
Xanthohumol-induced presynaptic reduction of glutamate release in the rat hippocampus
Yi
Chang
ab,
Tzu Yu
Lin
cd,
Cheng Wei
Lu
cd,
Shu Kuei
Huang
c,
Ying Chou
Wang
e and
Su Jane
Wang
*af
aSchool of Medicine, Fu Jen Catholic University, No. 510, Zhongzheng Rd., Xinzhuang Dist., New Taipei City, Taiwan 24205
bDepartment of Anesthesiology, Shin Kong Wu Ho-Su Memorial Hospital, Taipei, Taiwan 111
cDepartment of Anesthesiology, Far-Eastern Memorial Hospital, Pan-Chiao District, New Taipei City, Taiwan 22060
dDepartment of Mechanical Engineering, Yuan Ze University, Taoyuan 320, Taiwan
eDepartment of Clinical Psychology, Fu Jen Catholic University, No. 510, Zhongzheng Rd., Xinzhuang Dist., New Taipei City, Taiwan 24205
fGraduate Institute of Basic Medicine, Fu Jen Catholic University, No. 510, Zhongzheng Rd., Xinzhuang Dist., New Taipei City, Taiwan 24205. E-mail: med0003@mail.fju.edu.tw; Fax: +886-2-29052096; Tel: +886-2-29053465
First published on 15th December 2015
Abstract
This study examined whether xanthohumol, a hop-derived prenylated flavonoid present in beer, affects glutamate release in the rat hippocampus. In the rat hippocampal nerve terminals (synaptosomes), xanthohumol inhibited the release of 4-aminopyridine (4-AP)-evoked glutamate and the elevation of cytosolic Ca2+ concentration, whereas it had no effect on 4-AP-mediated depolarization. The inhibitory effect of xanthohumol on the evoked glutamate release was prevented by removing extracellular Ca2+, using the Cav2.2 (N-type) and Cav2.1 (P/Q-type) channel blocker ω-CgTX MVIIC, the calmodulin antagonists W7 and calmidazolium, and the protein kinase A inhibitor H89; however, no such effect was observed when the G-protein inhibitor N-ethylmaleimide was used. In addition, immunocytochemical data demonstrated that GABAA receptors are present in the hippocampal synaptosomes and that the xanthohumol effect on evoked glutamate release was antagonized by the GABAA receptor antagonist SR95531. Furthermore, in slice preparations, xanthohumol reduced the frequency of miniature excitatory postsynaptic currents without affecting their amplitude. We conclude that xanthohumol acts at GABAA receptors present in the hippocampal nerve terminals to decrease the Ca2+ influx through N- and P/Q-type Ca2+ channels, which subsequently suppresses the Ca2+-calmodulin/PKA cascade to decrease the evoked glutamate release.
Introduction
Glutamate is the most crucial excitatory neurotransmitter in the central nervous system (CNS) and is a crucial element in maintaining normal physiological brain functions.1,2 In addition to its physiological role, glutamate is a potent neurotoxin, and high levels in the synaptic cleft cause excessive glutamate receptor activation, leading to an overload of intracellular Ca2+ and subsequent neuronal cell death.3,4 This process is involved in several neuropathological conditions ranging from acute insults such as stroke, epileptic seizures, traumatic brain and spinal cord injury, to chronic neurodegenerative disorders such as Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD), and amyotrophic lateral sclerosis (ALS).1,5,6 Therefore, attenuating glutamate excitotoxicity might be an effective therapeutic approach for these diseases. Moreover, compounds that reduce glutamate release might counteract glutamate excitotoxicity and must be regarded as potential drug candidates.Growing evidence from in vitro and in vivo studies indicates that flavonoid-rich food aids in the prevention and halts the progression of many brain disorders.7–9 Xanthohumol (3′-[3,3-dimethyl allyl]-2,4,4′-trihydroxy-6′-methoxychalcone; Fig. 1) is one of the major prenylflavonoids present in hops (Humulus lupulus) used in beer production. Beer is the principal dietary source of xanthohumol.10 Xanthohumol has a good safety profile11 and exerts numerous beneficial effects, such as antioxidation, antiinflammation, antivirus, cardiovascular protection, anticancer, and immune modulation.12–16 In addition, the neuroprotective effect of xanthohumol in the CNS has been confirmed in numerous in vitro and animal studies. For example, xanthohumol attenuates hydrogen peroxide (H2O2)- or 6-hydroxydopamine (6-OHDA)-induced neurotoxicity,17 protects against ischemia-induced brain damage,18 promotes neuronal survival,19 and improves cognitive functions such as learning and memory.20 However, the mechanism by which xanthohumol mediates these effects is not completely understood.
 | ||
| Fig. 1 Xanthohumol inhibits 4-AP-evoked glutamate release in rat hippocampal synaptosomes. (A) Chemical structure of xanthohumol. (B) Glutamate release was evoked by 1 mM 4-AP in the absence (control) and presence of 10 μM xanthohumol (added 10 min before the addition of 4-AP). (C) Dose–response curve for xanthohumol inhibition of 4-AP-evoked glutamate release, showing percentage inhibition compared with controls. Results are the mean ± SEM of 4–7 independent experiments. ***, P < 0.001 vs. control group. | ||
Because the excitotoxicity caused by excessive glutamate is critical in the development of many brain diseases, reducing glutamate release may be an important mechanism for neuroprotective actions. Although xanthohumol has a neuroprotective activity, data describing the effect of xanthohumol on neuronal glutamate release in the CNS are unavailable. Therefore, the objectives of this study were as follows: (1) to determine whether xanthohumol inhibits glutamate release in rat hippocampal nerve terminals (synaptosomes), (2) to determine the signaling pathway involved in the xanthohumol-inhibited glutamate release from synaptosomes, and (3) to determine whether xanthohumol reduces miniature excitatory postsynaptic currents (mEPSCs) in rat hippocampal slices. mEPSCs are believed to arise from the spontaneous fusion of vesicles and the terminal membrane independent of the activation of presynaptic voltage-dependent ion channels.21 These studies clarify the mechanism of xanthohumol in inhibiting glutamate release in CNS.
Materials and methods
Materials
4-Aminopyridine (4-AP), ω-conotoxin MVIIC (ω-CgTX MVIIC), dantrolene, 7-chloro-5-(2-chloropheny)-1,5-dihydro-4,1-benzothiazepin-2(3H)-one (CGP37157), D(−)-2-amino-5-phosphonopentanoic acid (D-AP5), bisindolylmaleimide I (GF109203X), 2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one) (PD98059), isoguvacine, bicuculline, and SR95531 were purchased from Tocris Cookson (Bristol, UK). 3′,3′,3′-Dipropylthiadicarbocyanine iodide [DiSC3(5)] and fura-2-acetoxymethyl ester (Fura-2-AM) were purchased from Invitrogen (Carlsbad, CA, USA). Xanthohumol, ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), tetrodotoxin and all other reagents were purchased from Sigma-Aldrich (St Louis, MO, USA). Xanthohumol, DiSC3(5), and Fura-2 were dissolved in dimethylsulfoxide, with a final concentration in the medium of ≤0.1% (v/v).Animals
Adult male Sprague-Dawley rats (150–200 g) and 8- to 23-day-old male Sprague-Dawley rats were purchased from BioLASCO (Taiwan Co., Ltd, Taipei, Taiwan). Animals were housed at constant temperature (22 ± 1 °C) and relative humidity (50%) under a regular 12 h light–dark cycle (light off at 7 pm). Food and water were freely available. The experimental procedures were approved by the Fu Jen Institutional Animal Care and Utilization Committee, in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All efforts were made to minimize animal suffering and to use a minimum number of animals necessary to produce reliable results.Preparation of synaptosomes
Synaptosomes were purified from the hippocampus of rats on discontinuous Percoll gradients as described previously.22,23 Briefly, the tissue was homogenized in a medium containing 0.32 M sucrose (pH 7.4), the homogenate was centrifuged (10 min, 3000g at 4 °C) to remove the nuclei and cell debris, and the supernatant was centrifuged again (12 min, 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500g). The pellet was gently resuspended in 0.32 M sucrose (pH 7.4), and an aliquot of this synaptosomal suspension (2 ml) was placed onto a 3 ml Percoll discontinuous gradient containing 0.32 M sucrose, 1 mM EDTA, 0.25 mM DL-dithiothreitol, and 3%, 10%, and 23% Percoll (pH 7.4). After centrifugation at 32500g for 7 min at 4 °C, the layer between 10 and 23% Percoll (synaptosomal fraction) was collected, washed by centrifugation and resuspended in HEPES-buffered medium (HBM) having the following composition (mM): 140 NaCl, 5 KCl, 5 NaHCO3, 1 MgCl2·6H2O, 1.2 Na2HPO4, 10 glucose, and 10 HEPES, pH 7.4. The protein concentration was determined using a Bradford assay. Synaptosomes were centrifuged in a final wash to obtain synaptosomal pellets containing 0.5 mg of protein. The synaptosomal pellets were stored on ice and used within 4–6 h.
500g). The pellet was gently resuspended in 0.32 M sucrose (pH 7.4), and an aliquot of this synaptosomal suspension (2 ml) was placed onto a 3 ml Percoll discontinuous gradient containing 0.32 M sucrose, 1 mM EDTA, 0.25 mM DL-dithiothreitol, and 3%, 10%, and 23% Percoll (pH 7.4). After centrifugation at 32500g for 7 min at 4 °C, the layer between 10 and 23% Percoll (synaptosomal fraction) was collected, washed by centrifugation and resuspended in HEPES-buffered medium (HBM) having the following composition (mM): 140 NaCl, 5 KCl, 5 NaHCO3, 1 MgCl2·6H2O, 1.2 Na2HPO4, 10 glucose, and 10 HEPES, pH 7.4. The protein concentration was determined using a Bradford assay. Synaptosomes were centrifuged in a final wash to obtain synaptosomal pellets containing 0.5 mg of protein. The synaptosomal pellets were stored on ice and used within 4–6 h.
Glutamate release assay
Glutamate release from purified hippocampal synaptosomes was monitored online, with an assay that employed exogenous glutamate dehydrogenase (GDH) and NADP+ to couple the oxidative deamination of the released glutamate to the generation of NADPH detected fluorometrically.24 Synaptosomal pellets were resuspended in HBM that contained 16 μM BSA and incubated in a stirred and thermostated cuvette maintained at 37 °C in a Perkin-Elmer LS-55 spectrofluorometer (PerkinElmer Life and Analytical Sciences, Waltham, MA, USA). NADP+ (2 mM), GDH (50 units per ml) and CaCl2 (1 mM) were added after 3 min. In experiments that investigated Ca2+-independent efflux of glutamate, EGTA (200 μM) was added in place of CaCl2. Other additions before depolarization were made as described in the figure legends. Glutamate release was monitored by measuring the increase of fluorescence (excitation and emission wavelengths of 340 and 460 nm, respectively) caused by NADPH being produced by oxidative deamination of released glutamate by GDH. In the absence of axonal connectivity, synaptosomes are not amenable to electrical stimulation, so several biochemical secretagogues have been developed, including the use of depolarization protocols involving potassium channel blockers such as 4-AP or high external KCl.24,25 4-AP (1 mM) or high external KCl (15 mM) was added after 10 min of incubation to stimulate glutamate release. A standard of exogenous glutamate (5 nmol) was added at the end of each experiment, and the fluorescence response used to calculate the released glutamate was expressed as nanomoles of glutamate per milligram synaptosomal protein (nmol mg−1). Values quoted in the text represent levels of glutamate cumulatively released after 5 min of depolarization. Data were gathered at 2 s intervals. Cumulative data were analyzed using Lotus 1-2-3.Synaptosomal plasma membrane potential
The synaptosomal membrane potential can be monitored by using positively charged, membrane-potential-sensitive carbocyanine dyes such as DiSC3(5). Synaptosomes were preincubated and resuspended as described for the glutamate release experiments. After 3 min incubation, 5 μM DiSC3(5) was added and allowed to equilibrate before the addition of CaCl2 (1 mM) after 4 min incubation. 4-AP (1 mM) was added to depolarize the synaptosomes at 10 min, and DiSC3(5) fluorescence was monitored at excitation and emission wavelengths of 646 and 674 nm, respectively. Data were gathered at 2 s intervals. Cumulative data were analyzed using Lotus 1-2-3, and results are expressed in fluorescence units.Cytosolic free Ca2+ concentration ([Ca2+]C)
[Ca2+]C was measured using the Ca2+ indicator Fura-2-AM. Synaptosomes (0.5 mg ml−1) were preincubated in HBM with 16 μM BSA in the presence of 5 μM Fura-2-AM and 0.1 mM CaCl2 for 30 min at 37 °C in a stirred test tube. After Fura-2-AM loading, synaptosomes were centrifuged in a microcentrifuge for 30 s at 3000g (5000 rpm). The synaptosomal pellets were resuspended in HBM with BSA, and the synaptosomal suspension was stirred in a thermostated cuvette in a Perkin-Elmer LS-55 spectrofluorometer. CaCl2 (1 mM) was added after 3 min and further additions were made after an additional 10 min. Fura-2-Ca fluorescence was determined at excitation wavelengths of 340 and 380 nm (emission wavelength, 505 nm), and data were gathered at 2 s intervals. [Ca2+]C (nM) was calculated by using calibration procedures25 and equations described previously.26 Cumulative data were analyzed using Lotus 1-2-3.Immunocytochemistry
The synaptosomes were allowed to attach to coverslips (diameter 20 mm) precoated with poly-L-lysine for 40 min at 4 °C before being fixed with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) for 30 min. After rinsing with phosphate buffer 3 times, the synaptosomes were incubated in blocking buffer containing 3% normal goat serum and 0.2% Triton X-100 for 60 min. They were then incubated with a mixture of primary mouse monoclonal antibodies against synaptophysin (1:200; Abcam, Cambridge, UK) and rabbit monoclonal antibodies against GABAA receptor α1 (1:100; Abcam, Cambridge, UK) for 90 min at room temperature. After rinsing with blocking buffer, the synaptosomes were incubated with a mixture of goat anti-mouse DyLight 549- and goat anti-rabbit fluoresce in isothiocyanate (FITC)-conjugated secondary antibodies (1:200; Jackson ImmunoResearch Inc., West Grove, PA, USA) for 1 h at room temperature. The synaptosomes were then washed 3 times with phosphate buffer and 0.1 M carbonate buffer (pH 9.2), and coverslipped with fluorescence mounting medium (DAKO North America, Inc., CA, USA). Double immunofluorescence images were observed at a magnification of 400×, using upright fluorescence microscopy (LeicaDM2000 LED, Wetzlar, Germany), and images were captured using a CCD camera (SPOT RT3, Diagnostic Instruments, Sterling Heights, MI, USA).
Slice preparation and electrophysiological recordings
Hippocampal slices were prepared from 8- to 23-day-old male rats for whole-cell patch clamp recordings, using procedures described previously.27 Briefly, after decapitation, transverse brain slices (300 μm) were prepared using a vibratome (VT1000S, Leica, Germany). The slices were placed in a beaker of artificial cerebrospinal fluid continuously oxygenated with 95% O2–5% CO2 at room temperature (23–26 °C) for at least 1 h before use. Artificial cerebrospinal fluid was composed of (in mM): 117 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 MgCl2, 25 NaHCO3, 1.2 NaH2PO4, 11 glucose (pH 7.4, 300 mOsm).Slices were moved to a recording chamber mounted on a BX51W1 upright microscope (Olympus) equipped with infra-red differential interference contrast (IR-DIC). The chamber was continuously perfused with oxygenated artificial cerebrospinal fluid. Neurons were visualized with an Olympus Optical 40× water immersion objective. Whole-cell voltage-clamp recordings were performed in hippocampal CA3 neurons using pipettes with 2–5 MΩ resistance after files with an internal solution containing (in mM): 0.3 Na3GTP, 135 K-gluconate, 20 KCl, 0.1 EGTA, 2 CMgCl2, 4 Na2ATP, and 10 HEPES (pH 7.3, 280 mOsm). Pipettes were fabricated from borosilicate glass (1.5 mm outer diameter, 0.86 mm internal diameter) using a micropipette puller (P87, Sutter Instruments, USA). Positive pressure was applied to the recording pipette as it was lowered into the medium and approached the cell membrane. A constant negative pressure was applied to form the seal (>1 GΩ) when the recording pipette attached to the membrane, and then suck quickly to rupture the cell membrane and access the whole cell configuration. Neurons were voltage clamped at −70 mV using an Axopatch 200B amplifier (Axon Instruments, Foster City, CA, USA). Access resistance (8–30 MΩ) was regularly monitored during recordings and cells were rejected if it changed more than 15% during the experiments. Membrane currents were filtered at 2 kHz, digitized and stored on a computer using pCLAMP (Axon Instruments, Foster City, CA, USA). Data were analyzed offline using commercially available software.
Data analysis
Data were expressed as mean ± SEM. The data reported were analyzed by using the unpaired Student's t test or by using one-way ANOVA accompanied by Tukey's test for multiple comparisons. Analysis was completed via the software SPSS (17.0; SPSS Inc., Chicago, IL). P < 0.05 was considered to represent a significant difference.Results
Xanthohumol inhibits 4-AP-evoked glutamate release from rat hippocampal synaptosomes via an exocytotic mechanism
Synaptosomes were purified from the hippocampus of rats, and exposed to 4-aminopyridine (4-AP), a K+-channel blocker that opens voltage-dependent Ca2+ channels (VDCCs) and induces the release of a neurotransmitter,28 to assess glutamate release. Under control conditions, 4-AP (1 mM) evoked a glutamate release of 7.2 ± 0.1 nmol per mg per 5 min from synaptosomes incubated in the presence of 1 mM CaCl2. Application of xanthohumol (10 μM) produced an inhibition of 4-AP-evoked glutamate release to 4.3 ± 0.1 nmol per mg per 5 min (P < 0.001), without altering the basal release of glutamate (Fig. 1B). The xanthohumol-mediated inhibition of 4-AP-evoked glutamate release was concentration dependent, with 1, 5, 10, 30, 50, and 70 μM xanthohumol inhibiting glutamate release to 8.1 ± 1.9%, 24.7 ± 1.3%, 37.9 ± 2.0%, 64.5 ± 2.1%, 74.5 ± 2.7%, and 71.9 ± 2.4% of control values respectively. The IC50 value for the xanthohumol-mediated inhibition of evoked glutamate release, derived from a dose–response curve, was 15 μM (Fig. 1C).The 4-AP-evoked release of glutamate from synaptosomes can be sustained by different mechanisms, including exocytosis (Ca2+-dependent release) and reversal of the transporter (Ca2+-independent release).28,29 Thus, we examined the effect of xanthohumol on the Ca2+-independent component of 4-AP-evoked glutamate release that can be estimated in an extracellular Ca2+-free solution containing 300 μM EGTA. Fig. 2A shows that the Ca2+-independent release of glutamate evoked by 1 mM 4-AP was 2.9 ± 0.3 nmol per mg per 5 min (P < 0.001), and this release was not affected by xanthohumol at 10 μM (2.8 ± 0.2 nmol per mg per 5 min; P = 0.75). In addition, DL-threo-beta-benzyl-oxyaspartate (DL-TBOA), a glutamate reuptake inhibitor, or bafilomycin A1, a vesicular transporter inhibitor, was used to examine the effect of xanthohumol. DL-TBOA (10 μM) did not affect basal glutamate release, but increased the 4-AP-evoked glutamate release (P < 0.001). In the presence of DL-TBOA, however, xanthohumol (10 μM) still effectively inhibited the 4-AP-evoked glutamate release (P < 0.05; Fig. 2B). On average, xanthohumol resulted in a 35.1 ± 3.1% inhibition of 4-AP-evoked glutamate release in the presence of DL-TBOA, which was not different from the action of xanthohumol alone (41.2 ± 4.1%; P = 0.26; Fig. 2D). In contrast to DL-TBOA, bafilomycin A1 (0.1 μM) reduced 4-AP-evoked glutamate release (P < 0.001), and prevented the inhibitory effect of xanthohumol (10 μM) on 4-AP-evoked glutamate release (Fig. 2C). In the six synaptosomal preparations tested, xanthohumol produced a 5.5 ± 3.5% decrease in the 4-AP-evoked glutamate release after the application of bafilomycin A1, which was significantly different from the inhibition produced by xanthohumol alone (41.2 ± 4.1%; P < 0.05; Fig. 2D).
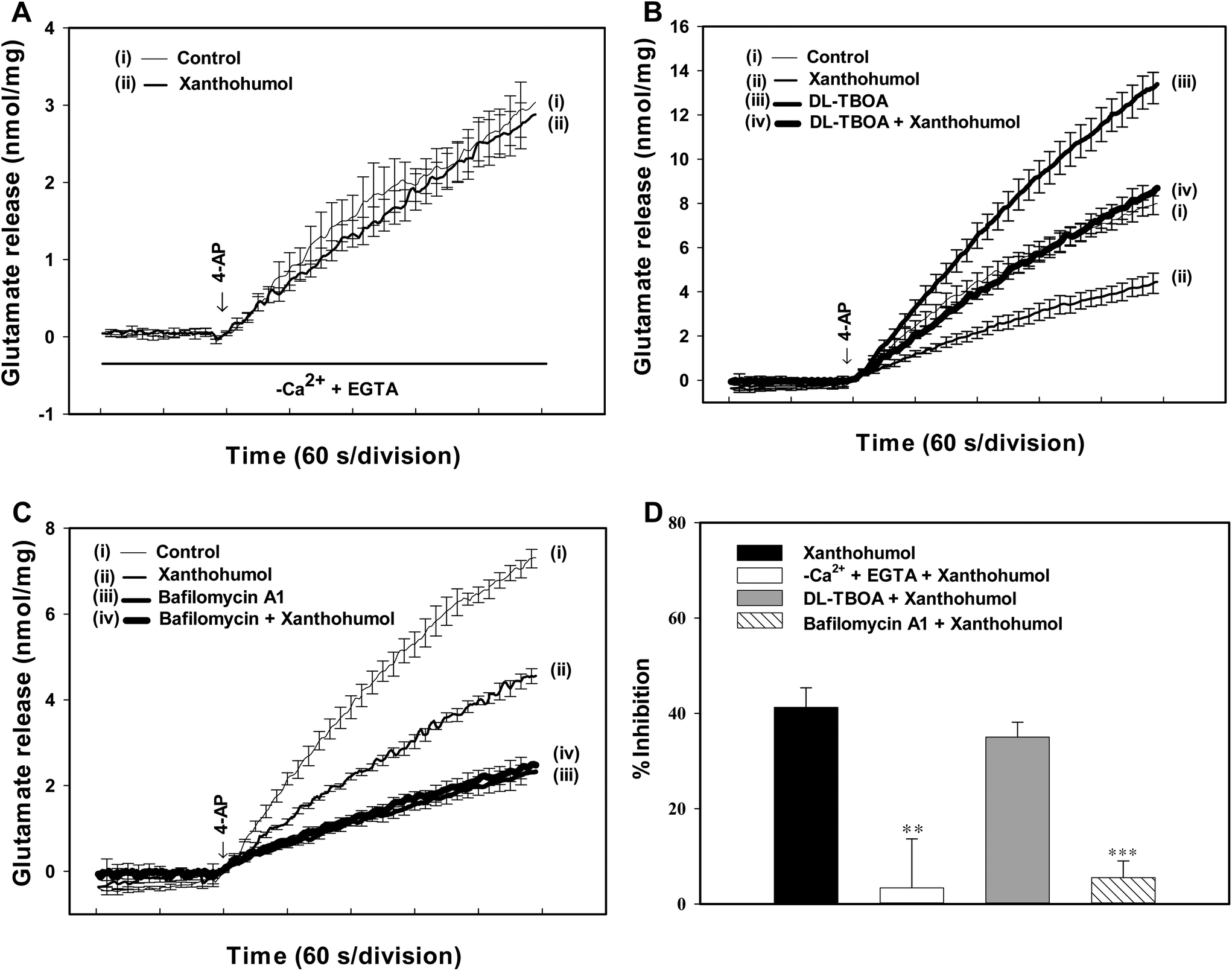 | ||
| Fig. 2 Xanthohumol-mediated inhibition of 4-AP-evoked glutamate release is blocked by chelating the extracellular Ca2+ and the vesicular transporter inhibitor bafilomycin A1, but not by the glutamate transporter inhibitor DL-TBOA. (A–C) Glutamate release was evoked by 1 mM 4-AP in the absence (control) and presence of 10 μM xanthohumol, and the absence and presence of an extracellular-Ca2+-free solution containing 300 μM EGTA, 0.1 μM bafilomycin A1, or 10 μM DL-TBOA. (D) Bar plots showing the average percent inhibition of 4-AP-evoked glutamate release produced by xanthohumol in the control group or in the presence of an extracellular-Ca2+-free solution containing 300 μM EGTA, DL-TBOA and bafilomycin A1. Results are the mean ± SEM of 5–6 independent experiments. **, P < 0.01; ***, P < 0.001 vs. the xanthohumol-treated group. | ||
Xanthohumol reduces intrasynaptosomal [Ca2+] but has no effect on synaptosomal membrane potential
To further understand the mechanism underlying the xanthohumol-mediated inhibition of glutamate release from synaptosomes, we used the Ca2+ indicator Fura-2 to assess the effect of xanthohumol on the 4-AP-induced increase in [Ca2+]C. As shown in Fig. 3A, after the addition of 4-AP (1 mM), [Ca2+]C in synaptosomes was elevated to a plateau level of 219.3 ± 6.9 nM. This 4-AP-evoked rise in [Ca2+]C was decreased by 43.7 nM with 10 μM xanthohumol (175.6 ± 6.6 nM; P < 0.001). The observed inhibitory effect of xanthohumol on 4-AP-evoked increase in [Ca2+]C might be attributed to the modulation of potassium channels and the consequently altered plasma membrane potential.30 To test this possibility, the effect of xanthohumol on the synaptosomal plasma membrane potential was determined using the membrane potential-sensitive dye DiSC3(5). Fig. 3B shows that 4-AP (1 mM) caused an increase in DiSC3(5) fluorescence of 13.8 ± 0.5 fluorescence units per 5 min. Preincubation of synaptosomes with xanthohumol (10 μM) before 4-AP addition did not alter the resting membrane potential and produced no significant change in 4-AP-mediated increase in DiSC3(5) fluorescence (P = 0.24). This indicates that the observed inhibition of evoked glutamate release by xanthohumol is unlikely to have been caused by a hyperpolarizing effect of the drug on the synaptosomal plasma membrane potential, or attenuation of the depolarization produced by 4-AP. Confirmation that the xanthohumol effect did not impinge on synaptosomal excitability was obtained with experiments using high external [K+]-mediated depolarization. Elevated extracellular KCl depolarizes the plasma membrane by shifting the K+ equilibrium potential above the threshold potential for the activation of voltage-dependent ion channels. While Na+ channels are inactivated under these conditions, VDCCs are activated nonetheless to mediate Ca2+ entry, which supports neurotransmitter release.31 Addition of 15 mM KCl evoked a controlled glutamate release of 6.7 ± 0.2 nmol per mg per 5 min, which was reduced to 3.6 ± 0.3 nmol per mg per 5 min in the presence of 10 μM xanthohumol (P < 0.001) (Fig. 3C). | ||
| Fig. 3 Xanthohumol reduces 4-AP-induced increase in cytosolic Ca2+ concentration ([Ca2+]C) but fails to alter the synaptosomal membrane potential. The [Ca2+]C (nM) (A) or synaptosomal membrane potential (B) was measured in the absence (control) and in the presence of 10 μM xanthohumol, added 10 min before depolarization with 1 mM 4-AP. (C) Inhibition of KCl (15 mM)-evoked glutamate release by xanthohumol. Results are the mean ± SEM of 5 independent experiments. ***, P < 0.001 vs. control group. | ||
A reduction in Ca2+ influx through the Cav2.2 (N-type) and Cav2.1 (P/Q-type) channels contributes to the effect of xanthohumol in hippocampal synaptosomes
A reduction in the Ca2+-dependent release of glutamate could be explained by the decreased entry of Ca2+ through Cav2.2 (N-type) and Cav2.1 (P/Q-type) channels which are coupled to glutamate exocytosis.32,33 To test whether the reduction of these Ca2+ channel activities was involved in the inhibitory action of xanthohumol on 4-AP-evoked glutamate release, we examined the effect of ω-conotoxin MVIIC (ω-CgTX MVIIC), a wide spectrum blocker of Cav2.2 (N-type) and Cav2.1 (P/Q-type) channels. Application of 2 μM ω-CgTX MVIIC reduced 4-AP-evoked glutamate release (P < 0.001; Fig. 4A). In the presence of ω-CgTX MVIIC, xanthohumol (10 μM) reduced glutamate release by a further 12.7 ± 4.6% (P < 0.05), which was significantly different from the inhibition produced by xanthohumol alone (45.2 ± 3.1%; P < 0.05; Fig. 4A and D). In addition to the Ca2+ influx through VDCCs, release of Ca2+ from internal stores is involved in the Ca2+ influx coupled to glutamate release.34 Thus, we tested the effect of dantrolene, an inhibitor of intracellular Ca2+ release from endoplasmic reticulum, and 7-chloro-5-(2-chloropheny)-1,5-dihydro-4,1-benzothiazepin-2(3H)-one (CGP37157), a membrane-permeant blocker of mitochondrial Na+/Ca2+ exchange, on the xanthohumol inhibition of glutamate release. Dantrolene (100 μM) reduced 4-AP (1 mM)-evoked release (P < 0.01). In the presence of dantrolene, however, xanthohumol (10 μM) still effectively inhibited 4-AP-evoked glutamate release (43.6 ± 2.1% inhibition; P < 0.05; Fig. 4B), which was similar to the inhibition produced by xanthohumol alone (45.2 ± 3.1%; P = 0.59; Fig. 4D). Similar results were also observed using 100 μM CGP37157 (Fig. 4C). In five synaptosomal preparations tested, xanthohumol (10 μM) produced a 44.9 ± 2.8% decrease in the 4-AP-evoked glutamate release after the application of CGP37157 (P < 0.05), which was not different from the inhibition produced by xanthohumol alone (45.2 ± 3.1%; P = 0.84; Fig. 4D). | ||
| Fig. 4 Xanthohumol-mediated inhibition of 4-AP-evoked glutamate release is prevented by the Cav2.2 (N-type) and Cav2.1 (P/Q-type) channel blocker ω-CgTX MVIIC and not by the intracellular Ca2+ release inhibitor dantrolene or the mitochondrial Na+/Ca2+ exchange blocker CGP37157. (A–C) Glutamate release was evoked by 1 mM 4-AP in the absence (control) and presence of 10 μM xanthohumol, and the absence and presence of 2 μM ω-CgTX MVIIC, 100 μM dantrolene, or 100 μM CGP37157. (D) Bar plots showing the average percent inhibition of 4-AP-evoked glutamate release produced by xanthohumol in the control group or in the presence of ω-CgTX MVIIC, dantrolene and CGP37157. Results are the mean ± SEM of 5–7 independent experiments. ***, P < 0.001 vs. the xanthohumol-treated group. | ||
The Ca2+-calmodulin/AC/cAMP/PKA signalling cascade is involved in the xanthohumol-mediated inhibition of glutamate release from hippocampal synaptosomes
To identify the intrasynaptosomal enzymatic pathways participating in the xanthohumol inhibition of glutamate release, we tested several enzyme inhibitors: bisindolylmaleimide I (GF109203X), a protein kinase C (PKC) inhibitor, N-[2-(p-bromocinnamylamino)-ethyl]-5-isoquinolinesulfonamide dihydrochloride (H89), a protein kinase A (PKA) inhibitor, and 2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one (PD98059), a mitogen-activated/extracellular signal-regulated kinase (MEK) inhibitor. Fig. 5A shows that GF109203X (1 μM) reduced 1 mM 4-AP-evoked glutamate release (P < 0.001). In synaptosomes pretreated with GF109203X, xanthohumol (10 μM) is still able to inhibit 4-AP-evoked glutamate release (44.2 ± 2.6% inhibition; P < 0.05), which was similar to the inhibition produced by xanthohumol alone (44.9 ± 1.8%; P = 0.87; Fig. 5A and E). Similar results were observed with PD98059 (50 μM) (Fig. 5B and E). Fig. 5C shows that H89 (100 μM) reduced 4-AP (1 mM)-evoked glutamate release (P < 0.001), and largely prevented the inhibition of glutamate release by xanthohumol (10 μM). In the presence of H89, therefore, xanthohumol induced a statistically insignificant inhibition of glutamate release of 2.6 ± 1.3% compared with the 44.9 ± 1.8% inhibition produced by xanthohumol alone (P < 0.05; Fig. 5E). | ||
| Fig. 5 Xanthohumol-mediated inhibition of 4-AP-evoked glutamate release involves Ca2+-calmodulin/AC/cAMP/PKA cascade but not G-protein action. (A–D) Glutamate release was evoked by 1 mM 4-AP in the absence (control) and presence of 10 μM xanthohumol, and the absence and presence of 1 μM GF109203X, 50 μM PD98059, 100 μM H89, or 25 μM W7. (E) Bar plots showing the average percent inhibition of 4-AP-evoked glutamate release produced by xanthohumol in the control group or in the presence of GF109203X, PD98059, H89, W7, calmidazolium, and N-ethylmaleimide. Results are the mean ± SEM of 5–6 independent experiments. ***, P < 0.001 vs. the xanthohumol-treated group. | ||
From the aforementioned results, it is clear that a decrease in the intrasynaptosomal Ca2+ levels is associated with the inhibition of glutamate release by xanthohumol; therefore, we hypothesize that the suppression of the PKA pathway possibly involves this regulation. Thus, the mechanism of PKA inhibition remains to be explored. It has been shown that Ca2+/calmodulin activates the adenylate cyclase (AC)/PKA signaling cascade through a G protein-independent mechanism in the hippocampus.35 This enhances the prospect of inhibition of PKA in the hippocampal nerve terminals by the decrease in Ca2+ activating calmodulin. To test this possibility, we examined the effect of inhibiting Ca2+/calmodulin by using the calmodulin antagonist W7 (N-(6-aminohexyl)-5-chloro-1-naphthalene-sulphonamide). W7 (25 μM) reduced 4-AP-evoked glutamate release (P < 0.001; Fig. 5D). In the presence of W7, xanthohumol (10 μM) produced a 2.7 ± 2.4% decrease in the 4-AP-evoked glutamate release, which was significantly different from the inhibition produced by xanthohumol alone (45.6 ± 2.3%; P < 0.05; Fig. 5E). As with W7, in synaptosome treated calmidazolium (CMZ), an alternative calmodulin antagonist, xanthohumol-mediated inhibition of 4-AP-evoked glutamate release was abolished (Fig. 5E). In addition, we examined the effect of xanthohumol in synaptosomes treated with N-ethylmaleimide (NEM), an alkylating agent that effects inactivation of G-proteins.35 In the presence of NEM (2 μM), however, the inhibitory effect of 4-AP-evoked glutamate release mediated by xanthohumol is still present (50.2 ± 5.3%, Fig. 5E). This result indicates that the observed inhibition of glutamate release by xanthohumol is not mediated by G-proteins.
Xanthohumol-inhibited glutamate release from synaptosomes is likely to involve interaction with GABAA receptors
It is known that xanthohumol modulates the function of GABAA receptors.36,37 The hippocampal synaptosomes are enriched in this receptor, as witnessed by co-labeling with antisera against the vesicle marker synaptophysin and the GABAA receptors (Fig. 6A and B). Indeed, among the synaptosomes that contained synaptophysin (1070 particles from ten fields), 36.5 ± 1.1% also contained GABAA receptor (Fig. 6B and C). To determine whether GABAA receptor was involved in the observed xanthohumol-mediated inhibition of glutamate release, the effect of isoguvacine (an agonist of GABAA receptor) was examined. Applying isoguvacine (300 μM) inhibited 4-AP-evoked glutamate release (P < 0.001). In the presence of isoguvacine, xanthohumol only reduced glutamate release by a further 2.9 ± 0.7%, indicating a significant reduction compared with that obtained when xanthohumol was applied alone (39.9 ± 4.9%; P < 0.05; Fig. 6D and F). Similar results were also observed using 100 μM SR95531, an antagonist of the GABAA receptor (Fig. 6E and F). 100 μM SR95531 had no significant effect on 4-AP-evoked glutamate release (P = 0.55; Fig. 6E). | ||
| Fig. 6 Nerve terminal GABAA receptor activity is involved in the xanthohumol-mediated inhibition of 4-AP-evoked glutamate release. (A) Synaptosomes were fixed onto polylysine-coated coverslips and double stained by immunocytochemistry using antisera against GABAA receptors and the vesicular marker synaptophysin. Example pictures of immunofluorescence staining demonstrate GABAA receptor (green in A), synaptophysin (red in B), and merged images (orange in C) of 2 proteins. The scale bar for A–C is 30 μm. (D and E) Glutamate release was evoked by 1 mM 4-AP in the absence (control) and presence of 10 μM xanthohumol, and the absence and presence of 300 μM isoguvacine or 100 μM SR95531. (F) Bar plots showing the average percent inhibition of 4-AP-evoked glutamate release produced by xanthohumol in the control group or in the presence of isoguvacine and SR95531. Results are the mean ± SEM of 5–6 independent experiments. ***, P < 0.001 vs. the xanthohumol-treated group. | ||
Xanthohumol reduces the frequency but not the amplitude of miniature excitatory postsynaptic currents (mEPSCs) in hippocampal slices
To further confirm the presynaptic site of action of xanthohumol, we used a whole-cell voltage clamp to analyze mEPSC frequency and amplitude in hippocampal slices. Analysis of mEPSC frequency and amplitude can provide useful indications of changes in the presynaptic release process and/or changes in the sensitivity of postsynaptic receptors, respectively.38,39Fig. 7A shows that mEPSCs were recorded at a holding potential of −70 mV and in the presence of the Na+ channel blocker tetrodotoxin (1 μM) and the GABAA receptor antagonist bicuculline (20 μM). No mEPSCs could be detected after the application of the non-N-methyl-D-aspartate (NMDA) receptor antagonist CNQX (20 μM) and the NMDA receptor antagonist D-AP5 (50 μM), demonstrating that they were glutamatergic events (data not shown). A typical example of traces recorded from a signal cell is shown in Fig. 7A, and xanthohumol (10 μM) decreased the occurrence of mEPSCs. Significant differences in cumulative interevent interval distributions were observed during xanthohumol application (Kolmogorov–Smirnov test, P < 0.001; Fig. 7B). However, no significant effect of xanthohumol (10 μM) was observed on the mEPSC amplitude. This can be observed from a lack of effect of xanthohumol on the cumulative probability plots (Kolmogorov–Smirnov test, P = 0.37; Fig. 7D). In the five neurons tested, xanthohumol reduced the mEPSC frequency (P < 0.01; Fig. 7C), without changing the amplitude (P = 0.87; Fig. 7E). Thus, these data indicated that xanthohumol acted presynaptically to inhibit glutamate release without changing the postsynaptic sensitivity to glutamate, which supported our synaptosomal findings.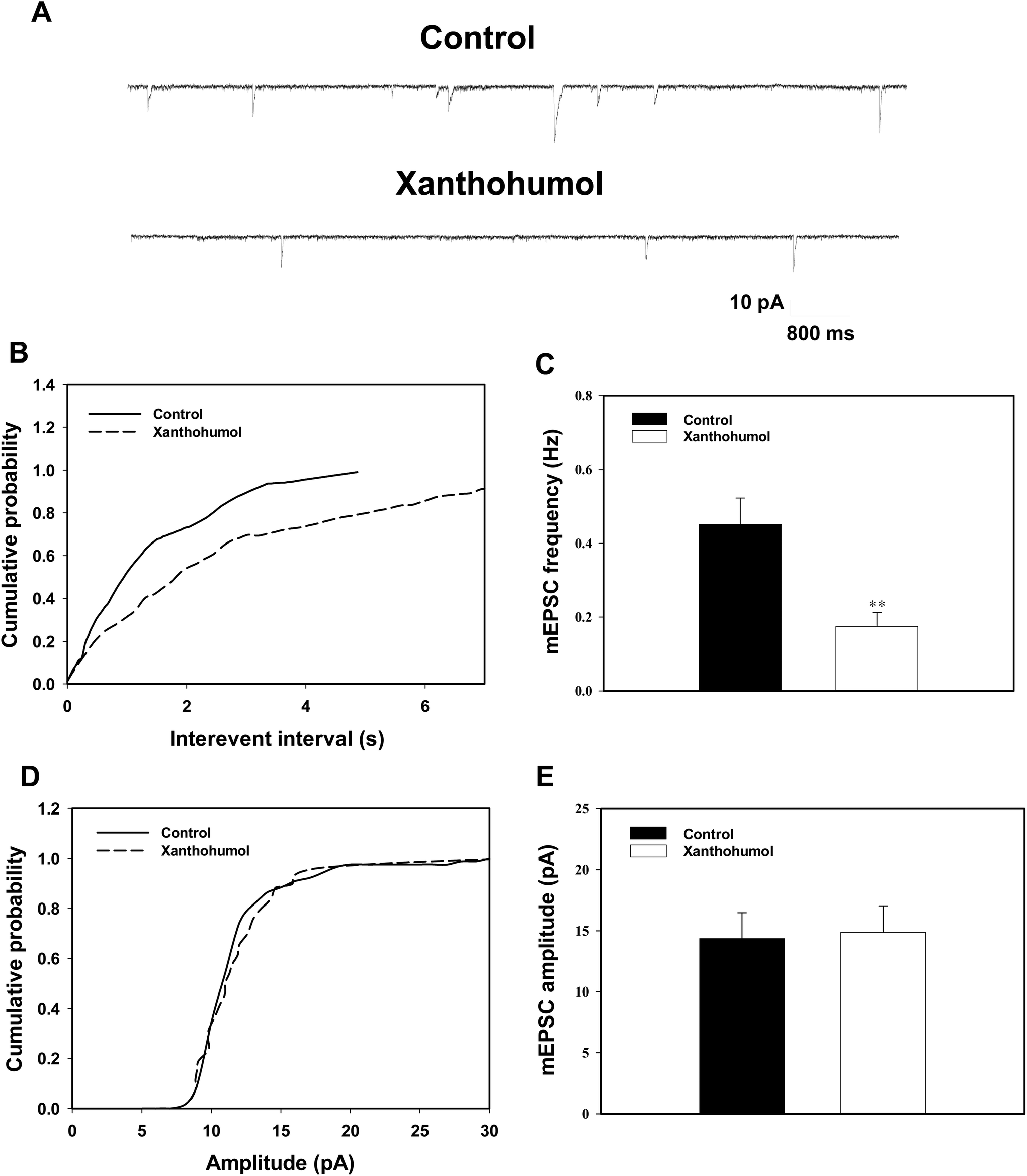 | ||
| Fig. 7 Xanthohumol decreases the frequency but not the amplitude of mEPSCs in hippocampal slices. (A) Sample traces of mEPSCs before (control, in the presence of 1 μM TTX) and after application of 10 μM xanthohumol. (B) Cumulative interevent interval distribution of mEPSCs before and after the application of xanthohumol. (D) Cumulative probability plot of mEPSC amplitude before and after the application of xanthohumol. (C and E) The average amplitude and frequency of mEPSCs in the control and xanthohumol treated groups. Results are the mean ± SEM of 6 independent experiments. **, P < 0.01 vs. the control group. | ||
Discussion
The health benefits of dietary flavonoids, particularly regarding their effects on improving brain dysfunction, have attracted increasing research interest. Although xanthohumol has been demonstrated as a neuroprotective agent,17–20 the underlying mechanisms in its neuroprotective role remain ambiguous. Considering that several neuroprotectants work by stabilizing glutamate release when its synaptic level becomes too high, a feature now considered part of the pathophysiology of numerous neurological disorders,1,5,6 we investigated the effect of xanthohumol on glutamate release. Our results from biochemical and electrophysiological studies in the rat hippocampal nerve terminals (synaptosomes) and slices revealed that xanthohumol at 10 μM decreases glutamate release and that this action involves a presynaptic mechanism. The use of synaptosomes, devoid of any functional postsynaptic element,40 confirms and emphasizes the presynaptic locus for xanthohumol action. Furthermore, we observed a clear decrease in the frequency and not the amplitude of mEPSCs in slice preparations, suggestive of a reduction in the release probability and definitively a presynaptic component in synaptic transmission and regulation.38,39 To our knowledge, no study has investigated the influence of xanthohumol on neuronal glutamate release in the CNS, thus making this report the first to present these findings.Glutamate exocytosis is a complex process involving the modulation of Na+, K+, and Ca2+ channels. The inhibition of Na+ channels and the activation of K+ channels result in presynaptic inhibition because of the stabilization of membrane excitability, which causes a subsequent decrease in the voltage-dependent Ca2+ entry and a consequent reduction in glutamate release.28,30 Therefore, a possible mechanism whereby xanthohumol inhibits glutamate release from synaptosomes entails a reduction in the synaptosomal excitability caused by the modulation of Na+ or K+ ion channels. However, this possibility was excluded because of two observations. First, xanthohumol inhibited the release of glutamate induced by 4-AP and KCl. This indicates that Na+ channels are not involved in the effect of xanthohumol on glutamate release, because the 4-AP-evoked glutamate release involves the action of Na+ and Ca2+ channels, whereas the 15 mM external KCl-evoked release involves only Ca2+ channels.28,31 Second, no considerable effect of xanthohumol on the synaptosomal plasma membrane potential appeared either under the resting conditions or on depolarization with 4-AP, indicating that xanthohumol does not affect K+ conductance. This observation is inconsistent with that of a previous electrophysiological study in which xanthohumol inhibited K+ channels in human leukemic Jurkat T cells.41 The inconsistency in these findings, although ambiguous, is attributable to the diverse experimental models.
If it is not the modulation of synaptosomal excitability, the locus of action of xanthohumol might lie downstream in the stimulus–exocytosis coupling cascade. By using fura-2, we have demonstrated here that a decrease in intrasynaptosomal Ca2+ levels is necessary for the observed xanthohumol-mediated decrease of glutamate release. In addition, we observed that xanthohumol failed to inhibit 4-AP-evoked glutamate release in the absence of extracellular Ca2+. Furthermore, the inhibitory effect of xanthohumol on 4-AP-evoked glutamate release was largely eliminated when synaptosomes were incubated with the Cav2.2 (N-type) and Cav2.1 (P/Q-type) channel blocker ω-conotoxin MVIIC. Dantrolene, an inhibitor of intracellular Ca2+ release from the endoplasmic reticulum ryanodine receptors, and CGP37157, a mitochondrial Na+/Ca2+ exchange blocker, had no effect on the action of xanthohumol. Therefore, these results demonstrate that a decrease in intrasynaptosomal Ca2+ levels involving Cav2.2 and Cav2.1 channel suppression is associated with the xanthohumol-mediated decrease of the 4-AP-evoked glutamate release observed, whereas a reduction of intracellular store Ca2+ release seems not to be involved. However, the blockade of Cav2.2 and Cav2.1 channel activity did not completely prevent the inhibitory effect of xanthohumol on the 4-AP-evoked glutamate release (∼12% of the activity remained), raising the possibility that the Cav2.2- and Cav2.1-resistant Ca2+ channel types or other presynaptic pathways are also involved in the action of xanthohumol.
In synaptic terminals, an increase in cytosolic Ca2+ either through a depolarization-dependent activation of Ca2+ channels or by direct receptor-mediated conduction affects a Ca2+/calmodulin-dependent activation of AC, including AC1 and AC8.42,43 The activation of these ACs increases cyclic adenosine monophosphate (cAMP) levels and activates PKA, which facilitates glutamate release.35,44,45 Thus, the inhibitory effect of xanthohumol on the intrasynaptosomal Ca2+ levels observed here may cause a reduction of the Ca2+-calmodulin/AC/cAMP/PKA cascade, thereby reducing glutamate release. Consistent with this hypothesis, we discovered that the inhibitory effect of xanthohumol on 4-AP-evoked glutamate release was prevented by the PKA inhibitor H89 and the calmodulin antagonists W7 or CMZ. Moreover, the inhibition of G-protein by NEM failed to affect the xanthohumol-mediated inhibition of 4-AP-evoked glutamate release, indicating that G-protein is not involved. However, suppression of PKA inhibiting the 4-AP-evoked glutamate release by xanthohumol remains to be investigated. PKA is present at the presynaptic level and phosphorylates numerous synaptic proteins, subsequently increasing glutamate release.46,47 Therefore, decreasing PKA-dependent phosphorylation of synaptic proteins to control glutamate release should be considered.
Another noteworthy finding of this study is that presynaptic GABAA receptors are involved in the xanthohumol-mediated inhibition of glutamate release from synaptosomes. This finding is based on the following results: (1) we observed that GABAA receptors were coexpressed with the presynaptic marker synaptophysin, confirming that GABAA receptors are present in the hippocampal synaptosomes, and (2) the inhibitory effect of xanthohumol on the 4-AP-evoked glutamate release was prevented by the GABAA receptor agonist and antagonist. Our finding, although discordant with that of a previous study,48 is consistent with experimental evidence supporting the partial agonist or antagonist activity of xanthohumol at GABAA receptors.37 GABAA receptors are present in the brain presynaptically and postsynaptically.49,50 At the presynaptic level, the activation of GABAA receptors has been shown to inhibit VDCCs and glutamate release.51 Therefore, we hypothesized that xanthohumol may act at the GABAA receptors present in the hippocampal nerve terminals to decrease the Ca2+ influx through N- and P/Q-type Ca2+ channels, which subsequently suppresses the Ca2+-calmodulin/AC/cAMP/PKA cascade to cause a decrease in the evoked glutamate release (Fig. 8). Nevertheless, the existence of a direct interaction between xanthohumol and presynaptic GABAA receptors remains to be explored. In fact, numerous neurotransmitter receptors, such as kainate receptors and Group II metabotropic glutamate receptors, are present at the presynaptic level, and their activation inhibits glutamate release through the suppression of the G-protein-coupled adenylate cyclase/protein kinase A pathway.52,53 In our study, these two presynaptic receptors are unlikely to underlie the modulation seen, given that the xanthohumol-mediated inhibition of glutamate release is not mediated by the activation of a G protein.
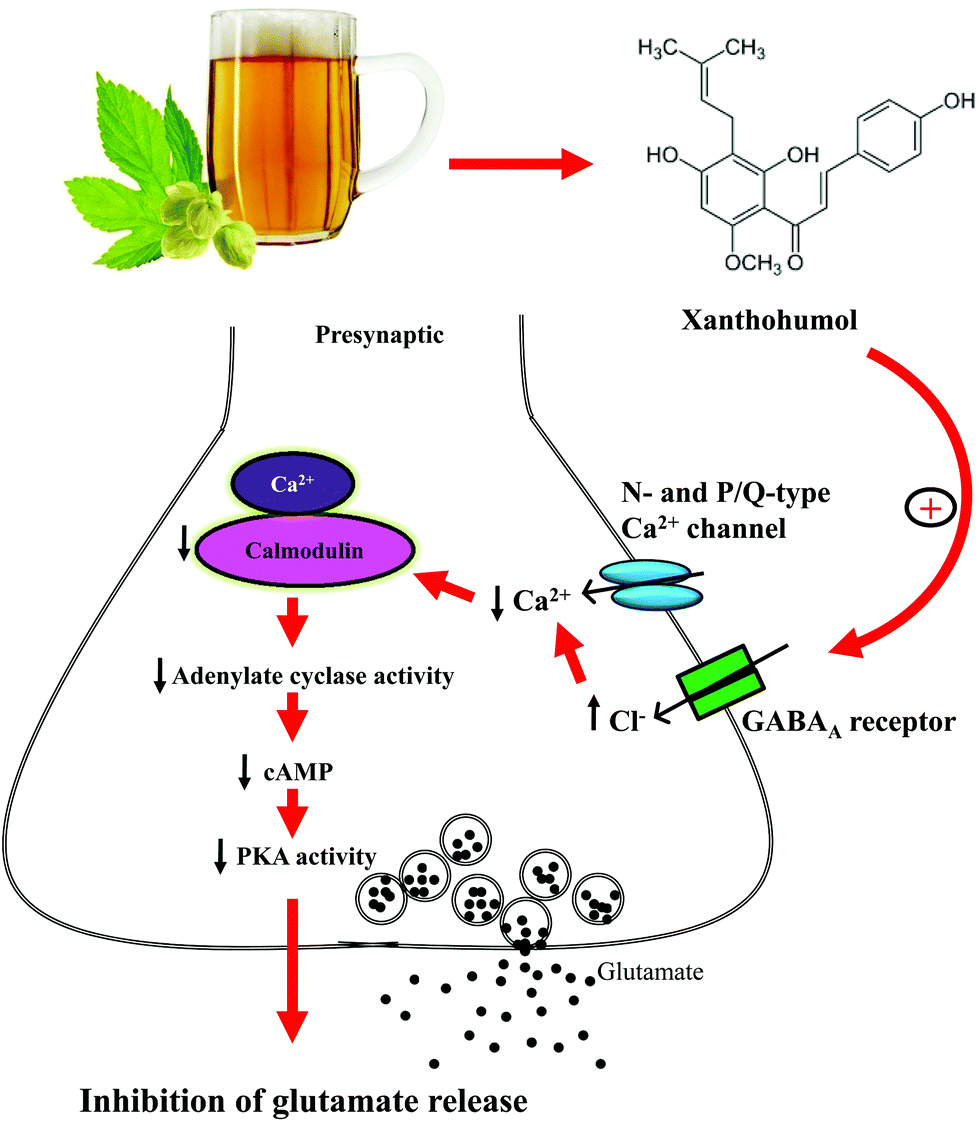 | ||
| Fig. 8 Potential mechanisms by which xanthohumol inhibits glutamate release. Xanthohumol may act at GABAA receptors present in hippocampal nerve terminals to decrease the Ca2+ influx through N- and P/Q-type Ca2+ channels, which subsequently reduces the Ca2+-calmodulin/AC/cAMP/PKA cascade to cause a decrease in glutamate release. | ||
Conversely, studies have confirmed the safety of xanthohumol usage. Oral administration of xanthohumol (700–1000 mg per kg per day) to mice for 3–4 weeks had no adverse effects on major organ functions.11,54 Furthermore, a recent study in healthy volunteers revealed that oral administration of a single xanthohumol dose (160 mg kg−1) is nontoxic.55 These results show that the dose of xanthohumol (10 μM) used to inhibit glutamate release in our study may be considered safe, although the concentration of xanthohumol we used was higher than that used in a previous study (0.5 μM).17
In summary, we conclude that xanthohumol, the dietary flavonoid in hops and beer, possesses glutamate release-inhibiting properties in the hippocampus. Considering glutamate excitotoxicity in the development of many brain diseases,1 our finding is of particular interest because it provides new insights into the mechanism of xanthohumol action in the brain.
Abbreviations
| 4-AP | 4-Aminopyridine |
| [Ca2+]C | Cytosolic free Ca2+ concentration |
| CNS | Central nervous system |
| DiSC3(5) | 3′,3′,3′-Dipropylthiadicarbocyanine iodide |
| VDCCs | Voltage-dependent Ca2+ channels |
| Cav2.2 and Cav2.1 channels | Cav2.2 (N-type) and Cav2.1 (P/Q-type) channels |
| DL-TBOA | DL-threo-β-Benzyloxy aspartate |
| Fura-2-AM | Fura-2-acetoxymethylester |
| GDH | Glutamate dehydrogenase |
| HBM | HEPES buffer medium |
| BSA | Bovine serum albumin |
| EGTA | Ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′acid |
| H89 | N-[2-(p-Bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide |
| SDS | Sodium dodecyl sulfate |
| CGP37157 | 7-Chloro-5-(2-chlorophenyl)-1,5-dihydro-4,1-benzothiazepin-2(3H)-one |
| ω-CgTX MVIIC | ω-Conotoxin MVIIC |
| MEK | Mitogen-activated/extracellular signal-regulated kinase |
| PKA | Protein kinase A |
| PKC | Protein kinase C |
| PD98059 | 2-(2-Amino-3-methoxyphenyl)-4H-1-benzopyran-4-one) |
| GF109203X | Bisindolylmaleimide I |
| TBS | Tris-buffered saline |
| AC | Adenylate cyclase |
| W7 | N-(6-Aminohexyl)-5-chloro-1-naphthalene-sulphonamide |
| CMZ | Calmidazolium |
| NEM | N-Ethylmaleimide |
| mEPSC | Miniature excitatory postsynaptic currents |
| TTX | Tetrodotoxin |
| NMDA | N-Methyl-D-aspartate |
Funding
This work was supported by grants from the Shin Kong Wu Ho-Su Memorial Hospital (103-SKH-FJU-01) and the Ministry of Science and Technology (MOST 103-2320-B-030-001 MY3).Conflict of interest
The authors declare no competing financial interest.References
- B. S. Meldrum, Glutamate as a neurotransmitter in the brain: review of physiology and pathology, J. Nutr., 2000, 130, 1007S–1015S CAS.
- G. Riedel, B. Platt and J. Micheau, Glutamate receptor function in learning and memory, Behav. Brain Res., 2003, 140, 1–47 CrossRef CAS PubMed.
- R. Sattler and M. Tymianski, Molecular mechanisms of glutamate receptor-mediated excitotoxic neuronal cell death, Mol. Neurobiol., 2001, 24, 107–129 CrossRef CAS PubMed.
- A. Lau and M. Tymianski, Glutamate receptors, neurotoxicity and neurodegeneration, Pflugers Arch., 2010, 460, 525–542 CrossRef CAS PubMed.
- T. P. Obrenovitch and J. Urenjak, Altered glutamatergic transmission in neurological disorders: from high extracellular glutamate to excessive synaptic efficacy, Prog. Neurobiol., 1997, 51, 39–87 CrossRef CAS PubMed.
- L. Raiteri, S. Stigliani, S. Zappettini, N. B. Mercuri, M. Raiteri and G. Bonanno, Excessive and precocious glutamate release in a mouse model of amyotrophic lateral sclerosis, Neuropharmacology, 2004, 46, 782–792 CrossRef CAS PubMed.
- T. Y. Lin, C. W. Lu, C. C. Chang, S. K. Huang and S. J. Wang, Luteolin inhibits the release of glutamate in rat cerebrocortical nerve terminals, J. Agric. Food Chem., 2011, 59, 8458–8466 CrossRef CAS PubMed.
- S. L. Hwang, P. H. Shih and G. C. Yen, Neuroprotective effects of citrus flavonoids, J. Agric. Food Chem., 2012, 60, 877–885 CrossRef CAS PubMed.
- N. Solanki, I. Alkadhi, F. Atrooz, G. Patki and S. Salim, Grape powder prevents cognitive, behavioral, and biochemical impairments in a rat model of posttraumatic stress disorder, Nutr. Res., 2015, 35, 65–75 CrossRef CAS PubMed.
- J. F. Stevens and J. E. Page, Xanthohumol and related prenylflavonoids from hops and beer: to your good health!, Phytochemistry, 2004, 65, 1317–1330 CrossRef CAS PubMed.
- C. Dorn, F. Bataille, E. Gaebele, J. Heilmann and C. Hellerbrand, Xanthohumol feeding does not impair organ function and homoeostasis in mice, Food Chem. Toxicol., 2010, 48, 1890–1897 CrossRef CAS PubMed.
- C. L. Miranda, Y. H. Yang, M. C. Henderson, J. F. Stevens, G. Santana-Rios, M. L. Deinzer and D. R. Buhler, Prenylflavonoids from hops inhibit the metabolic activation of the carcinogenic heterocyclic amine 2-amino-3-methylimidazo[4, 5-f]quinoline, mediated by cDNA-expressed human CYP1A2, Drug Metab. Dispos., 2000, 28, 1297–1302 CAS.
- E. Lupinacci, J. Meijerink, J. P. Vincken, B. Gabriele, H. Gruppen and R. F. Witkamp, Xanthohumol from hop (Humulus lupulus L.) is an efficient inhibitor of monocyte chemoattractant protein-1 and tumor necrosis factor-alpha release in LPS-stimulated RAW 264.7 mouse macrophages and U937 human monocytes, J. Agric. Food Chem., 2009, 57, 7274–7281 CrossRef CAS PubMed.
- P. J. Magalhães, D. O. Carvalho, J. M. Cruz, L. F. Guido and A. A. Barros, Fundamentals and health benefits of xanthohumol, a natural product derived from hops and beer, Nat. Prod. Commun., 2009, 4, 591–610 Search PubMed.
- Y. M. Lee, K. H. Hsieh, W. J. Lu, H. C. Chou, D. S. Chou, L. M. Lien, J. R. Sheu and K. H. Lin, Xanthohumol, a prenylated flavonoid from hops (Humulus lupulus), prevents platelet activation in human platelets, Evid. Based Complement. Alternat. Med., 2012, 2012, 852362 Search PubMed.
- M. Liu, P. E. Hansen, G. Wang, L. Qiu, J. Dong, H. Yin, Z. Qian, M. Yang and J. Miao, Pharmacological profile of xanthohumol, a prenylated flavonoid from hops (Humulus lupulus), Molecules, 2015, 20, 754–779 CrossRef PubMed.
- J. Yao, B. Zhang, C. Ge, S. Peng and J. Fang, Xanthohumol, a polyphenol chalcone present in hops, activating Nrf2 enzymes to confer protection against oxidative damage in PC12 cells, J. Agric. Food Chem., 2015, 63, 1521–1531 CrossRef CAS PubMed.
- T. L. Yen, C. K. Hsu, W. J. Lu, C. Y. Hsieh, G. Hsiao, D. S. Chou, G. J. Wu and J. R. Sheu, Neuroprotective effects of xanthohumol, a prenylated flavonoid from hops (Humulus lupulus), in ischemic stroke of rats, J. Agric. Food Chem., 2012, 60, 1937–1944 CrossRef CAS PubMed.
- E. Oberbauer, C. Urmann, C. Steffenhagen, L. Bieler, D. Brunner, T. Furtner, C. Humpel, B. Bäumer, C. Bandtlow, S. Couillard-Despres, F. J. Rivera, H. Riepl and L. Aigner, Chroman-like cyclic prenylflavonoids promote neuronal differentiation and neurite outgrowth and are neuroprotective, J. Nutr. Biochem., 2013, 24, 1953–1962 CrossRef CAS PubMed.
- D. R. Zamzow, V. Elias, L. L. Legette, J. Choi, J. F. Stevens and K. R. Magnusson, Xanthohumol improved cognitive flexibility in young mice, Behav. Brain Res., 2014, 275, 1–10 CrossRef CAS PubMed.
- A. Bouron, Modulation of spontaneous quantal release of neurotransmitters in the hippocampus, Prog. Neurobiol., 2001, 63, 613–635 CrossRef CAS PubMed.
- D. G. Nicholls and T. S. Sihra, Synaptosomes possess an exocytotic pool of glutamate, Nature, 1986, 321, 772–773 CrossRef CAS PubMed.
- T. Y. Lin, C. W. Lu and S. J. Wang, Astaxanthin inhibits glutamate release in rat cerebral cortex nerve terminals via suppression of voltage-dependent Ca2+ entry and mitogen-activated protein kinase signaling pathway, J. Agric. Food Chem., 2010, 58, 8271–8278 CrossRef CAS PubMed.
- D. G. Nicholls, The glutamatergic nerve terminal, Eur. J. Biochem., 1993, 212, 613–631 CrossRef CAS PubMed.
- T. S. Sihra, E. Bogonez and D. G. Nicholls, Localized Ca2+ entry preferentially effects protein dephosphorylation, phosphorylation, and glutamate release, J. Biol. Chem., 1992, 267, 1983–1989 CAS.
- G. Grynkiewicz, M. Poenie and R. Y. Tsien, A new generation of Ca2+ indicators with greatly improved fluorescence properties, J. Biol. Chem., 1985, 260, 3440–3450 CAS.
- A. Rodríguez-Moreno and T. S. Sihra, Presynaptic kainate receptor facilitation of glutamate release involves protein kinase A in the rat hippocampus, J. Physiol., 2004, 557, 733–745 CrossRef PubMed.
- D. G. Nicholls, Presynaptic modulation of glutamate release, Prog. Brain Res., 1998, 116, 15–22 CAS.
- D. G. Nicholls, T. S. Sihra and J. Sanchez-Prieto, Calcium-dependent and -independent release of glutamate from synaptosomes monitored by continuous fluorometry, J. Neurochem., 1987, 49, 50–57 CrossRef CAS PubMed.
- L. Wu and P. Saggau, Presynaptic inhibition of elicited neurotransmitter release, Trends Neurosci., 1997, 20, 204–212 CrossRef CAS PubMed.
- A. P. Barrie, D. G. Nicholls, J. Sanchez-Prieto and T. S. Sihra, An ion channel locus for the protein kinase C potentiation of transmitter glutamate release from guinea pig cerebrocortical synaptosomes, J. Neurochem., 1991, 57, 1398–1404 CrossRef CAS PubMed.
- E. Vazquez and J. Sanchez-Prieto, Presynaptic modulation of glutamate release targets different calcium channels in rat cerebrocortical nerve terminals, Eur. J. Neurosci., 1997, 9, 2009–2018 CrossRef CAS PubMed.
- C. Millan and J. Sanchez-Prieto, Differential coupling of N and P/Q-type calcium channels to glutamate exocytosis in the rat cerebral cortex, Neurosci. Lett., 2002, 330, 29–32 CrossRef CAS PubMed.
- M. J. Berridge, Neuronal calcium signaling, Neuron, 1998, 21, 13–26 CrossRef CAS PubMed.
- Y. Andrade-Talavera, P. Duque-Feria, J. V. Negrete-Díaz, T. S. Sihra, G. Flores and A. Rodríguez-Moreno, Presynaptic kainate receptor-mediated facilitation of glutamate release involves Ca2+-calmodulin at mossy fiber-CA3 synapses, J. Neurochem., 2012, 122, 891–899 CrossRef CAS PubMed.
- H. Aoshima, K. Takeda, Y. Okita, S. J. Hossain, H. Koda and Y. Kiso, Effects of beer and hop on ionotropic gamma-aminobutyric acid receptors, J. Agric. Food Chem., 2006, 54, 2514–2519 CrossRef CAS PubMed.
- O. Meissner and H. Häberlein, Influence of xanthohumol on the binding behavior of GABAA receptors and their lateral mobility at hippocampal neurons, Planta Med., 2006, 72, 656–658 CrossRef CAS PubMed.
- A. Malgaroli and R. W. Tsien, Glutamate-induced long-term potentiation of the frequency of miniature synaptic currents in cultured hippocampal neurons, Nature, 1992, 357, 134–139 CrossRef CAS PubMed.
- T. Manabe, P. Renner and R. A. Nicoll, Postsynaptic contribution to long-term potentiation revealed by the analysis of miniature synaptic currents, Nature, 1992, 355, 50–55 CrossRef CAS PubMed.
- P. R. Dunkley, P. E. Jarvie, J. W. Heath, G. J. Kidd and J. A. Rostas, A rapid method for isolation of synaptosomes on Percoll gradients, Brain Res., 1986, 372, 115–129 CrossRef CAS PubMed.
- J. Gąsiorowska, A. Teisseyre, A. Uryga and K. Michalak, Inhibition of Kv1.3 Channels in Human Jurkat T Cells by Xanthohumol and Isoxanthohumol, J. Membr. Biol., 2015, 1–7 Search PubMed.
- M. G. Weisskopf, P. E. Castillo, R. A. Zalutsky and R. A. Nicoll, Mediation of hippocampal mossy fiber long-term potentiation by cyclic AMP, Science, 1994, 265, 1878–1882 CAS.
- D. M. Cooper, Molecular and cellular requirements for the regulation of adenylate cyclases by calcium, Biochem. Soc. Trans., 2003, 31, 912–915 CrossRef CAS PubMed.
- A. Rodríguez-Moreno and T. S. Sihra, Presynaptic kainate receptor-mediated facilitation of glutamate release involves Ca2+-calmodulin and PKA in cerebrocortical synaptosomes, FEBS Lett., 2013, 587, 788–792 CrossRef PubMed.
- Y. Andrade-Talavera, P. Duque-Feria, T. S. Sihra and A. Rodríguez-Moreno, Pre-synaptic kainate receptor-mediated facilitation of glutamate release involves PKA and Ca2+-calmodulin at thalamocortical synapses, J. Neurochem., 2013, 126, 565–578 CrossRef CAS PubMed.
- E. M. Fykse, C. Li and T. C. Südhof, Phosphorylation of rabphilin-3A by Ca2+/calmodulin- and cAMP-dependent protein kinases in vitro, J. Neurosci., 1995, 15, 2385–2395 CAS.
- C. Risinger and M. K. Bennett, Differential phosphorylation of syntaxin and synaptosome-associated protein of 25 kDa (SNAP-25) isoforms, J. Neurochem., 1999, 72, 614–624 CrossRef CAS PubMed.
- T. E. Ceremuga, L. A. Johnson, J. M. Adams-Henderson, S. McCall and D. Johnson, Investigation of the anxiolytic effects of xanthohumol, a component of humulus lupulus (Hops), in the male Sprague-Dawley rat, J. Am. Assoc. Nurse Anesth., 2013, 81, 193–198 Search PubMed.
- D. M. Kullmann, A. Ruiz, D. M. Rusakov, R. Scott, A. Semyanov and M. C. Walker, Presynaptic, extrasynaptic and axonal GABAA receptors in the CNS: where and why?, Prog. Biophys. Mol. Biol., 2005, 87, 33–46 CrossRef CAS PubMed.
- W. Sieghart, Structure, pharmacology, and function of GABAA receptor subtypes, Adv. Pharmacol., 2006, 54, 231–263 CAS.
- P. Long, A. Mercer, R. Begum, G. J. Stephens, T. S. Sihra and J. N. Jovanovic, Nerve terminal GABAA receptors activate Ca2+/calmodulin-dependent signaling to inhibit voltage-gated Ca2+ influx and glutamate release, J. Biol. Chem., 2009, 284, 8726–8737 CrossRef CAS PubMed.
- J. V. Negrete-Díaz, T. S. Sihra, J. M. Delgado-García and A. Rodríguez-Moreno, Kainate receptor-mediated inhibition of glutamate release involves protein kinase A in the mouse hippocampus, J. Neurophysiol., 2006, 96, 1829–1837 CrossRef PubMed.
- J. V. Negrete-Díaz, T. S. Sihra, J. M. Delgado-García and A. Rodríguez-Moreno, Kainate receptor-mediated presynaptic inhibition converges with presynaptic inhibition mediated by Group II mGluRs and long-term depression at the hippocampal mossy fiber-CA3 synapse, J. Neural Transm., 2007, 114, 1425–1431 CrossRef PubMed.
- B. W. Vanhoecke, F. Delporte, E. Van Braeckel, A. Heyerick, H. T. Depypere, M. Nuytinck, D. De Keukeleire and M. E. Bracke, A safety study of oral tangeretin and xanthohumol administration to laboratory mice, In Vivo, 2005, 19, 103–107 CAS.
- L. Legette, C. Karnpracha, R. L. Reed, J. Choi, G. Bobe, J. M. Christensen, R. Rodriguez-Proteau, J. Q. Purnell and J. F. Stevens, Human pharmacokinetics of xanthohumol, an antihyperglycemic flavonoid from hops, Mol. Nutr. Food Res., 2014, 58, 248–255 CAS.
| This journal is © The Royal Society of Chemistry 2016 |