
Wild Roman chamomile extracts and phenolic compounds: enzymatic assays and molecular modelling studies with VEGFR-2 tyrosine kinase
Rafaela
Guimarães
ab,
Ricardo C.
Calhelha
ab,
Hugo J. C.
Froufe
a,
Rui M. V.
Abreu
a,
Ana Maria
Carvalho
a,
Maria João, R. P.
Queiroz
b and
Isabel C. F. R.
Ferreira
*a
aCentro de Investigação de Montanha, Escola Superior Agrária, Campus de Santa Apolónia, apartado 1172, 5301-854 Bragança, Portugal. E-mail: iferreira@ipb.pt; Fax: +351-273-325405; Tel: +351-273-303219
bCentro de Química da Universidade do Minho, Campus de Gualtar, 4710-057 Braga, Portugal
First published on 25th September 2015
Abstract
Angiogenesis is a process by which new blood vessels are formed from the pre-existing vasculature, and it is a key process that leads to tumour development. Some studies have recognized phenolic compounds as chemopreventive agents; flavonoids, in particular, seem to suppress the growth of tumor cells modifying the cell cycle. Herein, the antiangiogenic activity of Roman chamomile (Chamaemelum nobile L.) extracts (methanolic extract and infusion) and the main phenolic compounds present (apigenin, apigenin-7-O-glucoside, caffeic acid, chlorogenic acid, luteolin, and luteolin-7-O-glucoside) was evaluated through enzymatic assays using the tyrosine kinase intracellular domain of the Vascular Endothelium Growth Factor Receptor-2 (VEGFR-2), which is a transmembrane receptor expressed fundamentally in endothelial cells involved in angiogenesis, and molecular modelling studies. The methanolic extract showed a lower IC50 value (concentration that provided 50% of VEGFR-2 inhibition) than the infusion, 269 and 301 μg mL−1, respectively. Regarding phenolic compounds, luteolin and apigenin showed the highest capacity to inhibit the phosphorylation of VEGFR-2, leading us to believe that these compounds are involved in the activity revealed by the methanolic extract.
1. Introduction
Angiogenesis is a process by which new blood vessels are formed from the pre-existing vasculature, developing a hemovascular network.1 It is tightly controlled by a balance of angiogenesis factors and inhibitors, occurring in the embryonic development, wound healing and the female reproductive cycle. Angiogenic diseases result from new blood vessels growing either excessively (e.g. cancer, diabetic retinopathy and psoriasis) or insufficiently (e.g. chronic wounds and ischaemic heart disease).1,2During angiogenesis, endothelial cells degrade the basement membrane, migrate into the surrounding intercellular matrix, proliferate to form new blood vessels, and differentiate into contiguous tubular sprouts, which subsequently form functional capillary loops. Such cellular events are mediated by various intracellular signal transduction pathways.3,4 Angiogenesis happens in the body all the time. It occurs through a so-called angiogenesis “cascade” which involves a series of biochemical steps by which cells make and secrete molecules that initiate the growth of capillaries. After the process is over, certain other molecular “factors” turn off the angiogenesis process. Cancer cells use this normal process for another purpose, creating an imbalance of angiogenesis activators that overrides the inhibitors and gives the nearby tumour ready access to a blood supply.5 This explains why angiogenesis is essential for the growth, progression, and metastasis of solid tumours.6
In the mentioned pathophysiological processes, excessive angiogenesis occurs when diseased cells produce abnormally large amounts of angiogenesis factors [e.g. vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF)-2 and hepatocyte growth factor], overcoming the effects of natural angiogenesis inhibitors (e.g. angiostatin, endostatin and thrombospondin).1
VEGF is a secreted growth factor by tumor cells that plays a critical role in angiogenesis; low oxygen tension dramatically induces the expression of this major angiogenic factor.7 Its biological effects are mediated by two receptor tyrosine kinases namely VEGFR-1 (fms-like tyrosine kinase, Flt-1) and VEGFR-2 (kinase-insert domain-containing receptor, KDR), which differ considerably in their signalling characteristics.8,9 Although increasing evidence indicates that angiogenesis is a highly sophisticated and coordinated process, the activation of the VEGF/VEGFR pathway remains the key modulator of angiogenesis.10 Furthermore, VEGF is the leading angiogenic factor involved in tumoral angiogenesis.7,9
Of the primary receptors, VEGFR-2 is thought to mediate the majority of tumor angiogenic effects (Fig. 1a). Current clinical treatments against tumor antiangiogenesis that target VEGFR-2 include: monoclonal antibodies (e.g. bevacizumab) that target the VEGFR-2 extracellular VEGF binding domain and small tyrosine kinase inhibitors (TKIs) that target the VEGFR-2 intracellular tyrosine kinase domain (Fig. 1b). TKIs act by binding to the ATP binding pocket and to the adjacent pockets thus preventing the phosphorylation of this intracellular domain (e.g., sunitinib, sorafenib, ZD6474, erlotinib or thalidomide) and blocking the angiogenic signaling pathway (Fig. 1c), lowering blood tumoral irrigation, and improving chemotherapy distribution.9
 | ||
| Fig. 1 (a) Main angiogenesis signaling pathways mediated by VEGFR-2; (b) X-ray crystal structure of the VEGFR-2 intracellular tyrosine kinase domain (PDB: 2XIR), co-crystallized with a TKI; (c) detailed representation of the ATP binding pocket and adjacent binding pockets showing the main interactions between VEGF-2 and the TKI (PDB: 2XIR). | ||
Several polyphenolic compounds are recognized as cancer chemopreventive agents. Flavonoids are especially well known to suppress tumor cell growth via cell-cycle arrest and by the induction of apoptosis in several tumor cell lines.11,12 Moreover, flavonoids namely genistein inhibit endothelial cell cultures on collagen gels.13 The antiangiogenic effect of apigenin on tumor cells was also reported and related to a reduction in the expression of VEGF.12
Other plant-derived anticancer drugs (e.g. Taxol®, camptothecin and combretastatin) proved to be antiangiogenic. In traditional Chinese medicine, many herbs are used in the treatment of angiogenic diseases such as chronic wounds and rheumatoid arthritis.1 Furthermore, it has been reported that drinking green tea could inhibit VEGF-induced angiogenesis in vivo.5
In a previous study, we reported the antitumor activity of Roman chamomile (Chamaemelum nobile L.) methanolic extract and infusion in five different human tumour cells (non-small cell lung cancer, breast, colon, cervical and hepatocellular carcinomas). Furthermore, flavonoids such as flavonols and flavones, phenolic acids and derivatives were found in this wild herb.14 In the present work, the antiangiogenic activity of Roman chamomile (Chamaemelum nobile L.) extracts (methanolic extract and infusion) and main phenolic compounds (apigenin, apigenin-7-O-glucoside, caffeic acid, chlorogenic acid, luteolin, and luteolin-7-O-glucoside) were evaluated through enzymatic assays using the tyrosine kinase intracellular domain of VEGFR-2. To better understand the inhibition phosphorylation mechanism of the tyrosine kinase receptor by luteolin, apigenin and apigenin-7-O-glucoside, docking studies were performed.
2. Materials and methods
2.1 Biological material and sample preparation
C. nobile was gathered during the flowering season (June–July 2010) from wild populations located in grasslands in Bragança (Trás-os-Montes, Northeastern Portugal). Samples consist of pieces of about 8 cm, corresponding to terminal soft leafy stems and inflorescences with flowers fully open and functional, picked up in plants randomly selected in a meadow of about a hectare. The plant material was put together in a single sample for analysis. Voucher specimens are deposited in the Herbarium of the Escola Superior Agrária de Bragança (BRESA). The sample was lyophilized (FreeZone 4.5, Labconco, Kansas, USA), reduced to a fine dried powder (20 mesh) and mixed to obtain a homogenate sample.A methanolic extract was prepared from the lyophilized plant material. The sample (1 g) was extracted by stirring with 25 mL of methanol (25 °C at 150 rpm) for 1 h and subsequently filtered through Whatman No. 4 paper. The residue was then extracted with 25 mL of methanol (25 °C at 150 rpm) for 1 h. The combined methanolic extracts were evaporated at 40 °C (rotary evaporator Büchi R-210) to dryness and re-dissolved in DMSO to a final concentration of 400 μg mL−1.
An infusion was also prepared from the lyophilized plant material. The sample (1 g) was added to 200 mL of boiling distilled water and left to stand at room temperature for 5 min, and then filtered under reduced pressure. The obtained infusion was frozen, lyophilized and re-dissolved in DMSO to a final concentration of 400 μg mL−1.
2.2. Phenolic compounds
Apigenin, apigenin-7-O-glucoside, caffeic acid, chlorogenic acid, luteolin, and luteolin-7-O-glucoside were from Extrasynthese (Genay, France). Each phenolic compound was dissolved in DMSO to a final concentration of 40 μg mL−1.2.3. VEGFR-2 enzymatic inhibition assay
C. nobile methanolic extract and infusion, and the pure phenolic compounds were assessed for VEGFR-2 inhibition activity using the Z′-LYTE-Tyr1 Peptide assay kit (Invitrogen, Cat. PV3190) according to the procedures recommended by the manufacturer.15 Briefly, assays were performed in a total of 20 μL in 384-well plates using fluorescence resonance energy transfer technology. A Tyr1 substrate (coumarin-fluorescein double-labeled peptide) at 1 μM was incubated for 1 h with 4 μg mL−1 VEGFR-2, 50 μM ATP and the C. nobile methanolic extract/infusion (400 at 6.25 μg mL−1) or the pure phenolic compounds (40 at 0.04 μg mL−1) at room temperature in 50 mM Hepes/NaOH (pH 7.5), 10 mM MgCl2, 2 mM MnCl2, 2.5 mM DTT, 0.1 mM orthovanadate, and 0.01% bovine serum albumin. The wells were incubated at 25 °C for 1 h and 5 μL development reagents were added to each well. After a second incubation of 1 h a stop reagent was added to each well. Using a Biotek FLX800 micro-plate the fluorescence was read at 445 nm and 520 nm (excitation 400 nm), and Gen5™ Software was used for data analysis. Genistein (Extrasynthese, Genay, France) was used as positive control.The assays were performed in triplicate and the results were expressed as mean values ± standard deviation (SD). The results were analyzed using a Student's t-test with α = 0.05, to determine the significant difference among the two extracts. For the phenolic compounds, the analysis was performed using one-way analysis of variance (ANOVA) followed by Tukey's HSD test with α = 0.05. These treatments were carried out using the SPSS v. 22.0 program.
2.4 Docking simulations using AutoDockVina
The 2D structure of the compounds apigenin, apigenin-7-O-glucoside, luteolin and luteolin-7-O-glucoside was constructed using the ACD/ChemSketch Freeware 12.0 software. Open Babel16 was used to convert compounds from 2D to 3D and they were saved in the pdb format.A VEGFR-2 crystal structure (PDB: 2XIR) was extracted from the Protein Data Bank (PDB) (http://www.rcsb.org). The co-crystallized ligand was extracted from the PDB file, and AutoDockTools17 was used to assign polar hydrogens and Gasteiger charges to the compounds and VEGFR-2 protein. All structures were saved in the PDBQT file format required to use AutoDockVina.18 AutodockVina was used to perform docking in an area of 30 Å × 30 Å × 30 Å, centered on the co-crystallized ligand. The docking simulations were performed on a cluster of 6 AMD Opteron 6128 8 core 2.0 GHz by using MOLA software.19 All figures with structure representations were prepared using PyMOL (The PyMOL Molecular Graphics System, Version 1.3, Schrödinger, LLC). Available at: (http://www.pymol.org/). Accessed on 03 September, 2012.
2.5 Molecular dynamics simulation
The protein preparation wizard from Maestro (Schrodinger, LLC, Portland, OR) was used to prepare ligand/VEGFR-2 complexes and then used to perform explicit solvent molecular dynamics (MD) simulations. The parallelized Desmond Molecular Dynamics System v2.2 (D. E. Shaw Research, New York, NY) and associated analysis tools, available within the Schrodinger suite (Schrodinger, LLC, Portland, OR), were used for this purpose. The protocol used was described by Mukherjee et al.203. Results and discussion
According to previous studies of the authors, Roman chamomile is an equilibrated valuable species rich in carbohydrates and proteins, and poor in fat, providing tocopherols, carotenoids and essential fatty acids (C18:2n6 and C18:3n3). Moreover, the herb and its infusion are a source of phenolic compounds and organic acids with high bioactive potential.14 Herein, methanolic extract, infusion and phenolic compounds of Roman chamomile were evaluated for their ability to interact with the VEGFR-2 kinase domain, using an enzymatic (fluorescence resonance energy transfer) FRET-based assay. The results are shown in Table 1.| Chamaemelum nobile | VEGFR-2 IC50, μg mL−1 |
|---|---|
| IC50-concentration that provided 50% of VEGFR-2 inhibition. *Different letters mean significant differences between compounds (p < 0.05). | |
| Methanolic extract | 269.26 ± 8.74 |
| Infusion | 301.09 ± 13.07 |
| Student's t test; p-value | <0.001 |
| Phenolic compound | VEGFR-2 IC50, μg mL−1 |
| Luteolin | 0.60 ± 0.03c* |
| Apigenin | 1.29 ± 0.07b |
| Apigenin-7-O-glucoside | 19.21 ± 1.58a |
| Luteolin-7-O-glucoside | >40 |
| Caffeic acid | >40 |
| Chlorogenic acid | >40 |
| Genistein | 1.04 ± 0.06 |
The methanolic extract showed a lower IC50 value than the infusion, 269 and 301 μg mL−1, respectively. These results are in agreement with the higher phenolic compound amount, antioxidant and antitumor activities also previously reported for the methanolic extract.14
Regarding individual molecules, apigenin, apigenin-7-O-glucoside, caffeic acid, chlorogenic acid, luteolin and luteolin-7-O-glucoside were chosen because these compounds were the ones used to quantify all the phenolic compounds identified in Roman chamomile.14 Phenolic acids (caffeic and chlorogenic acids) and luteolin-7-O-glucoside did not show the VEGFR-2 inhibition activity (IC50 values higher than 40 μg mL−1), whereas apigenin-7-O-glucoside showed VEGFR-2 inhibition activity with the IC50 value = 19.21 μg mL−1. A drastic increase in the VEGFR-2 inhibition activity was observed for the corresponding aglycones (compounds without the glycosyl group) of the mentioned flavonoids: luteolin and apigenin (IC50 values = 0.60 and 1.29 μg mL−1, respectively). The active concentrations, corresponding to the last IC50 values, are easily provided by the Roman chamomile infusion, which contains 8.42 μg mL−1 and 9.28 μg mL−1 of luteolin and apigenin derivatives (compounds with glycosyl groups: luteolin-7-O-glucoside and apigenin-7-O-glucoside), respectively (values calculated from the ones reported previously by the authors and taking into account the extraction yields).14 It should be highlighted that the methanolic extract prepared from the herb would provide even higher amounts of those derivatives (21.31 and 13.50 μg mL−1, respectively14).
The possible VEGFR-2 inhibition mechanism of luteolin, apigenin and apigenin-7-O-glucoside (Fig. 2) was predicted using docking tools. A careful analysis of the predicted docking poses showed that apigenin and luteolin probably interact with the VEGFR-2 ATP binding site with a similar binding pose, stabilized by three predicted hydrogen bonds (Fig. 3): one H-bond between the CYS919 backbone and the carbonyl group at position 3 of the benzopyrone moiety; a second H-bond between the CYS919 backbone and the hydroxyl group at position 5 of the benzopyrone moiety; and a third H-bond between the amino group of the LYS868 side chain and the hydroxyl group at position 4 of the benzene ring. The higher VEGFR-2 inhibition capacity of luteolin compared to apigenin can probably be explained by the better occupation of the ATP binding site, accomplished by the lutein extra hydroxyl group occupation of a small pocket located inside the structure shown in Fig. 3. Furthermore, comparing the docking poses of apigenin and apigenin-7-O-glucoside, it was possible to observe that the presence of the glucoside moiety shifts the compound slightly away from the ATP binding site. This shift probably weakens the described H-bonds, explaining the lower VEGFR-2 inhibition capacity of apigenin-7-O-glucoside.
 | ||
| Fig. 2 Chemical structures of luteolin, apigenin and apigenin-7-O-glucoside. | ||
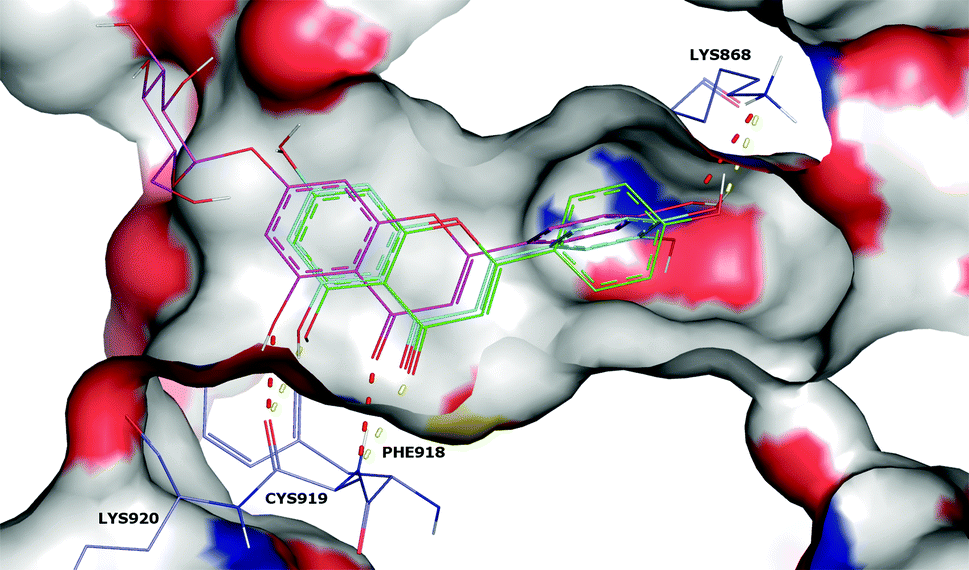 | ||
| Fig. 3 Surface representation of the VEGFR-2 ATP binding site docked with apigenin (green line), luteolin (blue line) and apigenin-7-O-glucoside (magenta line). Apigenin and luteolin hydrogen bonds are represented as yellow dashes, and apigenin-7-O-glucoside hydrogen bonds as red dashes. | ||
Moreover, the inability of AutodockVina to predict the binding pose of luteolin-7-O-glucoside similar to luteolin, apigenin and apigenin-7-O-glucoside seems to indicate that luteolin-7-O-glucoside probably cannot interact with the ATP binding site. This was experimentally proved by the high IC50 value obtained in the enzymatic assay (>100 μM).
MD (Molecular Dynamics) simulations were performed using the most active compounds, luteolin and apigenin, to verify whether both the predicted docking poses remain stable in a more physiologically relevant setting. The docking poses of both complexes were the starting points for 5 ns MD simulations, and the overall stability of each MD simulation was evaluated by plotting the receptor backbone (VEGFR-2) and ligands’ RMSD (Root Mean Square Deviation) as a function of time (Fig. 4).
 | ||
| Fig. 4 RMSD values obtained during the 5 ns MD simulation timeframe for: (a) VEGFR-2/apigenin and (b) VEGFR-2/luteolin complexes. | ||
After small adjustments in the first ns of the MD simulation, both apigenin and luteolin structures remained stable during the MD simulation with an average RMSD of 0.37 and 0.57 Å, respectively (Fig. 4). This is an indication that the predicted docking pose is reliable and is probably close to the experimental VEGFR-2 binding pose. In both MD simulations, the RMSD values for the VEGFR-2 backbone structure were also analyzed and it was observed that, after a normal adjustment of around 2 ns, the RMSD values also remained stable during rest of the MD simulation. This is the expected MD simulation behavior of the protein backbone indicating that the VEGFR-2 structure used is suitable for this type of molecular modeling study.
In general the MD simulations performed gave us further assurance that the predicted docking pose probably corresponds closely to the experimental binding pose although this can only be completely established by the elucidation of the VEGFR-2/apigenin or VEGFR2/luteolin complex structures, usually performed by X-ray crystallography.
The antiangiogenic effect of apigenin on tumor cells has already been reported but related to the reduction in the expression of VEGF12 and not to the inhibition of VEGFR activity, so it is demonstrated in the present work. Regarding luteolin, as far as we know this is the first report on antiangiogenic activity, and only its anticarcinogenic effects mainly by induction of apoptosis and cell cycle arrest by the action on critical molecular targets for cell survival such as p53, p21, cyclin dependent kinases and caspases in liver21 and non-small cell lung22 cancer cells are reported.
Acknowledgements
The authors are grateful to strategic projects PEst-OE/AGR/UI0690/2014 and PEst-C/QUI/UI0686/2013–2014 for financial support to the research centres. R. Guimarães, and R. Calhelha thank FCT, POPH-QREN and FSE for their grants (SFRH/BD/78307/2011 and SFRH/BPD/68344/2010).References
- T.-P. Fan, J.-C. Yeh, K. H. Leung, P. Y. K. Yue and R. N. S. Wong, Trends Pharmacol. Sci., 2006, 27, 297–309 CrossRef CAS PubMed.
- J. Folkman, Nat. Med., 1995, 1, 27–31 CrossRef CAS PubMed.
- H. K. Avraham, T. H. Lee, Y. Koh, T. A. Kim, S. Jiang, M. Sussman, A. M. Samarel and S. Avraham, J. Biol. Chem., 2003, 278, 36661–36668 CrossRef CAS PubMed.
- J. Jeon, J. Lee, C. Kim, Y. An and C. Choi, Microvasc. Res., 2010, 80, 303–309 CrossRef PubMed.
- T. K. Maiti, J. Chatterjee and S. Dasgupta, Biochem. Biophys. Res. Commun., 2003, 308, 64–67 CrossRef CAS.
- L. A. Liotta, P. S. Steeg and W. G. Stetler-Stevenson, Cell, 1991, 64, 327–336 CrossRef CAS.
- J. A. Forsythe, B. H. Jiang, N. V. Iyer, F. Agani, S. W. Leung, R. D. Koos and G. L. Semenza, Mol. Cell. Biol., 1996, 16, 4604–4613 CAS.
- Y. Huang, X. Chen, K. M. Dikov, S. V. Novitskiy, C. A. Mosse, L. Yang and D. P. Carbone, Blood, 2007, 110, 624–631 CrossRef CAS PubMed.
- J. F. Morère, J. M. Brechot and R. Etessami, Targeted Oncol., 2006, 1, 215–219 CrossRef.
- C.-M. Lin, H. Chang, Y.-H. Chen, S. Y. Li, I.-H. Wu and J.-H. Chiu, Int. Immunopharmacol., 2006, 6, 1690–1698 CrossRef CAS PubMed.
- C. Kandaswami, L. T. Lee, P. P. Lee, J. J. Hwang, F. C. Ke, Y. T. Huang and M. T. Lee, in Vivo, 2005, 19, 895–909 Search PubMed.
- M. Osada, S. Imaoka and Y. Funae, FEBS Lett., 2004, 575, 59–63 CrossRef CAS PubMed.
- T. Fotsis, M. Pepper, H. Adlercreutz, G. Fleischamann, T. Hase, R. Montesano and L. Scheweigerer, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 2690–2694 CrossRef CAS.
- R. Guimarães, L. Barros, M. Dueñas, R. C. Calhelha, A. M. Carvalho, C. Santos-Buelga, M. J. R. P. Queiroz and I. C. F. R. Ferreira, Food Chem., 2013, 136, 718–725 CrossRef PubMed.
- P. Soares, R. Costa, H. J. C. Froufe, R. C. Calhelha, D. Peixoto, I. C. F. R. Ferreira, R. M. V. Abreu, R. Soares and M. J. R. P. Queiroz, BioMed Res. Int., 2013, 154856 Search PubMed.
- N. M. O'Boyle1, M. Banck and C. A. James, http://www.jcheminf.com/content/3/1/33/-ins3; C. Morley, T. Vandermeersch and G. R. Hutchison, J. Cheminf., 2011, 3, 33 Search PubMed.
- M. F. Sanner, Structure, 2005, 13, 447–462 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- R. M. Abreu, H. J. Froufe, M. J. R. P. Queiroz and I. C. F. R. Ferreira, J Cheminf., 2010, 2, 10 Search PubMed.
- P. Mukherjee, F. Shah, P. Desai and M. Avery, J. Chem. Inf. Model., 2011, 51, 1376–1392 CrossRef CAS PubMed.
- D. Stagos, G. D. Amoutzias, A. Matakos, A. Spyrou, A. M. Tsatsakis and D. Kouretas, Food Chem. Toxicol., 2012, 50, 2155–2170 CrossRef CAS PubMed.
- X. Cai, T. Ye, C. Liu, W. Lu, M. Lu, J. Zhang, M. Wang and P. Cao, Toxicol. in Vitro, 2011, 25, 1385–1391 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |