
Towards multi-purpose biorefinery platforms for the valorisation of red grape pomace: production of polyphenols, volatile fatty acids, polyhydroxyalkanoates and biogas†
Gonzalo A.
Martinez
a,
Stefano
Rebecchi
a,
Deborha
Decorti
b,
Joana M. B.
Domingos
a,
Andrea
Natolino
b,
Daniele
Del Rio
 c,
Lorenzo
Bertin
*a,
Carla
Da Porto
b and
Fabio
Fava
a
c,
Lorenzo
Bertin
*a,
Carla
Da Porto
b and
Fabio
Fava
a
aDepartment of Civil, Chemical, Environmental and Materials Engineering (DICAM), University of Bologna, via Terracini, 28, I-40131 Bologna, Italy. E-mail: lorenzo.bertin@unibo.it; Fax: (+39) 051 20 90322; Tel: (+39) 051 20 90317
bDepartment of Food Science, University of Udine, via Sondrio, 2/a, I-33100 Udine, Italy
cDepartment of Food Science, University of Parma, Parco Area delle Scienze 47A, 43124 Parma, Italy
First published on 8th October 2015
Abstract
The development of a multi-purpose four step-cascading biorefinery scheme for the valorization of red grape pomace (GP) was proposed. The consecutive processes were respectively dedicated to (a) the recovery of polyphenols by supercritical CO2 extraction, (b) the production of volatile fatty acids (VFAs) by anaerobic acidogenic digestion, (c) the exploitation of produced VFAs as the precursors for the biotechnological production of polyhydroxyalkanoates (PHAs) and (d) the production of a CH4-rich biogas by the anaerobic digestion of solid leftovers from the acidogenic process. More than 2.7 g of total polyphenols (as gallic acid equivalents) per 100 g of dry matter were extracted. A high content of valuable proanthocyanidins occurred in the recovered polyphenolic fraction. The dephenolized GP was anaerobically digested under batch acidogenic conditions, obtaining about 20 g L−1 of total VFAs in the liquid effluent. The latter matrix was employed to feed a pure culture of a Cupriavidus necator strain, which was induced to produce and store PHAs under nitrogen-limiting conditions. The process was carried out in 0.5 L-shake flasks by using a two-step production approach. In particular, pre-grown biomass was fed with different concentrations of the acidic effluent (20 or 40% v/v) in two sequential batch processes. Poly(3-hydroxybutyrate) was accumulated up to 63% of the cells dry weight when pre-grown biomass was fed with 40% of the acidic effluent. No inhibitory effects due to non-VFA compounds occurring in the actual acidogenic effluent were observed. Finally, the anaerobic digestion of the exhausted solid fraction from the acidogenic process allowed obtaining 113 mL of biomethane per gram of fed volatile solids.
Introduction
According to an estimation reported by the OIV (International Organisation of Vine and Wine), 279 million hectolitres of wine were globally produced in 2014, 44.4 of which was produced in Italy.1 Winemaking processes lead to the generation of significant amount of solid and liquid residues. In particular, grape pomace (GP), which represents the main solid winery waste, consists of about 50% skin, 25% stem and 25% seed.2 Considering that 18 kg of GP is generated on average per 100 L of wine produced,3 about 5 million tons of such residue are annually spawned worldwide, 0.8 of which is in Italy. According to a previous regulation (EC Regulation 1493/99), GP and lees of winery waste had to be processed by distilleries within the EU. Nowadays, a recent European reform in the wine sector (EC Regulation 479/2008) promotes the gradual withdrawal of distillation subsidies and consequently revokes the compulsory distillation. This should drive the promotion of integrated, sustainable and standardized alternative protocols for the valorisation of solid winery waste.2In this frame, the development of multi-purpose cascading biorefinery schemes fed with GP appears to be of great interest. This approach allows obtaining different valuable products by applying consecutive modular processes, along with a more extensive exploitation of organic leftovers, thus minimizing the generation of waste.4,5
The extraction of bioactive compounds from GP can represent an option for valorising the residue. In particular, GP polyphenolic compounds can exert beneficial effects on human health6 and they were found in the grape skin and seeds after the fermentation process for the production of wine. Their extraction from GP was already proposed for recovering highly valuable substances for the cosmetics, food additives (nutraceuticals) and pharmaceutical industries.5 In particular, grape skins contain significant amounts of fibre (17–21%), fats (7–12%), tannins (16–27%) and other polyphenolic compounds (2–6.5%), including catechins, anthocyanins, proanthocyanidins, quercetin, ellagic acid and resveratrol. Grape seeds, in addition to oil, contain approximately 60% of the polyphenols occurring in grapes, with high concentrations of flavan-3-ols, catechins and epicatechins.2 However, the proposed recovery subtracts only a minor organic fraction.
An alternative valorisation of GP could be represented by the production of a methane-rich biogas by anaerobic digestion (AD) processes.7 However, low biomethanization performances were generally achieved. This was ascribed to the high content of lignin, which is not readily fermentable. Moreover, Fabbri et al.8 reported the detection of a significant lag phase during methane production. Inhibition by alcohols and phenols was proposed among possible explanations. Some preliminary GP anaerobic digestion tests, confirming scarce biomethanization of the waste, were carried out also in our labs. High volatile fatty acid (VFA) production, and therefore their accumulation, was supposed as a further inhibitory cause.9 On the other hand, VFAs, i.e., linear short chain (C2–C6) carboxylic acids, are functional molecules, which represent the precursors for the production of reduced added value chemicals (alcohols and aldehydes), polymers and biofuels in the frame of the carboxylate platform.10 Thus, the acidogenic anaerobic digestion (AAD) of GP for the production of VFAs can be considered an alternative low-cost valuable approach for the valorisation of the biowaste.
Besides, VFAs are suitable precursors for the biotechnological production of polyhydroxyalkanoates (PHAs), which are microbial aliphatic polyesters naturally produced by many microorganisms. PHAs can exhibit similar or even better physicochemical properties with respect to those of petrol-based polyolefins.11–14 Nowadays, PHAs are industrially produced by microbial pure cultures commonly fed with glucose.15 Nevertheless, this approach hardly allows an economically competitive polymer production16 when compared to that of petrol-based equivalent molecules, such as polypropylene. Alternative strategies based on the employment of mixed microbial culture (MMC) fed with VFA-rich effluents, which were obtained by digesting different biowastes under acidogenic conditions, were proposed with the aim of lowering the costs associated with the substrate and the process.17 However, even the best results18 showed that low PHA concentrations can be obtained in MMCs effluents, and this negatively affects downstream costs. In addition to this, the employment of MMCs leads to a mixture of polymers instead of a well-defined single polymer type. Therefore, the development of new PHA production processes mediated by pure cultures fed with VFA-rich effluents appears to be of great interest.19
Considering all this, the present work was dedicated to evaluate the technical feasibility of a multi-purpose cascading biorefinery scheme fed with a red GP for the obtainment of polyphenols, VFAs, PHAs and biomethane (Fig. 1). In particular, a supercritical CO2 extraction (SC-CO2) was applied for the recovery of polyphenols. The resulting dephenolised GP (GPDeph) was anaerobically digested under batch acidogenic conditions for the production of a VFA-rich liquid stream (GPAcidDeph). This liquid fraction was employed as the substrate for producing PHAs by a pure culture of Cupriavidus necator. Furthermore, the solid leftover from GPAcidDeph underwent a further methanogenic AD process dedicated to the production of a methane-rich biogas.
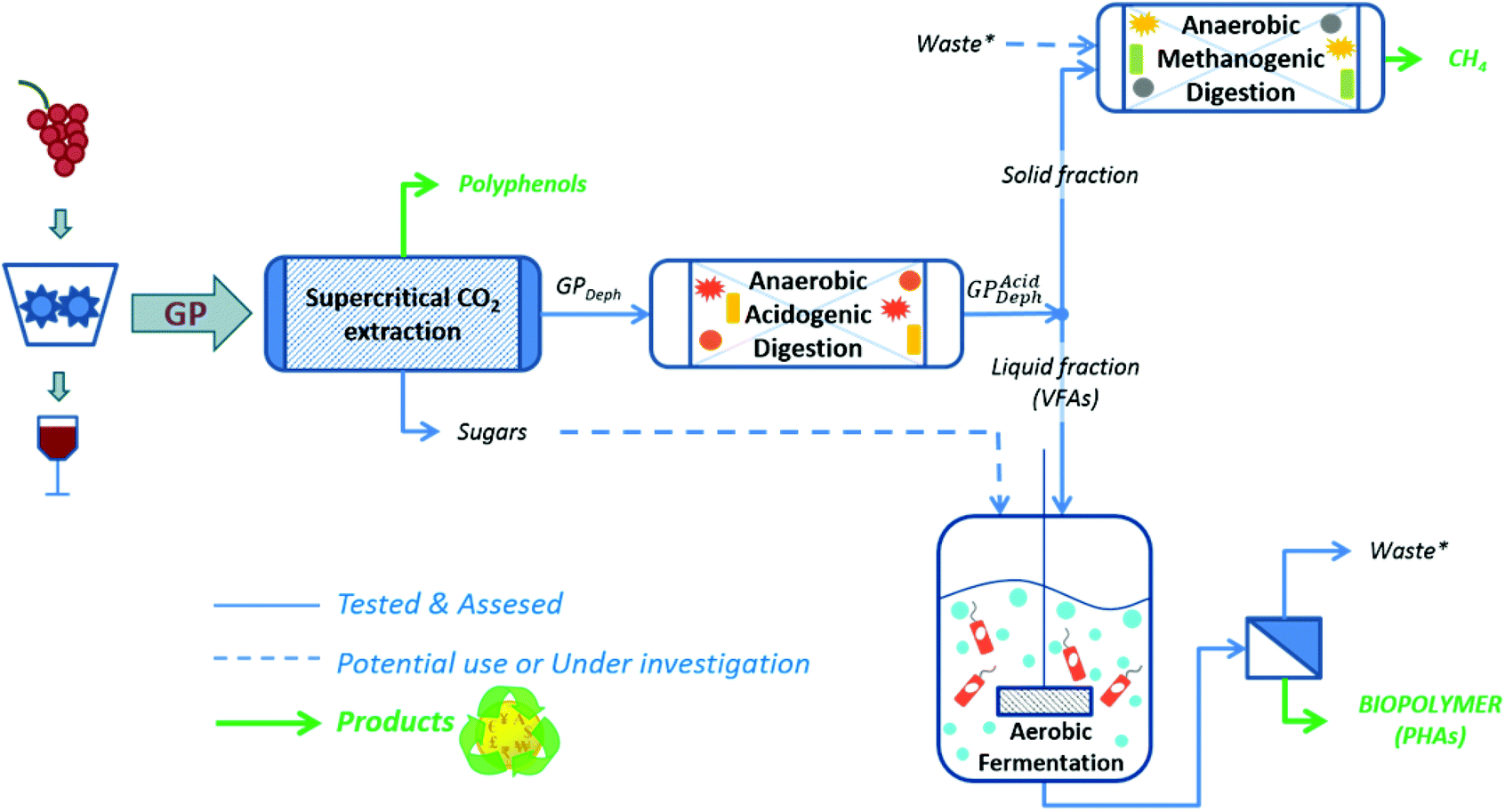 | ||
| Fig. 1 Multi-purpose biorefinery scheme for the obtainment of polyphenols and biopolymer from red grape pomace (GP). | ||
To the very best of our knowledge, this is the first study dedicated to develop an integrated GP valorisation scheme, and, in particular, it represents the first attempt to produce PHAs with a pure culture of C. necator by employing digested GP as an alternative carbon source.
Results and discussion
Several approaches dedicated to the valorisation of grape pomace were reported in the literature, as reviewed by Scoma et al.5 However, most of these processes would hardly be economically feasible at an industrial scale if singularly applied. Conversely, multi-purpose integrated biorefinery could generate some positive synergistic effects, such as (a) cost investment optimization by better exploiting the diverse equipment, (b) diversification of the incoming profits by covering multiple markets/niches, (c) sharing manpower, (d) minimizing waste generation and (e) reaching energy self-sufficiency (e.g. biogas production from waste streams). This strategy could lead to an overall economic sustainability of the employment of biowaste as an innovative renewable and low-cost feedstock.20 In this frame, olive pomace was recently proposed as a raw material for the integrated production of natural antioxidants and renewable energy.21 Moreover, the potential beneficial effects of multi-purpose biorefineries could be further enhanced if more than one waste is valorised. At the same time, this may also represent a solution for the valorisation of seasonal biowaste. As an example, the extraction of polyphenols from olive pomace and GP would allow the facility to run all over the year.According to the mentioned strategy, the present work represents an attempt to evaluate the possibility of valorising a red GP by the integrated production of natural antioxidants, biopolymers and biogas. The four processes included in the proposed GP biorefinery scheme were studied separately and sequentially, in agreement with the cascade approach. Experiments were performed at the bench-top/flask scale. Results are therefore presented according to the same processes and sequence order.
Polyphenol extraction
The extraction of polyphenols from GP was studied by using: (a) supercritical carbon dioxide (SC-CO2) containing 10% ethanol aqueous mixture at 57% (v/v) (SC-CO2 + 10% EtW) and (b) conventional methanol extraction. The results for both methods are reported in Table 1. The process efficiency is quantitatively related to extraction yield. No statistically significant difference (p ≤ 0.01) in the global yield of recovered dry matter (expressed as extracted mass per fed mass) obtained by SC-CO2 and by methanol extractions was highlighted (Table 1).| Extraction methods | ||
|---|---|---|
| Methanol | SC-CO2 + 10% EtW | |
| Each data represents the mean of three replicates ± SD.*Values with different letter within row indicate significant differences (p ≤ 0.05). | ||
| Global yield (% w/w) | 15.6 ± 1.2a* | 14.6 ± 1.5a |
| Total phenols (mgGAE per 100 gDM) | 2813 ± 10.8a | 2527 ± 11.5b |
| Phenolic yield (gGAE per kg extract) | 180.3 ± 0.4a | 173.1 ± 0.5b |
| Phenolic yield (% SC-CO2/methanolic yield) | 100 | 90 |
| Total antioxidant activity (mgα-tocopherol per 100 gDM) | 678 ± 15.5 | 8703 ± 17.5 |
| Proanthocyanidins (mgcatechin per 100 gDM) | ||
| Monomeric fraction | 1.2 ± 0.2 | 188.0 ± 3.8 |
| Oligomeric fraction | 4.1 ± 0.1 | 154.2 ± 5.8 |
| Polymeric fraction | 153.7 ± 0.2 | 361.5 ± 18.6 |
| Antioxidant activity (mgα-tocopherol per 100 gDM) | ||
| Monomeric fraction | 28.1 ± 1.2 | 808.7 ± 10.2 |
| Oligomeric fraction | 30.1 ± 2.4 | 545.8 ± 7.3 |
| Polymeric fraction | 600.5 ± 2.9 | 3675.5 ± 6.8 |
The application of the SC-CO2 extraction allowed recovering 90% of the total polyphenols recovered within the conventional solvent method. The yield was higher than that reported by Farías et al.22 (2200 mgGAE per 100 gDM), as well as the total antioxidant activity.
The obtained results indicate that the extracts recovered by the application of both methods contained a large number of soluble compounds, and that GP polyphenols included flavonoids and non-flavonoids.23 Among the former ones, catechins and their oligomeric and polymeric forms, and procyanidins (PCs), have been reported to exert potential health benefits in humans.24 The healthy properties of catechins and PCs may depend on their structure and on their degree of polymerization. Monomeric structures have been shown to be quite efficiently absorbed, while oligomers reach the large intestine where they are efficiently converted into smaller metabolites by the local colonic microbial community.25 In the present work, the amount of total catechins and PCs obtained by SC-CO2 was 703.7 mg of catechin equivalents per 100 gDM, and monomeric and oligomeric fractions together represented about half of total extracted flavan-3-ols. In particular, the small size oligomeric fraction was composed of several dimeric, trimeric and tetrameric B-type PCs (see ESI Table S1 and Fig. S1†).
The SC-CO2 polyphenol extraction from GP was recently demonstrated to allow better performances with respect to those of a conventional solvent-based approach.2 In fact, even if the total polyphenol extraction yields were nearly the same, the antioxidant activity was one order of magnitude higher when using the SC-CO2. Yet more important, the SC-CO2 extract presented a higher level of total proanthocyanidins (PAs) with monomeric and oligomeric fractions (Table 1). This suggests that supercritical CO2 extraction of PAs from GP is more selective in extracting proanthocyanidin fractions – beneficial for human health – than methanol extraction. Finally, it is worthy of note that about 60% of the total antioxidant activity resulted due to PAs in SC-CO2 + 10% EtW, and 97% in the conventional extraction. This evidence, together with the previous observation indicate that the supercritical operating conditions developed are able to extract not only selectively the PAs, but also a great amount of other antioxidant compounds, not extractable with the conventional method.
VFA production
After polyphenol extraction, the dephenolised leftover (GPDeph) contained 90% of total solids (TSs). Volatile solids (VSs) were 90% of the latter fraction. The application of a batch anaerobic acidogenic wet process onto such an organic matter allowed the accumulation of a mixture of VFAs in the liquid phase. The VFA concentration profile as a function of the experimental time is shown in Fig. 2. The whole AAD lasted 16 days, after which 22.2 ± 0.8 g L−1 of total VFAs were obtained, corresponding to 111 g of total VFAs per kilogram of GPDeph. Among the produced acids, acetic (15.5 g L−1) and butyric (4.3 g L−1) mainly accumulated in the medium. At the end of this process the measured COD of the dephenolised and acidified effluent (GPAcidDeph) was 35 ± 1 g COD L−1. Since the COD due to the occurrence of VFAs (according to stoichiometric calculations) was 28.5 ± 1.5 g COD L−1, more than 80% of the organic matter was represented by the target VFAs.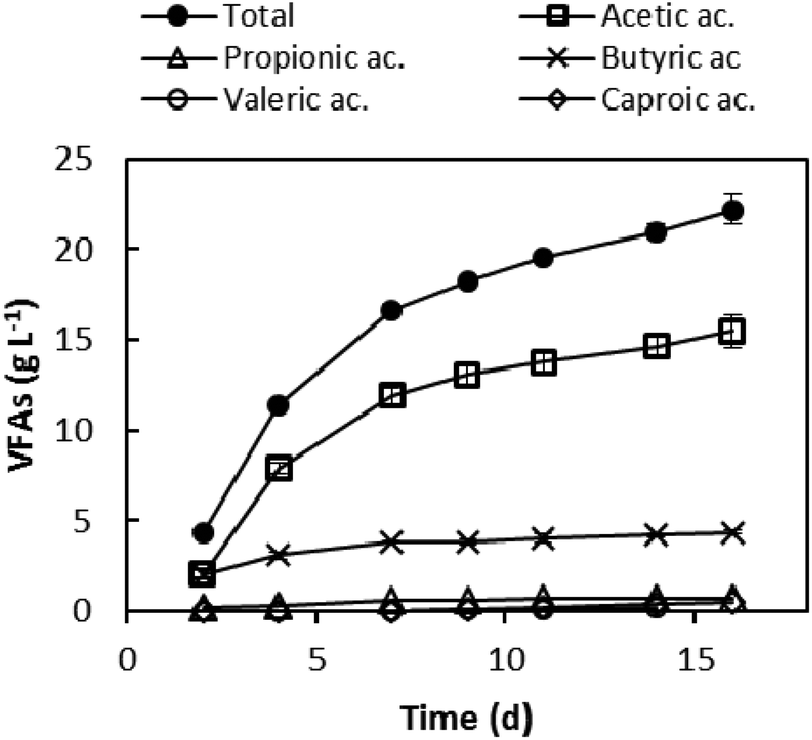 | ||
| Fig. 2 VFA production from GPDeph. Single and total VFA concentration trends. | ||
The final VFA concentration was comparable to that reported in a study where vinasse was used for VFA production (19 g L−1 of total VFAs).26 Furthermore, comparable VFAs’ overall concentration was obtained when the same process was carried out using non-dephenolised GP as the substrate (about 23 g L−1, see ESI Fig. S2†). Taken together, such evidence seems to demonstrate that the preliminary polyphenol extraction process did not significantly lower the potentialities of the acidogenic step, probably both because a large availability of readily biodegradable organics still occurred in the GPAcidDeph and the biological process is inhibited by higher overall VFA concentrations.
9 mL g VS−1 of biogas were produced all through the anaerobic acidogenic digestion. Importantly, no VFA-consuming methanogenic activity was detected, while the overall produced biogas was composed of H2 (35%) and CO2 (65%). The total polyphenol content of the VFA-rich liquid stream was 447 ± 39 mg L−1.
PHA production
Low cost substrates and high polymer amounts per cell dry weights are required in order to persecute economic sustainability of biotechnological PHA production. As a matter of fact, C. necator was found to grow and produce the biopolymer from diverse carbon sources.19,27–32 Among winery waste, wine lees were used as supplementary medium33 and enzyme pre-treated GP (saccharified) was used as a carbon source.34 However, acidified pre-treated GP was never tested as the substrate for the biotechnological production of PHAs. Recently, an effective two-step strategy for the production of PHAs from acidified olive mill wastewater by C. necator was proposed.19 In that work, the advantages of employing a two-stage production process (constituted by a preliminary balanced growth using glucose as the carbon source and a consecutive PHA accumulation step under NH4 limiting conditions by feeding grown cells with VFAs) were discussed. Briefly, the employment of a low-cost alternative carbon source for the accumulation phase would allow replacing a large majority of the costly sugar required by the conventional PHA production process. In fact, PHAs may represent over 80% of the total CDW of C. necator strain.15,35 Hence, the same approach was applied in this work, where grown cells of C. necator were fed (a) with different concentrations of the GPAcidDeph liquid fraction or (b) with aqueous solutions containing the same amount of VFAs occurring in the mentioned experimental VFA-rich substrates.During all experiments, the preliminary growth phase lasted 24.5 hours. The final cell concentration was 2.5 ± 0.3 g L−1 and the glucose consumption was 5.0 ± 0.1 g L−1. Thereafter, cells were harvested and re-suspended in the corresponding medium of each experimental test.
PHA accumulation was observed for all conditions as a linear increase of Abs600 (Fig. 3A). The VFA and PHA profiles as a function of the experimental time are shown in Fig. 3B and C.
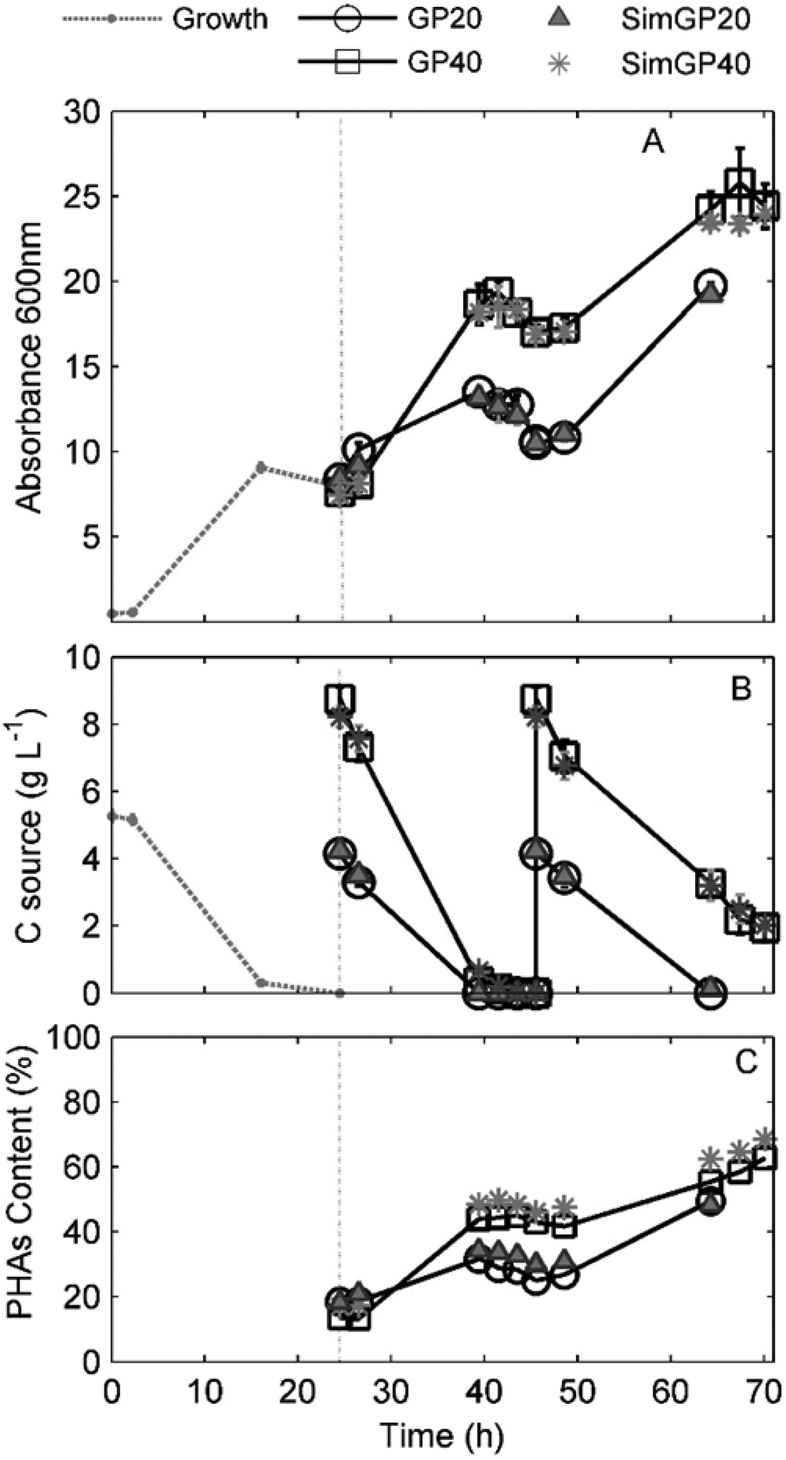 | ||
| Fig. 3 Responses of grown cells to 20% and 40% of GPAcidDeph and SimGPAcidDeph contents in the accumulation media (% v/v). (A) Absorbance (Abs600) values as a function of the time related to growth (–●–) and accumulation phase. (B) Total VFA consumption profiles. (C) PHA content (% on a cell dry weight basis) obtained from GC analyses. | ||
The complete consumption of the carbon sources was detected after 42 hours when GPAcidDeph represented 20% of the accumulation medium (Fig. 3B). Accordingly, a negative slope for biomass concentration, due to the consumption of accumulated PHAs (Fig. 3C), started after 42 h of observation (Fig. 3A). Similar evidence was observed for the 40% conditions, since VFAs were not detected anymore after 44 hours and absorbance started to decrease two hours later (46 h). Therefore, cells were harvested and re-suspended in fresh media for the application of the second accumulation batch process, which lasted 46 hours in all experimental conditions. The 20% conditions were monitored until VFAs were exhausted, which occurred after a complete experimental time of 64 h (Fig. 3B). The 40% conditions were stopped after 70 h since no further significant absorbance increase was detected. At that time, the overall VFA concentration was 2 g L−1. Final PHA content, PHA yields, accumulation rates and final pH values are shown in Table 2. PHA contents, which were measured according to GC analyses, were confirmed by TGA analyses (see ESI Fig. S3†).
| GPAcidDeph | SimGPAcidDeph | |||
|---|---|---|---|---|
| 20% | 40% | 20% | 40% | |
| a Considering only the real accumulation time. b Calculated for the whole second phase duration. | ||||
| PHAsCont (%) | 49 ± 1 | 63 ± 3 | 48 ± 1 | 68 ± 1 |
| Y PHAs/VFAs (g PHAs g VFAs−1) | 0.26 ± 0.06 | 0.25 ± 0.04 | 0.26 ± 0.06 | 0.27 ± 0.05 |
| Π Accum (h−1) | 0.0289 ± 0.0014a | 0.0645 ± 0.0019a | 0.0295 ± 0.0041a | 0.0607 ± 0.0035a |
| (0.0372 ± 0.0024)b | (0.0211 ± 0.0032)b | (0.0355 ± 0.0028)b | (0.0204 ± 0.0009)b | |
| ΔVFAs (g L−1) | 8.29 ± 0.12 | 15.53 ± 0.13 | 8.37 ± 0.11 | 15.17 ± 0.12 |
| pHf | 7.5 ± 0.1 | 7.9 ± 0.1 | 7.5 ± 0.1 | 8.0 ± 0.1 |
The highest PHA content in cells fed with the actual VFA-rich effluent (63%) was obtained for the 40% conditions as a consequence of the application of the two consecutive accumulation batch processes. This value represents an encouraging result for the design, set up and evaluation of the bioprocess at the bench-top scale. Moreover, the application of a cell-recycling culture system, as demonstrated elsewhere,36,37 would allow a continuous feeding together with an increase of the final cell concentration.
The comparison among results related to the employment of the actual effluent and the VFA solution suggests that no inhibition effects due to other organics in GPAcidDeph occurred. Indeed, GPAcidDeph tested concentrations were selected in order to avoid VFA inhibition,38,39 therefore it was important to exclude negative effects due to the effluent matrix. Polyphenols are well known anti-microbial agents. However, they probably did not inhibit the process both because of their low concentration in the GPAcidDeph (lower than the inhibitory concentration reported in a previous work19) and the fact that their antimicrobial activity is probably not significant for this case. This is in accordance with the wine fermentation process in which polyphenols do not cause inhibition.
The polymer production yields were lower than values previously published when pure acids were tested as the carbon source40,41 (YPHB/Acetic = 0.47 g g−1 and YPHB/Butyric = 0.65 g g−1, respectively). However, they were comparable to that obtained when pre-treated olive mill wastewater was employed.19 Furthermore, they resulted higher yields than reported when the palm oil mill effluent and a pure culture of Rhodobacter sphaeroides (0.22 g PHAs per g VFAs)42 or fermented organic waste and a pure culture of R. eutropha TF93 (0.16 g PHAs per g VFAs) were used.43
The lower calculated ΠAccum parameter related to both 20% conditions are concurrent with previous studies,38,39 reporting higher specific rates in response to higher VFA concentrations. This evidence was supposed to represent a kind of a mechanism for avoiding the toxic effects due to the acids.
On the other hand, the produced polymer was almost pure polyhydroxybutyrate (PHB). It is very well known that pure PHB has limited applicability, since it is highly crystalline and because its melting and degradation temperatures are close to each other.14,44 A possible perspective to persecute higher industrial interest for the proposed approach can be represented by the addition of a co-substrate such as propionic or valeric acids, these leading to the obtainment of the co-polymer poly(hydroxybutyrate-co-hydroxyvalerate), which is more flexible and stronger.44 Propionic and valeric acids are VFAs that can be easily obtained from other biowastes or by modifying the AAD conditions.45,46
To identify whether PHAs were produced only from VFAs or from other compounds occurring in the complex GPAcidDeph matrix, too, initial and final COD values were measured and COD depletions were compared with theoretical calculated COD decays. The measured decreases of COD were 9.7 ± 2.4 gCOD L−1 and 15.4 ± 2.6 gCOD L−1 for 20% and 40% conditions, respectively. The calculated theoretical COD decays were 10.20 ± 0.15 gCOD L−1 and 19.65 ± 0.20 gCOD L−1, respectively. These results suggested that other organics than VFAs did not significantly contribute to PHA accumulation.
Biogas production from GPAcidDeph solid fraction
The net cumulative biogas production profiles as a function of the experimental time are presented in Fig. 4. A rapidly increasing cumulative CH4 production was observed for about twelve days. After 31 days, 292 mL of biogas were produced. It was composed of methane (67.4%) and carbon dioxide (32.6%), while no hydrogen was detected. At the end of the experiment, 113 mL per g VS of biomethane were obtained.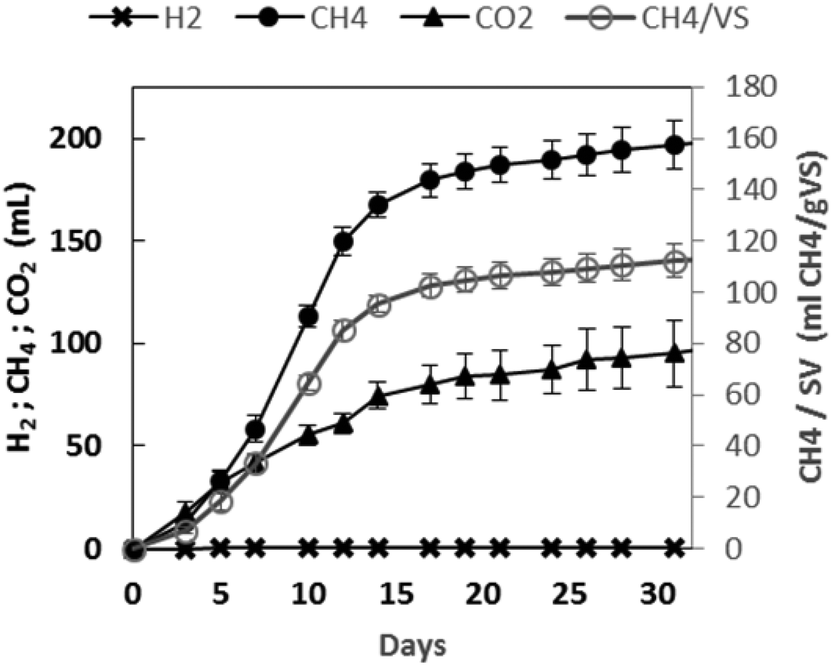 | ||
| Fig. 4 Effective biogas production using GPAcidDeph solid fraction. Accumulated hydrogen, methane and carbon dioxide production trends. | ||
Such a result did not represent a high biomethanization yield when compared to some evidence obtained with other biowastes.47 Furthermore, the AD of the same non-pretreated GP at the same inoculum to substrate ratio led to almost double biomethane production (data not shown). On the other hand, it was quite similar to the value reported by ref. 7 also with non-pretreated GP and a lower inoculum to substrate ratio (0.66). The yields obtained by ref. 8 were significantly higher than those obtained in the present work, but a shredding step was added for oil extraction from seeds. Therefore, the obtained results can be considered of interest in the perspective of developing effective continuous anaerobic methanogenic processes fed with the target leftover and with the potentiality of also including the residues from the PHA down-stream process.
Experimental
Chemicals and grape pomace
Folin–Ciocalteu reagent, gallic acid, (±)catechin, (+)-α-tocopherol, vanillin 99%, the standard volatile fatty acid (VFA) mixture (Supelco), poly(3-hydroxybutyric acid-co-3-hydroxyvaleric acid) (12 wt% PHV; natural origin), salts (BioReagent) for the mineral medium, single VFA and glucose (BioReagent) were purchased from Sigma Aldrich.GP from red grape (Vitis vinifera L.) varieties were collected during September 2012 in the Friuli Venezia-Giulia region (Italy). It was air dried at room temperature (moisture 14.3% ± 0.3 w/w) and stocked at 4 °C until use. It was ground with a domestic miller, with an average particle diameter of 0.83 ± 0.05 mm as calculated with Sauter's equation.48
Polyphenol extraction
The polyphenol recovery via supercritical CO2 extraction was carried out using a commercial pilot-plant (SCF100 series 3 PLC-GR-DLMP, Separeco s.r.l., Pinerolo, Italy) equipped with a 1 L extraction vessel, two 0.3 L separators in series and a tank for CO2 storage. The gas was recycled after the separation process. A simplified flow sheet of the SFE pilot plant is given in Fig. 5.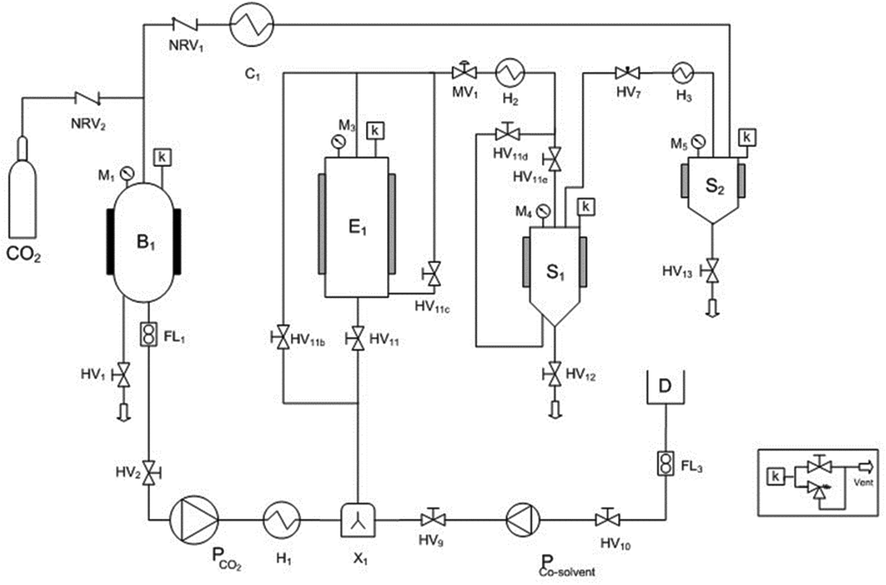 | ||
| Fig. 5 Schematic pilot-scale extractor: (B1) storage tank, (E1) extraction vessel, (S1, S2) separators, (H#) heat exchangers; (C1) condenser; (HV#) hand valves; (MV1) membrane valve; (NVR#) no return valves; (P) diaphragm pumps; (F1) flowmeter; (M#) manometers; (k) safety devices; (FL1) coriolis mass flowmeter; (D) co-solvent storage tank; (X#) mixer. | ||
Ground GP was fed to the extractor (0.480 kg; density 600 kg m−3) in order to be defatted by supercritical CO2. As suggested by Sovová et al.49 pressure was 28 MPa and temperature was 45 °C, while CO2 flow rate was 10 kg h−1 and the total extraction time was 3 h. Such conditions corresponded to 62.5 Q (kg CO2 per kg feed). Subsequently, a co-solvent was required for extracting polyphenols from the defatted GP, due to the polarity of polyphenols. Therefore, 0.1 kg of defatted GP were treated with supercritical CO2 containing 10% ethanol–water mixture (57%, v/v) (EtW) as a co-solvent at 8 MPa, 40 °C and CO2 flow rate of 6 kg h−1.2 Aliquots of grape extract were collected during extractions in volumetric flasks at intervals of about 30 min, to asses several data points for the overall extraction curves (OECs). The ethanol aqueous mixture was then removed from the extracts with a rotary evaporator (Buchi, B465, -Switzerland) at 45 °C. After solvent removal, extracts were weighed and analysed. All experiments were conducted in duplicate. The statistical significances of the differences between means were determined using Tukey's test with the level of significance set up at p ≤ 0.05.
Anaerobic acidogenic digestion
The anaerobic process was inoculated with an acidogenic microbial consortium, which was obtained from an anaerobic treatment of organic fraction of municipal solid waste and acclimated to the acidogenic digestion of GP and exhausted in terms of VFA production. The GPDeph coming from the extraction step was characterized in terms of total solid content (g TS g GPDeph−1) and volatile solid content (g VS g TS−1). Thereafter, a 1 L-Pyrex bottle (supplied with a tri-ports cap with silicone septum) was fed with water, GPDeph and a microbial inoculum (10%, v/v), so that final working volume and TS content were 560 mL and 20% (w/v), respectively. Incubation conditions were: pH 7, 37 °C and 150 rpm. The process was monitored every 2–3 days for biogas and VFA production. To the latter aim, 500 μL-liquid samples were withdrawn. pH was corrected to 7 by the addition of 10 M NaOH after each monitoring process. During such operation, nitrogen was flushed to maintain anaerobic conditions. VSs were determined at the end of the digestion in order to evaluate the amount of organic matter consumed during this step. The experiment was carried out in triplicate.PHA production
The experiments were performed according to a dual-phase process (reported above). In brief, PHA accumulation was induced after a preliminary phase, during which cells were grown under optimal conditions. A slightly modified Medium 81 from DSMZ was employed for the cell balanced growth (growth phase); it contained 3 g L−1 instead of 1 g L−1 of (NH4)2SO4. Glucose (5 g L−1) was added as the sole carbon source.
Conversely, an ammonia free-medium was employed for the subsequent PHA accumulation phase. It was prepared by combining two sterilized stock solutions, namely: (a) the VFA-rich effluent obtained by the acidogenic digestion of GPDeph (GPDephAcid), which was filtered (Whatman N11, 11 μm), amended with Medium 81-DSMZ salts (except for (NH4)2SO4) and autoclaved using special Beckman flasks allowing a subsequent centrifugation (8000 rpm, 4 °C and 25 minutes) under sterile conditions; and (b) distilled water amended with Medium 81-DSMZ salts (except for (NH4)2SO4) at the same concentrations they occur in such a medium. The accumulation culture media were prepared by mixing the two stock solutions at different proportions, namely: 20 and 40% v/v of GPDephAcid. In addition to this, a parallel control test was carried out using a simulated GPDephAcid (SimGPDephAcid), which was a VFA solution prepared by dissolving in distilled water the organic acids at the same concentrations as in GPDephAcid. The control test was aimed at verifying whether other compounds than VFAs occurring in GPDephAcid could affect PHA accumulation. Two sequential accumulation batch processes were carried out under all conditions with an initial pH of 7.2.
In this way, the possibility of using GPDephAcid as an alternative carbon source specifically only for PHA production was studied. The latter acid effluent constituted 20% and 40% of the accumulation phase media, as reported previously, in order to determine if GPDephAcid concentration could affect the PHA accumulation activity of grown cells. Each experiment was carried out in triplicate.
Biogas production
The solid leftover from the anaerobic acidogenic digestion step (GPDephAcid;Solid) was tested as a substrate for biogas production. The experiments were carried out in 100 mL Pyrex bottles (microcosms, 55 mL of working volume) tightly closed with a modified Pyrex-cap that allowed gas sampling. The inoculum to substrate ratio was 1 g of VS in the inoculum per g VS in the substrate, and the TS content was 8% (92% of which VS). The methanogenic microbial consortium employed as inoculum was obtained from a commercial biogas production plant located in the Emilia Romagna Region (Italy) fed with agro-industrial biowaste and zootechnical liquor. It was exhausted in terms of gas production before being employed. The incubation conditions were 37 °C and 150 rpm. The experiment was carried out in triplicate. A blank control experiment was set up by filling microcosms only with water and the inoculum, in order to calculate the effective biogas production by subtracting the amount of biogas eventually produced within control experiments to that produced within target test. Biogas production was measured every 2–3 days. After biogas sampling, the bottles were opened under nitrogen gas flux to keep anaerobiosis and pH was corrected to 7.5 by adding few drops of 10 M H2SO4. All the adopted experimental conditions were recommended by ref. 47, 50.Analytical procedures
The total phenolic content (TPC) of the extracts was measured using the Folin–Ciocalteu reagent, according to Yu et al.51 A calibration curve was obtained with standard solutions of gallic acid in the range 0.2–10 mg mL−1 and measurements were carried out at 765 nm (R2 = 0.99). Results were expressed as milligrams of equivalent gallic acid per 100 gram of dried matter (mgGAE per 100 gDM).
The fractionation of proanthocyanidins from the extracts was conducted as reported by ref. 52, as well as the total flavan-3-ol content that was determined by the vanillin assay. Results were expressed as milligrams of equivalent catechin acid per 100 g of dried matter (mgcatechin per 100 gDM).
The antioxidant activity of the phenolic extract and proanthocyanidin fraction was evaluated by the total free radical scavenger capacity (RSC) following the methodology described by ref. 53 with slight modification.2 The antioxidant activity of the samples was expressed as the milligrams of α-tocopherol per 100 g of dried matter (mgα-tocopherol per 100 gDM). A calibration curve was obtained with standard solutions of α-tocopherol in the range 5.8 × 10−5–2.3 × 10−3 mol L−1 (R2 = 0.98).
All analyses were performed in triplicate.
The qualitative characterization of polyphenolic extracts was carried out by UHPLC-MSn analyses as reported by Bresciani et al.54
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (7 mL min−1); injection volume was 1 μL. The temperature programme was: 80 °C for 0.5 min, then 20 °C min−1 to 150 °C for 1 min, then 20 °C min−1 to 240 °C for 2.5 min. Before the analyses, the samples were diluted with an equal amount of a 60 mM oxalic acid solution.
:1 (7 mL min−1); injection volume was 1 μL. The temperature programme was: 80 °C for 0.5 min, then 20 °C min−1 to 150 °C for 1 min, then 20 °C min−1 to 240 °C for 2.5 min. Before the analyses, the samples were diluted with an equal amount of a 60 mM oxalic acid solution.
At the end of the fermentation, organic matter content in the liquid phase was measured by determining chemical oxygen demand (COD) of the sample supernatant experimentally and theoretically, therefore obtaining the percentage of the total COD content that was ascribed to the occurrence of VFAs 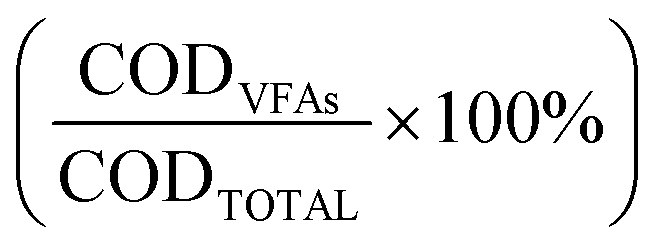 .
.
TPC in the GPAcidDeph was measured by colorimetry with a down-scaled procedure of the method reported elsewhere.55
When performing the Abs600 vs. cell dry weight (CDW) calibration curve, linear correlations were obtained for the growth and the accumulation phases (data not shown). PHA content was defined as gPHAs × gCDW−1 × 100%, on a cell dry weight basis.
Organic matter consumption during the accumulation phase was followed by measuring the sample supernatant COD and the theoretical COD variation was calculated.
TSs were determined by conventional gravimetric method exposing the sample to 105 °C overnight and VSs were determined by exposing the resulting dried sample to 600 °C for 1 hour.
Conclusions
In conclusion, the possibility of developing a multi-purpose biorefinery scheme for the valorisation of red grape pomace by obtaining natural antioxidants, volatile fatty acids, biopolymers and biomethane was demonstrated. The extracted polyphenolic fraction included significant amounts of bioactive compounds, which are readily adsorbed by the organisms. The acidification of the dephenolised residue was obtained by feeding the organic matrix to a biological anaerobic acidogenic process. The resulting VFA-rich liquid effluent was employed as the substrate for an effective biotechnological production of PHAs. Biomethane was obtained from the exhausted solid leftover, which was digested under anaerobic methanogenic conditions. To the very best of our knowledge, this study represents the first attempt of exploiting grape pomace for the integrated production of several industrial products. In particular, the target biowaste have never been tested before as an alternative low-cost substrate for the production of PHAs.Acknowledgements
The work was supported by the Italian National “AGER Project” (grant-making foundations) under the grant no. 2011-0283. Authors also thank very much Dr. Luca Calani (Department of Food Science, University of Parma) for his valuable help in the characterization of polyphenolic extracts.References
- OIV, State of Vitiviniculture World Market 2015, International Organisation of Vine and Wine - Intergovernmental Organisation, 2015 Search PubMed.
- C. Da Porto, A. Natolino and D. Decorti, J. Supercrit. Fluids, 2014, 87, 59–64 CrossRef CAS.
- I. I. Rockenbach, E. Rodrigues, L. V. Gonzaga, V. Caliari, M. I. Genovese, A. E. d. S. S. Gonçalves and R. Fett, Food Chem., 2011, 127, 174–179 CrossRef CAS.
- F. Cherubini, Energy Convers. Manage., 2010, 51, 1412–1421 CrossRef CAS.
- S. R. Alberto Scoma, L. Bertin and F. Fava, Crit. Rev. Biotechnol., 2014 DOI:10.3109/07388551.2014.947238.
- J. Yu and M. Ahmedna, Int. J. Food Sci. Technol., 2013, 48, 221–237 CrossRef CAS.
- E. Dinuccio, P. Balsari, F. Gioelli and S. Menardo, Bioresour. Technol., 2010, 101, 3780–3783 CrossRef CAS PubMed.
- A. Fabbri, G. Bonifazi and S. Serranti, Waste Manage., 2015, 36, 156–165 CrossRef CAS PubMed.
- L. B. Stefano Rebecchi, V. Vallini, G. Bucchi, F. Bartocci and F. Fava, Environ. Eng. Manag. J., 2013, 12, 105–108 Search PubMed.
- M. T. Agler, B. A. Wrenn, S. H. Zinder and L. T. Angenent, Trends Biotechnol., 2011, 29, 70–78 CrossRef CAS PubMed.
- K. Sudesh, H. Abe and Y. Doi, Prog. Polym. Sci., 2000, 25, 1503–1555 CrossRef CAS.
- C. S. K. Reddy, R. Ghai, Rashmi and V. C. Kalia, Bioresour. Technol., 2003, 87, 137–146 CrossRef CAS PubMed.
- Y. B. Kim and R. W. Lenz, Adv. Biochem. Eng./Biotechnol., 2001, 71, 51–79 CrossRef CAS PubMed.
- P. J. Barham, A. Keller, E. L. Otun and P. A. Holmes, J. Mater. Sci., 1984, 19, 2781–2794 CrossRef CAS.
- G. Q. Chen, Chem. Soc. Rev., 2009, 38, 2434–2446 RSC.
- J. Choi and S. Y. Lee, Appl. Microbiol. Biotechnol., 1999, 51, 13–21 CrossRef CAS.
- M. Majone, P. Massanisso, A. Carucci, K. Lindrea and V. Tandoi, Water Sci. Technol., 1996, 34, 223–232 Search PubMed.
- M. G. Albuquerque, S. Concas, S. Bengtsson and M. A. Reis, Bioresour. Technol., 2010, 101, 7123–7133 CrossRef CAS PubMed.
- G. A. Martinez, L. Bertin, A. Scoma, S. Rebecchi, G. Braunegg and F. Fava, Biochem. Eng. J., 2015, 97, 92–100 CrossRef.
- A. A. Koutinas, A. Vlysidis, D. Pleissner, N. Kopsahelis, I. Lopez Garcia, I. K. Kookos, S. Papanikolaou, T. H. Kwan and C. S. K. Lin, Chem. Soc. Rev., 2014, 43, 2587–2627 RSC.
- A. Schievano, F. Adani, L. Buessing, A. Botto, E. N. Casoliba, M. Rossoni and J. L. Goldfarb, Green Chem., 2015, 17, 2874–2887 RSC.
- A. M. Farías-Campomanes, M. A. Rostagno and M. A. A. Meireles, J. Supercrit. Fluids, 2013, 77, 70–78 CrossRef.
- J.-M. Souquet, V. Cheynier, F. Brossaud and M. Moutounet, Phytochemistry, 1996, 43, 509–512 CrossRef CAS.
- D. Del Rio, A. Rodriguez-Mateos, J. P. E. Spencer, M. Tognolini, G. Borges and A. Crozier, Antioxid. Redox Signaling, 2013, 18, 1818–1892 CrossRef CAS PubMed.
- N. C. Michael and R. DanieleDel, in Flavonoids and Related Compounds, CRC Press, 2012, DOI:10.1201/b11872-3.
- K. Lappa, P. Kandylis, N. Bastas, S. Klaoudatos, N. Athanasopoulos, A. Bekatorou, M. Kanellaki and A. Koutinas, Biotechnol. Biofuels, 2015, 8, 74 CrossRef PubMed.
- R. A. Verlinden, D. J. Hill, M. A. Kenward, C. D. Williams, Z. Piotrowska-Seget and I. K. Radecka, AMB Express, 2011, 1, 11 CrossRef PubMed.
- P. Kahar, T. Tsuge, K. Taguchi and Y. Doi, Polym. Degrad. Stab., 2004, 83, 79–86 CrossRef CAS.
- J. W. Holder, J. C. Ulrich, A. C. DeBono, P. A. Godfrey, C. A. Desjardins, J. Zucker, Q. Zeng, A. L. Leach, I. Ghiviriga, C. Dancel, T. Abeel, D. Gevers, C. D. Kodira, B. Desany, J. P. Affourtit, B. W. Birren and A. J. Sinskey, PLoS Genet., 2011, 7, 8 Search PubMed.
- Y. H. Yang, C. J. Brigham, C. F. Budde, P. Boccazzi, L. B. Willis, M. A. Hassan, Z. A. Yusof, C. Rha and A. J. Sinskey, Appl. Microbiol. Biotechnol., 2010, 87, 2037–2045 Search PubMed.
- A. Nickzad, A. Mogharei, A. Monazzami, H. Jamshidian and F. Vahabzadeh, Water Environ. Res., 2012, 84, 626–634 CrossRef CAS PubMed.
- S. L. Riedel, J. Bader, C. J. Brigham, C. F. Budde, Z. A. M. Yusof, C. Rha and A. J. Sinskey, Biotechnol. Bioeng., 2012, 109, 74–83 CrossRef CAS PubMed.
- C. Dimou, N. Kopsahelis, A. Papadaki, S. Papanikolaou, I. K. Kookos, I. Mandala and A. A. Koutinas, Food Res. Int., 2015, 73, 81–87 CrossRef CAS.
- S. Follonier, M. S. Goyder, A.-C. Silvestri, S. Crelier, F. Kalman, R. Riesen and M. Zinn, Int. J. Biol. Macromol., 2014, 71, 42–52 CrossRef CAS PubMed.
- S. Y. Lee, Biotechnol. Bioeng., 1996, 49, 1–14 CrossRef CAS PubMed.
- J. L. Ienczak, W. Schmidell and G. M. Aragao, J. Ind. Microbiol. Biotechnol., 2013, 40, 275–286 CrossRef CAS PubMed.
- W. Ahn, S. Park and S. Lee, Biotechnol. Lett., 2001, 23, 235–240 CrossRef CAS.
- J. H. Kim, B. G. Kim and C. Y. Choi, Biotechnol. Lett., 1992, 14, 903–906 CrossRef CAS.
- J. Wang and J. Yu, Process Biochem., 2000, 36, 201–207 CrossRef CAS.
- T. Yamane, Biotechnol. Bioeng., 1993, 41, 165–170 CrossRef CAS PubMed.
- H. Shi, M. Shiraishi and K. Shimizu, J. Ferment. Bioeng., 1997, 84, 579–587 CrossRef CAS.
- M. Ali Hassan, Y. Shirai, N. Kusubayashi, M. Ismail Abdul Karim, K. Nakanishi and K. Hasimoto, J. Ferment. Bioeng., 1997, 83, 485–488 CrossRef.
- K. J. Ganzeveld, A. van Hagen, M. H. van Agteren, W. de Koning and A. J. M. Schoot Uiterkamp, J. Cleaner Prod., 1999, 7, 413–419 CrossRef.
- J.-i. Choi and S. Y. Lee, Appl. Environ. Microbiol., 1999, 65, 4363–4368 CAS.
- S. Bengtsson, J. Hallquist, A. Werker and T. Welander, Biochem. Eng. J., 2008, 40, 492–499 CrossRef CAS.
- M. Monti, A. Scoma, G. Martinez, L. Bertin and F. Fava, New Biotechnol., 2015, 32, 341–346 CrossRef CAS PubMed.
- F. Raposo, M. A. De la Rubia, V. Fernández-Cegrí and R. Borja, Renewable Sustainable Energy Rev., 2012, 16, 861–877 CrossRef CAS.
- N. P. Povh, M. O. M. Marques and M. A. A. Meireles, J. Supercrit. Fluids, 2001, 21, 245–256 CrossRef CAS.
- H. Sovová, M. Zarevúcka, M. Vacek and K. Stránský, J.Supercrit. Fluids, 2001, 20, 15–28 CrossRef.
- F. Raposo, V. Fernández-Cegrí, M. A. De la Rubia, R. Borja, F. Béline, C. Cavinato, G. Demirer, B. Fernández, M. Fernández-Polanco, J. C. Frigon, R. Ganesh, P. Kaparaju, J. Koubova, R. Méndez, G. Menin, A. Peene, P. Scherer, M. Torrijos, H. Uellendahl, I. Wierinck and V. de Wilde, J. Chem. Technol. Biotechnol., 2011, 86, 1088–1098 CrossRef CAS.
- L. Yu, J. Perret, M. Harris, J. Wilson and S. Haley, J. Agric. Food Chem., 2003, 51, 1566–1570 CrossRef CAS PubMed.
- B. Sun, G. P. Belchior, J. M. Ricardo-da-Silva and M. I. Spranger, J. Chromatogr., A, 1999, 841, 115–121 CrossRef CAS.
- J. C. Espín, C. Soler-Rivas and H. J. Wichers, J. Agric. Food Chem., 2000, 48, 648–656 CrossRef.
- L. Bresciani, L. Calani, M. Cossu, P. Mena, M. Sayegh, S. Ray and D. Del Rio, PharmaNutrition, 2015, 3, 11–19 CrossRef CAS.
- V. L. Singleton, R. Orthofer and R. M. Lamuela-Raventós, in Methods in Enzymology, ed. P. Lester, Academic Press, 1999, vol. 299, pp. 152–178 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5gc01558h |
| This journal is © The Royal Society of Chemistry 2016 |