
Dose-related effects of ferric citrate supplementation on endoplasmic reticular stress responses and insulin signalling pathways in streptozotocin–nicotinamide-induced diabetes
Kai-Li
Liu
ab,
Pei-Yin
Chen
a,
Chi-Mei
Wang
c,
Wei-Yu
Chen
de,
Chia-Wen
Chen
f,
Eddy
Owaga
g and
Jung-Su
Chang
*f
aDepartment of Nutrition, Chung Shan Medical University, Taichung 40203, Taiwan, Republic of China
bDepartment of Dietitian, Chung Shan Medical University Hospital, Taichung 40203, Taiwan, Republic of China
cDepartment of Nutrition, MacKay Memorial Hospital, Hsinchu Branch, Taiwan, Republic of China
dDepartment of Pathology, School of Medicine, College of Medicine, Taipei Medical University, Taiwan, Republic of China
eDepartment of Pathology, Wan Fang Hospital, Taipei Medical University, Taiwan, Republic of China
fSchool of Nutrition and Health Sciences, College of Public Health and Nutrition, Taipei Medical University, Taipei, Taiwan, Republic of China. E-mail: susanchang@tmu.edu.tw; Fax: +886-(2)2737-3112; Tel: +886-(2)27361661 ext. 6542
gInstitute of Food and Bioresources Technology, Dedan Kimathi University of Technology, Nyeri, Kenya
First published on 18th November 2015
Abstract
Diabetic patients are at high risk of developing anemia; however, pharmacological doses of iron supplementation may vary greatly depending on diabetes-related complications. The aim of this study was to investigate the dose-dependent effect of iron on glucose disposal with a special focus on endoplasmic reticular (ER) stress, iron metabolism, and insulin signalling pathways. Diabetes was induced in overnight fasted rats by intraperitoneal (i.p.) injections of 40 mg kg−1 streptozotocin (STZ) and 100 mg kg−1 nicotinamide. Diabetic rats were fed a standard diet (36.7 mg ferric iron per kg diet) or pharmacological doses of ferric citrate (0.5, 1, 2, and 3 g ferric iron per kg diet). Ferric citrate supplementation showed a dose-related effect on hepatic ER stress responses and total iron levels, which were associated with increased hepcidin and decreased ferroportin expressions. Iron-fed rats had increased sizes of their pancreatic islets and hyperinsulinemia compared to rats fed a standard diet. A western blot analysis revealed that iron feeding decreased total insulin receptor substrate 1 (IRS1), phosphorylated IRS1ser307, and AS160 but increased phosphorylated GSK-3β. Iron supplementation inhibited the nuclear translocation of AKT but promoted FOXO1 translocation to nuclei. Ferric citrate supplementation showed a dose-related effect on ER stress responses, hepatic iron, and the insulin signaling pathway. Adverse effects were more evident at high iron doses (>1 g ferric iron per kg diet), which is equivalent to a 60 kg human male consuming >500 mg elemental iron per day.
1. Introduction
Iron deficiency is one of the most common nutrient deficiencies in the world. The prevalence rate of anemia is greater in diabetes-related complications such as chronic kidney disease (CKD), which can be up to 50%.1 For adult patients with iron deficiency, the usual therapeutic dose is 180 mg elemental iron per day in divided doses (ranging 100–200 mg elemental iron). CKD patients with anemia may receive doses of 900–975 mg elemental iron per day (300–325 mg elemental iron per tablet three times daily).1 Currently, ferric citrate has also been tested in phase 2 trials as a phosphate binder for dialysis patients who suffer from hyperphosphatemia.2 As a phosphate binder, doses of 210–2520 mg ferric iron per day (equivalent to 1–10 g ferric citrate per day) are recommended by the U.S. Food and Drug Administration (FDA). These data indicate that doses of pharmacological iron supplementation may vary greatly depending on the underlying pathophysiology of individual patients.Whether iron supplementation is beneficial or detrimental to diabetic patients is still controversial, and to what degree iron interferes with glucose metabolism remains to be clarified.3 Using a magnetic resonance imaging (MRI) T2 gradient-recalled echo technique, Zheng and colleagues observed a mild iron overload in patients with prediabetes and type 2 diabetes mellitus (T2DM) compared to controls.4 It is believed that persistently high hepcidin levels, a feature commonly seen in patients with chronic inflammatory disorders such as T2DM, are responsible for tissue iron overload and hypoferremia.3,5 Effects of iron therapy on glucose metabolism have been examined in experimental diabetic animal models, which usually employed one of two broad approaches. One approach is to use acute models involving direct injections of high doses of iron6–10 or an iron chelator11 into the animal. The other approach used by some researchers is to look at effects of high doses of iron supplementation6,9–11 or an iron-depleted diet12 on experimental diabetic animals. Briefly, these data suggest that a low-iron diet significantly ameliorates diabetes in obese mice11 and improves β-cell function and oxidative stress in mouse models of hemochromatosis.12 On the other hand, high doses of carbonyl iron supplementation or iron dextrin injections induce iron accumulation in the liver,6,9 kidneys,7 spleen,6 and adipose tissues.6,9 High-dose iron treatment also impairs glucose metabolism, possibly via targeting insulin receptor substrate (IRS)–AKT pathways9 or down-regulating adiponectin transcription via forkhead box protein O1 (FOXO1).10 However, most animal studies used high doses of iron supplementation and did not report the total doses of elemental iron used in the experiment. Hence, it remains unclear whether pharmacological doses of iron supplementation affect liver function and glucose metabolism.
Recent studies suggested that unresolved endoplasmic reticular (ER) stress may be associated with insulin resistance (IR).13–16 ER stress-associated pathways, also called unfolded protein responses (UPRs), are an important homeostatic device for the liver during stresses of metabolic processes.17 When cells experience metabolic stress, the glucose-regulated protein 78 (Grp78) dissociates from the ER-resident membrane receptors, ATF6, IRE1, and PERK, leading to activation of UPR signal transduction and selective gene expression mediated by downstream transcription factors, such as ATF4, ATF6, and XBP1.16 Prolonged ER stress is considered to be responsible for some of the adverse effects of pancreatic β cell dysfunction and plays an important role in the impairment of insulin biosynthesis.16 Nutrients such as iron are potent ER stress inducers.17 However, ER stress-associated mediators can also increase hepcidin transcription activity resulting in hypoferremia and tissue iron overload.18 Moreover, hepcidin also acts as a gluconeogenic sensor and may play a key role in glucose metabolism.3
Animal models have been extensively used in diabetes research. Administration of a high dose of streptozotocin (STZ) induces type 1 diabetes.19 Recently, administration of low doses of both STZ and nicotinamide or a low dose of STZ and a high-fat diet is increasingly being used to induce type 2 diabetes in rodents.20–23 STZ preferentially accumulates in pancreatic β cells via the glucose transporter 2 (GLUT2), where it causes β cell death due to DNA alkylating activity.19 Masiello and colleagues showed that nicotinamide protects against STZ-induced β cell toxicity via preventing β cell apoptosis and increasing β cell regeneration.20 Currently, it is still not clear whether pharmacological doses of iron supplementation affect ER stress-associated responses. In addition, studies of the effects of pharmacological doses of iron supplementation on glucose disposal in STZ/nicotinamide-induced diabetes are also scarce. The aim of this study was to investigate the dose-dependent effect of iron supplementation on glucose disposal with a special focus on liver (1) ER stress responses, (2) iron metabolism, and (3) insulin signaling pathways. To this end, human equivalent doses (HEDs) of ferric iron of 500, 1000, 2000, and 3000 mg Fe3+ per kg diet were administered to rats with STZ/nicotinamide-induced diabetes for 10 weeks.
2. Materials and methods
2.1 Animal, diets, and the experimental design
Twenty-five male albino Wistar rats (450 g ± 20 g) aged 20 weeks were purchased from BioLasco (Taipei, Taiwan) and kept at the animal facility of Taipei Medical University, under standard conditions. Diabetes was induced in overnight fasted rats by intraperitoneal (i.p.) injections of 40 mg kg−1 STZ and 100 mg kg−1 nicotinamide (both purchased from Sigma-Aldrich, St Louis, MO, USA). Albino rats with a fasting plasma glucose (FPG) level of >250 mg dL−1 were considered to be diabetic. Diabetic rats were fed a standard diet (36.7 mg ferric iron per kg diet) or an iron-enriched diet (0.5, 1, 2, and 3 g ferric iron per kg diet) (Sigma-Aldrich) for 10 weeks. This would be equivalent to a 60 kg human male consuming almost 266, 480, 960, and 1440 mg ferric iron per day. Rats were sacrificed at 10 weeks post-treatment. The experimental protocol was approved by the Animal Ethical Committee of Taipei Medical University (LAC-2013-0150).2.2 Histological assessment
A histological study was performed on liver and pancreas sections. Briefly, a fresh liver and pancreas were placed in 10% buffered formalin and subsequently embedded in paraffin. Tissue sections were stained with hematoxylin and eosin (H&E) using standard techniques. The pancreatic islet size was calculated by an image analysis of single measured islets and expressed as the area in μm2.2.3 Serum biochemistry measurement
Fasting plasma insulin was determined using a commercial kit (Mercodia AB, Sylveniusgatan 8A, Sweden). FPG, hemoglobin (Hb), and serum ferritin levels were determined using commercial kits (all products were purchased from Fortress Diagnosis, Antrim, UK). The total liver iron concentration was quantitated with an Iron Assay kit (Abcam, Cambridge, UK). A homeostatic model assessment was used to evaluate insulin resistance (HOMA-IR) and basal β cell function (HOMA-β). HOMA-IR was calculated according to the formula: fasting insulin (μU L−1) × fasting glucose (nmol L−1)/22.5. HOMA-β was calculated as: 20 × fasting plasma insulin (μU ml−1)/[fasting plasma glucose (mmol L−1) − 3.5].242.4 Western blot analysis
Liver samples (0.05–0.1 g) were lysed in RIPA buffer, and the total protein content was determined by the Bradford method (Carl Roth, Karlsruhe, Germany). Protein lysates were boiled in 1× sodium dodecylsulfate (SDS) buffer at 95 °C for 10 min. Equal amounts of protein (50 μg) were resolved on 10% SDS-polyacrylamide gels. Proteins were transferred by electroblotting to a nitrocellulose membrane. Membranes were probed with antibodies (Abs) and the specific signals were detected using an enhanced chemiluminescence (ECL) detection system (GE Healthcare, Barrington, UK). Rat liver nuclear and cytosolic protein fractions were isolated as described by Tian et al.25 Briefly, 0.1 g of frozen liver tissue was homogenized in ice-cold lysis buffer [10 mM HEPES, 10 mM KCl, 2 mM MgCl2, 0.1 mM EDTA, 1 mM dithiothreitol (DTT), and 0.5 mM phenylmethylsulfonyl fluoride] and centrifuged for 30 s at 500g and 4 °C. Nonidet P-40 (10%) was added to the supernatants and incubated on ice. The crude nuclear pellet was harvested after centrifugation, suspended in ice-cold buffer B, and incubated on ice for 30 min. The supernatants were collected as nuclear extracts after centrifugation and stored at 80 °C until used. Anti-rat Abs (hepcidin, ferroportin, IRS1, p-IRS1tyr612, p-IRS1ser307, pAS160, AKT, FoxO1a/3a, ATF6, XBP1, Grp78, phospho-glycogen synthase kinase-3β (pGSK-3β), phospho-FOXO1, superoxide dismutase (SOD1), glutathione peroxidase (Gpx1), H2B, GAPDH, and β-actin) were used at dilutions of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 or 1:2000 to detect immunoreactive signals. Antibodies were purchased from Cell Signaling (Beverly, MA, USA) except hepcidin-25, ferroportin (Abcam, Cambridge, UK), SOD1, and Gpx1 (GeneTex, Hsinchu City, Taiwan).
:1000 or 1:2000 to detect immunoreactive signals. Antibodies were purchased from Cell Signaling (Beverly, MA, USA) except hepcidin-25, ferroportin (Abcam, Cambridge, UK), SOD1, and Gpx1 (GeneTex, Hsinchu City, Taiwan).
2.5. High-performance liquid chromatographic (HPLC)/mass spectrometric (MS) analysis
Liver total glutathione (GSH) and glutathione disulfide (GSSG) concentrations were evaluated by the HPLC/MS system, as previously described.26 Briefly, samples were homogenized in buffer and centrifuged, and supernatants were analyzed for GSH and GSSG by HPLC. An Agilent Zorbax Eclipse XDB-C8 column was used. The mobile phase consisted of solvent A (10 mM ammonia acetate containing 0.5% formic acid) and solvent B (acetonitrile containing 0.5% formic acid). The gradient system was 10%–90% B (0–45 min), 90%–10% B (45–50 min), and 10% B (50–60 min). The flow rate was 0.6 ml min−1. Data acquisition was via selected ion monitoring. Ions representing negative species of the compounds were selected, and peak areas were measured. Calibration curves of authentic standards were linear over the concentration range of 0.005–40 g ml−1 with a correlation coefficient of r = 0.99.2.6 RNA isolation and quantitative reverse-transcription polymerase chain reaction (qPCR)
Total RNA was extracted from tissues (liver, pancreas, and muscle) using an RNeasy Mini Kit (Qiagen, Hilden, Germany). Total RNA at 1 μg was used for reverse transcription using M-MLV reverse transcriptase (Invitrogen, Carlsbad, CA, USA). Primer sequences were as follows: phosphoenolpyruvate carboxykinase (PEPCK) (forward: GGT ATT TGC CGA AGT TGT AG, reverse: CTT GTC TAT GAA GCC CTC AG), glucose transporter 2 (GLUT2) (forward: CTG TCT GTG TCC AGC TTT GCA, reverse: CAA GCC ACC CAC CAA AGA AC), Grp78 (forward: ACT GGA ATC CCT CCT GCT C, reverse: CAA ACT TCT CGG CGT CAT), hepcidin (forward: CTG AGC AGC ACC ACC TAT CTC, reverse: TGG CTC TAG GCT ATG TTT TGC), and GAPDH (forward: CCC TTC ATT GAC CTC AAC TAC ATG, reverse: CTT CTC CAT GGT GGT GAA GAC). Amplicon sizes ranged 50–150 base pairs (bp) to ensure high amplification efficiency. RNA expression profiles of both target and reference genes were performed using LightCycler 480 (Roche, Mannheim, Germany), as previously described.17 The relative induction of messenger (m) RNA expression was calculated using the following equation, Ecp (sample − control), and normalized to the expression of GAPDH.2.7 Statistical analysis
A one-way analysis of variance (ANOVA) with Bonferroni post-tests and correction was used to analyze differences between groups (n = 5 per group). Analyses were conducted using GraphPad Prism 4. Data are presented as the mean ± standard error of the mean (SEM). p < 0.05 was considered statistically significant.3. Results
3.1 Ferric citrate supplementation induces mild hepatic iron overload in the absence of systemic iron elevation
At week 10, no significant different was observed in body weight (A), Hb (B), or serum ferritin (C) concentrations between diabetic rats that received the standard and iron-enriched diets (Fig. 1). Diabetic rats that received the 2 and 3 g ferric iron per kg diets had an almost 2-fold increase in total hepatic iron concentrations compared to the controls (Fig. 1D). The western blot analysis showed that the 1, 2, and 3 g ferric iron per kg diets were associated with increased hepcidin and decreased ferroportin expressions (Fig. 1E–G). These data suggest that an altered hepcidin–ferroportin expression may contribute to mild liver iron overload, and liver iron accumulation may exist prior to changes in systemic iron levels.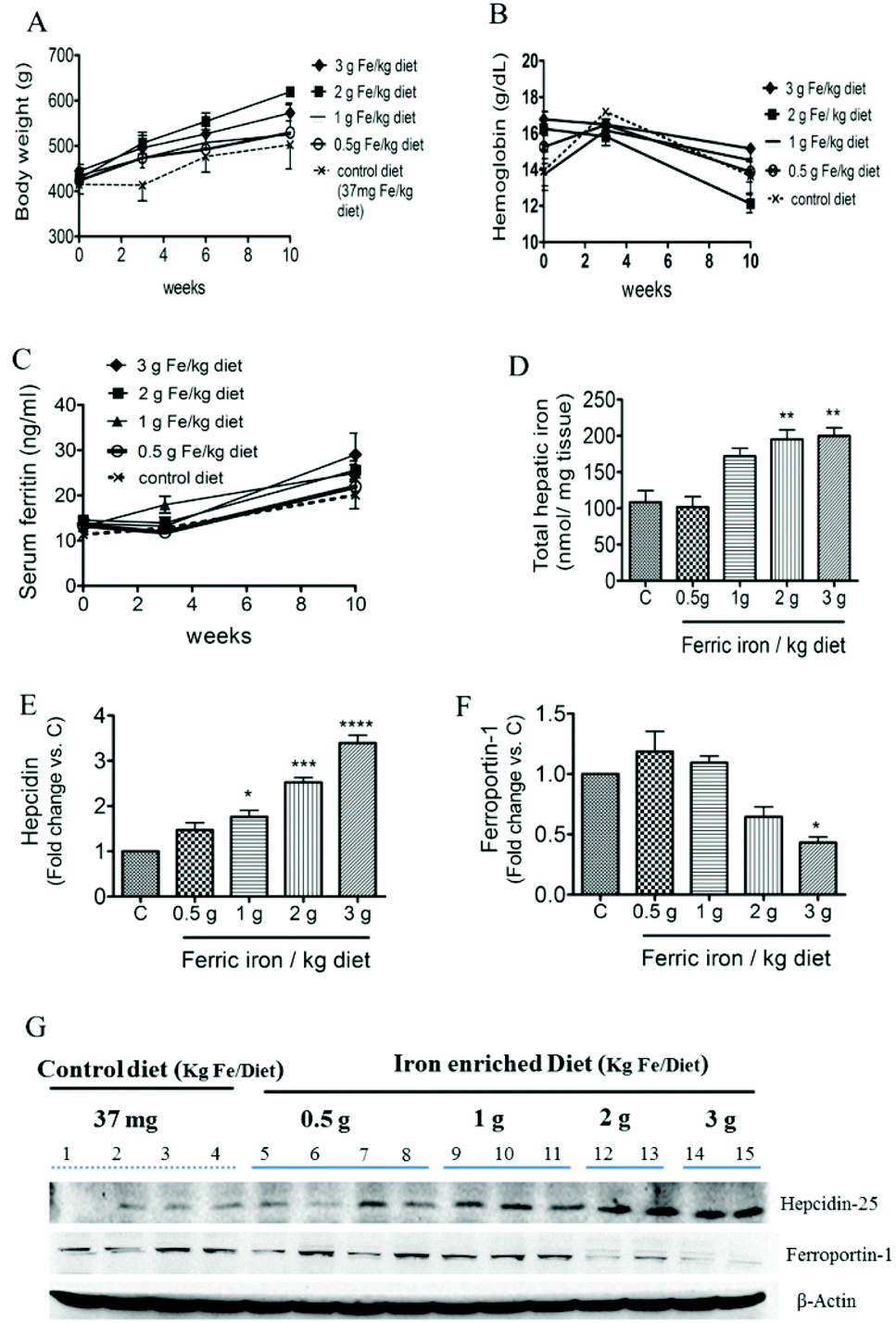 | ||
| Fig. 1 Effects of ferric citrate supplementation on the body weight and iron status. Body weight changes (A), hemoglobin (B), serum ferritin levels (C), and total hepatic iron concentrations (D) in control and iron-fed diabetic rats. Hepatic hepcidin (E and G) and ferroportin-1 (F and G) were detected by a Western blot analysis. Data are expressed as the mean ± SEM (n = 5 per group). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 vs. the controls by a one-way ANOVA with Bonferroni post-tests and correction. | ||
3.2. Ferric citrate, oxidative stress, and ER stress
Fig. 2A shows that 2 and 3 g of ferric iron supplementation resulted in small increases in the hepatic ratio of reduced to oxidized glutathione compared to the controls, but these did not reach statistical significance. The western blot analysis revealed that Gpx1 protein expression in the liver had decreased in iron-fed diabetic rats, whereas the SOD1 level did not significantly change (Fig. 2B and C). We next investigated the effects of pharmacological doses of ferric citrate supplementation on ER stress-associated pathways. Compared to the pancreas and muscle, the liver had relatively higher GRP78 (A) and hepcidin (B) mRNA expressions in diabetic rats fed the 3 g ferric iron per kg diet compared to the standard diet (Fig. 3). A correlation analysis revealed that the liver hepcidin level was positively correlated with Grp78 protein expression (Fig. 3C). The western blot analysis showed increased levels of Grp78 (Fig. 3E and G) and ATF6 (Fig. 3F and G) expressions in iron-fed rats. Although liver XBP1 (G) protein expression was abolished in response to >1 g ferric iron supplementation, the qPCR analysis revealed that spliced XBP1 mRNA (D) was upregulated in all iron-fed rats (Fig. 3).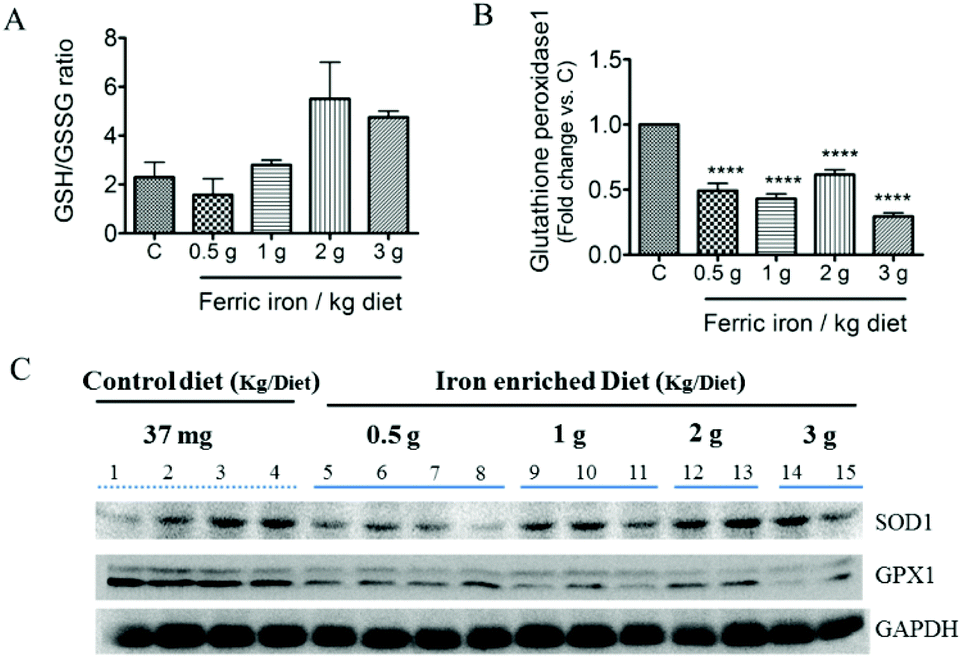 | ||
| Fig. 2 Effects of ferric citrate supplementation on the antioxidant status. Liver total glutathione (GSH) and glutathione disulfide (GSSG) concentrations were evaluated by an HPLC/MS system, and results are expressed as the ratio of GSH to GSSG (A). Liver glutathione peroxidase (Gpx1) (B and C) and superoxide dismutase (SOD1) (C) were detected by a Western blot analysis. Data are expressed as the mean ± SEM (n = 5 per group). *p < 0.05; **p < 0.01; ***p < 0.001 vs. the controls by a one-way ANOVA with Bonferroni post-tests and correction. | ||
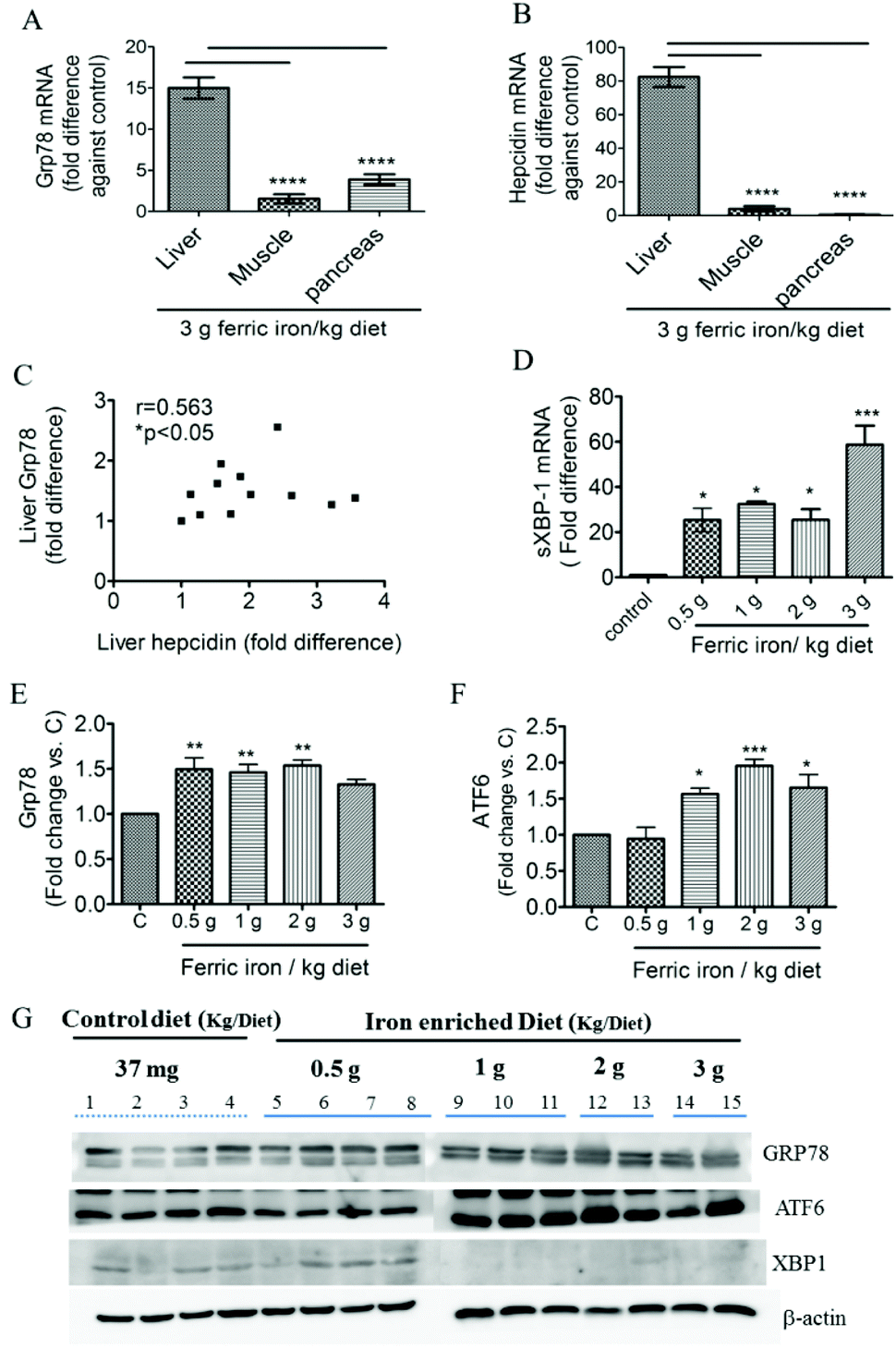 | ||
| Fig. 3 Effects of ferric citrate supplementation on endoplasmic reticular stress. Quantitative gene expression analysis of Grp78 (A) and hepcidin (B) in the liver, muscle, and pancreas. Gene expression data were normalized to the expression level of GAPDH and are presented as multiples of difference between the standard diet and the 3 g ferric iron per kg diet. Associations between hepatic Grp78 and hepcidin protein expressions were assessed using Spearman's rank correlation coefficient (C). Liver Grp78 (E and G), ATF6 (F and G), and XBP1 (G) were detected by a Western blot analysis. Data are expressed as the mean ± SEM (n = 3–5 per group). *p < 0.05; **p < 0.01; ***p < 0.001 vs. the controls by the Mann–Whitney U-test or by a one-way ANOVA with Bonferroni post-tests and correction. | ||
3.3 Ferric citrate supplementation induces hyperinsulinemia and pancreatic islet enlargement
At week 10, fasting plasma insulin levels were higher in diabetic rats that had received iron-enriched diets compared to the standard diet (Fig. 4A; all p < 0.01). Iron supplementation did not alter FPG levels during the treatment period (Fig. 4B). This resulted in a higher HOMA-IR in iron-fed diabetic rats compared to the controls (Fig. 4C). However, HOMA-β cell function did not change significantly (Fig. 4D). A histomorphometric examination of pancreatic islets showed that iron-fed rats had increased pancreatic islet sizes compared to those fed a standard diet (Fig. 4E–J).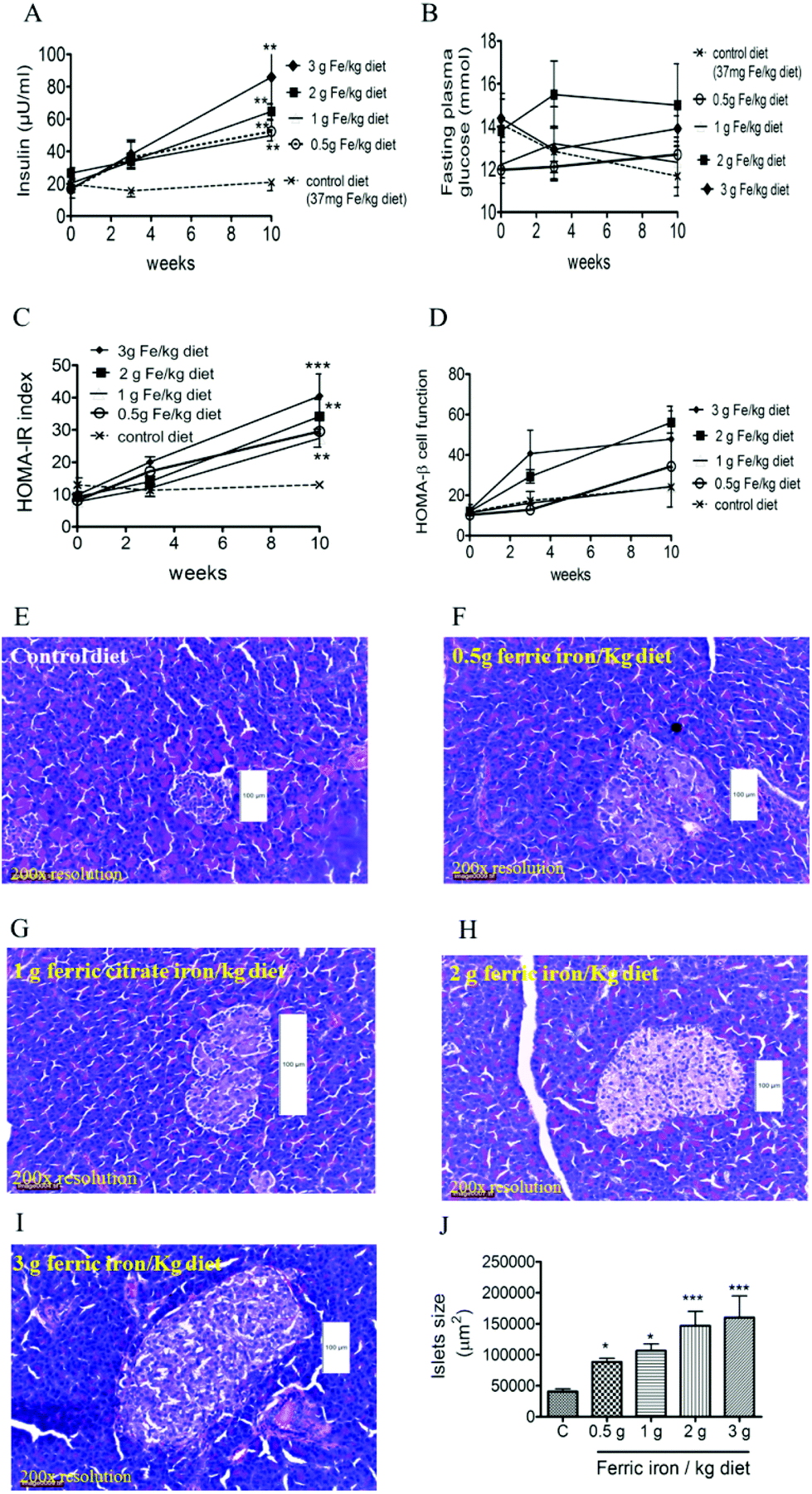 | ||
| Fig. 4 Effects of ferric citrate supplementation on blood glucose levels and pancreatic islets. Iron feeding increased insulin secretions (A) and homeostatic model assessment of insulin resistance (HOMA-IR) (C), but had no effect on fasting plasma glucose (B) or HOMA-β cell function (D). Representative photomicrographs of islets in pancreatic sections from diabetic rats fed a standard diet (E), 0.5 g ferric iron (F), 1 g ferric iron (G), 2 g ferric iron (H), and 3 g ferric iron(I) stained with H&E (original magnification, ×200). The islet size is expressed as the area of an individual islet, which was also greater in iron-fed than control rats (J). Data are expressed as the mean ± SEM (n = 5 per group). *p < 0.05; **p < 0.01; ***p < 0.001 vs. the controls by a one-way ANOVA with Bonferroni post-tests and correction. | ||
3.4. Ferric citrate supplementation exacerbates hepatic IR
Western blot analysis showed inhibition of both IRS1 and serine 307 phosphorylation of IRS1 in the liver of iron-fed rats (Fig. 5C). These resulted in a non-significant decrease in the ratio of pIRS1ser 307/IRS1 (Fig. 5A). Liver GLUT2 mRNA expression was down-regulated in diabetic rats that received the >1 g ferric iron per kg diet (Fig. 5B). The western blot analysis also revealed that nuclear translocation of AKT decreased in iron-fed rats even at the lowest dosage (Fig. 5D and F). Phosphorylation of AS160, the Akt substrate regulating glucose transport, was also markedly down-regulated in diabetic rats that received the >1 g ferric iron per kg diet (Fig. 5E and F).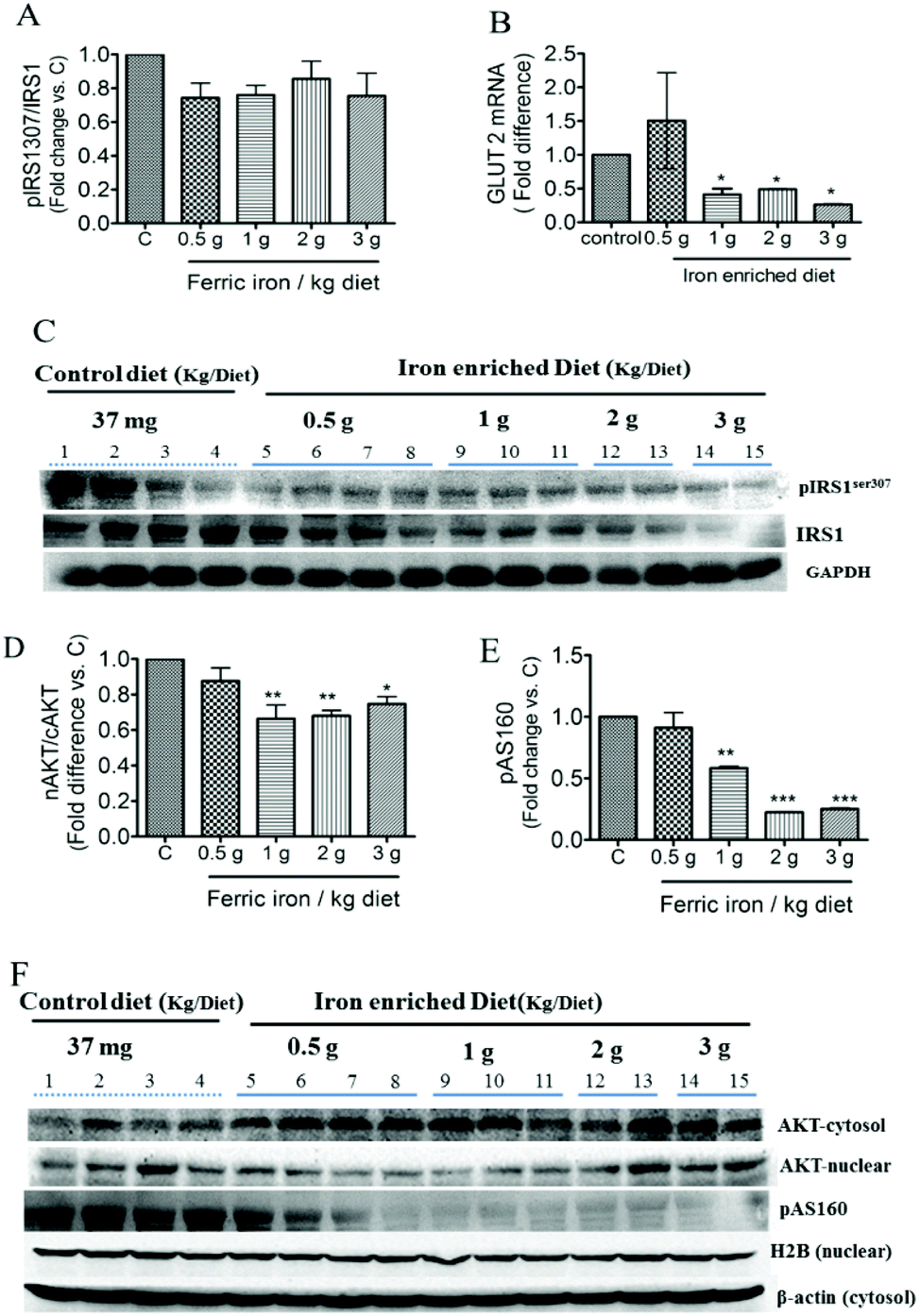 | ||
| Fig. 5 Effects of ferric citrate supplementation on hepatic insulin signaling pathways. Hepatic expressions of p-insulin receptor substrate 1 (IRS1), IRS1, p-AS160, and AKT in the cytosol and nuclear fractions were detected by a Western blot analysis (A, C–F). Hepatic GLUT2 mRNA was determined by a qPCR and normalized to GAPDH (B). Data are expressed as the mean ± SEM (n = 3–5 per group). *p < 0.05; **p < 0.01 vs. the controls by a one-way ANOVA with Bonferroni post-tests and correction. | ||
Increased hepatic gluconeogenesis and decreased glycogen synthesis are common features associated with IR; we next investigated the effects of ferric citrate supplementation on glucose utilization. The western blot analysis showed that ferric citrate increased the nuclear translocation of FOXO1 (Fig. 6A and C) and phosphorylation of GSK-3β (Fig. 6B and C). The mRNA expression of the hepatic gluconeogenic gene, PEPCK, significantly increased in diabetic rats that had received the >1 g ferric iron per kg diet (Fig. 6D).
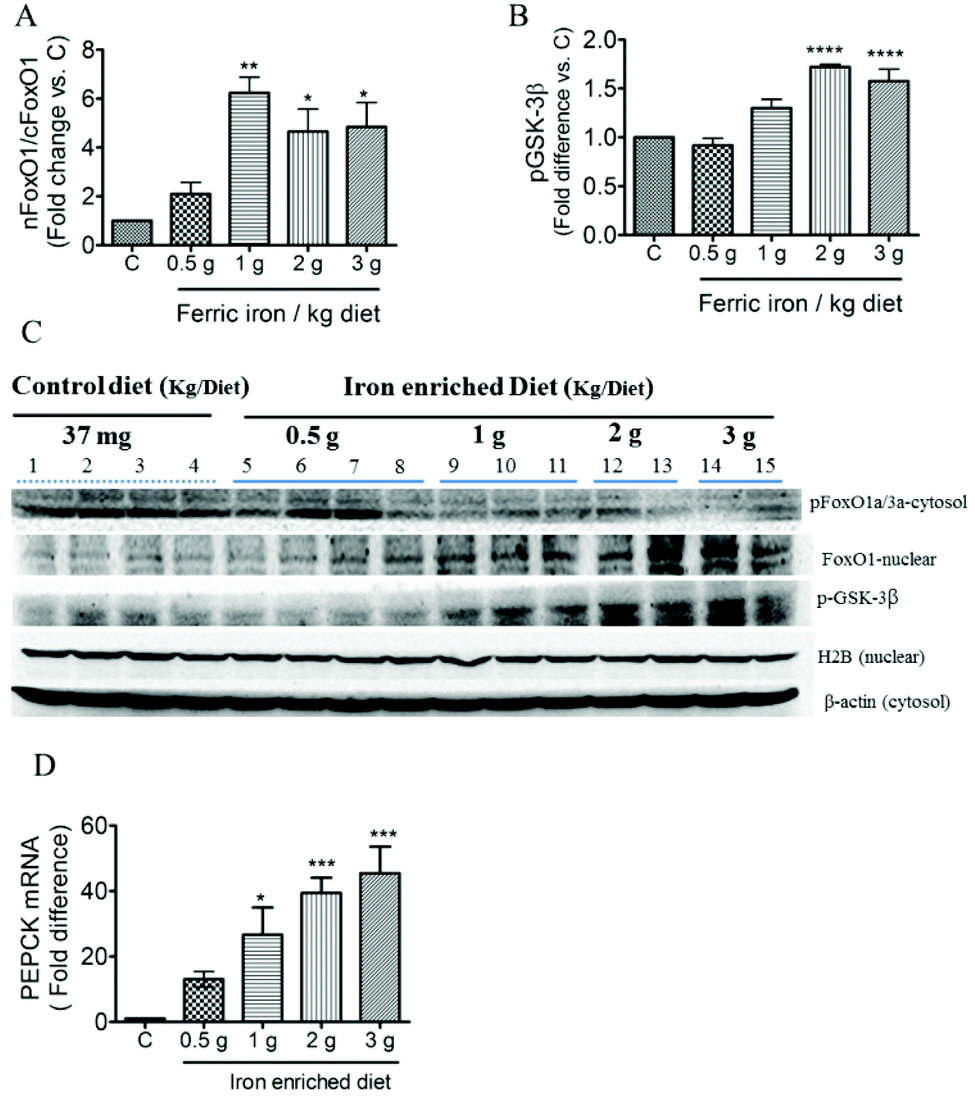 | ||
| Fig. 6 Effects of ferric citrate supplementation on gluconeogenesis. Expression of FOXO1 (A and C) and phosphorylated GSK-3β expression (B and C) in the liver was detected by a Western blot analysis. Hepatic mRNA of PEPCK was determined by a qPCR and normalized to GAPDH (D). Data are expressed as the mean ± SEM (n = 3–5 per group). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 vs. the controls by a one-way ANOVA with Bonferroni post-tests and correction. | ||
4. Discussion
Our study demonstrated that 10 weeks of pharmacological doses of ferric citrate supplementation induced pancreatic islet enlargement, mild liver iron overload, increased gluconeogenesis, and impaired insulin signaling pathways via down-regulating the IRS1-AS160-AKt signaling pathways. Adverse effects of ferric citrate supplementation on the pancreas and liver were more evident with high iron doses (>1 g ferric iron per kg diet). This would be the equivalent to a 60 kg human male consuming >500 mg elemental iron per day. Our study also suggests that ER stress-associated responses may play a role in ferric citrate-induced mild iron overload possibly via altering hepcidin–ferroprotin expression. Importantly, our experimental animal model suggests that mild hepatic iron overload, disrupted pancreatic islet size, and impaired glucose metabolism may occur without a rise in systemic iron levels.Many studies indicated that ER stress-associated responses play key roles in the pathogenesis of IR.16 Our study showed that high doses of ferric citrate supplementation (>1 g ferric iron per kg diet) induced ER stress responses, particularly the ATF6 and XBP1 axis. Unresolved ER stress is known to induce pancreatic β cell death through CHOP-mediated apoptosis.16 Our study showed that the liver, and to a lesser extent, the pancreas, is the primary organ subjected to iron-induced ER stress responses. In spite of the increasing pancreatic islet size in iron-fed diabetic rats, insulin levels did not decrease nor did the HOMA-β cell function. It is likely that pro-survival pathways are activated to alleviate ER stress in response to iron supplementation. Interestingly, we also observed that iron supplementation abolished both total IRS1 and pIRS1ser307 protein expressions in the liver of diabetic rats. It is likely that iron-induced upregulated ER stress responses may be involved in insulin biosynthesis.14 Phosphorylation of IRS1 on tyrosine residues is known to induce insulin-stimulated responses. In contrast, phosphorylation of IRS1 on serine residues can either increase or decrease insulin signaling. Inhibition of serine 307 phosphorylation by rapamycin was associated with reduced IRS1 tyrosine phosphorylation in response to insulin.27 Hepatic Grp78 overexpression resulted in decreased phosphorylation of IRS-1 on serine residues in the liver of ob/ob mice.14 Overexpression of ATF6 may decrease insulin transcription activity possibly via upregulation of the orphan nuclear receptor small heterodimer partner (SHP), which negatively regulates insulin production.16 Although our study did not measure ATF4 levels, the literature shows that ATF4 can induce translational attenuation which may affect insulin biosynthesis at post-translational levels.16 An increase in spliced XBP1 may also lead to attenuation of insulin transcription and mRNA stability.16
Our study indicates that Akt-AS160 may play an important role in iron-induced hepatic IR. Iron feeding disrupts the insulin-mediated signaling cascade involving the adaptor proteins, IRS1, Akt, and AS160, to control glucose disposal in the liver. AS160 is highly expressed in many tissues including muscles, adipocytes, and to a lesser extent, the liver.28 AS160 is a Rab GTPase-activating protein and plays an important role in insulin-mediated glucose disposal.29 Multiple Akt phosphorylation sites have been identified on AS160. It is well characterized that phosphorylation of AS160 activates translocation of GLUT4 to plasma membranes in adipocytes30 and skeletal muscle cells.29 Unphosphorylated AS160 acts as a restraint on GLUT4 translocation. However, the role of AS160 in hepatic glucose regulation is not clear, since glucose uptake in response to insulin in hepatocytes is mediated by GLUT2, not GLUT4. Recently, AS160-knockout mice exhibited whole-body IR in both muscles and the liver in a euglycemic clamp study.28 Taken together, these data suggest that ferric iron may influence the ability of insulin to mediate glucose disposal through Akt-AS160-GLUT2.
Our results are in agreement with previous findings that dietary iron modulates glucose and iron metabolism irrespective of the type of iron (e.g., carbonyl vs. ferric iron) or the route of iron supplementation (diet or intravenous). Our study suggests that the primary effects of HED of ferric citrate supplementation are on insulin signaling pathways and to a lesser extent on glucose synthesis. Our study showed that the HED of ferric iron supplementation did not increase FPG levels in spite of increased gluconeogenesis as indicated by enhanced FOXO1 nuclear translocation. Gao et al.7 also observed that an injection of 300 mg kg−1 iron dextran did not elevate blood glucose levels in rats with diabetes-induced kidney injury. Instead, diabetic nephropathy was in part due to iron-mediated oxidative kidney injury.7 Our study confirms the findings of Dongiovanni et al.9 who reported that an impaired insulin signaling pathway was at least partly due to a decrease in the phospho-AKT/AKT ratio. Our data further suggest that dietary iron may target AKT-AS160-GLUT2 pathways to influence glucose disposal in the liver. Overall, our study supports the view of Gao et al.7 who suggested that the combination of iron and diabetes damages the physiological functions more severely than either does alone.
5. Conclusions
Our data suggest that the effects of ferric iron are influenced by the dosage, and adverse health effects are more evident at high iron doses (>1 g ferric iron per kg diet).Acknowledgements
Dr Jung-Su Chang was supported by grants from Taipei Medical University Hospital (103TMU-TMUH-11 and 104TMU-TMUH-18) and the Ministry of Science and Technology, Taiwan (MOST 103-2320-B-038-015 and MOST 104-2311-B-038-005). The authors declare that no competing interest exists.References
- U. Mehdi and R. D. Toto, Diabetes Care, 2009, 32, 1320–1326 CrossRef PubMed.
- J. P. Dwyer, M. Sika, G. Schulman, I. J. Chang, M. Anger, M. Smith, M. Kaplan, S. Zeig, M. J. Koury, S. S. Blumenthal, J. B. Lewis and G. Collaborative Study, Am. J. Kidney Dis., 2013, 61, 759–766 CrossRef CAS PubMed.
- C. Vecchi, G. Montosi, C. Garuti, E. Corradini, M. Sabelli, S. Canali and A. Pietrangelo, Gastroenterology, 2014, 146, 1060–1069 CrossRef CAS PubMed.
- X. Zheng, T. Jiang, H. Wu, D. Zhu, L. Wang, R. Qi, M. Li and C. Ling, Am. J. Clin. Nutr., 2011, 94, 1012–1019 CrossRef CAS PubMed.
- T. Ganz, Blood, 2011, 117, 4425–4433 CrossRef CAS PubMed.
- A. Daba, K. Gkouvatsos, G. Sebastiani and K. Pantopoulos, J. Mol. Med., 2013, 91, 95–102 CrossRef CAS PubMed.
- W. Gao, X. Li, Z. Gao and H. Li, Biol. Trace Elem. Res., 2014, 160, 368–375 CrossRef CAS PubMed.
- M. Silva, F. Bonomo Lde, P. Oliveira Rde, W. Geraldo de Lima, M. E. Silva and M. L. Pedrosa, J. Clin. Biochem. Nutr., 2011, 49, 102–108 CrossRef CAS PubMed.
- P. Dongiovanni, M. Ruscica, R. Rametta, S. Recalcati, L. Steffani, S. Gatti, D. Girelli, G. Cairo, P. Magni, S. Fargion and L. Valenti, Am. J. Pathol., 2013, 182, 2254–2263 CrossRef CAS PubMed.
- J. S. Gabrielsen, Y. Gao, J. A. Simcox, J. Huang, D. Thorup, D. Jones, R. C. Cooksey, D. Gabrielsen, T. D. Adams, S. C. Hunt, P. N. Hopkins, W. T. Cefalu and D. A. McClain, J. Clin. Invest., 2012, 122, 3529–3540 CAS.
- R. C. Cooksey, D. Jones, S. Gabrielsen, J. Huang, J. A. Simcox, B. Luo, Y. Soesanto, H. Rienhoff, E. D. Abel and D. A. McClain, Am. J. Physiol.: Endocrinol. Metab., 2010, 298, E1236–E1243 CrossRef CAS PubMed.
- R. C. Cooksey, H. A. Jouihan, R. S. Ajioka, M. W. Hazel, D. L. Jones, J. P. Kushner and D. A. McClain, Endocrinology, 2004, 145, 5305–5312 CrossRef CAS PubMed.
- Y. Nakatani, H. Kaneto, D. Kawamori, K. Yoshiuchi, M. Hatazaki, T. A. Matsuoka, K. Ozawa, S. Ogawa, M. Hori, Y. Yamasaki and M. Matsuhisa, J. Biol. Chem., 2005, 280, 847–851 CrossRef CAS PubMed.
- H. L. Kammoun, H. Chabanon, I. Hainault, S. Luquet, C. Magnan, T. Koike, P. Ferre and F. Foufelle, J. Clin. Invest., 2009, 119, 1201–1215 CAS.
- H. J. Koo, Y. Piao and Y. K. Pak, NeuroSignals, 2012, 20, 265–280 CrossRef CAS PubMed.
- M. K. Kim, H. S. Kim, I. K. Lee and K. G. Park, Exp. Diabetes Res., 2012, 2012, 509437 Search PubMed.
- C. M. Wang, S. J. Li, C. H. Wu, C. M. Hu, H. W. Cheng and J. S. Chang, Asian Pac. J. Cancer Prev., 2014, 15, 605–610 CrossRef PubMed.
- C. Vecchi, G. Montosi, K. Zhang, I. Lamberti, S. A. Duncan, R. J. Kaufman and A. Pietrangelo, Science, 2009, 325, 877–880 CrossRef CAS PubMed.
- S. Lenzen, Diabetologia, 2008, 51, 216–226 CrossRef CAS PubMed.
- P. Masiello, C. Broca, R. Gross, M. Roye, M. Manteghetti, D. Hillaire-Buys, M. Novelli and G. Ribes, Diabetes, 1998, 47, 224–229 CrossRef CAS PubMed.
- S. L. Badole and S. L. Bodhankar, Eur. J. Pharmacol., 2010, 632, 103–109 CrossRef CAS PubMed.
- W. Pierre, A. J. Gildas, M. C. Ulrich, W. N. Modeste, N. T. Benoit and K. Albert, BMC Complementary Altern. Med., 2012, 12, 264 CrossRef PubMed.
- H. Wang, H. Li, X. Jiang, W. Shi, Z. Shen and M. Li, Diabetes, 2014, 63, 1506–1518 CrossRef CAS PubMed.
- D. R. Matthews, J. P. Hosker, A. S. Rudenski, B. A. Naylor, D. F. Treacher and R. C. Turner, Diabetologia, 1985, 28, 412–419 CrossRef CAS PubMed.
- J. Tian, X. Lin, R. Guan and J. G. Xu, Anesth. Analg., 2004, 98, 768–774 CAS , table of contents.
- C. C. Chen, C. S. Liu, C. C. Li, C. W. Tsai, H. T. Yao, T. C. Liu, H. W. Chen, P. Y. Chen, Y. L. Wu, C. K. Lii and K. L. Liu, Food Chem. Toxicol., 2013, 59, 610–617 CrossRef CAS PubMed.
- A. Danielsson, A. Ost, F. H. Nystrom and P. Stralfors, J. Biol. Chem., 2005, 280, 34389–34392 CrossRef CAS PubMed.
- H. Y. Wang, S. Ducommun, C. Quan, B. Xie, M. Li, D. H. Wasserman, K. Sakamoto, C. Mackintosh and S. Chen, Biochem. J., 2013, 449, 479–489 CrossRef CAS PubMed.
- H. K. Karlsson, J. R. Zierath, S. Kane, A. Krook, G. E. Lienhard and H. Wallberg-Henriksson, Diabetes, 2005, 54, 1692–1697 CrossRef CAS PubMed.
- H. Sano, S. Kane, E. Sano, C. P. Miinea, J. M. Asara, W. S. Lane, C. W. Garner and G. E. Lienhard, J. Biol. Chem., 2003, 278, 14599–14602 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |