
Oxidative esterification via photocatalytic C–H activation†
Sanny
Verma‡
a,
R. B. Nasir
Baig‡
a,
Changseok
Han
a,
Mallikarjuna N.
Nadagouda
b and
Rajender S.
Varma
*a
aSustainable Technology Division, National Risk Management Research Laboratory, U. S. Environmental Protection Agency, MS 443, Cincinnati, Ohio 45268, USA. E-mail: varma.rajender@epa.gov; Fax: +513-569-7677; Tel: +513-487-2701
bWQMB, WSWRD, National Risk Management Research Laboratory, U. S. Environmental Protection Agency, MS 443, Cincinnati, Ohio 45268, USA
First published on 2nd October 2015
Abstract
Direct oxidative esterification of alcohol via photocatalytic C–H activation has been developed using VO@g-C3N4 catalyst; an expeditious esterification of alcohols occurs under neutral conditions using visible light as the source of energy.
Introduction
Esters are important building blocks that are extensively used in the chemical industry and academic laboratories.1 Conventionally, esterification is accomplished using activated carboxylic acids or their acid-catalyzed condensation with alcohols.2 In basic media, the reaction can be achieved using nucleophilic substitution reactions with alkyl halides.3 The direct conversion of aldehydes into the corresponding esters is known, but most of these methods require stoichiometric amount of heavy metals and their derivatives4 that entail the use of homogenous metal complexes and metal salts in combination with an oxidant.5 Further, the recyclability and reusability of metal catalysts are not well exploited. In view of the emphasis on the development of environmentally benign and cost-effective procedures, the direct conversion of alcohols to the corresponding ester has garnered attention,6 because alcohols are easily accessible, as the fermentation product of renewable plant-derived materials, namely carbohydrates, sugars, cellulose and their derivatives.7Hitherto, most of the reported methods for the conversion of alcohols to the corresponding esters necessitate the use of precious metals (e.g. gold, palladium, iridium etc.) in basic media.8 In addition to finding alternatives to expensive noble metals, it is imperative to heterogenize the catalysts, which eventually can be efficiently recycled and reused.9 The elimination of basic media will be an advantage if it could be achieved in the direct esterification process. Engaged in the development of sustainable protocol in organic synthesis,10 herein, we report a simple and efficient method for the direct esterification of alcohols via C–H activation using oxo-vanadium–graphitic carbon nitride, VO@g-C3N4, under photochemical conditions. The use of graphitic surface not only heterogenized the oxo-vanadium complex, but also provided the activation energy for the esterification reaction. The in-built nitrogenous framework of g-C3N4 provides the milder basic environment required to accomplish the reaction without the need for an external base.
Synthesis and characterization
The graphitic carbon nitride support (g-C3N4) was synthesized by calcination of urea at 500 °C over a period of three hours and dispersed in aqueous methanol (50%) under sonication.11 A methanolic solution of vanadyl acetylacetonate [VO(acac)2] was added to a dispersed solution of g-C3N4 and stirred for 3 h at room temperature. The reaction mixture was centrifuged, washed with methanol and dried under vacuum at 50 °C to afford catalyst, VO@g-C3N4, as a pale yellow solid (Scheme 1).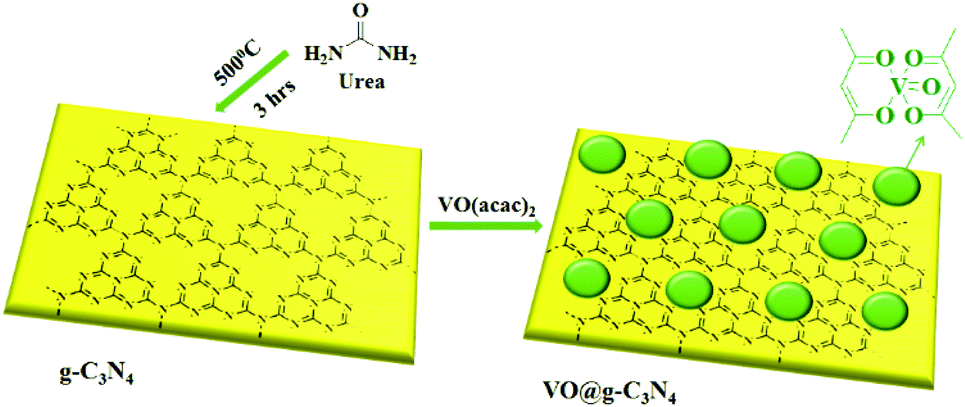 | ||
| Scheme 1 Synthesis of VO@g-C3N4 catalyst. | ||
The catalyst was characterized using scanning electron microscopy (SEM), energy-dispersive X-ray spectroscopy (EDX), transmission electron microscopy (TEM), X-ray diffraction (XRD), and inductive-coupled plasma atomic emission spectroscopy (ICP-AES). The SEM image of g-C3N4 support and the catalyst, VO@g-C3N4, clearly manifest the immobilization of vanadium over the graphitic carbon nitride surface (Fig. 1a and c). The EDX spectra of VO@g-C3N4 (Fig. 1d) indicates the presence of vanadium metal, whereas this peak is absent in the EDX of g-C3N4 (Fig. 1c).
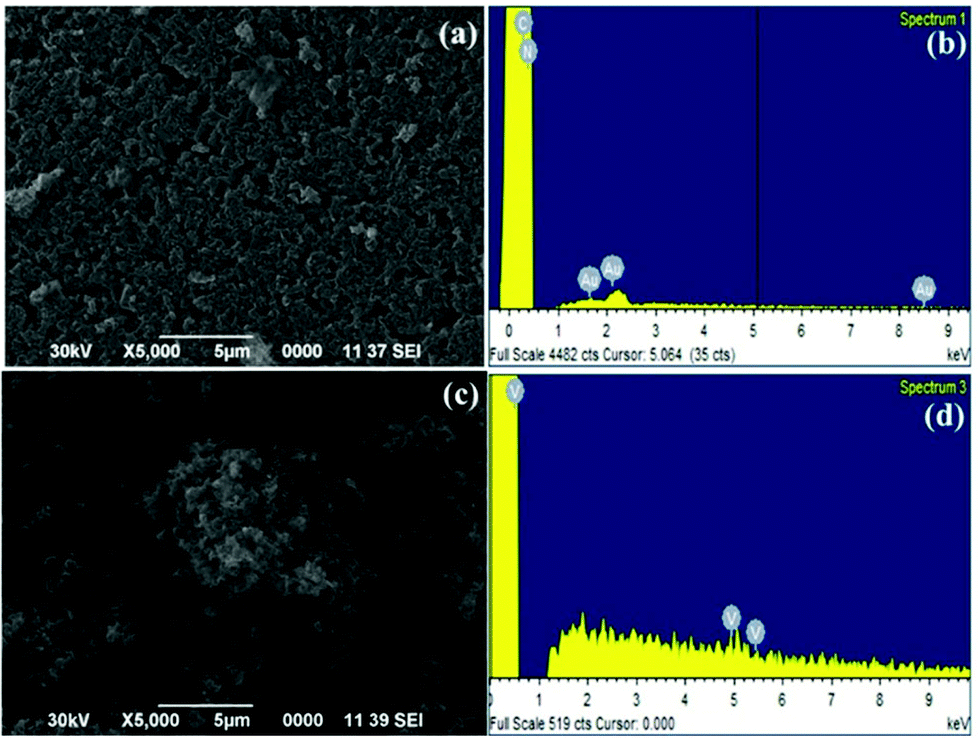 | ||
| Fig. 1 (a) SEM image of g-C3N4 support; (b) EDX image of g-C3N4 support; (c) SEM image of VO@g-C3N4 catalyst; (d) EDX image of VO@g-C3N4 catalyst. | ||
The TEM images of the catalyst (Fig. 2a and b) and g-C3N4 support (ESI, S1†) do not show morphological difference. EDX (Fig. 2c) and ICP-AES analysis confirm the presence of vanadium in the VO@g-C3N4 catalyst. The broad peaks in the XRD pattern (Fig. 3) of VO@g-C3N4 and the support (g-C3N4) do not give any additional information about vanadium may be due to strong complexation and amorphous nature. The weight percentage of vanadium was found to be 4.91% by ICP-AES.
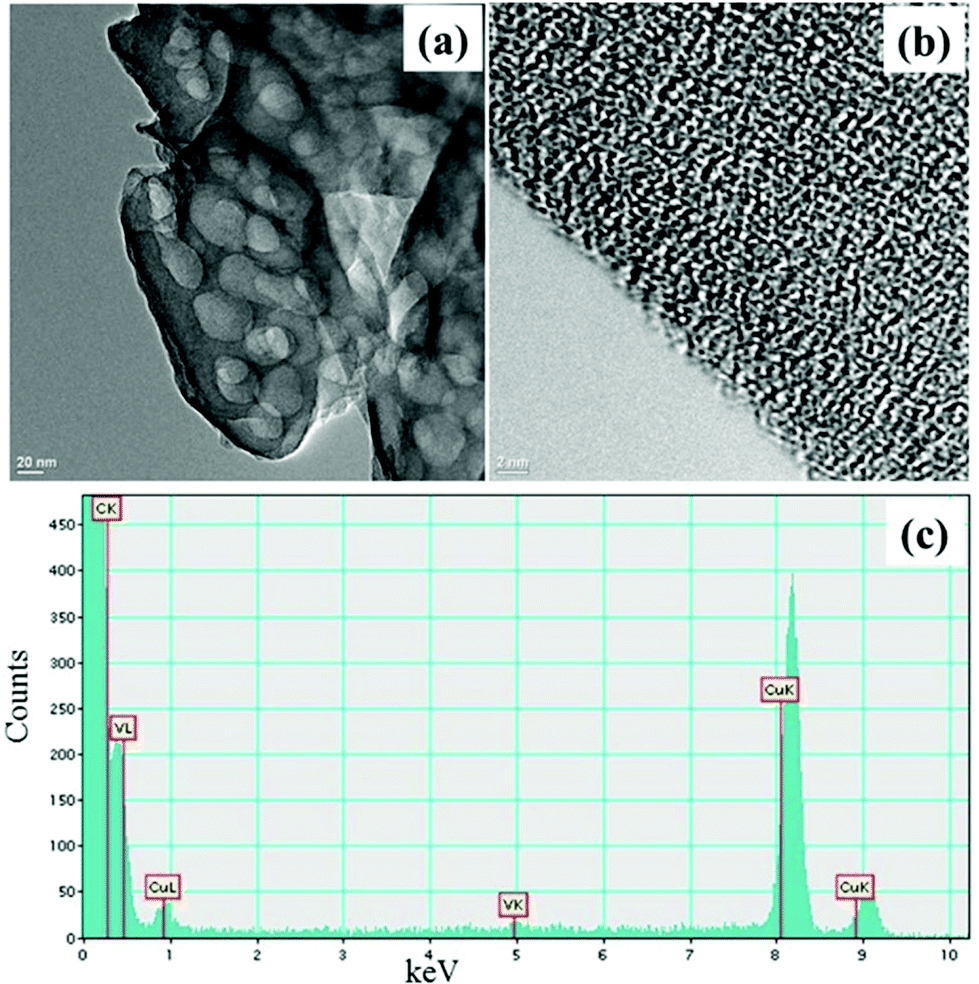 | ||
| Fig. 2 (a) TEM image of VO@g-C3N4 catalyst; (b) TEM image of VO@g-C3N4 catalyst at higher resolution; (c) EDX image of VO@g-C3N4 catalyst. | ||
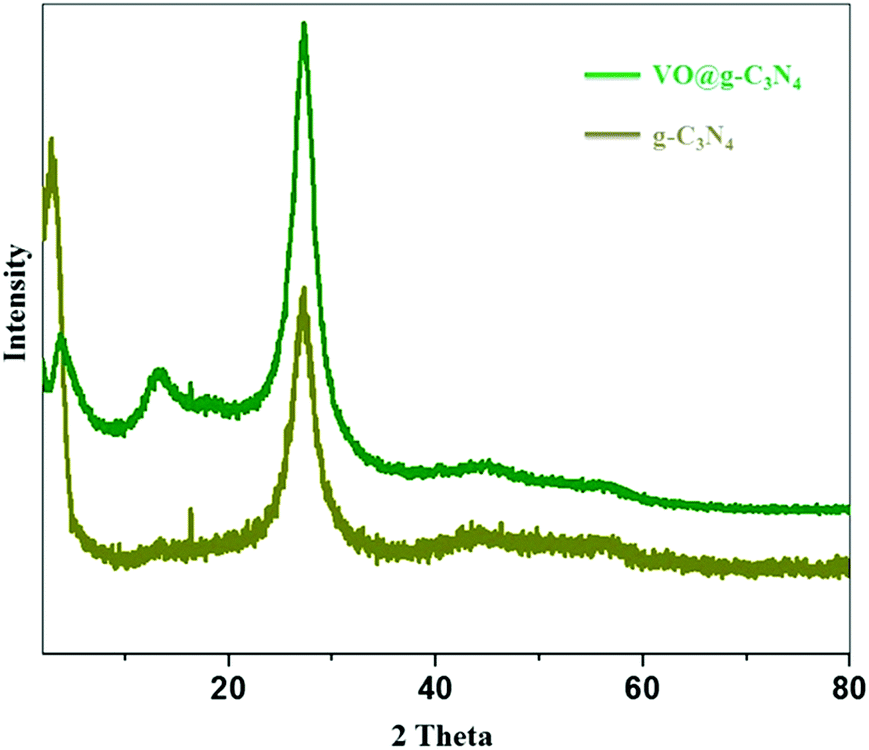 | ||
| Fig. 3 XRD spectra of g-C3N4 and VO@g-C3N4. | ||
Results and discussion
The present study focuses on the direct oxidative esterification of alcohols using an array of metal catalysts supported over graphitic carbon nitride surfaces under visible light irradiation. It was envisioned that g-C3N4 would provide a basic surface to accelerate C–H activation and esterification. The built-in photoactive chromophore would absorb energy and assist in crossing the activation energy barrier to accomplish esterification. Thus, we prepared a series of supported catalysts, namely Fe3O4@g-C3N4, Pd@g-C3N4, Cu@g-C3N4, Ag@g-C3N4, V2O5@g-C3N4, VO@g-C3N4 and V(II)@g-C3N4. The catalysts were evaluated for oxidative C–H activation esterification of benzyl alcohol with methanol using H2O2 as an oxidant. The reaction with Fe3O4@g-C3N4 (Table 1; entry 1) gave only benzaldehyde, even after extending the reaction time to 24 hours. The change of metal from iron to copper (Cu@g-C3N4, Table 1; entry 2) and silver (Ag@g-C3N4, Table 1; entry 3) did not alter the reaction outcome. Pd@g-C3N4 (Table 1; Entry 4) gave 35% of the desired product and the remaining benzyl alcohol was converted into aldehyde and the corresponding acid. The V2O5@g-C3N4 (Table 1; entry 5) proved to be a better choice as it gave 64% yield of ester when exposed to visible light. In contrast, the control reaction using pure V2O5 offered only trace amount of oxidative esterification product, indicating that g-C3N4 is not only heterogenizing the V2O5, but also acting as a promoter under photochemical conditions. The immobilization of VCl2 over g-C3N4 (Table 1; entry 6) was not as effective as V2O5. Changing vanadium complex to VO(acac)2 gave a highly active VO@g-C3N4 catalyst (Table 1; entry 7), which accomplished the oxidative esterification reaction in 3 hours to afford methyl benzoate in 98% yield. The control experiment (Table 1; entry 8) clearly manifest that it proceeds with the absorption of visible light.|
|
|||
|---|---|---|---|
| Entry | Catalyst | Time | Yielda,b |
| a Reaction condition: 1 mmol of benzyl alcohol; methanol 2 mL; 1.5 mmol H2O2; catalyst 25 mg; 40 watt domestic bulb. b Isolated yield. c Benzaldehyde formation was observed. d Reaction performed under dark. | |||
| 1c | Fe3O4@g-C3N4 | 24 h | — |
| 2c | Cu@g-C3N4 | 24 h | — |
| 3c | Ag@g-C3N4 | 24 h | — |
| 4c | Pd@g-C3N4 | 12 h | 35% |
| 5 | V2O5@g-C3N4 | 12 h | 64% |
| 6 | V(II)@g-C3N4 | 12 h | 37% |
| 7 | VO@g-C3N4 | 3 h | 98% |
| 8c,d | VO@g-C3N4 | 12 h | — |
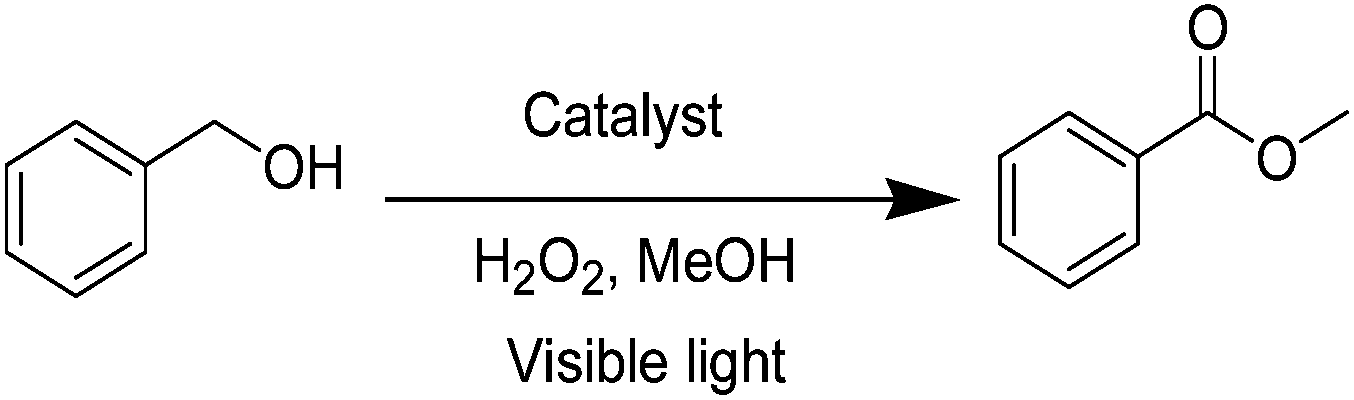
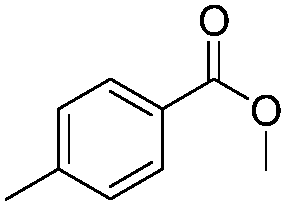
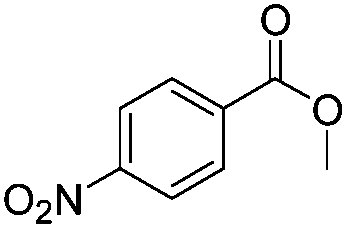
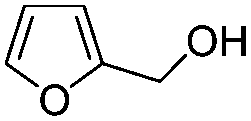
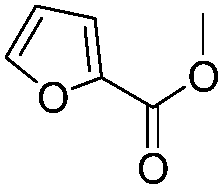
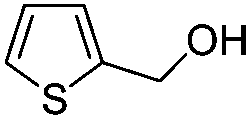
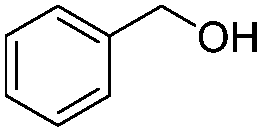
After discovering the active catalyst, the general reactivity towards the esterification of alcohols was examined using range of benzyl alcohols. The substituents on the benzene ring did not show any peculiar effect over the product outcome and reaction rate. Substrates with electron withdrawing and electron donating substituents were efficiently converted into the corresponding methyl esters (Table 2; entries 1–4). Furfuryl alcohol and 2-thiophenemethanol were efficaciously converted into methyl ester derivatives with excellent yield (Table 2; entries 5 and 6). The change of solvent from methanol to ethanol and the introduction of nucleophilic functional groups, such as amines and hydroxyls, did not interfere with the product formation. 3,4-Dihydroxy benzyl alcohol, 4-amino benzyl alcohol, and 2-amino benzyl alcohol efficiently formed the corresponding ethyl esters using the VO@g-C3N4 catalyst (Table 2; entries 8–10). The presence of a double bond is tolerated well in the formation of the corresponding esters. Cinnamyl alcohol was converted into ethyl cinnamate with 93% yield (Table 2; entry 11). The change of solvent from methanol/ethanol to isopropanol and n-propanol afforded the corresponding isopropyl benzoate and n-propyl benzoate (Table 2; entries 12 and 13) with 76% and 84% yield, respectively. The longer reaction time for these transformations may be due to the slow reaction of alcohols bearing extended alkyl chains.
| Entry | Substrate | Product | Time | Yieldb |
|---|---|---|---|---|
| a Reaction condition: 1 mmol of alcohol; 2 mL alcoholic solvent; 1.5 mmol H2O2; catalyst 25 mg; 40 watt domestic bulb. b Isolated yield. | ||||
| 1 |
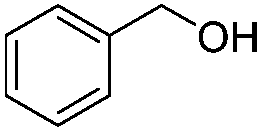
|
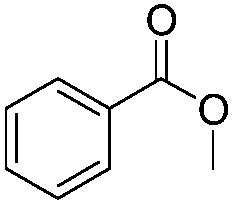
|
3 h | 98% |
| 2 |
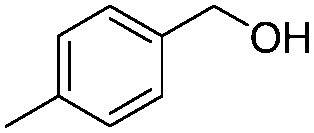
|
|
3 h | 97% |
| 3 |
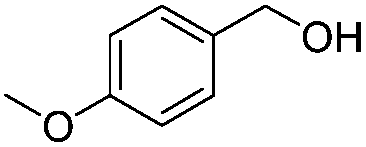
|
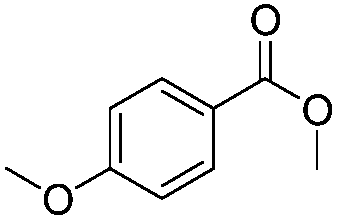
|
3 h | 98% |
| 4 |
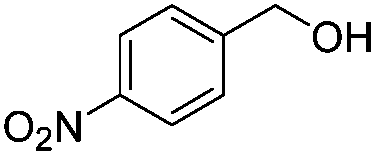
|
|
3 h | 95% |
| 5 |
|
|
3 h | 96% |
| 6 |
|
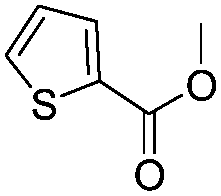
|
3 h | 96% |
| 7 |
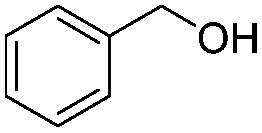
|
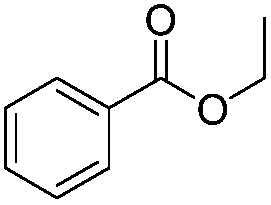
|
3 h | 99% |
| 8 |
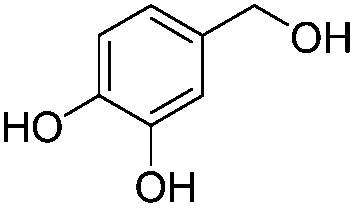
|
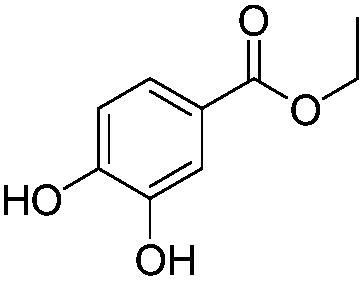
|
3 h | 83% |
| 9 |
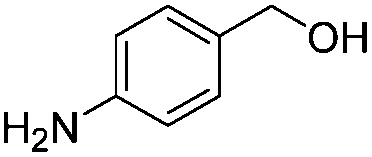
|
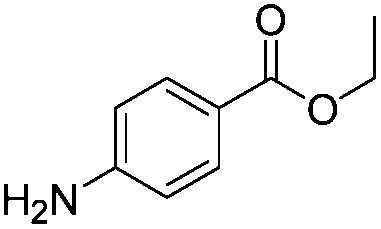
|
3 h | 85% |
| 10 |
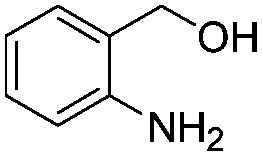
|
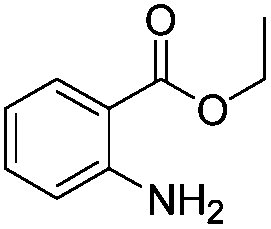
|
3 h | 82% |
| 11 |
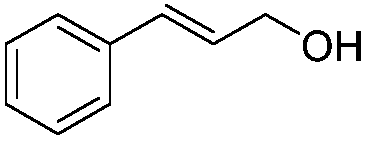
|
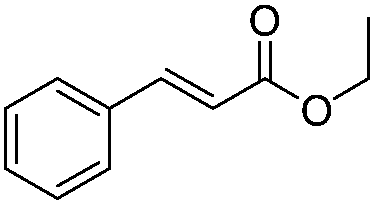
|
3 h | 93% |
| 12 |
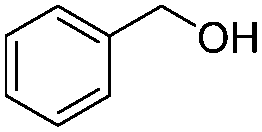
|
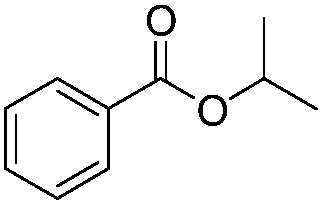
|
8 h | 76% |
| 13 |
|
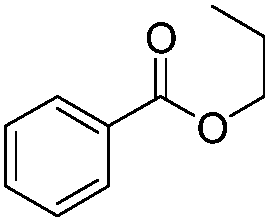
|
5 h | 84% |
The success in industrial applications of the catalyst depends on its shelf life and recyclability. To establish its reuse in industrial context, a set of experiments was conducted for the oxidative esterification of benzyl alcohol in methanol using VO@g-C3N4. The reaction was monitored by TLC. After reaction completion, the catalyst was recovered using centrifuge, washed with acetone, and reused for the oxidative esterification of fresh reactants. The catalyst could be used up to 8 cycles without losing its activity. Metal leaching was studied by inductive-coupled plasma atomic emission spectroscopic analysis of the catalyst both before and after the reaction. The concentration of vanadium was found to be 4.91% before the reaction and 4.88% after the 8th cycle of the reaction. The SEM image of the catalyst after the 8th cycle of the reaction did not show any change in the morphology of the catalyst (ESI, Fig. S2†).The ICP-AES of reaction solvent does not show traces of vanadium, thus asserting that g-C3N4 holds the oxo-vanadium complex tightly, which precludes vanadium leaching and promotes the effective recycling of the catalyst.
Conclusions
A simple and sustainable protocol for the oxidative esterification of alcohol has been developed via photocatalytic C–H activation using VO@g-C3N4 catalyst. The expeditious reaction proceeds under neutral conditions due to the photoactive graphitic carbon nitride surface and its strong interaction with vanadium metal. The support stimulates the metal towards the oxidation of alcohol followed by C–H activation–esterification while the in-built nitrogenous framework provides an adequate mild, basic environment and energy through visible light absorption.Disclaimer
The U.S. Environmental Protection Agency, through its Office of Research and Development, funded and managed the research described herein. It has been subjected to the Agency's administrative review and has been approved for external publication. Any opinions expressed in this paper are those of the author(s) and do not necessarily reflect the views of the Agency, therefore, no official endorsement should be inferred. Any mention of trade names or commercial products does not constitute endorsement or recommendation for use.Acknowledgements
SV, RBNB and CH were supported by the Postgraduate Research Program at the National Risk Management Research Laboratory administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy and the U.S. Environmental Protection Agency.Notes and references
- J. Otera, Esterification: Methods, Reactions, and Applications, Wiley-VCH, Weinheim, 2003 Search PubMed.
- R. C. Larock, Comprehensive Organic Transformations: A Guide to Functional Group Preparations, Wiley-VCH, New York, 2nd edn, 1999 Search PubMed.
- (a) W. Mägerlein, M. Beller and A. F. Indolese, J. Mol. Catal. A: Chem., 2000, 156, 213 CrossRef; (b) A. Brennfuhrer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114 CrossRef PubMed.
- D. Talukdar, K. Sharma, S. K. Bharadwaj and A. J. Thakur, Synlett, 2013, 963 CAS.
- R. Gopinath and B. K. Patel, Org. Lett., 2000, 2, 577 CrossRef CAS PubMed.
- (a) R. V. Jagadeesh, H. Junge, M. M. Pohl, J. Radnik, A. Brückner and M. Beller, J. Am. Chem. Soc., 2013, 135, 10776 CrossRef CAS PubMed; (b) S. Verma, D. Verma, A. K. Sinha and S. L. Jain, Appl. Catal., A, 2015, 489, 17 CrossRef CAS.
- S. Sreekumar, Z. C. Baer, P. Anbarasan, G. Gunbas, A. Grippo, H. W. Blanch, D. S. Clark and F. D. Toste, Nat. Protocols, 2015, 10, 528 CAS.
- (a) M. Nielsen, H. Junge, A. Kammer and M. Beller, Angew. Chem., Int. Ed., 2012, 51, 5711 CrossRef CAS PubMed; (b) C. Liu, J. Wang, L. Meng, Y. Deng, Y. Li and A. Lei, Angew. Chem., Int. Ed., 2011, 50, 5144 CrossRef CAS PubMed; (c) C. Liu, S. Tang and A. Lei, Chem. Commun., 2013, 49, 1324 RSC; (d) N. Yamamoto, Y. Obora and Y. Ishii, J. Org. Chem., 2011, 76, 2937 CrossRef CAS PubMed; (e) S. Gowrisankar, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 5139 CrossRef CAS PubMed; (f) H. Miyamura, T. Yasukawa and V. Kobayashi, Green Chem., 2010, 12, 776 RSC.
- (a) R. V. Jagadeesh, T. Stemmler, A. E. Surkus, M. Bauer, M. M. Pohl, J. Radnik, K. Junge, H. Junge, A. Brückner and M. Beller, Nat. Protocols, 2015, 10, 916 CrossRef PubMed; (b) H. P. Mungse, S. Verma, N. Kumar, B. Sain and O. P. Khatri, J. Mater. Chem., 2012, 22, 5427 RSC; (c) S. Verma, M. Nandi, A. Modak, S. L. Jain and A. Bhaumik, Adv. Synth. Catal., 2011, 353, 1897 CrossRef CAS.
- (a) R. S. Varma, Green Chem., 2014, 16, 2027 RSC; (b) R. B. Nasir Baig and R. S. Varma, Chem. Commun., 2013, 49, 752 RSC; (c) R. B. Nasir Baig and R. S. Varma, Green Chem., 2013, 15, 1839 RSC; (d) R. B. Nasir Baig and R. S. Varma, ACS Sustainable Chem. Eng., 2014, 2, 2155 CrossRef CAS; (e) S. Verma, R. Singh, D. Tripathi, P. Gupta, G. M. Bahuguna and S. L. Jain, RSC Adv., 2013, 3, 4184 RSC; (f) R. B. Nasir Baig, M. N. Nadagouda and R. S. Varma, Green Chem., 2014, 16, 4333 RSC.
- (a) F. Dong, L. Wu, Y. Sun, M. Fu, Z. Wu and S. C. Lee, J. Mater. Chem., 2011, 21, 15171 RSC; (b) S. Verma, R. B. Nasir Baig, C. Han, M. N. Nadagouda and R. S. Varma, Chem. Commun., 2015, 51 10.1039/C5CC05895C.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5gc02025e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |