DOI:
10.1039/C5FO00871A
(Paper)
Food Funct., 2016,
7, 183-193
Naringenin ameliorates renal and platelet purinergic signalling alterations in high-cholesterol fed rats through the suppression of ROS and NF-κB signaling pathways
Received
16th July 2015
, Accepted 20th October 2015
First published on 30th October 2015
Abstract
Naringenin (NGEN) is a natural flavonoid aglycone of naringin that has been reported to have a wide range of pharmacological properties, such as antioxidant activity and free radical scavenging capacity. The aim of this study was to investigate the protective effect of NGEN on oxidative and inflammatory parameters, as well as to evaluate the hydrolysis of adenine nucleotides in kidney and platelet membranes of rats exposed to a hypercholesterolemic diet (HCD) for 90 days. Kidney oxidative stress and mRNA expression of the ectonucleoside triphosphate diphosphohydrolases (NTPDases), ecto-5′-nucleotidase (CD73), inducible NO synthase (iNOS), tumor necrosis factor-α (TNF-α), interleukin 6 (IL-6) and the nuclear factor kappa B (NF-κB) genes were evaluated by real time RT-PCR. The co-administration of NGEN (50 mg kg−1) for 90 days significantly prevented renal failure in HCD rats as indicated by an improvement of renal markers. Histopathological observation findings are also consistent with these effects. Moreover, NGEN (50 mg kg−1) significantly decreased the lipid profile and inhibited pro-oxidant and inflammation marker levels in the kidney of HCD rats. Furthermore, the NTPDase activities were significantly decreased in platelets and kidney membranes of HCD-treated rats and these alterations were improved by NGEN. In conclusion, this study suggests that naringenin can potentially improve the renal failure and platelet alterations observed in rats fed a hypercholesterolemic diet probably through its antioxidant effects.
1. Introduction
Hypercholesterolemia-related metabolic syndrome increases cholesterol and triglyceride levels and remains one of the major risk factors for the development and progression of Chronic Kidney Disease (CKD).1 The incidence and prevalence of CKD are increasing worldwide; however, overweight or obese individuals have an increased risk (40%) of kidney disease compared to individuals of normal weight.2 Further, obese individuals have a reduced chance of successful kidney transplants and an increased tendency for progression to end stage renal failure.3 Dyslipidaemia in renal disease has been demonstrated in animal studies; however, 10- to 12-week diet-induced hypercholesterolemia can be used to generate a valid rodent model accompanied by the development of glomerulosclerosis and disbalance of tissue redox activity partly mediated by increased oxidative stress.4 In this context, recent studies have linked the relationship of oxidative stress and mitochondrial dysfunction as well as the development of glomerulosclerosis and tubulo-interstitial damage leading to the destruction of the kidney tissue.5 The mechanisms by which hyperlipidemia contributes to systemic oxidative stress in CKD remain unclear. Inflammatory mediators, including TNFα and IL-1β, are reactive oxygen species (ROS)-activating factors in the kidney and may induce oxygen radical production by mesangial cells (MCs).6,7 Bakker et al.8 have shown that excess FFAs bound to albumin are filtered through glomeruli and reabsorbed into the renal proximal tubules, which causes interstitial inflammation in the kidney. Moreover, Solini et al.9 demonstrated that pro-inflammatory cytokine IL-1β also inhibits ATP-binding cassette A1 (ABCA1)-mediated cholesterol efflux from MCs leading to an imbalance of cholesterol uptake, synthesis, and cholesterol efflux, which disrupts cholesterol homeostasis and causes foam cell formation. The purinergic system consists of cell surface purinergic receptors, which fall into the P1 or adenosine receptor family and the P2 or ATP/ADP receptor family, as well as metabolic enzymes, which convert extracellular purines to each other and nucleoside transporters, which terminate purinergic receptor signaling by purine uptake into the cell.10 In this regard, control of circulating nucleotide levels is noteworthy in the maintenance of physiological nucleotide-mediated signaling processes and ATP is an important extracellular signal in the regulation of many intracellular processes in normal tubular cells as well as in the progression to chronic renal failure.11 It is also important to consider that ATP acts as a short range/short timing signaling molecule because the ATP concentration is rapidly declining around the source of the release due to fast degradation by numerous ectonucleotidases.12,13 In this context, the specific pattern of ATP hydrolysis in plasma likely has significant implications on the role of ATP in platelet aggregation, ADP promotes platelet aggregation and the nucleoside adenosine is an endogenous inhibitor of platelet aggregation and limits the degree of vascular injury by reducing the release of ROS.14 Four families of ectonucleotidases exist: ectonucleoside triphosphate diphosphohydrolases (NTPDases), ectonucleotide pyrophosphatase phosphodiesterases (NPPs), ecto-5′-nucleotidase and alkaline phosphatases can modulate platelet function through the hydrolysis of extracellular adenine nucleotides and all are found in the kidney.10 NTPDase1 (CD39, apyrase) is the major ecto-nucleotidase in the renal vasculature and can be shown to efficiently bind and hydrolyze extracellular ADP (and ATP) to AMP.15 NTPDase2 and NTPDase3 are localised in Bowman's capsules and in most nephron segments.10 The expression of CD39 and CD73 ectonucleotidases on either endothelial or immune cells allows for homeostatic integration and control of vascular inflammatory and immune cell reactions at sites of injury.16 Nevertheless, these cellular processes and nucleotide-triggered events are further modulated during the development of atherosclerosis.10 A recent study has shown that the protective effects of CD39 in various inflammatory diseases under such episodes of stress conditions, such as palmitoylation status and cholesterol content of membrane lipid rafts, can be ameliorated by statins or by incorporation of saturated fatty acids into cell membranes and by limitation of lipid peroxidation responses by antioxidants.12 Naringenin is a flavanon that is naturally produced by several plants, especially the skin of grapefruit.17 The biological functions of naringenin were widely investigated and include anti-inflammation, anti-oxidation, anti-cancer, cardiovascular protection, anti-diabetes, renal protection, protection against Alzheimer's disease and antihyperuricemic activities.17 Moreover, Xiao et al.18 have shown that naringenin inhibits platelet aggregation and release by reducing the blood cholesterol levels and the cytosolic free calcium concentration in hyper-lipidemic rabbits. However, the detailed mechanisms are unclear. Hence, the present study aimed to investigate the protective effects of naringenin on proinflammatory cytokines, pro/antioxidant markers, mitochondrial dysfunction as well as the NTPDase, 5′-nucleotidase activities and its expression in renal tissue and platelets in high-cholesterol fed rats.
2. Materials and methods
2.1. Animals and reagents
Forty 2-month-old adult male Wistar rats (220–250 g) were purchased from the Central Pharmacy (SIPHAT, Tunisia). All animal procedures were conducted in strict conformation to the General Guidelines on the Use of Living Animals in Scientific Investigations19 and were approved by the Ethical Committee of Sciences Faculty, Sfax University. Animals were kept in an air conditioned room (22 ± 2 °C; 45% humidity) and housed in individual cages and subjected to a 12 h light and dark cycle. They were given a regular rodent chow with free access to food and water for two weeks to acclimatize them to the new environment. Unless otherwise specified, naringenin and all other chemicals used in this study were obtained from Sigma Chemicals Co. (St. Louis, France).
2.2. Experimental protocol
Rats were treated in the same manner as in the animal model described and recently published by us.20 Briefly, rats were randomized into four groups: the first group (C) served as the control, and received ad libitum distilled water, a standard diet supplied by the Company of Animal Nutrition, Sfax, Tunisia, and 0.5 ml vehicle solution of naringenin (NGEN) by gavage. The second group had a hypercholesterolemia diet (HCD) for three months by adding 10 g cholesterol per kg + 1 g cholic acid per kg to the standard diet. Animals in the third group (HCD + NGEN) were fed a cholesterol-enriched diet supplemented with NGEN (50 mg per kg body weight) administered by oral gavage. The fourth group (NGEN) received normal pellets and also received 0.5 ml of naringenin (50 mg per kg body weight) administered by oral gavage. Urinary samples were obtained from each animal housed in a specially designed metabolic cage within 24 h cycles. The volume of each sample was recorded and centrifuged at 3000g for 5 min. At the end of the experimental period (3 months), the animals of different groups were sacrificed by cervical decapitation to avoid stress conditions. Total blood was collected with citrate (3.8%; 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9, v/v) as an anticoagulant for platelet-rich plasma preparations. The kidney was quickly excised, rinsed in ice-cold physiological saline, weighed (to calculate the ratio of the kidney weight to the body weight (%)) and then divided into parts for homogenization in the appropriate buffer as indicated in the procedures for the measurement of each parameter.
:9, v/v) as an anticoagulant for platelet-rich plasma preparations. The kidney was quickly excised, rinsed in ice-cold physiological saline, weighed (to calculate the ratio of the kidney weight to the body weight (%)) and then divided into parts for homogenization in the appropriate buffer as indicated in the procedures for the measurement of each parameter.
2.3. Hematological study
White blood cells (WBCs), red blood cells (RBCs), hematocrit (HCT) and platelets were analyzed using standard methods with the fully automated Coulter MAXM (Beckman Coulter, Inc., Fullerton, CA).
2.4. Assessment of renal function and platelet-rich plasma preparations (PRP)
Blood samples were collected in EDTA tubes from the eye vein of rats 24 h before sacrifice and were centrifuged for 10 min at 3000g to obtain clear plasma, which was stored at −20 °C for the subsequent measurement of renal functions. The levels of urea, uric acid and creatinine in plasma and urine were estimated spectrophotometrically using commercial diagnostic kits, respectively (ref. 20151, 20143, and 20091), purchased from Biomagreb (Ariana, Tunisia). Creatinine clearance (μl min−1), an index of the glomerular filtration rate, was calculated by using the UV/P equation, where U is the urinary creatinine level, V the volume of the urine sample collected within 24 h and P the plasma creatinine concentration. Platelets were prepared by a method modified by Lunkes et al.21 Total blood was collected with citrate (3.8%; 1:9, v/v) as the anticoagulant. Platelet rich plasma was obtained after centrifugation at 800g for 15 min to remove residual blood cells and the remaining blood was further centrifuged at 3500g for 10 min and washed twice by centrifugation at 1400g with 3.5 mM (HEPES). The washed platelets were resuspended in HEPES isosmolar buffer and adjusted to 0.4–0.6 mg of protein per ml.
2.5. Assessment of lipid contents from kidney tissues
Total lipids were extracted from 100 mg of tissue by the method of Folch et al.22 Total cholesterol and triglyceride contents were determined using enzymatic kits available from Biomaghreb (Ariana, Tunisia, ref. 20111 and 20131, respectively). Phospholipid (PL) content was determined by the method of Davis.23 Free fatty acids (FFA) were estimated by the method of Falholt et al.24 Values were expressed as mg per g tissue.
2.6. Plasma and renal measurements of oxidant stress markers
Hydrogen peroxide (H2O2) was measured using the ferrous ion oxidation xylenol orange version-2 (FOX-2) method of Gay et al.25 Concentrations were calculated at 560 nm using a standard curve prepared with H2O2. Nitric oxide production was determined based on the Griess reaction.26 Absorbance was measured spectrophotometrically at 550 nm using a microplate reader. The nitrite (NO2−) concentration was determined from a standard nitrite curve generated using NaNO2. The results were expressed as μM.
ROS was measured as described in our previous report, based on the oxidation of 2′,7′-dichlorofluorescein diacetate (DCFH-DA) in a 96-well plate assay.20 Protein samples (25 μg) from kidney or plasma of all animals were diluted in PBS and incubated with 5.0 μM DCFH-DA in the dark for 15 min at 37 °C. Fluorescence was measured every 15 min for 1 h with excitation and emission wavelengths of 488 and 525 nm, respectively, using a CFX96 (Bio-Rad Laboratories, Inc., Hercules, CA, USA) fluorescence plate reader. Results were expressed as pmol of DCF per mg of protein, using a standard curve with DCF.
The extent of lipid peroxidation by measuring thiobarbituric acid reactive substances (TBARS) in terms of malondialdehyde (MDA) formation was measured according to the method of Draper and Hadley.27 The MDA values were calculated using 1,1,3,3-tetraethoxypropane as the standard and expressed as nmol of MDA per mg of protein.
2.7. Determination of non-enzymatic and enzymatic antioxidants in kidney tissue
Glutathione (GSH) in the kidney was determined by the method of Ellman modified by Jollow et al.28 Vitamin C (Vit C) measurement was performed as described by Jacques-Silva et al.29 Vitamin E (Vit E) level was assayed by the extraction method of Katsanidis and Addis.30 Catalase (CAT) activity was assayed by the method of Aebi.31 Superoxide dismutase (SOD) activity was estimated according to the method of Marklund.32 Glutathione peroxidase (GPx) activity was measured according to the method of Flohe and Gunzler.33
2.8. Assays for mitochondrial complex activities
Kidney mitochondria were isolated as previously described.34 NADH dehydrogenase (complex I), succinate-cytochrome c oxidoreductase (complexes II–III) and cytochrome c reductase (complex IV) activities were measured spectrophotometrically using microplate (Jenway 6300) conventional assays as previously described.34
2.9. Kidney membranes and platelet-rich plasma preparations
Kidney membranes were isolated as described previously by Fürstenau et al.35 Briefly, about 1.0 g of both right and left kidneys was dissected on ice, washed and gently homogenized in 10 volumes of an ice-cold medium (0.32 M sucrose, 0.1 mM EDTA, and 5 mM HEPES–Tris, pH 7.4) with a Teflon-glass homogenizer, using a discontinuous percoll gradient. The pellet was resuspended in an isosmotic solution and the final protein concentration was adjusted to 0.5–0.8 mg ml−1.35
2.10. Assay of NTPDase and 5′-nucleotidase activities
NTPDase and ecto-5′-nucleotidase activities in the kidney membranes were assayed by a method described previously.21,35 The ectonucleoside triphosphate diphosphohydrolase (NTPDase) enzymatic assay was carried out in a reaction medium containing 5 mM KCl, 1.5 mM CaCl2, 0.1 mM EDTA, 10 mM glucose, 225 mM sucrose and 45 mM Tris–HCl buffer, pH 8.0, in a final volume of 200 μl. The ecto-5′-nucleotidase (CD73) activity in the kidney and platelets was determined in a reaction medium containing 10 mM MgSO4 and 100 mM Tris–HCl buffer, pH 7.5, in a final volume of 200 μl. The reaction was initiated by the addition of the substrate (ATP or ADP) at a final concentration of 2.0 mM and was stopped with 200 μl of 10% trichloroacetic acid (TCA). The released inorganic phosphate (Pi) molybdate complex was determined by using a spectrophotometer at 690 nm. All samples were run in triplicate and the specific activity is expressed as nmol of Pi released per min per mg of protein.
2.11. Analysis of gene expression by quantitative RT-PCR (qRT-PCR)
The expressions of the ectonucleoside triphosphate diphosphohydrolase (NTPDase1-3), ecto-5′-nucleotidase (CD73), inducible NO synthase (iNOS), tumor necrosis factor-α (TNF-α), interleukin 6 (IL-6) and nuclear factor kappa B (NF-κB) genes in the kidney tissues of experimental rats (n = 4 per group) were measured using a reverse transcriptase qRT-PCR technique. Total RNA was extracted using the iScript™ RT-qPCR Sample Preparation Reagent according to the manufacturer's instructions (170-8898, Bio-Rad). RNA concentrations and purity were determined by measuring the absorbance A260/A280 ratios by using a NanoPhotometer™ (Implen GmBH). The cDNA was produced from 2 μg of total mRNA by reverse transcription with superscript reverse transcriptase (Invitrogen, France) using oligo(dT)18 as a primer in a total volume of 20 μl. After incubation for 50 min at 42 °C, the reaction was terminated by a denaturing enzyme for 10 min at 70 °C. cDNA (2 μl) was used as a template for PCR according to the recommended protocol using the 2× SYBR Permix Ex Taq™ (TaKaRa) and the primer sequences used for the gene amplification are given in Table 1.20,36 The 2−ΔΔCt method was used to analyze the relative changes in gene expression from real-time quantitative PCR experiments. Data were normalized to mRNA levels of the housekeeping gene, β-actin, and results were expressed relative to the average values for the control group, which was set to 1.
Table 1 Primer sequences used for quantitative RT-PCR reactions
| Gene name |
Forward primer (5′–3′) |
Reverse primer (5′–3′) |
| Abbreviations: β-actin, beta actin; ectonucleoside triphosphate diphosphohydrolase, NTPDase; ecto-5′-nucleotidase, CD73; IL-6, interleukin 6; TNF-α, tumor necrosis factor alpha; NF-κB, nuclear factor kappa B. |
| β-Actin |
GGAGATTACTGCCCTGGCTCCTA |
GACTCATCGTACTCCTGCTTGCTG |
| NTPDase1 |
GATCATCACTGGGCAGGAGGAAGG |
AAGACACCGTTGAAGGCACACTGG |
| NTPDase2 |
GCTGGGTGGGCCGGTGGATACG |
GGGTTCCATGGTGAAGTCAAC |
| NTPDase3 |
CGGGATCCTTGCTGTGCGTGGCATTTCTT |
TCTAGAGGTGCTCTGGCAGGAATCAGT |
| CD73 |
CCCGGGGGCCACTAGCACCTCA |
GCCTGGACCACGGGAACCTT |
| IL-6 |
TCCTACCCCAACTTCCAATGCTC |
TTGGATGGTCTTGGTCCTTAGCC |
| TNF-α |
AAATGGGCTCCCTCTCATCAGTTC |
TCTGCTTGGTGGTTTGCTACGAC |
| iNOS |
AGCATCACCCCTGTGTTCCACCC |
TGGGGCAGTCTCCATTGCCA |
| NF-κB |
GCCGTGGAGTACGACAACATC |
TTTGAGAAGAGCTGCCAGCC |
2.12. Histopathological analysis
Kidney tissues from the normal and experimental rats (n = 3 per group) were fixed in 10% buffered formalin and have been processed for paraffin sectioning. Sections of 5 μm thickness were stained with hematoxylin and eosin (H&E), and examined with a Leica® microscope fitted with a Sony® digital camera to capture images for histological studies.
2.13. Quantitative protein determination
The protein concentration in kidney homogenates was measured by the Bradford method using bovine serum albumin as the standard.
2.14. Statistical analysis
Results were expressed as the means ± standard deviation (mean ± SD). All analyses were carried out using GraphPad Prism 4.02 for Windows (GraphPad Software, San Diego, CA). Significant differences between treatment effects were determined by using one-way ANOVA, followed by a Tukey's post-hoc test for multiple comparisons with statistical significance of p < 0.05.
3. Results
3.1. NGEN ameliorates HCD-induced renal markers and hematological parameters
Results from the biochemical plasma and urine study revealed that HCD for a successive 12 weeks significantly (p < 0.001) impaired kidney function as assessed by an increase in uric acid and urea and a decrease in creatinine clearance in comparison to the control rats (Table 2). Oral co-administration of NGEN significantly improved (p < 0.001) these parameters induced by cholesterol treatment.
Table 2 Hematological parameters (white blood cells (WBCs), red blood cells (RBCs), hematocrit (HCT)), plasma and urinary levels of urea, uric acid and creatinine clearance of the control and rats treated with high cholesterol diet (HCD), naringenin (NGEN) and their combination (HCD + NGEN)
| Parameters & treatments |
C |
HCD |
HCD + NGEN |
NGEN |
| Values are expressed as the mean ± SD of 8 rats per group. HCD group vs. control group: ***p < 0.001, *p < 0.05. HCD + NGEN group vs. HCD group: ¥¥¥p < 0.001, ¥¥p < 0.01. |
| Urea (mg dL−1) |
| Plasma |
35.44 ± 1.77 |
52.30 ± 4.80*** |
41.80 ± 3.0¥¥¥ |
34.70 ± 2.50 |
| Urine |
45.80 ± 1.50 |
49.40 ± 1.90* |
44.28 ± 1.80¥¥ |
45.40 ± 2.10 |
| |
| Uric acid (mg dL−1) |
| Plasma |
1.7 ± 0.12 |
3.0 ± 0.28*** |
2.28 ± 0.14¥¥¥ |
1.76 ± 0.10 |
| Urine |
6.4 ± 0.28 |
8.54 ± 0.70*** |
7.14 ± 0.20¥¥¥ |
6.30 ± 0.25 |
| Creatinine clearance (μl min−1) |
340 ± 14.50 |
183 ± 12.00*** |
264 ± 18.00¥¥¥ |
348 ± 13.00 |
| |
| Hematological parameters |
| WBC (103 μL−1) |
9.55 ± 0.28 |
15.40 ± 0.84*** |
12.55 ± 0.50¥¥¥ |
9.40 ± 0.30 |
| Platelets (103 μL−1) |
680 ± 21.70 |
1045 ± 32.80*** |
848 ± 40.00¥¥¥ |
684 ± 20.80 |
| RBCs (106 μL−1) |
8.34 ± 0.16 |
7.35 ± 0.23*** |
7.85 ± 0.13¥¥ |
8.20 ± 0.11 |
| HCT (%) |
43.50 ± 0.38 |
44.74 ± 0.80 |
44.52 ± 0.40 |
44.26 ± 0.50 |
Upon an examination of hematological parameters (Table 2), a decrease in total red blood cell (RBC) number as well as an increase in the platelet concentration and total white blood cell (WBC) number (p < 0.001) were observed in rats exposed to HCD as compared to the control group. Hematocrit (HCT) was found to be normal in all the experimental groups. Oral co-administration of NGEN significantly (p < 0.001) ameliorated these alterations induced by cholesterol treatment. No changes were observed in the rats treated with NGEN alone when compared to the control rats (Table 2).
3.2. NGEN restores relative kidney weight and lipid profile changes in high-cholesterol fed rats
As shown in Table 3, the present data showed that consuming a HCD for 3 months induced a significant increase (p < 0.001) in the ratio of kidney weight to body weight (%), which was prevented (p < 0.01) by NGEN supplementation. Furthermore, a significant increase (p < 0.001) of renal lipid profiles such as TC, TG, PL and FFAs was observed in HCD fed rats as compared with the control group. Co-administration of NGEN significantly (p < 0.001) reduced the ratio of kidney weight and attenuated the tissue lipid composition as compared to HCD rats (Table 3). Comparably, no significant changes were observed between the control rats and rats treated with NGEN alone.
Table 3 Ratio of the kidney weight to the body weight (%) and renal lipid profile (total cholesterol (TC), triglycerides (TG), free fatty acids (FFA) and phospholipids (PL) of the control and rats treated with High cholesterol diet (HCD), naringenin (NGEN) and their combination (HCD + NGEN)
| Parameters & treatments |
C |
HCD |
HCD + NGEN |
NGEN |
| Values are expressed as the mean ± SD of 8 rats per group. HCD group vs. control group: ***p < 0.001. HCD + NGEN group vs. HCD group: ¥¥¥p < 0.001. |
| Relative kidney weights (%) |
0.69 ± 0.026 |
0.87 ± 0.04*** |
0.75 ± 0.032¥¥¥ |
0.68 ± 0.02 |
| TC (mg per g tissue) |
2.20 ± 0.14 |
3.26 ± 0.19*** |
2.43 ± 0.21¥¥¥ |
2.06 ± 0.11 |
| TG (mg per g tissue) |
2.54 ± 0.20 |
5.23 ± 0.16*** |
3.35 ± 0.22¥¥¥ |
2.45 ± 0.30 |
| FFA (mg per g tissue) |
4.10 ± 0.15 |
7.04 ± 0.3*** |
4.88 ± 0.31¥¥¥ |
3.90 ± 0.29 |
| PL (mg per g tissue) |
13.6 ± 1.5 |
24.2 ± 1.58*** |
18.4 ± 1.4¥¥¥ |
13.2 ± 2.3 |
3.3. Effects of NGEN treatment on renal oxidative markers and the activities of mitochondrial enzyme markers in high-cholesterol fed rats
As shown in Table 4, HCD treatment markedly increased ROS, H2O2, NO2 and TBARS levels as compared with those of the controls in the plasma and kidney tissue (p < 0.001). However, treatment with NGEN significantly (p < 0.001) alleviated these changes. Interestingly, rats treated with NGEN alone do not present any difference with the control group (Table 4).
Table 4 Effect of High cholesterol diet (HCD), naringenin (NGEN) and their combination (HCD + NGEN) in plasma and tissues hydrogen peroxide (H2O2, nitrite (NO2−), reactive oxygen species (ROS) and lipid peroxidation (TBARS)
| Parameters & treatments |
C |
HCD |
HCD + NGEN |
NGEN |
| Values are expressed as the mean ± SD of 8 rats per group. HCD group vs. control group: ***p < 0.001, *p < 0.05. HCD + NGEN group vs. HCD group: ¥¥¥p < 0.001, ¥¥p < 0.01. |
| H2O2 |
| Plasma (μmol L−1) |
22.30 ± 2.90 |
46.00 ± 2.40*** |
32.50 ± 1.90¥¥¥ |
21.00 ± 2.00 |
| Kidney (μmol per mg tissue) |
1.48 ± 0.19 |
4.04 ± 0.30*** |
2.64 ± 0.28¥¥¥ |
1.30 ± 0.26 |
| |
| NO2− |
| Plasma (μM) |
0.67 ± 0.03 |
1.35 ± 0.11*** |
1.02 ± 0.10¥¥ |
0.60 ± 0.15 |
| Kidney (μM) |
1.56 ± 0.07 |
2.50 ± 0.16*** |
1.91 ± 0.14¥¥ |
1.40 ± 0.16 |
| |
| ROS |
| Plasma (pmol DCF per mg protein) |
63.1 ± 3.90 |
94.0 ± 4.30*** |
70.00 ± 3.80¥¥¥ |
58.8 ± 5.16 |
| Kidney (pmol DCF per mg protein) |
33.50 ± 2.16 |
79.3 ± 7.20*** |
55.20 ± 7.60¥¥¥ |
33.30 ± 3.20 |
| |
| TBARS |
| Plasma (μmol ml−1) |
0.14 ± 0.010 |
0.52 ± 0.06*** |
0.35 ± 0.03¥¥¥ |
0.15 ± 0.025 |
| Kidney (nmol per mg protein) |
14.17 ± 0.90 |
31.0 ± 3.40*** |
22.60 ± 2.07¥¥¥ |
14.05 ± 1.60 |
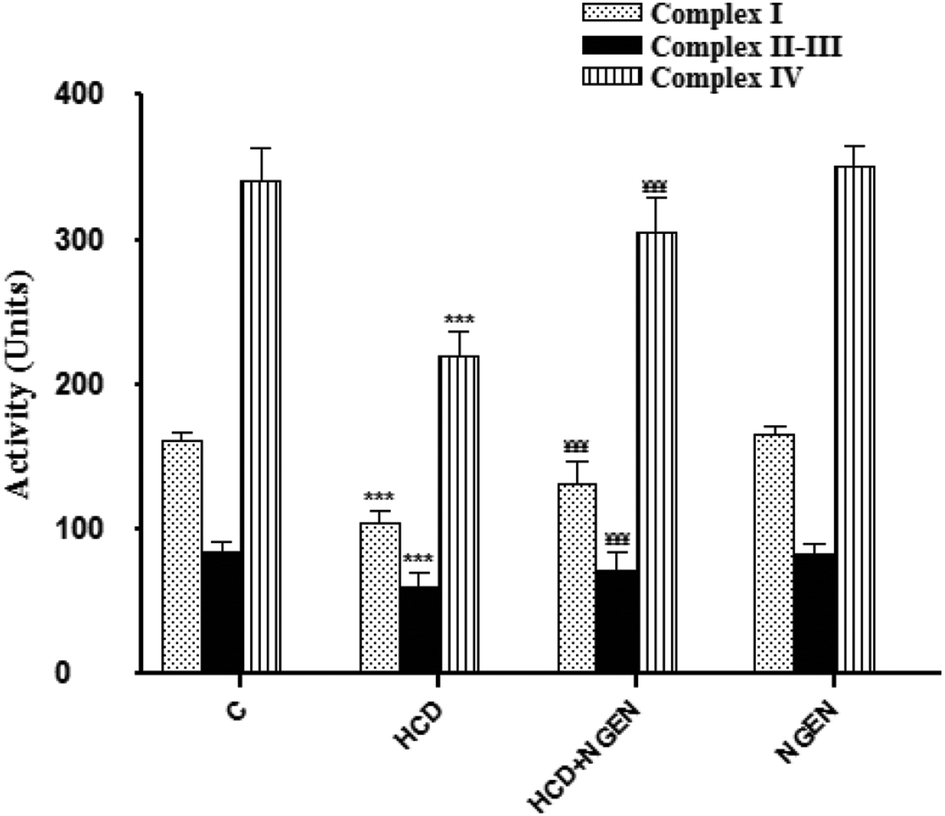
In order to determine whether impaired mitochondria may play a role in lipid accumulation, we measured mitochondrial activity. The renal mitochondrial enzyme activities such as NADH dehydrogenase (complex I), succinate-cytochrome c oxidoreductase (complexes II–III) and cytochrome c reductase (complex IV) (Fig. 1) significantly (p < 0.001) decreased in the kidney mitochondria of HCD-treated rats. Co-administration of NGEN along with HCD significantly (p < 0.001) increased the activities of these enzymes when compared with HCD-treated rats. The activities of these renal mitochondrial enzymes in NGEN treated rats alone were close to the control rats (Fig. 1).
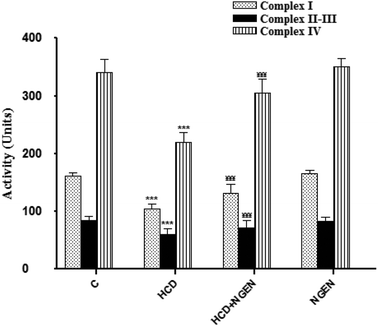 |
| | Fig. 1 Effect of naringenin (NGEN) co-administration on the activities of NADH dehydrogenase (complex I), succinate-cytochrome c oxidoreductase (complexes II–III) and cytochrome c reductase (complex IV) mitochondrial enzymes in kidney of high cholesterol diet (HCD)-induced rats. Values are expressed as the means ± SD of 6 rats per group. HCD group vs. control group: ***p < 0.001. HCD + NGEN group vs. HCD group: ¥¥¥p < 0.001, ¥¥p < 0.01, ¥¥p < 0.05. | |
3.4. NGEN restores the renal antioxidant status
Vit C, Vit E, GSH, GPx, SOD and catalase levels were significantly (p < 0.001) decreased in the HCD group as compared to the control group (Table 5). Co-administration of NGEN and HCD restored partially (p < 0.01) the antioxidant status in the renal tissue compared with HCD-treated rats. No appreciable change in any of these parameters was observed in the group treated with NGEN alone compared to the control group, indicating no adverse effect of NGEN in healthy rats.
Table 5 Effect of high cholesterol diet (HCD), naringenin (NGEN) and their combination (HCD + NGEN) on non-enzymatic and enzymatic antioxidants status in kidney tissue: catalase (CAT); glutathione peroxidase (GPx); superoxide dismutase (SOD); glutathione (GSH); vitamin C (Vit C) and vitamin E (Vit E)
| Parameters & treatments |
C |
HCD |
HCD + NGEN |
NGEN |
| Values are expressed as the mean ± SD of 8 rats per group. HCD group vs. control group: ***p < 0.001. HCD + NGEN group vs. HCD group: ¥¥¥p < 0.001, ¥¥p < 0.01. |
| CAT (U per mg protein) |
66.25 ± 0.11 |
43.20 ± 1.70*** |
51.75 ± 2.50¥¥ |
62.6 ± 2.20 |
| SOD (U per mg protein) |
6.54 ± 0.3 |
4.32 ± 0.19*** |
5.56 ± 0.18¥¥¥ |
6.3 ± 0.25 |
| GPx (U per mg protein) |
12.84 ± 1.02 |
6.06 ± 0.50*** |
8.70 ± 0.4¥¥¥ |
12.3 ± 0.7 |
| GSH (μg per mg protein) |
8.67 ± 0.22 |
5.64 ± 0.56*** |
6.92 ± 0.44¥¥ |
8.54 ± 0.37 |
| Vit C (μg per mg protein) |
2.52 ± 0.13 |
1.44 ± 0.16*** |
2.14 ± 0.11¥¥¥ |
2.55 ± 0.12 |
| Vit E (μg per mg protein) |
4.20 ± 0.20 |
2.05 ± 0.13*** |
3.45 ± 0.35¥¥¥ |
4.40 ± 0.45 |
3.5. NGEN down-regulated the expression of proinflammatory-mediated genes in kidney
To investigate the role of pro-inflammatory markers on the NGEN-mediated renoprotective effect, mRNA expression levels of iNOS, TNF-α and IL-6 and the activation of the nuclear factor kappa B (NF-κB) were determined using qRT-PCR (Table 6). The results show concomitant increases (p < 0.001) in the mRNA expression levels of TNF-α, IL-6, iNOS and NF-κB in kidneys derived from HCD-treated rats as compared to the control. Importantly, gene expression was significantly (p < 0.001) down-regulated in the HCD + NGEN group compared with the HCD group. The administration of NGEN alone did not show any significant effects when compared with the control group.
Table 6 Effect of high cholesterol diet (HCD), naringenin (NGEN) and their combination (HCD + NGEN) on renal gene expression of proinflammatory markers iNOS, TNF-α, and IL-6, the activation of the nuclear factor kappa B (NF-κB) and ecto-nucleotidases: NTPDase1, NTPDase2, NTPDase3 and 5′-nucleotidase (CD73)
| Parameters & treatments |
C |
HCD |
HCD + NGEN |
NGEN |
| Values are expressed as the mean ± S.D. of 4 rats per group. HCD group vs. control group: ***p < 0.001, **p < 0.01, *p < 0.05. HCD + NGEN group vs. HCD group: ¥¥¥p < 0.001, ¥¥¥p < 0.01, ¥p < 0.05. |
| Proinflammatory markers |
| IL-6 |
1.00 ± 0.12 |
1.8.0 ± 0.25*** |
1.40 ± 0.08¥ |
1.00 ± 0.05 |
| TNF-α |
1.00 ± 0.11 |
2.90.0 ± 0.20*** |
2.10 ± 0.30¥¥0 |
1.00 ± 0.20 |
| iNOS |
1.00 ± 0.13 |
3.5 ± 0.18*** |
2.32 ± 0.16¥¥¥ |
1.00 ± 0.10 |
| NF-κB |
1.00 ± 0.07 |
1.94 ± 0.12*** |
1.4 ± 0.13¥¥ |
1.00 ± 0.15 |
| |
| Ecto-nucleotidase |
| NTPDase1 |
1.00 ± 0.10 |
0.80 ± 0.04* |
0.90 ± 0.03 |
1.00 ± 0.09 |
| NTPDase2 |
1.00 ± 0.08 |
0.6 ± 0.12*** |
0.9 ± 0.07¥¥¥ |
1.00 ± 0.10 |
| NTPDase3 |
1.00 ± 0.05 |
0.85 ± 0.03 |
0.85 ± 0.13 |
1.00 ± 0.08 |
| CD73 |
1.00 ± 0.11 |
0.74 ± 0.10*** |
0.92 ± 0.05¥¥ |
1.00 ± 0.11 |
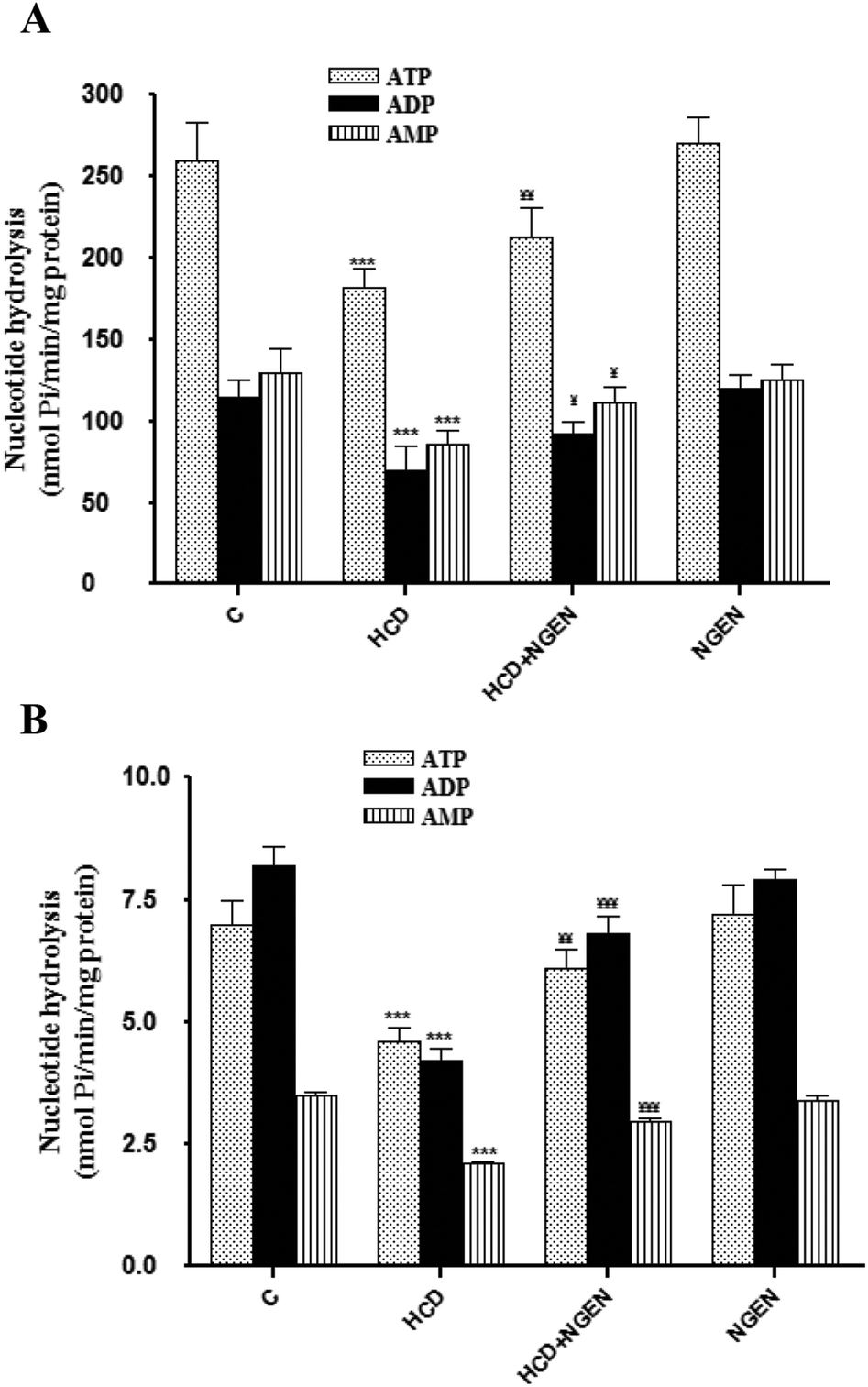
3.6. Effects of NGEN on ectonucleotidase activities and gene expression in the platelets and kidney of HCD-treated rats
Fig. 2 shows the results obtained for ectonucleotidase (NTPDases, 5′-nucleotidase) activities in the renal membranes (Fig. 2A) and platelets (Fig. 2B) from rats exposed to HCD or NGEN or exposed simultaneously to HCD + NGEN. As can be seen, a significant decrease of ectonucleotidase activities in HCD-treated rats when compared with the control group (p < 0.001) was observed when ATP, ADP or AMP was used as substrate and the decrease induced by HCD was abolished (p < 0.001) by NGEN co-administration. The administration of NGEN alone did not have any significant effects when compared with the control group.
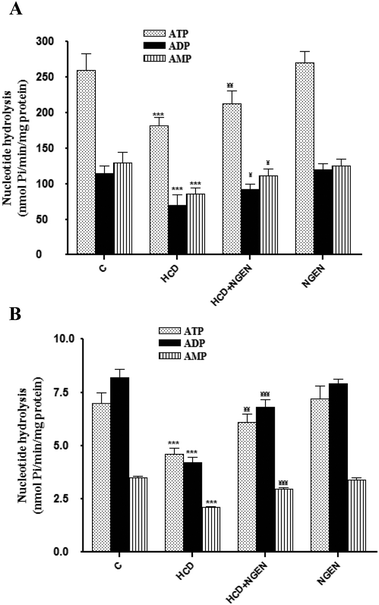 |
| | Fig. 2 Effect of high-cholesterol diet (HCD), high-cholesterol diet plus naringenin (HCD + NGEN) and naringenin alone (NGEN) on ectonucleotidase activity (nmol of Pi per mg of protein per min) using ATP, ADP and AMP as substrate in kidney membranes (A) and platelets (B). Values are expressed as the means ± SD of 6 rats per group. HCD group vs. control group: ***p < 0.001. HCD + NGEN group vs. HCD group: ¥¥¥p < 0.001, ¥¥p < 0.01, ¥¥p < 0.05. | |
To identify related genes that may be regulated on HCD and NGEN exposure, the expression analysis of NTPDase1–3 and 5′-nucleotidase was carried out by the qRT-PCR assay (Table 6). Results revealed specific signals accompanied by the down regulation corresponding to mRNA for NTPDase1, NTPDase2 and CD73 in the HCD-treated group when compared to the control group. There were no differences in mRNA transcript levels for NTPDase3. On the other hand, gene expression was significantly (p < 0.05) up-regulated in the HCD + NGEN group compared with the HCD-treated group (Table 6). The administration of NGEN alone did not have any significant effects when compared with the control group.
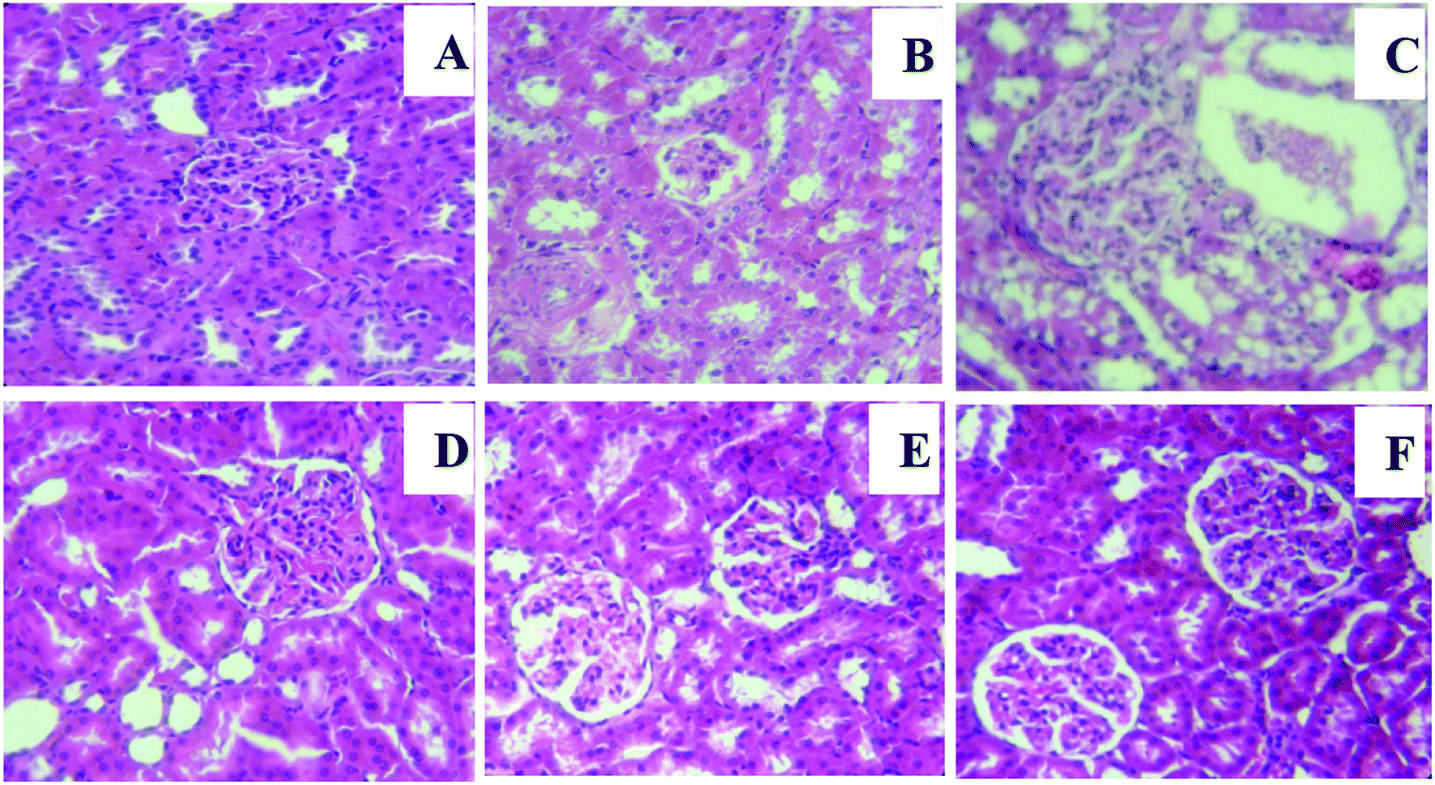
3.7. Effects of NGEN on histological injury of kidney in experimental animals
A light microscopy evaluation of kidneys in the control and NGEN groups showed normal renal histology with apparent glomerulus with a constant tuft and normal tubular architecture (Fig. 3A and F). While HCD-treated rats showed a distinctive glomerular basement membrane accompanied by an enlarged Bowman's space and vascular congestion outside fragmented glomeruli and between tubules with mild tubular degeneration (Fig. 3B and C), NGEN co-treatment induced a marked improvement in the histopathological changes in the kidneys of HCD rats (Fig. 3D and E).
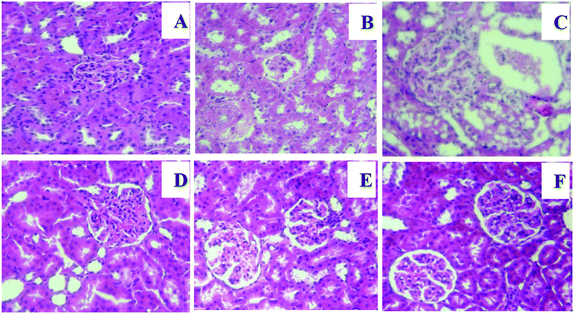 |
| | Fig. 3 Effect of naringenin (NGEN) on kidney histopathological images of hematoxylin and eosin (H&E) staining of high-cholesterol diet (HCD) induced rats (400×). (A) and (F) Sections from the control and NGEN (50 mg kg−1)-treated groups show normal histological structure of renal parenchyma. The HCD group showed a gradually progressing increase in glomerular tuft size and Bowman's space expansion (B, C). Kidney tissue from the (HCD + NGEN) group shows slight congestion of glomerular tufts (D, E). | |
4. Discussion
A high-cholesterol diet (HCD) is a well-known model for excess calorie intake, which will contribute to the development of obesity and metabolic syndrome.7 Feeding for three months by adding 10 g cholesterol per kg + 1 g cholic acid per kg to the standard diet in this model is characterized by increased renal oxidative/nitrosative damage and is well suited for the study of potential preliminary preventive or therapeutic compounds against hyperlipidemia-induced kidney injury.4 In this study, we showed that dietary (NGEN) supplementation had a beneficial effect against hypercholesterolemia kidney injury as evidenced by the alleviation of histopathological changes and the improvement of serum biochemical markers such as urea, uric acid and creatinine clearance and renal lipid profile. NGEN has the potential to restore kidney mitochondrial dysfunction and oxidative stress, improve renal antioxidant defense capacity, and inhibit NF-κB signaling and is able to modulate the expression of proinflammatory cytokines responsible for the aetiology of renal complications. Phosphohydrolysis of extracellular ATP and ADP in the kidney is an essential step in purinergic signaling that regulates key pathophysiological processes such as those linked to inflammation. The major new findings of this study are that treatment with NGEN was suitable for modulating the adenine nucleotide hydrolysis alterations induced by a HCD diet as indicated by the biochemical and qRT-PCR analyses in the platelets and renal tissue. Since hypercholesterolemia-induced oxidative stress plays an essential role in the development and progression of early nephropathy, as shown in a previous study, elevated serum creatinine, urea and uric acid as a result of both increased generation and decreased excretion is a well known characteristic of renal injury.38,39 In the current study, feeding HCD for three months was evidenced to cause elevated urea and uric acid levels while creatinine clearance was decreased, which were taken as a direct in vivo index of nephropathy in HCD-treated rats (Table 2). One would have expected that NGEN would have ameliorated kidney function parameters in HCD rats. We are tempted to speculate that the prevention of HCD-mitochondrial oxidative stress by NGEN, documented in this study, may be involved, at least in part, in the prevention of renal oxidative stress. Corroborating these results, several studies have reported that supplementation with NGEN attenuated negative physiological sequels induced by oxidative stress in high fat diet fed streptozotocin-diabetics due to its direct free radical scavenger activity and its indirect antioxidant properties.40 Additionally, Tsai et al.41 demonstrated that treatment of diabetic mice with NGEN provided antioxidative and antihyperlipidemic activities in the kidney tissue.
Administration of a cholesterol-rich diet results in increased organ lipid profile, which is well supported by many previous studies.39 In accordance with this, in our present study, we found that there was a significant elevation in the levels of renal total cholesterol (TC), triglycerides (TG), free fatty acids (FFA) and phospholipids (PL) in HCD fed rats (Table 3) as compared with normal rats, an increase that was prevented by oral administration of NGEN. The approach for lowering lipid nephrotoxicity must be multifaceted and regardless of the specific sources of intracellular ROS production, the close relationship between ROS and hypercholesterolemia has been confirmed by a number of recent studies.7,20,37 Mitochondrial cholesterol accumulation not only sensitizes to oxidative stress but also it can contribute to the metabolic reprogramming. The present study endeavored to determine the in vivo protective effect of NGEN against the mitochondrial dysfunction and oxidative stress induced by high cholesterol levels in the renal tissue. In the present study, our results demonstrated that oral co-administration of NGEN significantly reduced the levels of HCD-induced overproduction of ROS, H2O2, NO2- and TBARS in the plasma and renal tissue (Table 4). The effect of ROS is balanced by the antioxidant action of non-enzymatic antioxidants as well as by antioxidant enzymes. In this regard, under HCD conditions, the activities of renal antioxidant enzymes, the first line of defense against ROS, namely SOD, CAT and GPx, and non-enzymic antioxidants, namely glutathione, vitamin C and vitamin E, were substantially suppressed (Table 5). The ability of NGEN to increase renal reduced glutathione, vitamin C and vitamin E levels and catalase, SOD and GPx activities is in agreement with the finding of Hermenean et al.43 who found that NGEN corrects oxidative disturbances on carbon tetrachloride-induced acute nephrotoxicity in mice. It is increasingly evident that elevated levels of ROS can adversely affect cell function by impairing the renal mitochondria electron transport chain complex function. However, Mershiba et al.42 detected a reduction in mitochondrial respiratory function using the free fatty acid (FFA) bound BSA-overload nephropathy model to induce renal lipotoxicity/ROS-related inflammation. In agreement with this view, we observed a marked change in mitochondrial complexes such as NADH dehydrogenase, succinate cytochrome c oxidoreductase and cytochrome c reductase activities were significantly decreased in the kidney of HCD-treated rats (Fig. 1). On the other hand, this study reveals that the treatment with NGEN is able to prevent excessive ROS production and was effective in maintaining the mitochondrial complex activities. A recent study showed that naringenin is better as compared to hesperidin and hesperetin (its related citrus flavanones) as far as interaction with intracellular enzymes, mitochondrial and cellular membrane is concerned.43 Moreover, Fouad et al.44 showed that naringin, the glycosidic form of NGEN, at a dose of 100 mg kg−1 was found to offer protection and restored the mitochondrial function by maintaining mitochondrial respiratory enzyme activity and thereby preventing the mitochondrial membrane potential (Δψm). A large number of inflammatory mediators, including the cytokines TNF-α and IL-6 and the reactive nitrogen intermediate NO, may induce mitochondrial damage.7 TNFα and IL-6 decrease the activity of MRC complex I, ATP production, and mitochondrial membrane potential. These mediators also induce the accumulation of significant amounts of ROS. Activation and nuclear translocation of nuclear factor kappa B (NF-κB), in response to oxidative stress, are thought to be the key factors in the renal inflammatory process by regulating the gene expression of cytokines.20 In the present study, NGEN co-treatment along with HCD significantly down-regulated HCD induced inducible nitric oxide synthase (iNOS) and proinflammatory cytokines TNF-α and IL-6 in the kidney by the inhibition of the NF-κB expressions analyzed by qRT-PCR. Our results are consistent with previous ones showing that NGEN reduces cholesterol-induced hepatic inflammation by modulating TNF-α and IL-6 via the down-regulation of NF-κB. During inflammation, nitric oxide (NO) and derived oxidants are formed by inflammatory cells. NO plays a number of physiological roles in the regulation of renal function and a marked increase in iNOS expression along the nephron characterizes experimental renovascular disease, and leads to decreases in tubular filtrate concentration capacity, a measure of intrinsic renal damage.35 Moreover, NO is a potent inhibitor of LDL oxidation and may be an important antioxidant for LDL in vivo but is rapidly inactivated by superoxide anions to form peroxynitrite (ONOO–) as a potent oxidant.45 Overproduction of NO was also argued to be beneficial in attenuating lipid peroxidation and has been implicated in the regulation of platelet function. Indeed, it has been proposed that the majority (60%) of blood nitrite, in humans, is carried by erythrocytes followed by white blood cells (WBC) and activated platelets which are frequently used as an indicator of systemic inflammation.45 In the current study, WBC and platelet counts were all significantly higher in HCD-treated rats than in the control rats (Table 2) and were partially alleviated by NGEN. Recently, Annadurai et al.46 reported that naringenin exhibited lower platelet counts, neutrophil and monocyte mean percentages with a higher mean percentage of lymphocytes in diabetic treated-rats.
Purinergic signaling mechanisms mediated by nucleotides and nucleosides have a complex influence on inflammatory processes and adenosine receptor activation strongly attenuates the production of several proinflammatory cytokines. In the present study, we observed a decrease of NTPDase and 5′-nucleotidase platelet activities associated with an increase of NO levels in whole blood. An important aspect to be discussed is that the decrease in the NTPDase and 5′-nucleotidase activities can be associated with an increased content of fats in the diet, which is related to the deleterious effects attributed to the increase in LDL-c and triglycerides, reduced circulating levels of HDL-c, and a rise in ROS-induced lipid peroxidation in HCD fed animals.47 Several mechanisms can regulate ectonucleotidase activities, including transcriptional and/or post-translational modifications.35 Quantitative RT-PCR analysis of the patterns of ectonucleotidase gene expression after HCD treatment showed a significant decrease in mRNA transcript levels of NTPDase2 and 3 and CD73 (Table 6). Previous studies have already demonstrated that the enzymes able to hydrolyze extracellular nucleotides can be regulated at the transcriptional level following drug treatment.10 At the time, the mechanism responsible for this effect was unknown. A number of studies have demonstrated that NGEN is a compound that presents anti-aggregant properties related to hydrolysis of nucleotides such as ATP and ADP.19 In platelets, concentrations of NGEN inhibit platelet activation in vitro as well as granular release reaction and platelet adhesion.19 Results obtained in this study may present a potential mechanism of inhibition of platelet aggregation by NGEN through ectonucleotidase modulation, which plays an important role in regulating both vascular and thrombotic factors. The increase of ATP, ADP, and AMP hydrolysis caused by NGEN is likely to decrease the ADP levels, the main promoter of platelet aggregation, and increase adenosine, a powerful inhibitor of platelet aggregation.48 Interestingly, we also observed that NGEN co-treatment prevented the increase in NO, proinflammatory cytokines and the decrease in NTPDase and 5′-nucleotidase activities in kidney tissue of HCD-treated rats. This effect is interesting because the increase in ATP, ADP, and AMP hydrolysis can maintain the levels of extracellular ATP molecules.
5. Conclusion
The results of the present study indicate that naringenin provides a significant protective effect against HCD-induced renal and platelet injury. The antioxidant, anti-inflammatory and anti-aggregant properties can be considered the main factors responsible for the beneficial effect of naringenin. Therefore, naringenin may represent a potential therapeutic option to prevent HCD-induced renal injury and dysfunction.
Abbreviation
| NF-κB | Nuclear factor-kappa B |
| TIMPs | Tissue inhibitors of metalloproteinases |
| ROS | Reactive oxygen species |
| NTPDases | Triphosphate diphosphohydrolases |
| iNOS | Inducible NO synthase |
| TNF-α | Tumor necrosis factor-α |
| IL6 | Interleukin 6 |
Conflict of interest
None of the authors have any conflicts of interest.
Acknowledgements
The present work was supported by the grants from DGRST (Appui a la Recherche Universitairede base, ARUB, UR11ES70), Tunisia.
References
- Z. A. Massy and D. de Zeeuw, Kidney Int., 2013, 84, 451–456 CrossRef PubMed.
- Y. Wang, X. Chen, Y. Song, B. Caballero and L. J. Cheskin, Kidney Int., 2008, 73, 19–33 CrossRef CAS PubMed.
- M. Afkarian, M. C. Sachs, B. Kestenbaum, I. B. Hirsch, K. R. Tuttle, J. Himmelfarb and I. H. de Boer, J. Am. Soc. Nephrol., 2013, 24, 302–308 CrossRef CAS PubMed.
- H. J. Kim, H. Moradi, J. Yuan, K. Norris and N. D. Vaziri, Am. J. Physiol.: Renal. Physiol., 2009, 296(6), 297–306 CrossRef PubMed.
- S. S. Al-Rejaie, H. M. Abuohashish, O. A. Alkhamees, A. M. Aleisa and A. S. Alroujayee, Lipids Health Dis., 2012, 11, 41 CrossRef CAS PubMed.
- A. E. Declèves, Z. Zolkipli, J. Satriano, L. Wang, T. Nakayama, M. Rogac, T. P. Le, J. L. Nortier, M. G. Farquhar, R. K. Naviaux and K. Sharma, Kidney Int., 2014, 85(3), 611–623 CrossRef PubMed.
- C. Ruggiero, M. Ehrenshaft, E. Cleland and K. Stadler, Am. J. Physiol.: Endocrinol. Metab., 2011, 300(6), E1047–E1058 CrossRef CAS PubMed.
- P. J. Bakker, L. M. Butter, L. Kors, G. J. Teske, J. Aten, F. S. Sutterwala, S. Florquin and J. C. Leemans, Kidney Int., 2014, 85(5), 1112–1122 CrossRef CAS PubMed.
- A. Solini, S. Menini, C. Rossi, C. Ricci, E. Santini, C. Blasetti Fantauzzi, C. Iacobini and G. Pugliese, J. Pathol., 2013, 231(3), 342–353 CrossRef CAS PubMed.
- G. Burnstock, L. C. Evans and M. A. Bailey, Purinergic Signalling, 2014, 10(1), 71–101 CrossRef CAS PubMed.
- K. Scholz-Pedretti, J. Pfeilschifter and M. Kaszkin, Br. J. Pharmacol., 2001, 132, 37–46 CrossRef CAS PubMed.
- K. Chen, J. Zhang, W. Zhang, J. Zhang, J. Yang, K. Li and Y. He, Int. J. Biochem. Cell Biol., 2013, 45, 932–943 CrossRef CAS.
- G. E. Garcia, L. D. Truong, J. F. Chen, R. J. Johnson and L. Feng, Kidney Int., 2011, 80, 378–388 CrossRef CAS PubMed.
- H. Xiao, H. Y. Shen, W. Liu, R. P. Xiong, P. Li, G. Meng, N. Yang, X. Chen, L. Y. Si and Y. G. Zhou, PLoS One, 2013, 8, e60173 CAS.
- R. M. Vekaria, D. G. Shirley, J. Sevigny and R. J. Unwin, Am. J. Physiol.: Renal. Physiol., 2006, 290, F550–F560 CrossRef CAS PubMed.
- K. Enjyoji, J. Sevigny, Y. Lin, P. S. Frenette, P. D. Christie, J. S. Esch, M. Imai, J. M. Edelberg, H. Rayburn, M. Lech, D. L. Beeler, E. Csizmadia, D. D. Wagner, S. C. Robson and R. D. Rosenberg, Nat. Med., 1999, 5, 1010–1017 CrossRef CAS PubMed.
- I. A. Mir and A. B. Tiku, Nutr. Cancer, 2015, 67(1), 27–42 CrossRef CAS PubMed.
- Y. Xiao, L. L. Li, Y. Y. Wang, J. J. Guo, W. P. Xu, Y. Y. Wang and Y. Wang, Exp. Ther. Med., 2014, 8(3), 968–972 CAS.
- Council of European Communities, Off. J. Eur. Communities, 1986, 358, 1–18 Search PubMed.
- Y. Chtourou, H. Fetoui, R. Jemai, A. Ben Slima, M. Makni and R. Gdoura, Eur. J. Pharmacol., 2015, 746, 96–105 CrossRef CAS PubMed.
- G. I. Lunkes, D. Lunkes, C. Mazzanti, A. Morsch, V. Morsch and M. Schetinger, Diabetes Res. Clin. Pract., 2004, 65, 1–6 CrossRef CAS PubMed.
- J. Folch, M. Lees and G. H. Sloane Stanly, J. Biol. Chem., 1957, 226(1), 497–509 CAS.
- D. B. Zilversmit and A. K. Davis, J. Lab. Clin. Med., 1950, 35(1), 155–160 CAS.
- K. Falholt, B. Lund and W. Falholt, Clin. Chim. Acta, 1973, 46(2), 105–111 CrossRef CAS.
- C. Gay, J. Collins and J. M. Gebicki, Anal. Biochem., 1999, 273, 149–155 CrossRef CAS PubMed.
- L. C. Green, D. A. Wagner, J. Glogowski, P. L. Skipper, J. S. Wishnok and S. R. Tannenbaum, Anal. Biochem., 1982, 126, 131–138 CrossRef CAS PubMed.
- H. H. Draper and M. Hadley, Methods Enzymol., 1990, 186, 421–431 CAS.
- D. J. Jollow, J. R. Mitchell, N. Zampaglione and J. R. Gillete, Pharmacology, 1974, 11(3), 151–169 CrossRef CAS PubMed.
- M. C. Jacques-Silva, C. W. Nogueira, L. C. Broch, E. M. Flores and J. B. T. Ro-cha, Pharmacol. Toxicol., 2001, 88, 119–125 CAS.
- E. Katsanidis and P. B. Addis, Free Radicals Biol. Med., 1999, 27(11–12), 1137–1140 CrossRef CAS PubMed.
- H. Aebi, Methods Enzymol., 1984, 105, 121–126 CAS.
- S. Marklund and G. Marklund, Eur. J. Biochem., 1974, 47, 469–474 CrossRef CAS PubMed.
- L. Flohe and W. A. Gunzler, Methods Enzymol., 1984, 105, 114–121 CAS.
- A. Barrientos, F. Fontanesi and F. Díaz, Curr. Protoc. Hum. Genet., 2009 Search PubMed , Chapter 19, Unit 19.3.
- C. R. Fürstenau, D. B. Ramos, F. C. Vuaden, E. A. Casali, P. S. Monteiro, D. S. Trentin, A. N. Gossenheimer, M. R. Bogo, C. D. Bonan, M. L. Barreto-Chaves, J. J. Sarkis and S. T. Wofchuk, Life Sci., 2010, 87(9–10), 325–332 CrossRef.
- G. P. Cognato, F. C. Vuaden, L. E. Savio, B. Bellaver, E. Casali, M. R. Bogo, D. O. Souza, J. Séviny and C. D. Bonan, Neuroscience, 2011, 180, 191–200 CrossRef CAS PubMed.
- Y. Chtourou, A. Ben Slima, M. Makni and R. Gdoura, Pharmacol. Rep., 2015, 67(6), 1090–1097 CrossRef PubMed.
- V. Kamesh and T. Sumathi, Pharm. Biol., 2014, 52(10), 1327–1334 CrossRef PubMed.
- L. Liu, P. Liao, B. Wang, X. Fang, W. Li and S. Guan, Mol. Med. Rep., 2015, 11(5), 3976–3980 CAS.
- D. H. Priscilla, M. Jayakumar and K. Thirumurugan, J. Funct. Foods, 2015, 14, 363–373 CrossRef CAS.
- S. J. Tsai, C. S. Huang, M. C. Mong, W. Y. Kam, H. Y. Huang and M. C. Yin, J. Agric. Food Chem., 2012, 60(1), 514–521 CrossRef CAS PubMed.
- S. D. Mershiba, M. V. Dassprakash and S. D. Saraswathy, Mol. Biol. Rep., 2013, 40(5), 3681–3691 CrossRef CAS PubMed.
- A. Hermenean, A. Ardelean, M. Stan, H. Herman, C. V. Mihali, M. Costache and A. Dinischiotu, Chem. – Biol. Interact., 2013, 205(2), 138–147 CrossRef CAS PubMed.
- A. A. Fouad, W. H. Albuali, A. Zahran and W. Gomaa, Environ. Toxicol. Pharmacol., 2014, 38(2), 420–429 CrossRef CAS PubMed.
- J. W. Park, B. Piknova, P. L. Huang, C. T. Noguchi and A. N. Schechter, PLoS One, 2013, 8(2), e55699 CAS.
- T. Annadurai, P. A. Thomas and P. Geraldine, Free Radicals Res., 2013, 47(10), 793–803 CrossRef CAS PubMed.
- M. R. Schetinger, V. M. Morsch, C. D. Bonan and A. T. Wyse, Biofactors, 2007, 31(2), 77–98 CrossRef CAS.
- S. Kanno, A. Tomizawa, T. Ohtake, K. Koiwai, M. Ujibe and M. Ishikawa, Toxicol. Lett., 2006, 166(2), 131–139 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.