene sulfonate as the substrate. Since we have carried out all the reactions at pH = 1.0, the latter is protonated forming 4-styrene sulfonic acid (4-HSS).39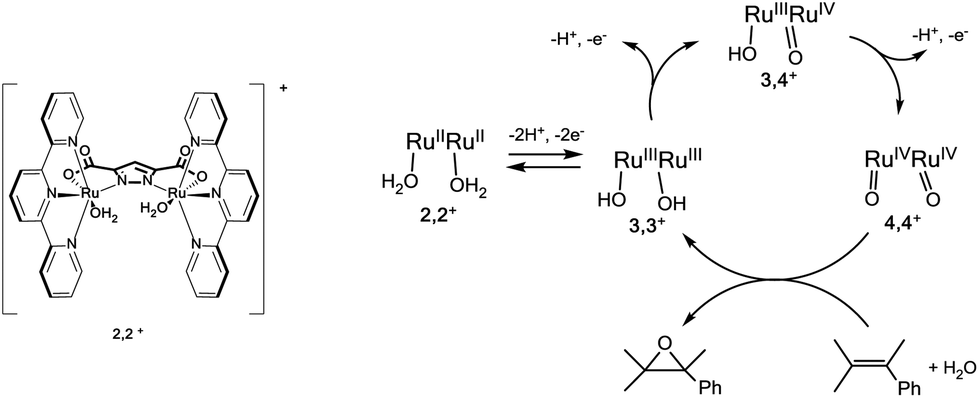 | ||
| Fig. 1 Drawn structure for the 2,2+ complex and the catalytic scheme proposed for the epoxidation of styrene by the active species derived from 2,2+. In the scheme the 2,2′:6′,2′′-terpyridine (trpy) and pyrazolate-3,5-dicarboxylate (pdc3−) ligands are not shown. | ||
A deoxygenated solution containing 2,2+ 0.01 mM/4-HSS 10 mM/[Ru(bpy)3]2+ 0.1 mM/CoIII 10 mM in 4 mL of a D2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CF3SO3D mixture (pD 1.0) was irradiated with visible light for 30 min and generated 1.1 mM 4-(1,2-dihydroxyethyl)-benzenesulfonic acid. The latter is a known phenomenon due to the ring-opening hydrolysis of the epoxide under acidic conditions, and was already reported by Fukuzumi et al. for the chemical oxidation of epoxides using a Ru catalyst.40 The conversion of the initial substrate reached 30% that represents a TOF of 0.17 s−1 (see Fig. S1 for details†). This result shows that [Ru(bpy)3]3+ is capable of activating the catalyst to its active state, 4,4+ (see Fig. 1 and eqn (4)), and that the latter is capable of oxidizing the styrene substrate to styrene oxide that is finally hydrolyzed to the corresponding diol (eqn (4–6)). The reaction mechanism for this catalyst was previously studied in our group.38 If no 4,4+ had been produced, only the oxidation by [Ru(bpy)3]3+ would have been observed. In that case, the reaction without a catalyst is not selective and produced only the aldehyde derivative (see Fig. S1†).
:CF3SO3D mixture (pD 1.0) was irradiated with visible light for 30 min and generated 1.1 mM 4-(1,2-dihydroxyethyl)-benzenesulfonic acid. The latter is a known phenomenon due to the ring-opening hydrolysis of the epoxide under acidic conditions, and was already reported by Fukuzumi et al. for the chemical oxidation of epoxides using a Ru catalyst.40 The conversion of the initial substrate reached 30% that represents a TOF of 0.17 s−1 (see Fig. S1 for details†). This result shows that [Ru(bpy)3]3+ is capable of activating the catalyst to its active state, 4,4+ (see Fig. 1 and eqn (4)), and that the latter is capable of oxidizing the styrene substrate to styrene oxide that is finally hydrolyzed to the corresponding diol (eqn (4–6)). The reaction mechanism for this catalyst was previously studied in our group.38 If no 4,4+ had been produced, only the oxidation by [Ru(bpy)3]3+ would have been observed. In that case, the reaction without a catalyst is not selective and produced only the aldehyde derivative (see Fig. S1†).
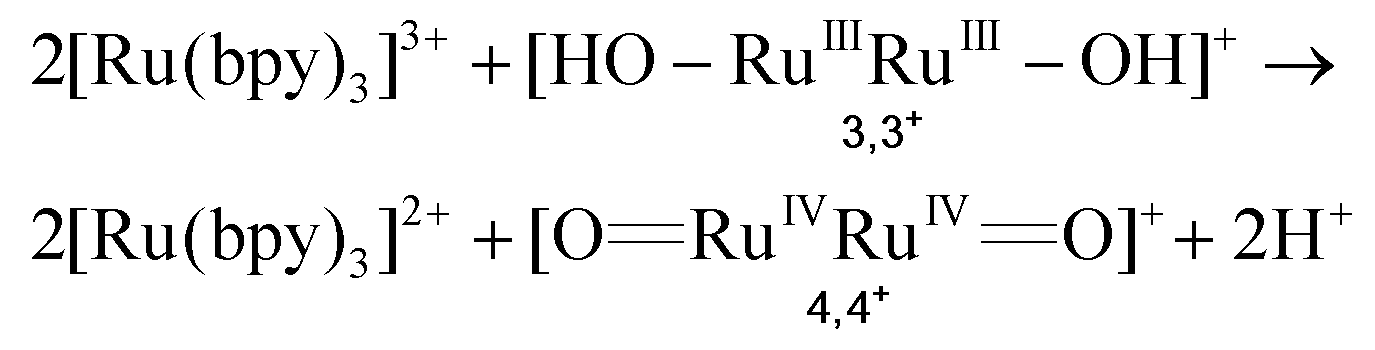 | (4) |
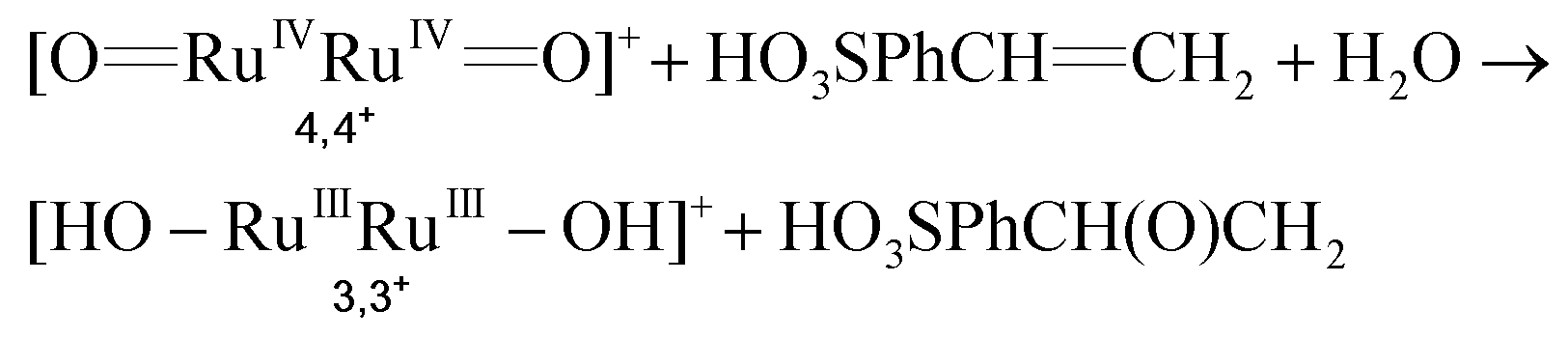 | (5) |
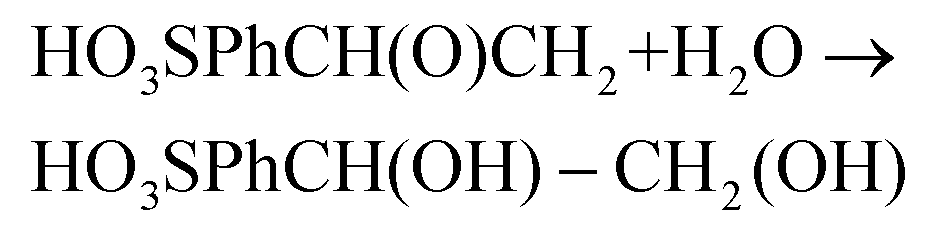 | (6) |
The performance of the photoanode
As a photoanode we use FTO coated with TiO2 containing both P-bpy-RuII,2+ and tert-butylphosphonic acid (P-tBu) (see Fig. S3† for UV-vis spectroscopic characterization). The latter minimizes interactions of the catalyst with the TiO2 surface and thus avoids undesirable back electron transfer (ET) reactions, improving the yield of the productive reactions. The same strategy has been used in dye sensitized solar cells (DSCs) with phosphinate amphiphiles.41,42 We label this photoanode as FTO/TiO2-P-bpy-Ru, TiO2-P-tBu/ and a schematic drawing of its structure is presented in Fig. 2. Transient absorption spectroscopy (TAS) was carried out in 0.1 M LiClO4/HClO4 aqueous solution at pH = 1.0 with this photoanode in the presence of the catalyst and the substrate in order to check the capacity of this anode to activate the catalyst with light and to kinetically characterize the reactions involved.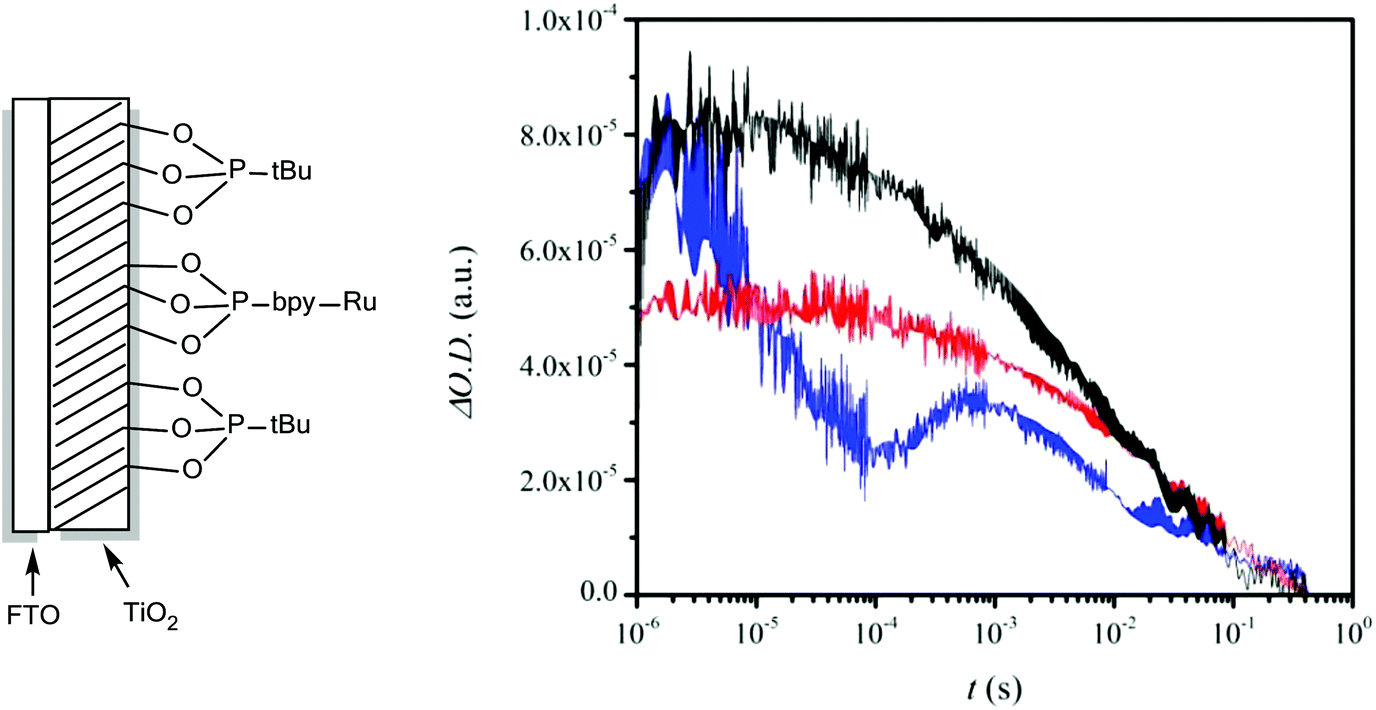 | ||
| Fig. 2 Left, schematic drawing of a photoanode. Right, transient absorption spectroscopy (TAS) kinetics of FTO/TiO2-P-bpy-Ru, TiO2-P-tBu photoanodes in 0.1 M LiClO4/HClO4 aqueous solution (pH 1.0) in the presence of 42.5 mM 4-HSS. Black, in the absence of the catalyst; red, with the addition of 30 μM 3,3+; blue, with the addition of 220 μM 3,3+. TAS kinetics were recorded under 10 mW cm−2, using laser excitation pulses at 450 nm and recorded at 650 nm. | ||
Fig. 2 shows the results obtained upon illumination under 10 mW cm−2, using laser excitation pulses at 450 nm and recording at 650 nm where the anchored [Ru(bpy)3]3+ moiety has a broad absorption band. In black is shown the kinetics in the absence of the catalyst whereas the red trace contains 30 μM 3,3+ and the blue one 220 μM 3,3+. We added the catalyst in the oxidation state 3,3 since, as we have shown previously,38 the lower oxidation states are a precursor to the real catalytic species that are in oxidation states 3,4+ and 4,4+ as indicated on the right hand side of Fig. 1.
In Fig. 3 is depicted a scheme that summarizes the most significant reactions that can occur at the photoanode compartment and that will be analyzed in detail here. In the absence of a catalyst the system decays with a half lifetime of about 4.1 ms which is attributed to the recombination reaction shown in eqn (7) (red arrow 6 in Fig. 3) once the electron is injected into the TiO2 conduction band. The latter is known to happen within the picosecond time scale.43
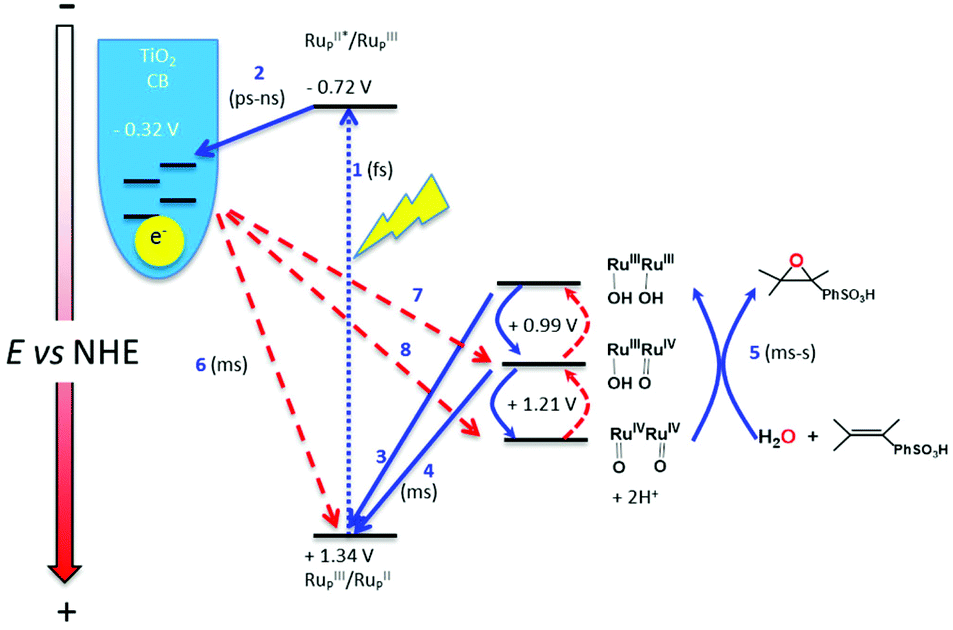 | ||
| Fig. 3 Scheme showing the most significant reactions that can occur at the photoanode compartment. Red arrows indicate unproductive reactions whereas blue arrows are the desired reactions. | ||
In the presence of a low concentration of catalyst 3,3+ the bpy-RuIII,3+ signal decays at a similar rate. However as the catalyst concentration is increased from 30 to 220 μM the decay of the bpy-RuIII,3+ signal becomes faster by almost an order of magnitude with a t1/2 ≈ 0.02 ms. A dependence of dye regeneration on redox couple concentration is also not uncommon in DSCs.44,45 The notable increase in signal intensity at slower timescales (0.1–1 ms) is attributed to the formation of a new species which we ascribe to the 4,4+ state of the catalyst according to eqn (8) and (9) (blue arrows 3 and 4 in Fig. 3). The decay of the 4,4+ state is due to the oxidation of the 4-HSS substrate concomitant with the generation of the catalyst, 3,3+ species (blue arrow 5 in Fig. 3). Due to the low signal-to-noise ratio of the kinetic traces we were not able to record the transient spectra of the long-lived species; however, we note that the 4,4+ state of the catalyst (generated in solution by bulk electrolysis at 1.12 V) also has an absorption band at 650 nm (see Fig. S2†).
| (−)TiO2-P-bpy-RuIII,3+ → TiO2-P-bpy-RuII,2+ | (7) |
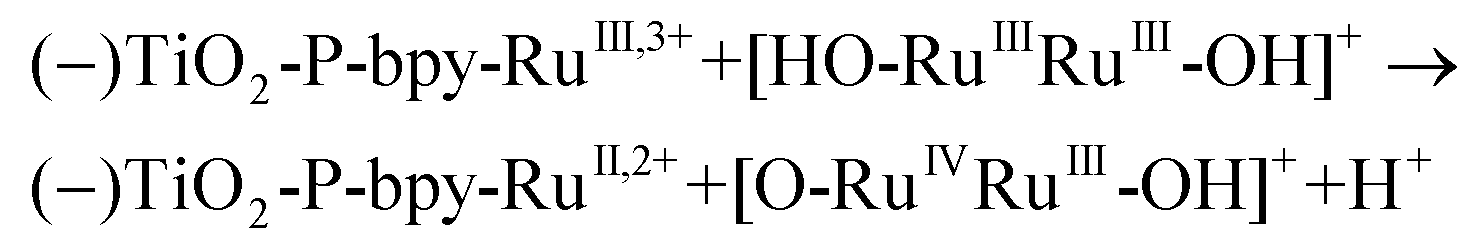 | (8) |
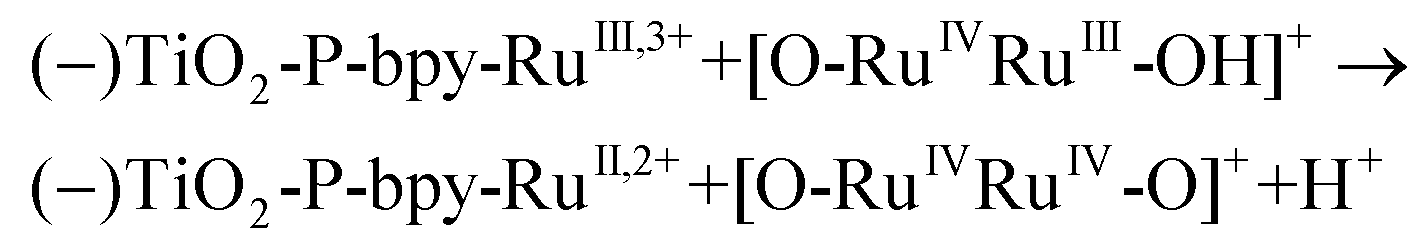 | (9) |
The fast kinetics observed in the catalyst oxidation all the way to 4,4+ together with the generation of the oxidized substrate (vide infra) indicates that the undesired back electron transfer from the (−)TiO2 and the catalyst (red arrows 7 and 8 in Fig. 3) takes place on longer than ms timescales. On the other hand this fast forward kinetics enables the use of this setup for the construction of a DSPEC, as described in the next section.
The photoelectrosynthesis cell
As a proton exchange membrane, Nafion® has been widely used in fuel cells.11,46 However, in our case, catalytic cationic species present in solution would be adsorbed on the surface of the Nafion® film, blocking the membrane and sequestering the catalyst. To avoid this, we have deposited a film of poly(3,4-ethylenedioxythiophene) (PEDOT)47 on the surface of Nafion® and generated a new membrane with practically the same conductivity and proton transfer kinetics as Nafion® and where the catalyst is not adsorbed (see ESI and Fig. S3† for details). With this membrane we have built a two compartment DSPEC using FTO/TiO2-P-bpy-Ru, TiO2-P-tBu/ as a photoanode and a Pt mesh as the cathode. The anodic compartment contains a 0.1 M solution of LiClO4 and 2,2+ in a 0.1 M perchloric acid solution whereas the cathodic compartment contains the same solution in the absence of the catalyst. LiClO4 is added to enhance current densities since it decreases recombination events by stabilizing the TiIII trap sites (see Fig. S5 and S6† for photocurrent enhancement using LiClO4).48,49For this type of cell based on TiO2 as a semiconductor, a small external electrical bias is generally needed.50 In the present case we apply a 0.30 V vs. NHE bias (all redox potentials in this work are reported vs. NHE), which should be a sufficient bias as confirmed by LSV voltammetry shown in Fig. 4 (up) in the presence and absence of sun simulated light irradiation. Once all the parameters have been optimized, the DSPEC is run for 24 h until no more hydrogen is produced under 1.5 AMG (Xe lamp, λ > 400 nm, 100 mW cm−2) with the above-mentioned 0.30 V applied external bias. Both photocurrent intensities and hydrogen evolution are monitored and are depicted in Fig. 4 (down).
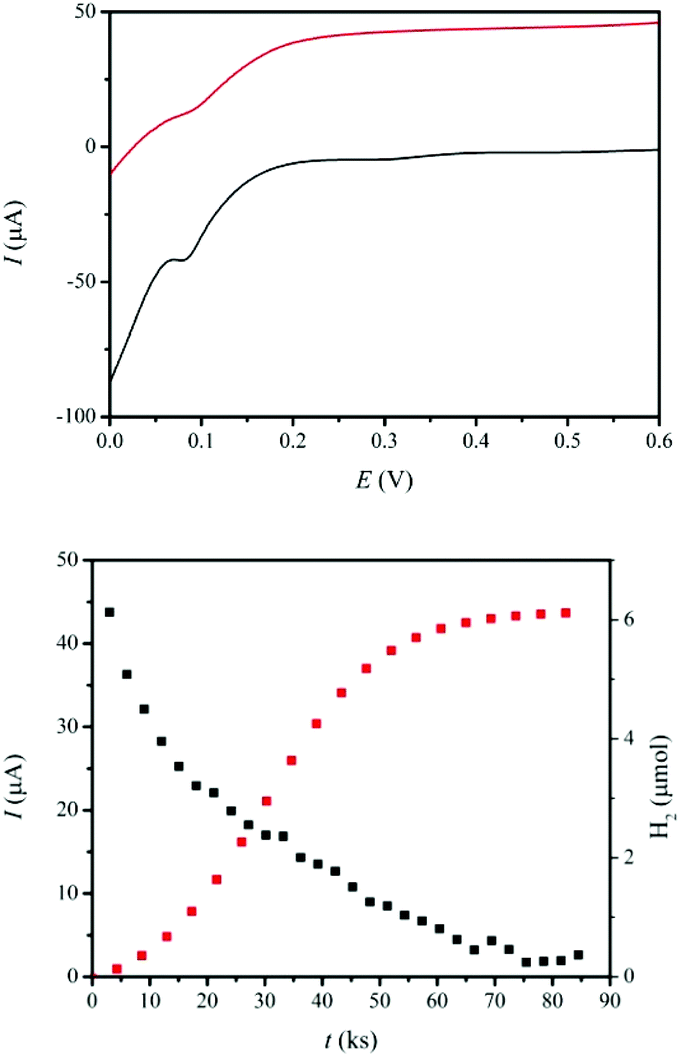 | ||
| Fig. 4 Up, linear sweep voltammetry or the I–V curve of the DSPEC upon 1.5 AMG irradiation with a Xe lamp (λ > 400 nm, 100 mW cm−2) (red line) or without light (black line) at a scan rate of 10 mV s−1. The anodic half-cell contains FTO/TiO2-P-bpy-Ru, TiO2-P-tBu/ (Γ = 8.8 × 10−8 mol cm−2) as the working electrode, 0.01 mM 2,2+ as the catalyst and 4-HSS 20 mM as the substrate. The cathodic half-cell consists of a Pt mesh as the auxiliary electrode. In both cases the supporting electrolyte solution is made of a 0.1 M LiClO4/HClO4 aqueous solution (pH 1.0) up to a total volume of 9 mL in each compartment. Down, hydrogen evolution and current intensity measured at 0.3 V vs. NHE applied bias for the same DSPEC under exactly the same conditions. | ||
After 24 h, 1.28 C (13.2 μmol of electrons) have passed through the external circuit together with the formation of 6.1 μmol of H2 at the cathodic compartment that corresponds to a faradaic efficiency of 92%. In addition, 0.5 mM of the 4-HSS substrate have been oxidized to 4-(1,2-dihydroxyethyl)-benzenesulfonic acid (see Fig. S7†) at the anodic compartment. Overall, 7% conversion yield was found from the disappearance of the starting material, corresponding to 70% selectivity towards diol formation, which is in accordance with the selectivity of the catalyst 2,2+ in chemical oxidation reactions.38 This represents 70 TON with a TOF of 0.8 ks−1 for the oxidation of the organic substrate and 68 TON with a TOF of 0.8 ks−1 for the production of hydrogen. In the absence of the organic substrate or Ru-catalyst or Ru-photosensitizer, the currents obtained were negligible in all cases (see Fig. S6†).
As can be observed in Fig. 4 (down), as the photocatalytic reaction proceeds, the current intensity decreases up to practically zero after 24 h. The main deactivation pathway is de-attachment of the Ru-phosphonate dye from the surface of the TiO2 due to the hydration of the phosphonate bipyridyl ligands (see Fig. S8†). It can thus be inferred that the current intensity decrease over time shown in Fig. 4 (down) basically correlates with Ru leaching. This low stability of the dye on the TiO2-surface is a phenomenon that has been reported earlier51 and that needs to be dramatically minimized in order to build more stable devices. Promising solutions include the use of atomic layer deposition (ALD),52 electropolymerization of alkene functionalized dyes53 or dip-coating of polymers,54 all of which have proven to improve the stability of photosensitizers in DSPEC.
This model DSPEC represents the first example of a molecular device that performs the epoxidation of an alkene and the production of hydrogen simultaneously using the most abundant and green reagents available: sunlight and water. It is also the first example where the catalysts operates in the homogeneous phase and thus gives a large flexibility to the system since the catalyst can be easily changed. The configuration used here in turn also allows foreseeing a large scope, of not only different types of catalytic oxidative reactions but also of substrates to be converted.
The literature related to using water as an oxygen source and at the same time generating dihydrogen is extremely scarce and is limited solely to the oxidation of alcohols.9,36,55 An interesting example is described for the oxidation of benzyl alcohol to benzaldehyde and hydrogen generation but with much lower operation times and much lower faradaic efficiencies.36
Conclusions
The use of water as an oxygen source in combination with sunlight for the oxidation of organic substrates is one of the hot topics that green oxidation chemistry is facing today. In the present paper we present for the first time a model DSPEC that transforms alkenes into the corresponding diols via epoxidation catalysis with simulated sunlight and a small potential applied bias. In addition the byproduct is hydrogen that can be used as a solar fuel. Here we establish the thermodynamics and kinetics of the main reactions involved in the cell. We also underline the unproductive reactions as well as the decomposition pathways that need to be improved to come up with a technologically useful device. It is also important to highlight that the present model DSPEC configuration possesses a high degree of chemical versatility since the catalyst is used in the homogeneous phase and thus it can be adapted to other substrates provided the right catalyst is used.Acknowledgements
We thank MINECO (CTQ-2013-49075-R, SEV-2013-0319, and CTQ2014-52974-REDC), the EU COST actions CM1202 and CM1205, and “LaCaixa Foundation”. PG-B thanks “LaCaixa Foundation” for a PhD grant. CDG is grateful for an FPI grant from MINECO. EP and JC would like to thank ICIQ and ICREA for their financial support.References
- G. Grigoropoulou, J. H. Clark and J. A. Elings, Green Chem., 2003, 5, 1–7 RSC.
- K. Bauer, D. Garbe and H. Surburg, in Common Fragrance and Flavor Materials, Wiley-VCH, New York/Weinheim, 1997 Search PubMed.
- G. Sienel, R. Rieth and K. T. Rowbottom, in Ullmann's Encyclopedia of Organic Chemicals, Wiley-VCH, Weinheim, 1999 Search PubMed.
- K. Sato, M. Aoki, M. Ogawa, T. Hashimoto and R. Noyori, J. Org. Chem., 1996, 61, 8310 CrossRef CAS PubMed.
- K. Sato, M. Aoki, M. Ogawa, T. Hashimoto, D. Panyella and R. Noyori, Bull. Chem. Soc. Jpn., 1997, 70, 905 CrossRef CAS.
- R. R. L. Martins, M. G. P. M. S. Neves, A. J. D. Silvestre, M. M. Q. Simões, A. M. S. Silva, A. C. Tomé, J. A. A. Cavaleiro, P. Tagliatesta and C. Crestini, J. Mol. Catal., 2001, 172, 33 CrossRef CAS.
- R. I. Kureshy, N. H. Khan, S. H. R. Abdi, S. T. Patel and R. V. Jasra, Tetrahedron: Asymmetry, 2001, 12, 433 CrossRef CAS.
- R. H. Holm and J. P. Donahue, Polyhedron, 1993, 12, 571 Search PubMed.
- E. Balaraman, E. Khaskin, G. Leitus and D. Milstein, Nat. Chem., 2013, 5, 122–125 CrossRef CAS PubMed.
- J. H. Choi, L. E. Heim, M. Ahrens and M. H. Prechtl, Dalton Trans., 2014, 43, 17248–17254 RSC.
- P. Farràs, C. Di Giovanni, J. N. Clifford, E. Palomares and A. Llobet, Coord. Chem. Rev., 2015, 304, 202–208 CrossRef.
- D. Kalita, B. Radaram, B. Brooks, P. P. Kannam and X. Zhao, ChemCatChem, 2011, 3, 571–573 CrossRef CAS.
- L. Zang, in Energy Efficiency and Renewable Energy Through Nanotechnology, Springer, Berlin, 2011 Search PubMed.
- Ethylene Oxide, WP Report, SRI Consulting. January 2009. Retrieved 2009-09-29 Search PubMed.
- S. Fukuzumi, T. Kishi, H. Kotani, Y. M. Lee and W. Nam, Nat. Chem., 2011, 3, 38–41 CrossRef CAS PubMed.
- F. Li, M. Yu, Y. Jiang, F. Huang, Y. Li, B. Zhang and L. Sun, Chem. Commun., 2011, 47, 8949–8951 RSC.
- S. Ohzu, T. Ishizuka, Y. Hirai, S. Fukuzumi and T. Kojima, Chem. – Eur. J., 2013, 19, 1563–1567 CrossRef CAS PubMed.
- X. Zhou, F. Li, X. Li, H. Li, Y. Wang and L. Sun, Dalton Trans., 2014, 44, 475–479 RSC.
- P. Farràs and A. C. Benniston, Tetrahedron Lett., 2014, 55, 7011 CrossRef.
- W. Chen, F. Rein and R. Rocha, Angew. Chem., Int. Ed., 2009, 121, 9852–9855 CrossRef.
- W. Chen, F. Rein, B. Scott and R. Rocha, Chem. – Eur. J., 2011, 17, 5595–5604 CrossRef CAS PubMed.
- M. Hajimohammadi, N. Safari, H. Mofakham and F. Deyhimi, Green Chem., 2011, 13, 991–997 RSC.
- O. Hamelin, P. Guillo, F. Loiseau, M. Boissonnet and S. Menage, Inorg. Chem., 2011, 50, 7952–7954 CrossRef CAS PubMed.
- P. Farràs, S. Maji, J. Benet-Buchholz and A. Llobet, Chem. – Eur. J., 2013, 19, 7162–7172 CrossRef PubMed.
- T. T. Li, F. M. Li, W. L. Zhao, Y. H. Tian, Y. Chen, R. Cai and W. F. Fu, Inorg. Chem., 2015, 54, 183–191 CrossRef CAS PubMed.
- Z. Han and R. Eisenberg, Acc. Chem. Res., 2014, 47, 2537–2544 CrossRef CAS PubMed.
- S. Berardi, S. Drouet, L. Francàs, C. Gimbert-Suriñach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501–7519 RSC.
- W. Youngblood, S. Lee, Y. Kobayashi, E. Hernandez-Pagan, P. Hoertz, T. Moore, A. Moore, D. Gust and T. Mallouk, J. Am. Chem. Soc., 2009, 131, 926–927 CrossRef CAS PubMed.
- R. Brimblecombe, A. Koo, G. Dismukes, G. Swiegers and L. Spiccia, J. Am. Chem. Soc., 2010, 132, 2892–2289 Search PubMed.
- L. Li, L. Duan, Y. Xu, M. Gorlov, A. Hagfeldt and L. Sun, Chem. Commun., 2010, 46, 7307 RSC.
- G. F. Moore, J. D. Blakemore, R. L. Milot, J. F. Hull, H. E. Song, L. Cai, C. A. Schmuttenmaer, R. H. Crabtree and G. W. Brudvig, Energy Environ. Sci., 2011, 4, 2389 CAS.
- Y. Gao, X. Ding, J. Liu, L. Wang, Z. Lu, L. Li and L. Sun, J. Am. Chem. Soc., 2013, 135, 4219–4222 CrossRef CAS PubMed.
- L. Alibabaei, M. K. Brennaman, M. R. Norris, B. Kalanyan, W. Song, M. D. Losego, J. J. Concepcion, R. A. Binstead, G. N. Parsons and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20008 CrossRef CAS PubMed.
- Y. Gao, L. Zhang, X. Ding and L. Sun, Phys. Chem. Chem. Phys., 2014, 16, 12008 RSC.
- X. Ding, Y. Gao, L. Zhang, Z. Yu, J. Liu and L. Sun, ACS Catal., 2014, 4, 2347 CrossRef CAS.
- W. Song, A. K. Vannucci, B. H. Farnum, A. M. Lapides, M. K. Brennaman, B. Kalanyan, L. Alibabaei, J. J. Concepcion, M. D. Losego, G. N. Parsons and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 9773 CrossRef CAS PubMed.
- Z. Yu, F. Li and L. Sun, Energy Environ. Sci., 2015, 8, 760–775 CAS.
- C. Di Giovanni, A. Poater, J. Benet-Buchholz, L. Cavallo, M. Solà and A. Llobet, Chem. – Eur. J., 2014, 20, 3898 CrossRef CAS PubMed.
- L. Li, L. Feng, Y. Wei, C. Yang and H. F. Ji, J. Colloid Interface Sci., 2012, 381, 11–16 CrossRef CAS PubMed.
- Y. Hirai, T. Kojima, Y. Mizutani, Y. Shiota, K. Yoshizawa and S. Fukuzumi, Angew. Chem., Int. Ed., 2008, 47, 5772–5776 CrossRef CAS PubMed.
- M. Wang, X. Li, H. Lin, P. Pechy, S. M. Zakeeruddin and M. Grätzel, Dalton Trans., 2009, 10015–10020 RSC.
- D. V. Pogozhev, M. J. Bezdek, P. A. Schauer and C. P. Berlinguette, Inorg. Chem., 2013, 52, 3001–3006 CrossRef CAS PubMed.
- F. Lakadamyali, A. Reynal, M. Kato, J. R. Durrant and E. Reisner, Chem. – Eur. J., 2012, 18, 15464–15475 CrossRef CAS PubMed.
- I. Montanari, J. Nelson and J. R. Durrant, J. Phys. Chem. B, 2002, 106, 12203–12210 CrossRef CAS.
- J. N. Clifford, E. Palomares, M. K. Nazeeruddin, M. Grätzel and J. R. Durrant, J. Phys. Chem. C, 2007, 111, 6561–6567 CAS.
- Z. Qi, in Proton Exchange Membrane Fuel Cells, CRC Press, 2013 Search PubMed.
- L. Li, J. Drillet, R. Dittmeyer and K. Jüttner, J. Solid State Electrochem., 2006, 10, 708–713 CrossRef CAS.
- M. K. Brennaman, A. P. T. Patrocinio, W. Song, J. W. Jurss, J. J. Concepcion, P. G. Hoertz, M. C. Traub, N. Y. Murakami Iha and T. J. Meyer, ChemSusChem, 2011, 4, 216–227 CrossRef CAS PubMed.
- J. E. Benedetti, M. A. de Paoli and A. F. Nogueira, Chem. Commun., 2008, 1121–1123 RSC.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- K. Hanson, M. K. Brennaman, H. Luo, C. R. Glasson, J. J. Concepcion, W. Song and T. J. Meyer, ACS Appl. Mater. Interfaces, 2012, 4, 1462–1469 CAS.
- K. Hanson, M. D. Losego, B. Kalanyan, D. L. Ashford, G. N. Parsons and T. J. Meyer, Chem. Mater., 2013, 25, 3–5 CrossRef CAS.
- A. M. Lapides, D. L. Ashford, K. Hanson, D. A. Torelli, J. L. Templeton and T. J. Meyer, J. Am. Chem. Soc., 2013, 135, 15450–15458 CrossRef CAS PubMed.
- K.-R. Wee, M. K. Brennaman, L. Alibabaei, B. H. Farnum, B. Sherman, A. M. Lapides and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 13514–13517 CrossRef CAS PubMed.
- T. P. A. Ruberu, N. C. Nelson, I. I. Slowing and J. Vela, J. Phys. Chem. Lett., 2012, 3, 2798–2802 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section containing the preparation of photoanodes and modified membranes, cell construction and additional spectroscopic, electrochemical, and catalytic data. See DOI: 10.1039/c5gc01589h |
| This journal is © The Royal Society of Chemistry 2016 |