
Nutritional marginal zinc deficiency disrupts placental 11β-hydroxysteroid dehydrogenase type 2 modulation
Y. L.
Huang
a,
S.
Supasai
a,
H.
Kucera
a,
N. W.
Gaikwad
a,
A. M.
Adamo
b,
P.
Mathieu
b and
P. I.
Oteiza
*a
aDepartments of Nutrition and Environmental Toxicology, University of California, Davis, One Shields Avenue, Davis, CA 95616, USA. E-mail: poteiza@ucdavis.edu; Fax: +530-752-8966; Tel: +530-754-6074
bDepartment of Biological Chemistry and IQUIFIB (UBA-CONICET), School of Pharmacy and Biochemistry, University of Buenos Aires, Buenos Aires, Argentina
First published on 18th November 2015
Abstract
This paper investigated if marginal zinc nutrition during gestation could affect fetal exposure to glucocorticoids as a consequence of a deregulation of placental 11βHSD2 expression. Placenta 11β-hydroxysteroid dehydrogenase type 2 (11βHSD2) plays a central role as a barrier protecting the fetus from the deleterious effects of excess maternal glucocorticoids. Rats were fed control (25 μg zinc per g diet) or marginal (10 μg zinc per g diet, MZD) zinc diets from day 0 through day 19 (GD19) of gestation. At GD19, corticosterone concentration in plasma, placenta, and amniotic fluid was similar in both groups. However, protein and mRNA levels of placenta 11βHSD2 were significantly higher (25% and 58%, respectively) in MZD dams than in controls. The main signaling cascades modulating 11βHSD2 expression were assessed. In MZD placentas the activation of ERK1/2 and of the downstream transcription factor Egr-1 was low, while p38 phosphorylation and SP-1-DNA binding were low compared to the controls. These results point to a central role of ERK1/Egr-1 in the regulation of 11βHSD2 expression under the conditions of limited zinc availability. In summary, results show that an increase in placenta 11βHSD2 expression occurs as a consequence of gestational marginal zinc nutrition. This seems to be due to a low tissue zinc-associated deregulation of ERK1/2 rather than to exposure to high maternal glucocorticoid exposure. The deleterious effects on brain development caused by diet-induced marginal zinc deficiency in rats do not seem to be due to fetal exposure to excess glucocorticoids.
Introduction
Marginal zinc deficiency can occur during gestation, not only as a primary nutritional deficiency, but also secondary to conditions such as disease states (e.g. diabetes), maternal infections, and exposure to certain drugs and toxicants.1 Maternal severe zinc deficiency causes dramatic effects on maternal and fetal outcomes, including major teratogenicity affecting most organs.2 Gestational marginal zinc deficiency is not teratogenic but is associated with maternal complications (reviewed in ref. 3) and fetal/offspring adverse effects involving the brain,4–7 bones,8 and glucose homeostasis.9 In the developing brain, gestational marginal zinc nutrition impairs the proliferation of neural progenitor cells,10 which could have long-term and irreversible consequences extending into infancy and adulthood.During pregnancy, maternal and fetal glucocorticoid levels increase to advance not only the structural development of organs but also their functional maturation.11–13 On the other hand, excess glucocorticoids may bring an adverse environment to the fetus and lead to detrimental effects on fetal development and predispose to disease later in life.14 Elevated glucocorticoid levels during pregnancy are associated with stress situations both in humans and rodents, pointing to a connection between stress and an abnormal activity of the hypothalamic–pituitary–adrenal (HPA) axis.15,16 Prenatal exposure to high glucocorticoid levels is associated with an increased risk of small for gestational age infants.17In utero, overexposure to glucocorticoids could also affect brain development/function. In this regard, prenatal anxiety and depression contribute to an altered cognitive development,18 high sympathetic nervous system reactivity,19 low fractional and axial diffusivity in the right amygdala,20 and low synaptic responses and long term potentiation.15
11β-hydroxysteroid dehydrogenase type 2 (11βHSD2) catalyzes the NAD+-dependent oxidation of cortisol in humans (corticosterone in rats) to inactive metabolites (cortisone and 11-dehydrocorticosterone in humans and rats, respectively). Placental 11βHSD2 plays a key role as a barrier protecting the fetus from excess exposure to maternal glucocorticoids.21 Thus, conditions that decrease 11βHSD2 expression in placenta could expose the fetus to the negative effects of excess maternal glucocorticoids. The regulation of 11βHSD2 expression is still not completely understood. However, mitogen activated kinases (MAPK), extracellular signal-regulated kinases 1/2 (ERK1/2) and p38 have been observed to downregulate and upregulate, respectively, 11βHSD2 expression. In addition, the HSD11B2 gene promoter sequence contains several binding sites for the zinc finger transcriptional factor, specificity protein 1 (Sp1).22
The activation of ERK1/2 and p38 MAPK is affected by zinc deficiency both in vivo and in vitro. In fetal brain and neuronal cells, zinc deficiency causes ERK1/2 inhibition,4,23,24 and a redox-dependent activation of p38.4,24,25 Based on the above, this work investigated the hypothesis that a marginal zinc nutrition during gestation in rats affects fetal exposure to corticosterone as a consequence of a deregulation of placental 11βHSD2 expression. Fetal exposure to excess glucocorticoids could explain the negative consequences of maternal marginal zinc deficiency on fetal brain development.
Materials and methods
Materials
TRIzol reagent was obtained from Invitrogen/Life Technologies (Grand Island, NY). Primary antibodies for 11βHSD2 (H-145) (sc-20176), p38 (sc-7149), β-actin (sc-47778), HRP-conjugated secondary antibodies, and the oligonucleotide containing the consensus sequences for Sp-1 and Egr-1 were from Santa Cruz Biotechnology (Santa Cruz, CA). Primary antibodies for p-ERK1/2 (T202/Y204) (4370), ERK1/2 (4695), p-p38 (T180/Y182) (9211), and streptavidin-HRP (3999) were from Cell Signaling Technology (Danvers, MA). Polyclonal goat anti-rabbit immunoglobulins/biotinylated (E0432) was from Dako Denmark A/S (Demark). PVDF membranes were from Bio-Rad (Hercules, CA). High-capacity cDNA reverse transcriptase was from Applied Biosystems/Life Technologies (Grand Island, NY). An absolute blue qPCR SYBR Green Fluorescein mix (AB-4220/A) was from Fisher Scientific Inc. (Waltham, MA).Animal studies
All procedures were in agreement with standards for the care of laboratory animals as outlined in the NIH Guide for the Care and Use of Laboratory Animals. All procedures were administered under the auspices of the Animal Resource Services of the University of California, Davis, which is accredited by the American Association for the Accreditation of Laboratory Animal Care. Experimental protocols were approved before implementation by the University of California, Davis Animal Use and Care Administrative Advisory Committee, and were administered by the Office of the Campus Veterinarian. Adult Sprague–Dawley rats (Charles River, Wilmington, MA) (200–225 g) were housed individually in suspended stainless steel cages in a temperature (22–23 °C)- and photoperiod (12 h light per d)-controlled room. An egg white protein based diet with adequate zinc (25 μg zinc per g) was the standard control diet.26 Animals were fed the control diet for one week before breeding. Males and females were caged together overnight and in the following morning the presence of a sperm plug confirmed successful breeding. On gestation day (GD) 0, rats were divided into two groups (8 animals per group) and fed ad libitum a control diet (25 μg zinc per g diet) or a diet containing a marginal concentration of zinc (10 μg zinc per g diet) until GD19. Food intake was recorded daily, and body weight was measured at 5 d intervals. At GD19, dams were anesthetized with isoflurane (2 mg per kg body weight), and laparotomies were performed. The gravid uterus was removed and fetuses, amniotic fluid and placentas identified for position in the uterus and collected. The zinc concentration in plasma and in the diets was measured by ICP-AES (inductively coupled plasma atomic emission spectroscopy) following procedures described previously.27Ultra-performance liquid chromatography tandem mass spectrometry (UPLC/MS-MS) analysis
UPLC-MS/MS was used to measure glucocorticoids, their metabolites and other steroids in the maternal plasma and placenta. After sample extraction, UPLC analyses of extracts were carried out with a Waters Acquity UPLC system connected to a high performance Xevo-TQ mass spectrometer. Analytical separations on the UPLC system were conducted using an Acquity UPLC BEH C18 or phenyl 1.7 μm column (1 × 100 mm) at a flow rate of 0.15 ml min−1. The gradient was started with 80% A (0.1% formic acid in H2O) and 20% B (0.1% formic acid in CH3CN), changed to 79% A over 4 min, followed by a 6 min linear gradient to 45% A, resulting in a total separation time of 10 min. The elutions from the UPLC column were introduced into the Xevo-TQ mass spectrometer. All MS experiments were performed by electrospray ionization (ESI) in positive ion (PI) and negative ion (NI) modes, with an ESI-MS capillary voltage of 3.0 kV, an extractor cone voltage of 2 V, and a detector voltage of 650 V. The following MS conditions were used: desolvation gas at 600 l h−1, cone gas flow at 60 l h−1, desolvation temperature of 200 °C and source temperature of 100 °C. Pure standards were used to optimize the UPLC-MS/MS conditions before analysing and performing calibration curves. Elutions from the UPLC column were analyzed in the MRM mode, and resulting data were processed by using TargetLynx 4.1 software (Waters).Western blot analysis
Placenta homogenates4,28 were prepared as previously described. The protein concentration was measured29 and aliquots containing 25–50 μg protein were separated using reducing 10% (w/v) polyacrylamide gel electrophoresis and electroblotted onto PVDF membranes. Colored (Bio-Rad Laboratories, Hercules, CA) and biotinylated (Cell Signaling Technologies, Danvers, MA) molecular weight standards were run simultaneously. Membranes were blocked for 1 h in 5% (w/v) non-fat milk, incubated overnight in the presence of the corresponding antibodies (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 dilution) in 5% (w/v) bovine serum albumin in TBS buffer (50 mM Tris, 150 mM NaCl, pH 7.6, containing 0.1% (v/v) Tween-20). After incubation for 90 min at room temperature in the presence of the secondary antibody (HRP-conjugated) (1:10000 dilution) the conjugates were visualized by chemiluminescence detection in a Phosphoimager 840 (Amersham Pharmacia Biotech, Inc., Piscataway, NJ).
:1000 dilution) in 5% (w/v) bovine serum albumin in TBS buffer (50 mM Tris, 150 mM NaCl, pH 7.6, containing 0.1% (v/v) Tween-20). After incubation for 90 min at room temperature in the presence of the secondary antibody (HRP-conjugated) (1:10000 dilution) the conjugates were visualized by chemiluminescence detection in a Phosphoimager 840 (Amersham Pharmacia Biotech, Inc., Piscataway, NJ).
Real time quantitative RT-PCR
Total RNA was extracted from placentas using TRIzol reagent. cDNA was generated using high-capacity cDNA reverse transcriptase. mRNA levels of 11βHSD2 were assessed by reverse transcription PCR (iCycler, Bio-Rad, Hercules, CA). The housekeeping gene β-actin (F: 5′-ACC CGC GAG TAC AAC CTT CT-3′; R: 5′-ATG GCT ACG TAC ATG GCT GG-3′) was used as an endogenous control. For RT-PCR, the Absolute blue SYBR green qPCR mix was mixed with the HSD11B2 primers (F: 5′-GCC CTG GTG CTC TAG AAC TG-3′; R: 5′-AGT TCC ACA TCG GCC ACT AC-3′).Immunohistochemistry for 11βHSD2
Placentas were dissected out and fixed in 4% (w/v) solution of paraformaldehyde in PBS overnight. Cryoprotection was then performed in 15% (w/v) sucrose in PBS for 24 h and 30% (w/v) sucrose for 3 days, after which tissues were submerged in Cryoplast (Biopack, Buenos Aires, Argentina), frozen, cut into 18 μm sections on a Leica CM 1850 cryotome (Leica Microsystems, Nussloch, Germany) and mounted on positively charged microscope slides. Sections were processed for antigen retrieval by incubation in 10 mM sodium citrate buffer (pH 6.0) at 95 °C, washed twice with PBS and then blocked for 45 min in 1% donkey serum, 0.1% Triton in 0.1 M PBS and incubated overnight at 4 °C with mouse anti-11βHSD2 (1:200) primary antibody. Sections were washed in PBS and incubated for 2 h at room temperature with Cy3-conjugated donkey anti-mouse IgG (1:500) (Jackson Immuno Research Co. Laboratories, West Grove, PA). After immunostaining, cell nuclei were stained with Hoechst 3334230 and sections were imaged using an Olympus FV 300 laser scanning confocal microscope (Olympus, Japan). Olympus Fluoview version 4.0 software was used to merge images. Four slices per animal and four animals from each group were analyzed.
Electrophoretic mobility shift assay (EMSA)
Nuclear fractions were prepared as previously described,31 with minor modifications.4 Sp-1-and Egr-1-DNA binding was assessed in nuclear fractions by EMSA. For the EMSA, the oligonucleotide containing the consensus sequences for Sp1 and Egr-1 was end-labelled with [γ-32P]ATP using T4 polynucleotide kinase, and purified using Chroma Spin-10 columns. Samples were incubated with the labelled oligonucleotide (20000–30000 cpm) for 20 min at room temperature in 1× binding buffer [5× binding buffer: 50 mM Tris-HCl buffer, pH 7.5, containing 20% (v/v) glycerol, 5 mM MgCl2, 2.5 mM EDTA, 2.5 mM DTT, 250 mM NaCl, and 0.25 mg ml−1 poly(dI-dC)]. The products were separated by electrophoresis in a 6% (w/v) non-denaturing polyacrylamide gel using 0.5× TBE (45 mM Tris/borate, 1 mM EDTA) as the running buffer. The gels were dried and the radioactivity quantified in a Phosphoimager 840.
Statistical analysis
Data were analyzed by a unpaired t-test, one-way analysis of variance (ANOVA) or simple linear regression using Statview 5.0 (SAS Institute Inc., Cary, NC, USA). The Fisher least significant difference test was used to examine differences between group means. A p value < 0.05 was considered to be statistically significant. Data are shown as mean ± SEM.Results
Animal outcome
Pregnant rats were fed control (25 μg zinc per g) or marginal (10 μg zinc per g) (MZD) zinc diets from GD0 until GD19. Given that zinc deficiency can be associated with anorexia, food intake and body weight gain were recorded throughout pregnancy. Both daily (data not shown) and cumulative food intake, and GD9 maternal body weight (Table 1) were similar in control and MZD dams. At GD19, the number of live fetuses per litter, gravid uterine weight, placenta and fetal body weight were similar in control and MZD groups (Table 1). At GD19, the plasma zinc concentration in the MZD dams was 34% lower (p < 0.05) compared to controls (Table 1).| Parameters | Control | MZD |
|---|---|---|
| Dams were fed the control (25 μg zinc per g) or marginal zinc (10 μg zinc per g) (MZD) diets from GD0 until GD19. Table shows data collected at GD19. Results are shown as means ± S.E.M (9 and 7 dams/litters for control and MZD groups, respectively). *Significantly different from control (p < 0.05, unpaired t-test). | ||
| Cumulative food intake (g) | 465 ± 52 | 497 ± 50 |
| Dam body weight (g) | 404 ± 18 | 417 ± 34 |
| Gravid uterine weight (g) | 68.7 ± 7.7 | 69.3 ± 7.4 |
| Fetal weight (mg) | 2387 ± 158 | 2313 ± 103 |
| Live fetuses per dam | 15.9 ± 2.1 | 15.8 ± 2.0 |
| Placental weight (mg) | 450 ± 68 | 458 ± 60 |
| Plasma zinc (μM) | 14.6 ± 0.9 | 9.7 ± 0.9* |
Effects of gestational marginal zinc nutrition on placental 11βHSD2 expression and distribution
The expression of 11βHSD2 was evaluated in placenta by measuring the levels of 11βHSD2 mRNA by RT-PCR and those of 11βHSD2 protein by western blotting. Placental 11βHSD2 mRNA and protein levels were significantly (p < 0.05) higher (25% and 58%, respectively) in the MZD group than in controls (Fig. 1A and B).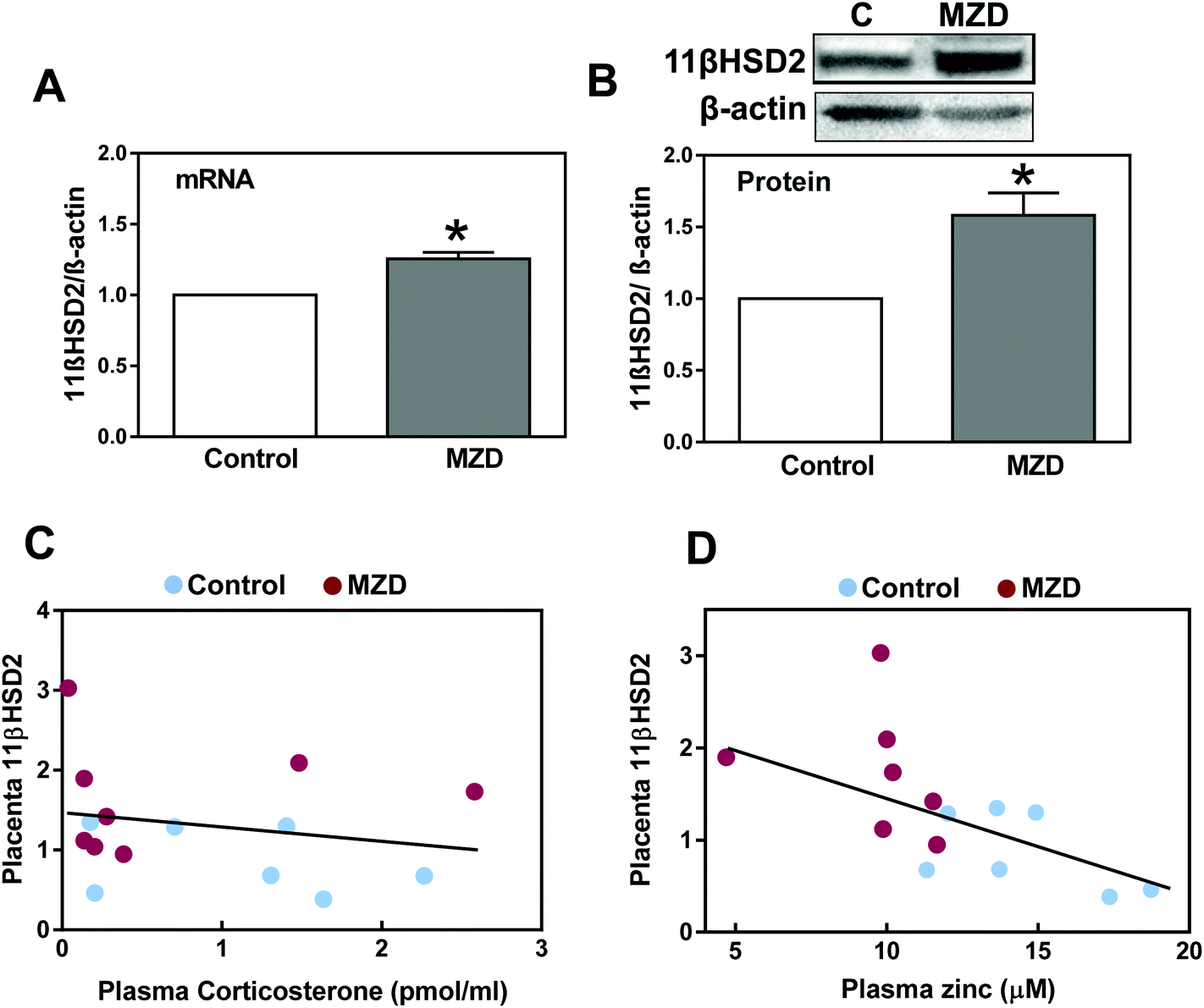 | ||
| Fig. 1 11βHSD2 mRNA and protein levels in placenta from dams fed control or marginal zinc diets during gestation. Dams were fed control (25 μg zinc per g) or marginal (10 zinc μg per g) (MZD) zinc diets from GD0 until GD19. (A) Placenta 11βHSD2 mRNA levels were measured by RT-PCR, and results are expressed as the ratio of 11βHSD2/β-actin mRNA. (B) Placenta 11βHSD2 protein levels were measured by western blotting and results are expressed as the ratio of 11βHSD2/β-actin protein. All results were normalized to control values. Data are shown as means ± S.E.M (8 and 7 litters for control and MZD groups, respectively). *Significantly different from the control (p < 0.05, unpaired t-test). (C, D) Correlations between placenta 11βHSD2 protein levels and the maternal plasma (C) corticosterone or (D) zinc concentration. | ||
The distribution of 11βHSD2 in the placenta was investigated by immunohistochemistry (Fig. 2). As previously observed by western blotting, the 11βHSD2 immunofluorescence was higher in the MZD placentas than in controls. The distribution of 11βHSD2 fluorescence throughout the placenta was similar in both groups (Fig. 2A and B). Fig. 2C and D show confocal microscopy images at a higher magnification for 11βHSD2 (red fluorescence) and nuclear (blue) fluorescence in the labyrinth zone of the placenta. Images show the cytosolic location of the enzyme, and high levels of 11βHSD2 in MZD compared to control placentas.
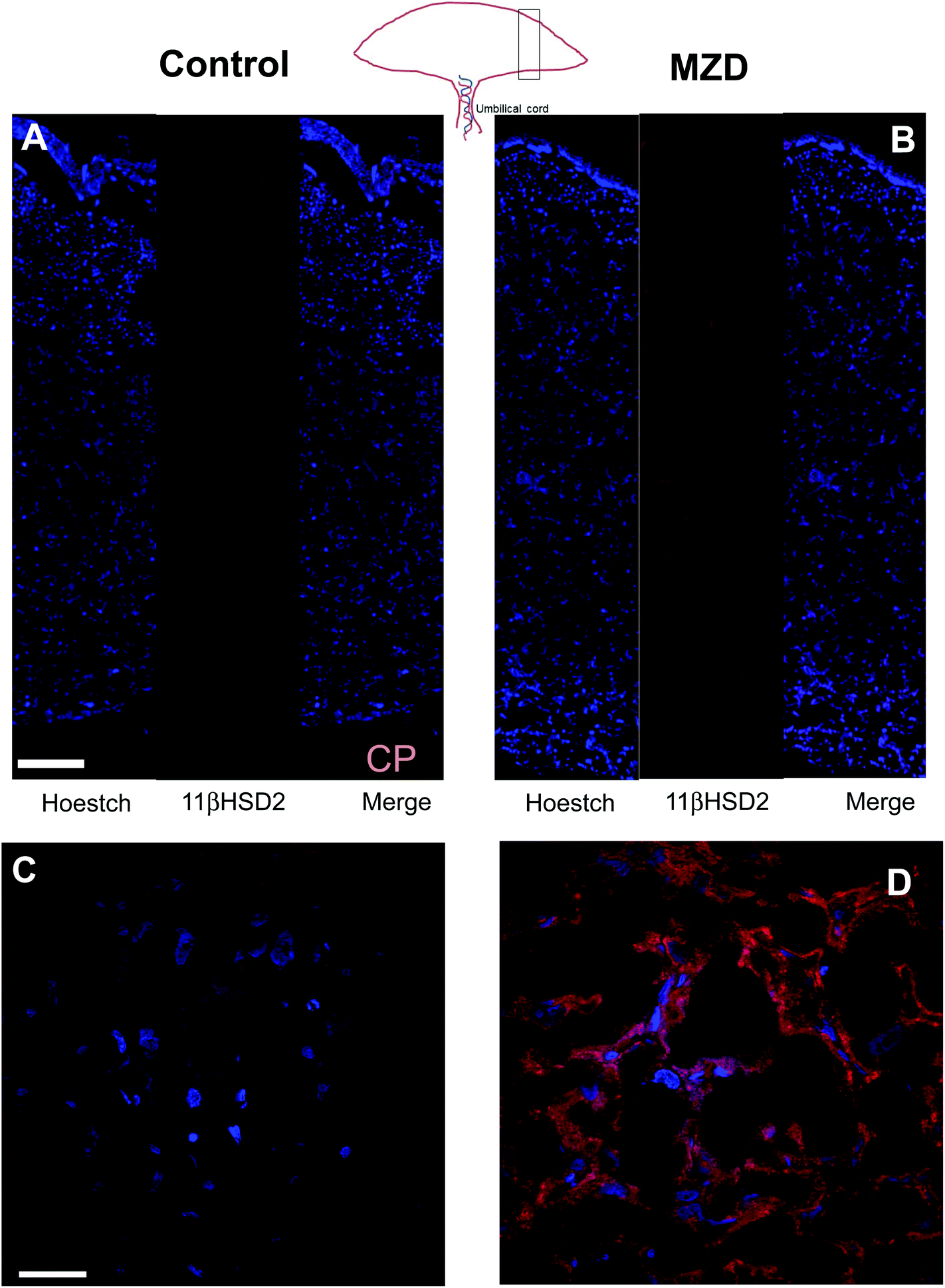 | ||
| Fig. 2 Immunohistochemistry for 11βHSD2 levels in placenta from dams fed control or marginal zinc diets during gestation. Dams were fed control (25 μg zinc per g) or marginal (10 μg zinc per g) (MZD) zinc diets from GD0 until GD19. Immunohistochemistry was performed as described in the Materials and methods section. (A and B) Nuclear Hoechst (blue), 11βHSD2 red fluorescence and the merge of both (scale bar: 250 μm). The analyzed section is shown in the placenta diagram. (C, D) Magnification of the labyrinth zones (scale bar: 30 μm). CP: chorionic plate. | ||
Effects of gestational marginal zinc nutrition on placenta glucocorticoid levels
We subsequently investigated the effects of gestational marginal zinc deficiency on the concentration of placenta and amniotic fluid glucocorticoids, including corticosterone, cortisol and their metabolites, 11β-dehydrocorticosterone and cortisone (Table 2). Corticosteroid concentrations in placenta and amniotic fluid were similar in both groups. Plasma corticosterone (251 ± 30 and 219 ± 53 pmol ml−1 for control and MZD groups, respectively) and cortisol (0.007 ± 0.07 and 0.125 ± 0.08 pmol ml−1 for control and MZD groups, respectively) concentrations were not significantly different between the groups. Maternal plasma corticosterone concentration did not correlate with placenta 11βHSD2 protein expression (Fig. 1C) nor placenta corticosterone with placenta 11βHSD2 protein expression. As a parameter of fetal exposure, glucocorticoids were also measured in amniotic fluid (Table 2). Glucocorticoid concentrations in amniotic fluid were similar in both groups. Only for the MZD group a significant correlation between placenta and amniotic fluid corticosterone concentration was found (r: 0.81, p < 0.03). Corticosterone concentration in maternal plasma correlated with that in amniotic fluid (r: 0.62, p < 0.01). A significant negative correlation (r: 0.56, p < 0.03) was observed between plasma zinc content and placenta 11βHSD2 protein expression (Fig. 1D). This suggests a potential direct regulatory action of zinc on 11βHSD2 expression.| Steroid hormones | Control | MZD |
|---|---|---|
| Dams were fed control (25 μg zinc per g) or marginal (10 μg zinc per g) (MZD) zinc diets from GD0 until GD19. Placentas were tested for steroid hormone levels by UPLC–MS/MS. Data are shown as means ± S.E.M (8 and 7 litters from control and MZD groups, respectively). | ||
| Placenta (pmol g−1 wt−1) | ||
| Pregnenolone | 7.61 ± 1.66 | 6.55 ± 1.47 |
| 17α-Hydroxypregnenolone | 1.90 ± 0.51 | 1.36 ± 0.36 |
| Aldosterone | 1.10 ± 0.38 | 0.82 ± 0.09 |
| Progesterone | 7.03 ± 0.62 | 6.47 ± 1.12 |
| 17α-Hydroxyprogesterone | 0.029 ± 0.008 | 0.022 ± 0.003 |
| 11-Deoxycortisol | 17.60 ± 2.55 | 14.39 ± 2.78 |
| Cortisol | 0.35 ± 0.22 | 0.14 ± 0.09 |
| Deoxycorticosterone | 5.58 ± 1.21 | 2.82 ± 0.69 |
| Corticosterone | 81.37 ± 9.21 | 71.19 ± 11.40 |
| Cortisone | 0.04 ± 0.02 | 0.12 ± 0.07 |
| 11-Dehydrocorticosterone | 137.70 ± 20.11 | 106.58 ± 28.15 |
| Amniotic fluid (pmol g−1 wt−1) | ||
| Pregnenolone | 15.02 ± 2.62 | 14.56 ± 3.14 |
| 17α-Hydroxypregnenolone | 1.84 ± 1.01 | 3.68 ± 1.47 |
| Aldosterone | 8.69 ± 0.04 | 0.32 ± 0.22 |
| Progesterone | 2.11 ± 0.21 | 1.68 ± 0.22 |
| 17α-Hydroxyprogesterone | 1.45 ± 0.18 | 1.06 ± 0.18 |
| 11-Deoxycortisol | 0.64 ± 0.26 | 0.96 ± 0.25 |
| Cortisol | 0.007 ± 0.007 | 1.23 ± 0.08 |
| Deoxycorticosterone | 1.75 ± 0.441 | 1.16 ± 0.20 |
| Corticosterone | 250.76 ± 29.59 | 211.05 ± 53.07 |
| Cortisone | 0.040 ± 0.020 | 0.007 ± 0.004 |
Effects of marginal zinc deficiency on the signaling cascades regulating placenta 11βHSD2 expression
The MAPK ERK1/2 and p38 differentially regulate 11βHSD2 expression.32,33 ERK1/2 is a negative regulator, while p38 is a positive regulator of 11βHSD2 expression. Thus, we investigated the activation (evaluated as phosphorylation levels) of ERK1/2 (phosphorylation in Tyr202 and Y204) and p38 (phosphorylation in T180 and Y182) in placentas from MZD and control dams. In MZD placentas, phosphorylation of ERK1/2 was lower (26%, p < 0.05) (Fig. 3A), while the total expression of ERK1/2 was higher (40%, p < 0.05) compared to controls. Levels of p38 phosphorylation were lower (29%, p < 0.05) (Fig. 3C), while the total p38 protein content was higher (50%, p < 0.05) in the MZD group compared to controls.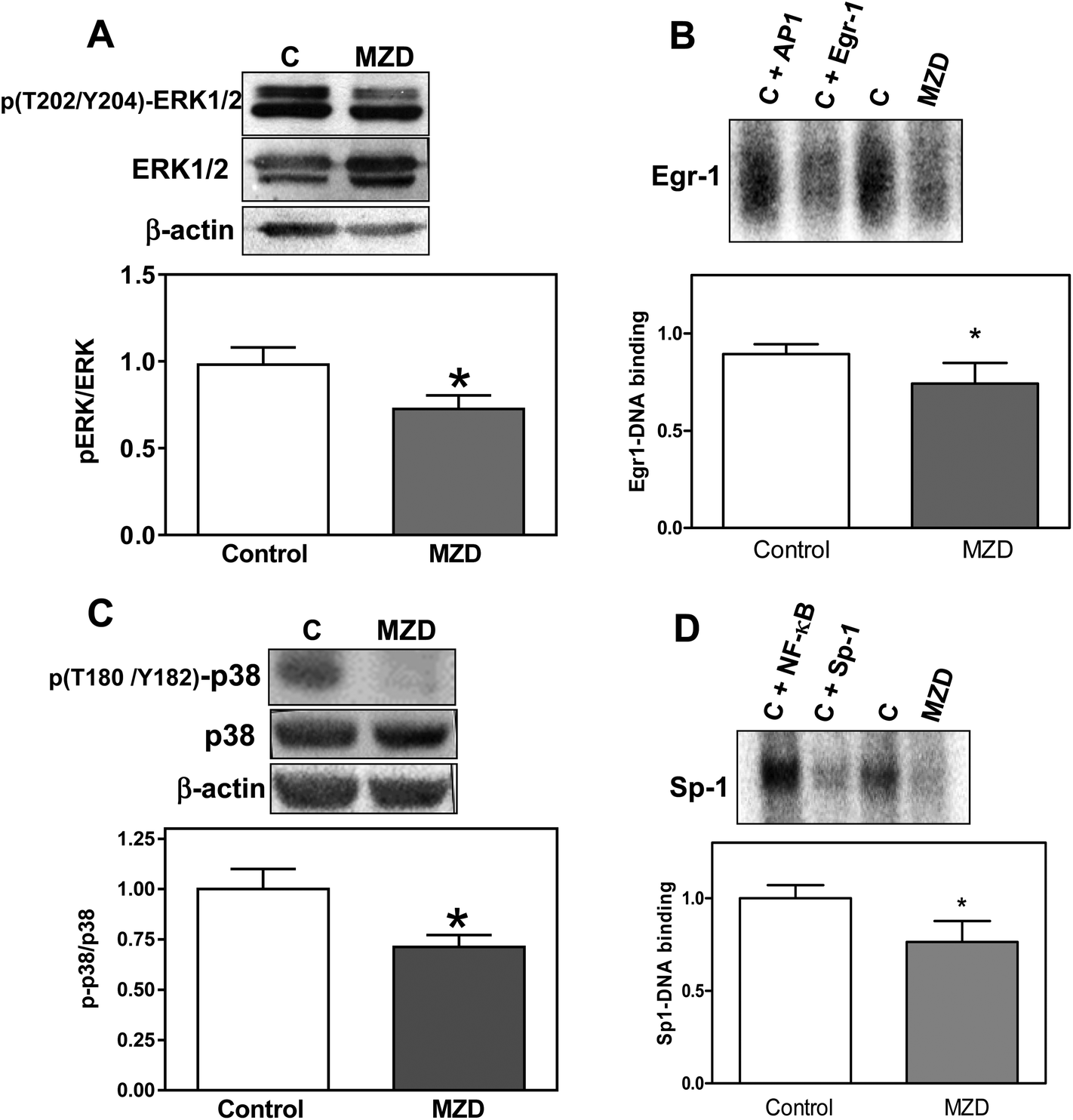 | ||
| Fig. 3 Effects of marginal zinc nutrition in pregnant rats on cell signals that regulate 11βHSD2 expression in placenta. Dams were fed control (25 μg zinc per g) or marginal (10 μg zinc per g) (MZD) zinc diets from GD0 until GD19. Protein levels of (A) p(T202/Y204)-ERK1/2, total ERK1/2, and β-actin; (C) p(T180/Y182)-p38, p38, and β-actin were measured in placenta total homogenates by Western blotting. One representative image is shown. After quantification, results were expressed as the ratios of phosphorylated/total protein and referred to control values. (B) Egr-1 and (D) Sp-1 binding to DNA in placental nuclear fractions was analyzed by EMSA. To determine the specificity of the Egr-1- or Sp-1-DNA complex, a control (C) sample was incubated in the presence of a 100-fold molar excess of unlabeled oligonucleotides containing the consensus sequence for either Egr-1/Sp-1 (specific competition) or NF-κB/AP-1 (non-specific competition) prior to the binding assay. Representative EMSA images are shown. After quantification, results were referred to control values. Data are shown as means ± S.E.M of 9 and 7 litters for the control and MZD groups, respectively. *Significantly different from controls (p < 0.05, unpaired t-test). | ||
We next investigated two zinc finger transcription factors, Sp-1 and Egr-1, which are also involved in the regulation of 11βHSD2. Using EMSA assays, we observed that in nuclear fractions from MZD placentas, Sp-1- and Egr-1 DNA bindings were lower (24% and 17%, respectively) compared to controls (p < 0.05) (Fig. 3B and D).
Discussion
This work shows that gestational marginal zinc deficiency causes an upregulation of placenta 11βHSD2. This upregulation does not seem to be associated with an increased exposure to maternal glucocorticoids but to decreased zinc availability. Placenta 11βHSD2 upregulation is linked to marginal zinc deficiency-associated changes in signaling regulation.Marginal zinc deficiency is a condition that can frequently happen in human populations, pregnant women being at particular risk. The deleterious effects of zinc deficiency on prenatal and early postnatal development depend on the severity of the deficiency (reviewed in ref. 1). Both, a defect of the zinc transporter Zip434 and severe nutritional zinc deficiency,2 cause major teratogenicity. On the other hand, maternal marginal zinc deficiency is not teratogenic but can affect brain development and the risk of the offspring for neurological/behavioral disorders later in life.35–37 In developing rats, marginal zinc deficiency during gestation affects neural progenitor cell proliferation,10 the brain expression of receptors (NMDA) and growth factors, oxidative stress, and deregulation of signaling pathways that play central roles in brain development and function.4,7,38
Similar to observations on gestational marginal zinc deficiency, an increased fetal exposure to high glucocorticoid concentrations, either due to an increased maternal production or to a downregulation of placenta 11βHSD2, can affect brain structure and behavior.14 In this study, corticosterone concentrations in maternal plasma, placenta and amniotic fluid at GD19 were not affected by dietary marginal zinc deficiency. The positive correlation between plasma and placenta/amniotic fluid corticosterone concentrations points to the relevance of the placenta in the relationship between maternal corticosterone production and fetal exposure to corticosterone.
An increased exposure of the fetus to excess glucocorticoids could also be caused by a downregulation of placenta 11βHSD2, given its capacity to metabolize cortisol/corticosterone to inactive compounds. In this regard, fetal glucocorticoid levels are about 5–10-fold lower than maternal glucocorticoid levels due to the action of 11βHSD2.39 Both protein and mRNA levels of placenta 11βHSD2 were high in the placentas from MZD dams. When the distribution of 11βHSD2 was investigated by immunohistochemistry, 11βHSD2 was mainly localized in the labyrinth zone of the placenta. Although higher levels of fluorescence corresponding to 11βHSD2 were observed in MZD placenta, 11βHSD2 tissue distribution was similar in both groups.
The finding of a significant and negative correlation between plasma zinc and placenta 11βHSD2 levels suggests a role of zinc per se in the modulation of 11βHSD2 expression. Thus, we next investigated the effects of zinc deficiency on the activation of signaling cascades that are involved in 11βHSD2 regulation. The MAPKs p38 and ERK are positive and negative regulators of 11βHSD2 expression, respectively. In human primary trophoblast cells p38 upregulates 11βHSD2 expression through the stabilization of 11βHSD2 mRNA.33 Also in human primary trophoblast cells, a downregulation of ERK1/2 leads to an increase in 11βHSD2 protein levels and activity.40,41 In renal cells, hypoxia causes a downregulation of 11βHSD2 as a consequence of ERK1/2-mediated induction of transcription factor Egr-1.42 In terms of Sp-1, footprinting of the human HSD11B2 gene promoter showed the presence of Sp-1 binding motifs.22 In human placenta, Sp-1 activation during syncytialization is central to the upregulation of 11βHSD2 expression.43 Gestational marginal zinc deficiency caused a decreased activation of ERK1/2/Egr-1, p38 and SP-1. Considering the pattern of signaling activation in placenta under conditions of marginal zinc deficiency, ERK1/2/Egr-1 seems to be the major signaling pathway regulating 11βHSD2 expression in placenta. However, future in vitro studies are necessary to further support this mechanism of 11βHSD2 modulation by zinc.
Although exposure to excess glucocorticoid at some time during the day, associated with circadian rhythm variations, cannot be discarded, current results point to a major role of zinc per se in the modulation of placenta 11βHSD2 expression. In conclusion, an increased exposure of the fetus to maternal glucocorticoids does not seem to contribute to the previously observed alterations in brain development and behavioral abnormalities associated with a marginal zinc nutrition during gestation and early development.4,7,44,45
Author contributions
Y.L.H. played a major role in the acquisition and analysis of the data, performed data collection, assisted in the interpretation of the data, edited the manuscript, and approved the final manuscript to be submitted. S.S. and H.K. carried out the acquisition and analysis of the data, performed data collection, assisted in the interpretation of the data, edited the manuscript, and approved the final manuscript to be submitted. N.W.G. assisted with the study design, supervised H.K. in the acquisition and analysis of the data, assisted in the interpretation of the data, edited the manuscript, and approved the final manuscript to be submitted. A.M.A. and P.M. performed the immunohistochemistry experiments and analyzed the corresponding data in the frame of the obtained results. P.I.O. designed the study, supervised Y.L.H. and S.S. in the acquisition and analysis of data, assisted in the interpretation of the data, drafted the manuscript, and approved the final manuscript to be submitted.Conflicts of interest
The authors declare no direct conflict of interest.Abbreviations
| Egr-1 | Early growth response protein 1 |
| ERK1/2 | Extracellular signal-regulated kinases 1/2 |
| GD | Gestation day |
| HPA | Hypothalamic–pituitary–adrenal axis |
| 11βHSD2 | 11β-hydroxysteroid dehydrogenase type 2 |
| MAPK | Mitogen activated kinases |
| SP-1 | Specificity protein 1 |
Acknowledgements
This work was supported by grants from NIFA-USDA (CA-D*-XXX-7244-H), the NIH West Coast Metabolomics Center, and the PackerWentz Endowment. S. Supasai was supported by a fellowship from the Thailand Government.References
- J. Y. Uriu-Adams and C. L. Keen, Birth Defects Res., Part B, 2010, 89, 313–325 CrossRef CAS PubMed.
- L. S. Hurley and H. Swenerton, Proc. Soc. Exp. Biol. Med., 1966, 123, 692–696 CrossRef CAS PubMed.
- C. L. Keen, J. Y. Uriu-Adams, A. Skalny, A. Grabeklis, S. Grabeklis, K. Green, L. Yevtushok, W. W. Wertelecki and C. D. Chambers, BioFactors, 2010, 36, 125–135 CAS.
- L. Aimo, G. G. Mackenzie, A. H. Keenan and P. I. Oteiza, J. Nutr. Biochem., 2010, 21, 1069–1075 CrossRef CAS PubMed.
- Y. Omata, G. A. Salvador, S. Supasai, A. H. Keenan and P. I. Oteiza, Toxicol. Sci., 2013, 133, 90–100 CrossRef CAS PubMed.
- G. G. Mackenzie, G. A. Salvador, C. Romero, C. L. Keen and P. I. Oteiza, Free Radical Biol. Med., 2011, 51, 480–489 CrossRef CAS PubMed.
- W. Chowanadisai, S. L. Kelleher and B. Lonnerdal, J. Neurochem., 2005, 94, 510–519 CrossRef CAS PubMed.
- M. Nagata, M. Kayanoma, T. Takahashi, T. Kaneko and H. Hara, Biol. Trace Elem. Res., 2011, 142, 190–199 CrossRef CAS PubMed.
- M. Y. Jou, B. Lonnerdal and A. F. Philipps, Pediatr. Res., 2012, 71, 228–234 CrossRef CAS PubMed.
- J. R. Nuttall, S. Supasai, J. Kha, B. M. Vaeth, G. G. Mackenzie, A. M. Adamo and P. I. Oteiza, J. Nutr. Biochem., 2015, 26, 1116–1123 CrossRef CAS PubMed.
- Y. Dong, L. Poellinger, J. A. Gustafsson and S. Okret, Mol. Endocrinol., 1988, 2, 1256–1264 CrossRef CAS PubMed.
- K. Chapman, M. Holmes and J. Seckl, Physiol. Rev., 2013, 93, 1139–1206 CrossRef CAS PubMed.
- A. L. Fowden, J. Li and A. J. Forhead, Proc. Nutr. Soc., 1998, 57, 113–122 CrossRef CAS PubMed.
- V. G. Moisiadis and S. G. Matthews, Nat Rev. Endocrinol., 2014, 10, 391–402 CrossRef CAS PubMed.
- M. Barzegar, F. S. Sajjadi, S. A. Talaei, G. Hamidi and M. Salami, Hippocampus, 2015, 25, 187–196 CrossRef PubMed.
- T. Field, M. Diego, M. Hernandez-Reif, O. Deeds, V. Holder, S. Schanberg and C. Kuhn, Infant. Behav. Dev., 2009, 32, 10–16 CrossRef PubMed.
- A. S. Khashan, C. Everard, L. M. McCowan, G. Dekker, R. Moss-Morris, P. N. Baker, L. Poston, J. J. Walker and L. C. Kenny, Psychol. Med., 2014, 44, 2799–2810 CrossRef CAS PubMed.
- K. Bergman, P. Sarkar, V. Glover and T. G. O'Connor, Biol. Psychiatry, 2010, 67, 1026–1032 CrossRef CAS PubMed.
- K. Vedhara, C. Metcalfe, H. Brant, A. Crown, K. Northstone, K. Dawe, S. Lightman and G. D. Smith, J. Neuroendocrinol., 2012, 24, 999–1011 CrossRef CAS PubMed.
- A. Rifkin-Graboi, J. Bai, H. Chen, W. B. Hameed, L. W. Sim, M. T. Tint, B. Leutscher-Broekman, Y. S. Chong, P. D. Gluckman, M. V. Fortier, M. J. Meaney and A. Qiu, Biol. Psychiatry, 2013, 74, 837–844 CrossRef PubMed.
- N. A. Rao, M. T. McCalman, P. Moulos, K. J. Francoijs, A. Chatziioannou, F. N. Kolisis, M. N. Alexis, D. J. Mitsiou and H. G. Stunnenberg, Genome Res., 2011, 21, 1404–1416 CrossRef CAS PubMed.
- A. R. Nawrocki, C. E. Goldring, R. M. Kostadinova, F. J. Frey and B. M. Frey, J. Biol. Chem., 2002, 277, 14647–14656 CrossRef CAS PubMed.
- A. M. Adamo, M. P. Zago, G. G. Mackenzie, L. Aimo, C. L. Keen, A. Keenan and P. I. Oteiza, Neurotoxic. Res., 2010, 17, 1–14 CrossRef CAS PubMed.
- M. P. Zago, G. G. Mackenzie, A. M. Adamo, C. L. Keen and P. I. Oteiza, Antioxid. Redox Signaling, 2005, 7, 1773–1782 CrossRef CAS PubMed.
- G. G. Mackenzie, M. P. Zago, A. G. Erlejman, L. Aimo, C. L. Keen and P. I. Oteiza, Free Radical Res., 2006, 40, 75–84 CrossRef CAS PubMed.
- C. L. Keen, J. M. Peters and L. S. Hurley, J. Nutr., 1989, 119, 607–611 CAS.
- M. S. Clegg, C. L. Keen, B. Lonnerdal and L. S. Hurley, Biol. Trace Elem. Res., 1981, 3, 107–115 CrossRef CAS PubMed.
- M. Da Silva, G. K. Jaggers, S. V. Verstraeten, A. G. Erlejman, C. G. Fraga and P. I. Oteiza, Free Radical Biol. Med., 2012, 52, 151–159 CrossRef CAS PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- F. Oberhammer, G. Fritsch, M. Schmied, M. Pavelka, D. Printz, T. Purchio, H. Lassmann and R. Schulte-Hermann, J. Cell Sci., 1993, 104(Pt 2), 317–326 CAS.
- L. Osborn, S. Kunkel and G. J. Nabel, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 2336–2340 CrossRef CAS.
- D. V. Kratschmar, A. Vuorinen, T. Da Cunha, G. Wolber, D. Classen-Houben, O. Doblhoff, D. Schuster and A. Odermatt, J. Steroid Biochem. Mol. Biol., 2011, 125, 129–142 CrossRef CAS PubMed.
- A. Sharma, H. Guan and K. Yang, Endocrinology, 2009, 150, 4278–4286 CrossRef CAS PubMed.
- J. Dufner-Beattie, S. J. Langmade, F. Wang, D. Eide and G. K. Andrews, J. Biol. Chem., 2003, 278, 50142–50150 CrossRef CAS PubMed.
- E. S. Halas, G. M. Reynolds and H. H. Sandstead, Physiol. Behav., 1977, 19, 653–661 CrossRef CAS PubMed.
- S. Grabrucker, L. Jannetti, M. Eckert, S. Gaub, R. Chhabra, S. Pfaender, K. Mangus, P. P. Reddy, V. Rankovic, M. J. Schmeisser, M. R. Kreutz, G. Ehret, T. M. Boeckers and A. M. Grabrucker, Brain, 2014, 137, 137–152 CrossRef PubMed.
- B. L. Summers, C. M. Henry, A. M. Rofe and P. Coyle, Behav. Brain. Res., 2008, 186, 230–238 CrossRef CAS PubMed.
- J. Warkany and H. G. Petering, Teratology, 1972, 5, 319–334 CrossRef CAS PubMed.
- A. L. Tomat, L. Costa Mde and C. T. Arranz, Nutrition, 2011, 27, 392–398 CrossRef CAS PubMed.
- H. Guan, K. Sun and K. Yang, Biol. Reprod., 2013, 89, 92 CrossRef PubMed.
- Y. Shang, X. Yang, R. Zhang, H. Zou and R. Zhao, Amino Acids, 2012, 42, 1879–1887 CrossRef CAS PubMed.
- C. D. Heiniger, R. M. Kostadinova, M. K. Rochat, A. Serra, P. Ferrari, B. Dick, B. M. Frey and F. J. Frey, FASEB J., 2003, 17, 917–919 CAS.
- J. N. Li, Y. C. Ge, Z. Yang, C. M. Guo, T. Duan, L. Myatt, H. Guan, K. Yang and K. Sun, J. Clin. Endocrinol. Metab., 2011, 96, E899–E907 CrossRef CAS PubMed.
- M. S. Golub, C. L. Keen, M. E. Gershwin and A. G. Hendrickx, J. Nutr., 1995, 125, 2263S–2271S CAS.
- M. S. Golub, P. T. Takeuchi, C. L. Keen, A. G. Hendrickx and M. E. Gershwin, Am. J. Clin. Nutr., 1996, 64, 908–915 CAS.
| This journal is © The Royal Society of Chemistry 2016 |