
In vitro chemo-preventive activities of hydroxytyrosol: the main phenolic compound present in extra-virgin olive oil
Patrizia
Rosignoli
*,
Raffaela
Fuccelli
,
Maria Vittoria
Sepporta
and
Roberto
Fabiani
Department of Chemistry, Biology and Biotechnologies, University of Perugia, Italy. E-mail: patrizia.rosignoli@unipg.it; Fax: +39 075 585 7328; Tel: +39 075 585 7336
First published on 5th October 2015
Abstract
The co-incubation in the culture medium with hydroxytyrosol [3,4-dihydroxyphenyl ethanol (3,4-DHPEA)], the main phenolic compound present in extra-virgin olive oil, and H2O2 reduces the oxidative DNA damage in peripheral blood mononuclear cells (PBMC). In this study we investigate, by the comet assay, the ability of 3,4-DHPEA to inhibit the H2O2 induced DNA damage when pre-incubated with PBMC and then removed before the exposure of cells to H2O2. Low doses of 3,4-DHPEA (10–100 μM) pre-incubated for 30 min with PBMC reduced the DNA damage induced by the treatment with H2O2 200 μM for 5 min at 4 °C. Prolonging the exposure time up to 6 h completely prevented the DNA damage. Furthermore we extensively analysed, by the MTT assay, the anti-proliferative activities of 3,4-DHPEA on breast (MDA and MCF-7), prostate (LNCap and PC3) and colon (SW480 and HCT116) cancer cell lines and correlated these effects with the H2O2 accumulation. The concentration of H2O2 in the culture medium was measured by the ferrous ion oxidation–xylenol orange method. The proliferation of all the cell lines was inhibited but at different levels: the prostate cancer cells were more resistant to the growth inhibition with respect to breast and colon cancer cells. The ability of the different cell lines to remove H2O2 from the culture medium was inversely correlated with their sensitivity to the anti-proliferative effect of 3,4-DHPEA. Therefore, 3,4-DHPEA may act as a chemopreventive agent acting on both initiation and promotion/progression phases of carcinogenesis.
1. Introduction
Many observational and clinical epidemiological studies have shown an inverse correlation between the consumption of olive oil and chronic-degenerative disease risk.1–3 Among the substances found in olive oil, phenols may be responsible for its preventive effects, in particular for the anti-tumor activity. Olive oil phenols constitute a class of very heterogeneous compounds which includes hydroxytyrosol (3,4-DHPEA) and its secoiridoid derivatives.4 The cancer preventive activity of olive oil phenolic compounds may act on both initiation and promotion/progression phases. One of the possible anti-initiation mechanisms is represented by the antioxidant ability of these compounds. Reactive oxygen species react with DNA and lead to mutations and cancer initiation.5In vitro induction of oxidative DNA damage by hydrogen peroxide is a widely validated experimental model for the study of the anti-genotoxic effects of various natural and/or synthetic antioxidant substances. The ability of 3,4-DHPEA to decrease the oxidative DNA damage induced by H2O2 has been reported in different human cells: chondrocytes,6 breast and prostate tumour cells,7,8 blood monocytes and neuroblastoma cell lines9 and Jurkat cells.10 This antigenotoxic ability of 3,4-DHPEA has been previously investigated also in our laboratories. We have demonstrated that this phenol, when co-incubated with H2O2, significantly decreased the oxidative damage at concentrations as low as 1 μM in human peripheral blood mononuclear cells (PBMC) and in human promyelocytic leukaemia cells (HL60).11 It should be noted that most of the above reported studies have been performed co-incubating the cells with 3,4-DHPEA and H2O2. On the other hand, much less is known about the ability of this phenol to prevent the oxidative DNA damage if removed from the culture medium before the H2O2 treatment.One of the possible anti-promotion/progression mechanisms by which olive oil phenols inhibit the carcinogenic process is represented by their ability to reduce the proliferation of cancer cells. Indeed, the anti-proliferative activity of 3,4-DHPEA has been widely investigated in several tumour cell types.12–16 However, the results obtained on tumour cells derived from different organs disagree with one other.14,17–21 We have recently investigated the anti-proliferative action of 3,4-DHPEA in different tumour cell lines and we found that the medium used for cell culture was responsible for the different sensitivities observed by various authors in different cell lines. In fact, 3,4-DHPEA induced accumulation of different amounts of H2O2 in the culture medium depending on the type of medium used and also on the ability of different cells to eliminate this peroxide.22,23 Therefore the variability of anti-proliferative results may depend upon both the capability of cells to eliminate H2O2 by specific enzymes such as catalase and glutathione peroxidase, and the effectiveness by which 3,4-DHPEA releases H2O2 in different cell culture media.
Based on these considerations, in this study we have investigated the potential oxidative DNA damage preventive activity of 3,4-DHPEA on PBMC pre-incubation with this compound and then exposure to H2O2. The oxidative DNA damage was measured by the highly sensitive comet assay.24
In addition, the anti-proliferative effects of 3,4-DHPEA on different cell lines and the role played by the accumulation of H2O2 in the culture medium have been deeply investigated.
2. Experimental
2.1 Materials
RPMI 1640 (catalogue no. 21875), D-MEM (catalogue no. 31885), heat-inactivated foetal calf serum (FCS), low melting-point and normal melting-point agarose were from Gibco (Gibco BRL, Life Technologies, Paisley, Scotland). 3,4-DHPEA was obtained from Cayman Chemicals Ltd (USA), supplied as ethanol solution at a concentration of 320 mM and stored at −20 °C in the dark. The phenol compound was diluted in RPMI 1640 medium to the desired concentration just before use. All the solutions were sterilised by filtration on 0.22 μm filters (Celbio S.r.l., Milan, Italy). All other reagents were purchased from Sigma unless differently specified (Sigma-Aldrich CO. Ltd, Irvine, UK). Conventional microscope slides were from Knittel Glaser (Steroglass, Perugia, Italy).2.2 Peripheral blood mononuclear cell isolation and treatment
Peripheral blood mononuclear cells (PBMC) were isolated from leucocyte enriched human peripheral blood on a density gradient.25 Briefly, blood samples (2 ml) diluted to 10 ml with RPMI 1640 without serum were layered over 2 ml of Histopaque 1077 and centrifuged at 1600 rpm for 20 min. The layer containing the mononuclear cells at the interface between the plasma and the Histopaque was recovered and washed twice with RPMI 1640. The viable PBMC obtained were counted by using the trypan-blue exclusion technique and the density was adjusted to 1 × 106 cells per ml with complete RPMI 1640. The cells were exposed to different concentrations of 3,4-DHPEA for 0.5 h, 6 h and 24 h. At the end the cells were centrifuged to 1200 rpm for 6 min, the supernatant was discarded and fresh complete RPMI medium with H2O2 200 μM was added for 5 min at 4 °C. After incubation, the cells were recovered and we evaluated the viability and the DNA damage using the trypan blue exclusion technique and the comet assay, respectively.2.3 Single cell gel electrophoresis (SCGE or “comet assay”)
The single cell gel electrophoresis assay was performed as previously described.26 After treatment, aliquots of the cell suspension (50–100 μl, 0.5–1.0 × 105 cells) were transferred to 1.5 ml Eppendorf tubes and centrifuged at 1200 rpm for 5 min. The supernatant was discarded and the pellet was mixed with 75 μl of low-melting agarose [0.7% in phosphate buffer (PBS)] and distributed onto conventional microscope slides. The slides were precoated with normal-melting agarose (0.5% in PBS) and dried at 50 °C. After the agarose was solidified (4 °C for 5 min), a second layer of low-melting agarose was applied similarly to the first. The slides were then immersed in the lysis solution (2.5 M NaCl, 100 mM Na2EDTA, 10 mM Tris-HCl pH 10, containing freshly added 1% Triton X100 and 10% DMSO) for 1 h at 4 °C and then placed into a horizontal electrophoresis apparatus filled with freshly made buffer (1 mM Na2EDTA. 300 mM NaOH, pH 13.0). After 20 min of preincubation (unwinding of DNA), the electrophoresis was run for 20 min at a fixed voltage of 25 V and 300 mA, which was adjusted by raising or lowering the level of the electrophoresis buffer in the tank. At the end of the electrophoresis, the slides were washed 3 times with the neutralization buffer (Tris-HCl 0.4 M, pH 7.5), stained with 50 μl ethidium bromide (20 mg ml−1), and kept in a moisture chamber in the dark at 4 °C until analysis. All the above reported steps were carried out under red light to prevent any additional DNA damage.2.4 Comet detection and statistical analysis
The cells were analyzed 24 h after staining at 400× magnification using a fluorescence microscope (Zeiss, R.G.) equipped with a 50 W mercury lamp. The microscopy images revealed circular shapes (undamaged DNA) and “comet-like” shapes in which the DNA had migrated from the head to form a tail (damaged DNA). The extension of each comet was analyzed by a computerized image analysis system (comet assay II, Perceptive Instruments, UK) that, among several other parameters, gave the “tail moment” that is considered to be the parameter most directly related to DNA damage. In fact, the tail moment is defined as the product of DNA in the tail and the mean distance of its migration in the tail. Calculation of the extent of DNA damage, which was not homogeneous, was based on the analysis of 100 randomly selected comets from each slide, divided into 5 classes according to the tail moment (t.m.) values as follows: class 0 (t.m. < 1; no damage), class 1 (t.m. 1–5; slightly damaged), class 2 (t.m. 5–10; medium damage), class 3 (t.m. 10–20; highly damaged) and class 4 (t.m. > 20; completely damaged). The overall score expressed in arbitrary units (A.U.) for each slide ranged from 0 (100% of the comets in class 0) to 400 (100% of the comets in class 4).272.5 Adherent cell line culture and treatments
Human breast (MDA and MCF-7), prostate (LNCap and PC3) and colon (SW480 and HCT116) cancer cell lines were obtained from the American Type Culture Collection (Rockville, MD, USA). The cells were cultured in complete medium (D-MEM supplemented with 10% FCS, 2.0 mM L-glutamine, 100 U per mL penicillin and 100 U per mL streptomycin). The cells were maintained at 37 °C and 5% CO2 under a humidified atmosphere and seeded every 4–5 days at a suitable density as suggested by the supplier. The cell lines were seeded on 96-well plates (5000 per well). After 24 h of incubation at 37 °C and 5% CO2, the medium was replaced with a fresh medium containing 3,4-DHPEA at the established concentrations and then proliferation was determined by the MTT assay (24-48-72-96-120-144 h).2.6 Cell proliferation
The proliferation of the cells was measured by the MTT [3-(4,5-dimethylthiazol-2-yl)-3,5-diphenyltetrazolium bromide] assay carried out essentially as previously described.28 This assay measures the production of highly coloured formazan as a result of the reduction of MTT by metabolically active cells due to the dehydrogenase enzymes. After exposure to 3,4-DHPEA, the medium was removed and the cells were incubated at 37 °C with MTT solution (0.5 mg per mL of MTT in Hank's balanced salt solution). After 2 h, the MTT solution was substituted by a lysing solution (100 μl, isopropanol containing 0.1 M HCl), and the plates were incubated for 10 min at 37 °C to dissolve the blue formazan crystals. The absorbance of the released formazan was then determined at 565 nm.2.7 Measurement of H2O2 concentration in the culture medium
The concentration of H2O2 in the culture medium was measured by the ferrous ion oxidation–xylenol orange method29 as follows: the medium (20 μl) was mixed with a reaction solution (200 μl) containing 250 μM ammonium iron(II) sulphate, 25 mM H2SO4, 100 mM sorbitol and 125 μM xylenol orange and incubated at room temperature for 30 min. The absorbance was then read at 595 nm, and the concentration of H2O2 was derived from a standard curve obtained by adding different concentrations of H2O2 in the RPMI medium just before the assay.2.8 Statistical analysis
The results of each experiment carried out on PBMC obtained from different donors, repeated at least 3 times, were compared using the Student's t-test. The experiments carried out on tumour cell lines were run in triplicate for each experimental condition, and each experiment was repeated at least five times; the results are reported as mean ± SD. Significant differences among treatments were assessed using Student's t test.3. Results
3.1 Pre-incubation of PBMC with 3,4-DHPEA reduced oxidative DNA damage
The basal DNA damage of freshly isolated human PBMC (without any treatment) decreased over time from a value of 20.8 UA after 30 min from isolation to 4.8 UA after 24 h. Treatment of cells with increasing concentrations of 3,4-DHPEA did not significantly modify the DNA damage (Fig. 1A–C). The exposure of PBMC to H2O2 (200 μM, 5 min, 4 °C) induced an evident DNA damage which was reduced by the pre-incubation of cells with 3,4-DHPEA. In particular, after 30 min of pre-incubation, excluding the lower concentration (10 μM), all the other doses of 3,4-DHPEA were able to reduce the DNA damage (Fig. 1A). The increment of the pre-incubation time up to 6 h caused a more evident DNA damage prevention by 3,4-DHPEA which was significant also at 10 μM. The maximum effect was reached at 75 μM of 3,4-DHPEA (Fig. 1B). After 24 h of pre-incubation of PBMC with 3,4-DHPEA we observed a weak reduction of DNA damage only at 10 μM while higher concentrations of 3,4-DHPEA failed to protect the DNA from oxidative damage. In addition, at 100 μM 3,4-DHPEA significantly increased the DNA damage over the level induced by H2O2 (Fig. 1C).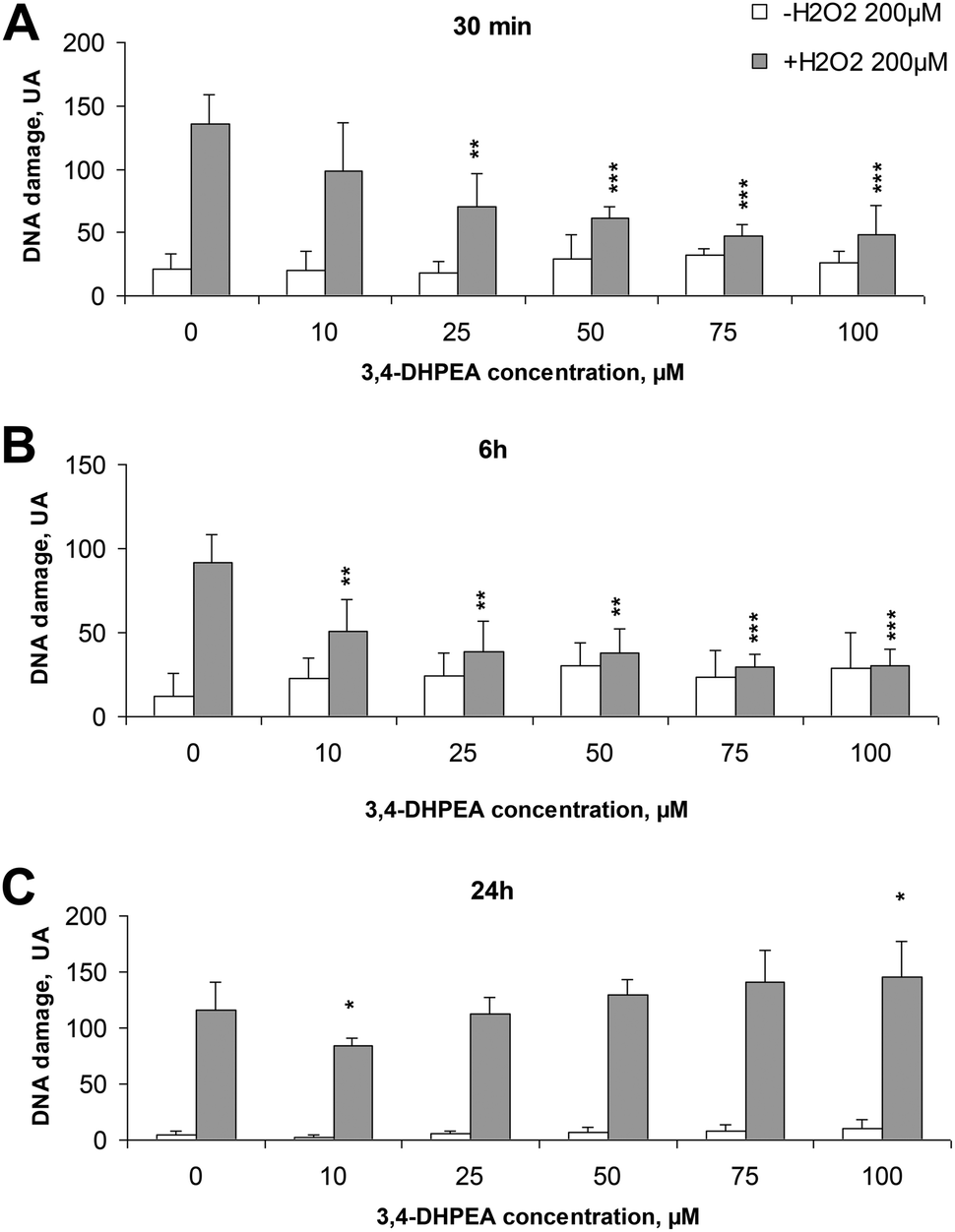 | ||
| Fig. 1 Effect of increasing concentrations of 3,4-DHPEA on DNA damage of PBMC induced by exposure to H2O2 (200 μM) pre-incubated for different times: (A) 0.5 h; (B) 6 h; (C) 24 h. The results are the mean ± SD of the arbitrary units (A.U.) values of 3 experiments performed on different cell preparations. *P < 0.05; **P < 0.01; ***P < 0.001 compared to the control. | ||
3.2 3,4-DHPEA inhibits the proliferation of different human tumour cell lines
The anti-proliferative activity of 3,4-DHPEA at 100 μM on tumour cells derived from breast (MDA and MCF-7), prostate (PC3 and LNCap) and colon (SW480 and HCT116) is shown in Fig. 2. The growth inhibition was evident as soon as 24 h after treatment with 3,4-DHPEA and continued for the subsequent incubation time up to 144 h. The cell lines showed a different response to the olive oil phenol treatment. In particular, the inhibition of proliferation by 3,4-DHPEA on colon and breast cancer cells was more evident in comparison with that observed on prostate cells. To further investigate the different sensitivities of cell lines to 3,4-DHPEA, a dose-dependent effect was measured at a fixed time of 72 h (Fig. 3). The results indicate that treatment of different human cancer cells with 25 μM of 3,4-DHPEA did not significantly affect proliferation while the higher doses induced a dose-dependent reduction of cell growth. In particular, the HCT116, MDA and MCF-7 cells were more sensitive to 3,4-DHPEA than the LNCap, PC3 and SW480 cells. Indeed, at 50 μM the proliferation was inhibited by 38%, 44% and 34% in HCT116, MDA and MCF-7 cells, respectively, while for the other cells the proliferation remained above 80%. The increment of 3,4-DHPEA concentration up to 75 and 100 μM reduced the proliferation in all cell types. Again, the LNCap, PC3 and SW480 were less sensitive to the anti-proliferative activity of 3,4-DHPEA while the most sensitive cell type was the breast cancer cell line MDA (Fig. 3).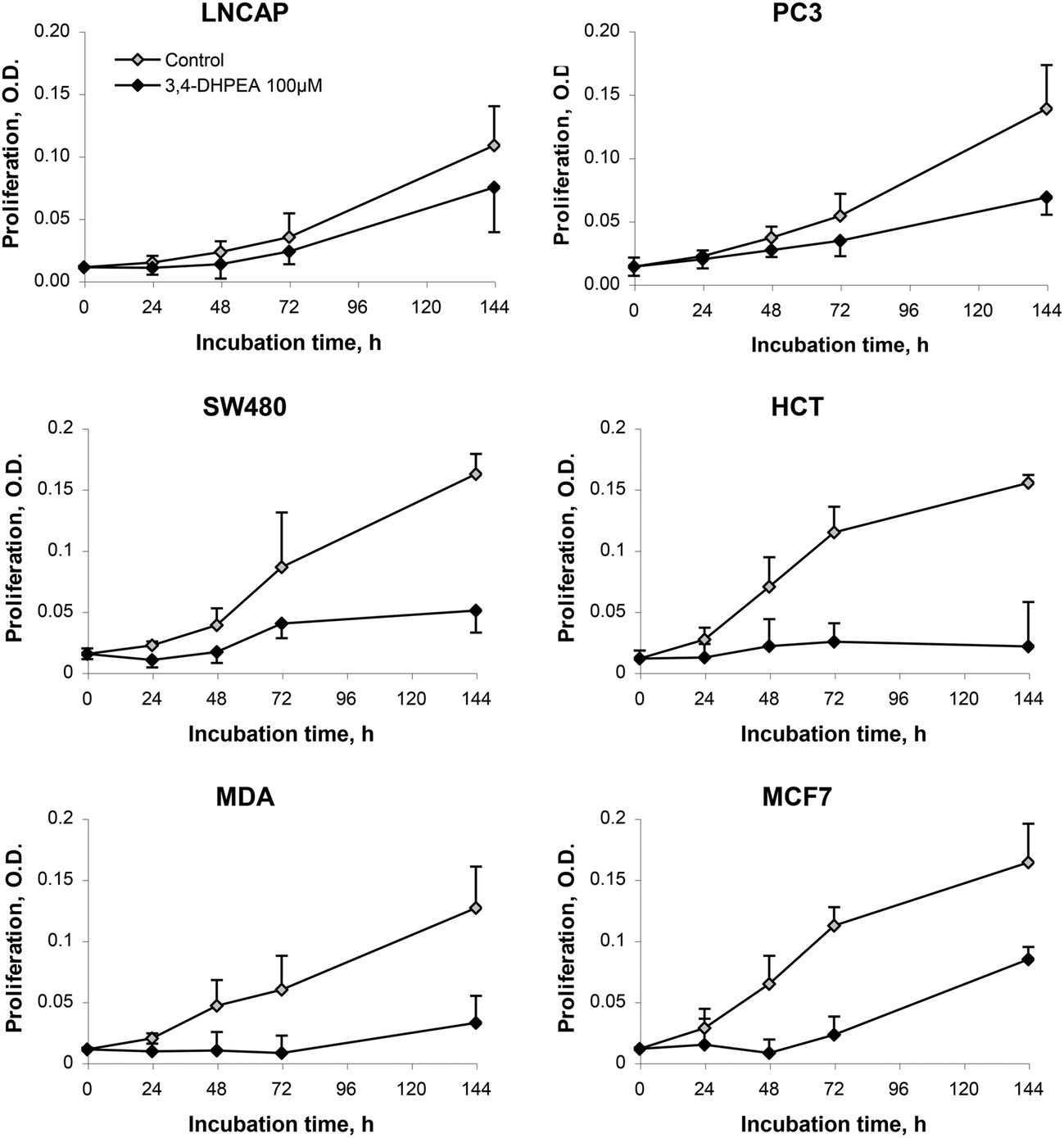 | ||
| Fig. 2 Effect of 3,4-DHPEA (100 μM) on proliferation of different cell lines. The results are the mean ± SD of optical density (O.D.) values of 5 experiments. | ||
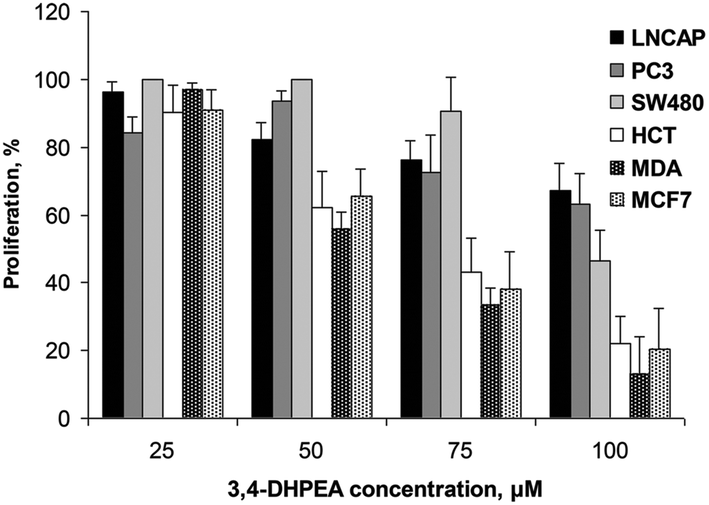 | ||
| Fig. 3 Effect of increasing concentrations of 3,4-DHPEA on proliferation of different cell lines after 72 h of incubation. The results are the mean ± SD of proliferation values of 5 experiments. | ||
3.3 3,4-DHPEA induced H2O2 accumulation in the culture medium
The accumulation of H2O2 in the D-MEM medium was also measured in the presence of different cell lines after 24 h of incubation with increasing concentrations of 3,4-DHPEA (Fig. 4). We observed that the lower concentrations of 3,4-DHPEA (25 and 50 μM) did not affect the level of H2O2 in the medium compared to the control. A slight increment of H2O2 concentration was induced by 75 μM of 3,4-DHPEA which was more evident in the presence of SW480 cells. At 100 μM of 3,4-DHPEA high amounts of H2O2 in the culture medium were detected in the presence of colon and breast cancer cells. Instead, in the presence of prostate cells the concentrations of H2O2 were about 50% reduced with respect to other cells (Fig. 4).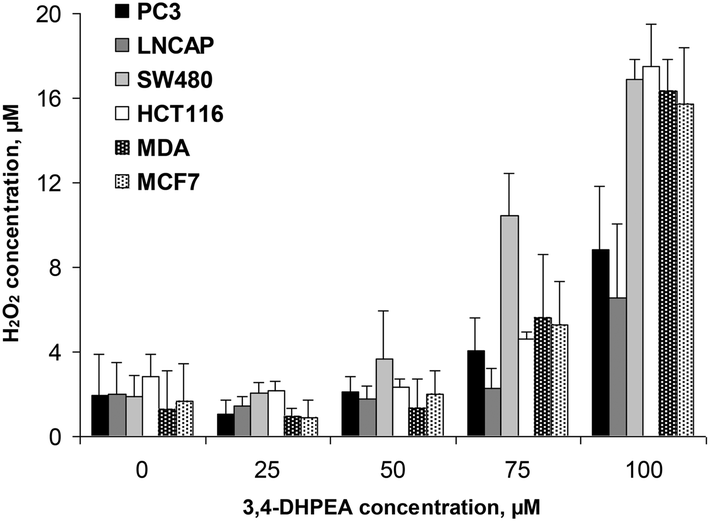 | ||
| Fig. 4 Effect of increasing concentrations of 3,4-DHPEA on hydrogen peroxide (H2O2) accumulation in the culture medium in the presence of different cell lines after 24 h of incubation. The results are the mean ± SD of H2O2 concentration values of 5 experiments. | ||
4. Discussion
In this study we have investigated the effect of the main phenolic compound of olive oil, 3,4-DHPEA, on both the initiation and promotion/progression phases of carcinogenesis. Regarding the preventive effect on initiation, several previous studies have demonstrated the ability of 3,4-DHPEA to reduce the DNA damage induced by H2O2 on various cell types.7,8 However, it should be noted that generally these effects have been observed when the cells were co-incubated with 3,4-DHPEA and H2O2. Therefore, the most plausible explanation for these effects was ascribed to the fact that this compound, acting as a potent scavenger of reactive oxygen species (ROS)30,31 prevents their interaction with DNA. Instead, in the present study we demonstrated that 3,4-DHPEA was able to prevent the H2O2 induced DNA damage in PBMC also when it was removed from the culture medium. These results suggest that 3,4-DHPEA is able to penetrate inside the cells where it may exert its effects. The observation that the increment of incubation time from 0.5 to 6 h increased the DNA preventive effect further supports the hypothesis. Noteworthy is the observation that on prolonging the incubation time up to 24 h the preventive effect was lost at the higher doses while it was maintained at the lower dose (10 μM) of 3,4-DHPEA. This phenomenon may be explained by considering that 3,4-DHPEA, in addition to its antioxidant activity, may also exert a pro-oxidant effect at high doses by inducing the production of H2O2 in the culture medium. This pro-oxidant activity has been firstly described by our research group,22 later confirmed by other investigators7,32 and further demonstrated in the present study. Indeed, we observed a growth inhibitory effect of 3,4-DHPEA on different tumour cell lines only at relatively high doses (75–100 μM) and this effect was related to the amount of H2O2 accumulated in the culture medium. Since the culture medium was the same for all the cell lines, the different amounts of H2O2 found are dependent upon the ability of the cells to metabolise this compound. Therefore we may suggest that the ability of the different cell lines to remove H2O2 from the medium influences their sensitivity to the anti-proliferative effect of 3,4-DHPEA. This conclusion is supported by a previous study where it was shown that the inclusion of catalase, an enzyme able to remove H2O2 from the culture medium, drastically reduced the anti-proliferative effect of 3,4-DHPEA.22 Among the cell lines tested we found that those derived from prostate cancer (LNCap and PC3) were less sensitive to the growth inhibitory effect of 3,4-DHPEA than those derived from breast and colon cancer. Several epidemiological studies have been performed to investigate the relationship between the olive oil consumption and cancer risk in different organs such as prostate, breast and colon. In a meta-analysis it was shown that olive oil consumption was associated with lower odds of developing both breast (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) OR = −0.45, 95% CI −0.78 to −0.12) and digestive system (logOR = −0.36, 95% CI −0.50 to −0.21) cancer.2 Much less is known about the effect of olive oil on prostate cancer. However, it should be underlined that these epidemiological studies have never considered the influence of phenols. In other words, so far no attention has been paid to the chemical composition of different olive oils and to their phenolic content in epidemiological studies. This may be an important task for further studies considering that the presence of phenols in olive oil may be widely varying from a few mg L−1 up to several hundreds mg L−1.33
OR = −0.45, 95% CI −0.78 to −0.12) and digestive system (logOR = −0.36, 95% CI −0.50 to −0.21) cancer.2 Much less is known about the effect of olive oil on prostate cancer. However, it should be underlined that these epidemiological studies have never considered the influence of phenols. In other words, so far no attention has been paid to the chemical composition of different olive oils and to their phenolic content in epidemiological studies. This may be an important task for further studies considering that the presence of phenols in olive oil may be widely varying from a few mg L−1 up to several hundreds mg L−1.33
5. Conclusions
The cancer preventive activity of olive oil may be mediated by the presence of phenols. They may act on both initiation and promotion/progression phases of carcinogenesis. Further epidemiological studies, focusing on the chemical olive oil composition, are necessary to ascertain and quantify the role played by these phenols in cancer risk.References
- M. A. Martínez-González, L. J. Dominguez and M. Delgado-Rodríguez, Olive oil consumption and risk of CHD and/or stroke: a meta-analysis of case-control, cohort and intervention studies, Br. J. Nutr., 2014, 112, 248–259 CrossRef PubMed.
- T. Psaltopoulou, R. I. Kosti, D. Haidopoulos, M. Dimopoulos and D. B. Panagiotakos, Olive oil intake is inversely related to cancer prevalence: a systematic review and a meta-analysis of 13,800 patients and 23,340 controls in 19 observational studies, Lipids Health Dis., 2011, 10, 127 CrossRef CAS PubMed . Review.
- C. Pelucchi, C. Bosetti, E. Negri, L. Lipworth and C. La Vecchia, Olive oil and cancer risk: an update of epidemiological findings through 2010, Curr. Pharm. Des., 2011, 17, 805–812 CrossRef CAS PubMed . Review.
- R. Selvaggini, M. Servili, S. Urbani, S. Esposto, A. Taticchi and G. F. Montedoro, Evaluation of phenolic compounds in virgin olive oil by direct injection in high-performance liquid chroma-tography with fluorometric detection, J. Agric. Food Chem., 2006, 54, 2832–2838 CrossRef CAS PubMed.
- M. S. Cooke, M. D. Evans, M. Dizdaroglu and J. Lunec, Oxidative DNA damage: mechanisms, mutation, and disease, Lancet J., 2003, 17, 1195–1214 CAS.
- A. Facchini, S. Cetrullo, S. D'Adamo, S. Guidotti, M. Minguzzi, A. Facchini, R. M. Borzì and F. Flamini, Hydroxytyrosol Prevents Increase of Osteoarthritis Markers in Human Chondrocytes Treated with Hydrogen Peroxide or Growth-Related Oncogene α, PLoS One, 2014, 9, e109724, DOI:10.1371/journal.pone.0109724.
- F. Warleta, C. S. Quesada, M. Campos, Y. Allouche, G. Beltrán and J. J. Gaforio, Hydroxytyrosol protects against oxidative DNA damage in human breast cells, Nutrients, 2011, 3, 839–857 CrossRef CAS PubMed.
- J. L. Quiles, A. J. Farquharson, D. K. Simpson, I. Grant and K. W. J. Wahle, Olive oil phenolics: effects on DNA oxidation and redox enzyme mRNA in prostate cells, Br. J. Nutr., 2002, 28, 225–234 CrossRef PubMed.
- J. Young, K. W. J. Wahle and S. P. Boyle, Cytoprotective effects of phenolic antioxidants and essential fatty acids in human blood monocyte and neuroblastoma cell lines: Surrogates for neurological damage in vivo, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2008, 78, 45–59 CrossRef CAS PubMed.
- L. Nousis, P. T. Doulias, N. Aligiannis, D. Bazios, A. Agalias, D. Galaris and S. Mitakou, DNA protecting and genotoxic effects of olive oil related components in cells exposed to hydrogen peroxide, Free Radical Res., 2005, 39, 787–795 CrossRef CAS PubMed.
- R. Fabiani, P. Rosignoli, A. De Bartolomeo, R. Fuccelli, M. Servili, G. F. Montedoro and G. Morozzi, Oxidative DNA Damage Is Prevented by Extracts of Olive Oil, Hydroxytyrosol, and Other Olive Phenolic Compounds in Human Blood Mononuclear Cells and HL60 Cells, J. Nutr., 2008, 138, 1411–1416 CAS.
- R. Bernini, F. Crisante, N. Merendino, R. Molinari, M. C. Soldatelli and F. Velotti, Synthesis of a novel ester of hydroxytyrosol and a-lipoic acid exhibiting an antiproliferative effect on human colon cancer HT-29 cells, Eur. J. Med. Chem., 2011, 46, 439–446 CrossRef CAS PubMed.
- S. D'Angelo, D. Ingrosso, V. Migliardi, A. Sorrentino, G. Donnarumma, A. Baroni, L. Masella, M. A. Tufano, M. Zappia and P. Galletti, Hydroxytyrosol, a natural antioxidant from olive oil, prevents protein damage induced by long-wave ultraviolet radiation in melanoma cells, Free Radicals Biol. Med., 2005, 38, 908–919 CrossRef PubMed.
- R. Fabiani, A. De Bartolomeo, P. Rosignoli, M. Servili, G. F. Montedoro and G. Morozzi, Cancer chemoprevention by hydroxytyrosol isolated from virgin olive oil through G1 cell cycle arrest and apoptosis, Eur. J. Cancer Prev., 2002, 11, 351–358 CrossRef CAS PubMed.
- R. Fabiani, P. Rosignoli, A. De Bartolomeo, R. Fuccelli and G. Morozzi, Inhibition of cell cycle progression by hydroxytyrosol is associated with the up-regulation of CDK inhibitors p21WAF1/Cip1 and p27Kip1 and with the induction of differentiation in HL60 cells, J. Nutr., 2008, 138, 42–48 CAS.
- J. Han, T. P. N. Talorete, P. Yamada and H. Isoda, Anti-proliferative and apoptotic effects of oleuropein and hydroxytyrosol on human breast cancer MCF-7 cells, Cytotechnology, 2009, 59, 45–53 CrossRef CAS PubMed.
- F. D. Della Ragione, V. Cucciolla, A. Borriello, V. D. Pietra, G. Pontoni, L. Racioppi, C. Manna, P. Galletti and V. Zappia, Hydroxytyrosol, a natural molecule occurring in olive oil, induces cytochrome c-dependent apoptosis, Biochem. Biophys. Res. Commun., 2000, 278, 733–739 CrossRef CAS PubMed.
- J. A. Menendez, A. Vazquez-Martin, R. Colomer, J. Brunet, A. Carrasco-Pacombo, R. Garcia-Villalba, A. Fernandez-Gutierrez and A. Segua-Carretero, Olive oil's bitter principle reverses acquired autoresistance to trastuzumab (Herceptin) in HER2-overexpressing breast cancer cells, Eur. J. Nutr., 2012, 51, 455–464 CrossRef PubMed.
- V. Goulas, V. Exarchou, A. N. Troganis, E. Psomiadou, T. Fotsis, E. Briasoulis and P. Gerothanassis, Phytochemicals in olive oil leaf extracts and their antiproliferative activity against cancer and endothelial cells, Mol. Nutr. Food Res., 2009, 53, 600–608 CAS.
- H. K. Obied, P. D. Prenzler, I. Konczak, A. Rehman and K. Robards, Chemistry and bioactivity of olive biophenols in some antioxidant and antiproliferative in vitro bioassay, Chem. Res. Toxicol., 2009, 22, 227–234 CrossRef CAS PubMed.
- G. Corona, M. Deiana, A. Incani, D. Vauzour, M. A. Dessı` and J. P. Spencer, Hydroxytyrosol inhibit the proliferation of human colon adenocarcinoma cells through inhibition of ERK1/2 and cyclin D1, Mol. Nutr. Food Res., 2009, 53, 897–903 CAS.
- R. Fabiani, R. Fuccelli, F. Pieravanti, A. De Bartolomeo and G. Morozzi, Production of hydrogen peroxide is responsible for then induction of apoptosis on HL60 cells, Mol. Nutr. Food Res., 2009, 53, 887–896 CAS.
- R. Fabiani, M. V. Sepporta, P. Rosignoli, A. De Bartolomeo, M. Crescimanno and G. Morozzi, Anti-proliferative and pro-apoptotic activities of hydroxytyrosol on different tumour cells: the role of extracellular production of hydrogen peroxide, Eur. J. Nutr., 2012, 51, 455–464 CrossRef CAS PubMed.
- A. R. Collins, The comet assay for DNA damage and repair, Mol. Biotechnol., 2004, 26, 249–261 CrossRef CAS PubMed.
- A. Boyum, Isolation of mononuclear cells and granulocytes from human blood, Scand. J. Clin. Lab. Invest., 1968, 21, 77–89 CrossRef CAS.
- N. P. Sing, M. T. McCoy, R. R. Tice and E. L. Schneider, A simple technique for quantitation of low levels of DNA damage in individual cells, Exp. Cell Res., 1988, 175, 184–191 CrossRef.
- A. R. Collins, M. Ai-guo and S. J. Duthie, The kinetics of repair of oxidative DNA damage (strand breaks and oxidised pyrimidines) in human cells, Mutat. Res., 1995, 336, 69–77 CAS.
- R. F. Hussain, A. M. Nouri and R. T. Oliver, A new approach for measurement of cytotoxicity using colorimetric assay, J. Immunol. Methods, 1993, 160, 89–96 CrossRef CAS PubMed.
- J. Nourooz-Zadeh, J. Tajaddini-Sarmadi and S. P. Wolff, Measurement of plasma hydroperoxide concentration by the ferrous oxidation-xylenol orange assay in conjunction with triphenylphosphine, Anal. Biochem., 1994, 220, 403–409 CrossRef CAS PubMed.
- S. K. Niture, C. S. Velu, Q. R. Smith, G. J. Bhat and K. S. Srivenugopal, Increased expression of the MGMT repair protein mediated by cysteine prodrugs and chemopreventive natural products in human lymphocytes and tumor cell lines, Carcinogenesis, 2007, 28, 378–389 CrossRef CAS PubMed.
- I. Casaburi, F. Puoci, A. Chimento, R. Sirianni, C. Ruggiero, P. Avena and V. Pezzi, Potential of olive oil phenols as chemopreventive and therapeutic agents against cancer: a review of in vitro studies, Mol. Nutr. Food Res., 2013, 57, 71–83 CAS.
- E. M. Odiatou, A. L. Skaltsounis and A. I. Constantinou, Identification of the factors responsible for the in vitro pro-oxidant and cytotoxic activities of the olive polyphenols oleuropein and Hydroxytyrosol, Cancer Lett., 2013, 330, 113–121 CrossRef CAS PubMed.
- M. Servili, S. Esposto, R. Fabiani, S. Urbani, A. Taticchi, F. Mariucci, R. Selvaggini and G. F. Montedoro, Phenolic compounds in olive oil: antioxidant, health and organoleptic activities according to their chemical structure, Inflammopharmacology, 2009, 17, 76–84 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |