
Induction of ROS-independent DNA damage by curcumin leads to G2/M cell cycle arrest and apoptosis in human papillary thyroid carcinoma BCPAP cells
Li
Zhang
ab,
Xian
Cheng
b,
Yanyan
Gao
b,
Jiandong
Bao
b,
Haixia
Guan
c,
Rongrong
Lu
d,
Huixin
Yu
*b,
Qiang
Xu
*a and
Yang
Sun
*a
aState Key Laboratory of Pharmaceutical Biotechnology, School of Life Sciences, Nanjing University, Nanjing, Jiangsu, China. E-mail: yangsun@nju.edu.cn; molpharm@163.com; Fax: +86 25 89687620; Tel: +86 25 89687620
bKey Laboratory of Nuclear Medicine, Ministry of Health, Jiangsu Key Laboratory of Molecular Nuclear Medicine, Jiangsu Institute of Nuclear Medicine, Wuxi, Jiangsu, China. E-mail: yuhuixin@jsinm.org
cDepartment of Endocrinology & Metabolism and Institute of Endocrinology, the First Hospital of China Medical University, Shenyang, Liaoning, China
dState Key Laboratory of Food Science and Technology, School of Food Science and Technology, Jiangnan University, Wuxi, Jiangsu, China
First published on 25th September 2015
Abstract
Previously we found that curcumin, the active constituent of dietary spice turmeric, showed potent inhibitory effects on the cell growth of thyroid cancer cells. However, the detailed anti-cancer mechanism of curcumin is still unknown. In this study, we have reported that curcumin induces significant DNA damage in human papillary thyroid carcinoma BCPAP cells in a dose-dependent manner as evidenced by the upregulated phosphorylation of H2A.X at Ser139, which was further confirmed by the long tails in the comet assay and the increase in the number of TUNEL-positive cells. Subsequently, curcumin treatment caused a significant accumulation of cells at the G2/M phase that eventually resulted in a caspase-dependent apoptosis in BCPAP cells. DNA agarose gel electrophoresis revealed that curcumin-induced DNA damage in BCPAP cells was independent of DNA conformational change. Pretreatment with reactive oxygen species (ROS) scavengers failed to block the phosphorylation of H2A.X, suggesting the non-involvement of ROS in curcumin-mediated DNA damage. Interestingly, ATM/ATR activation by curcumin induced phosphorylation of Chk2 (Thr68) followed by that of Cdc25C (Ser216) and Cdc2 (Tyr15), and Cyclin B1 accumulation. In addition, the ATM-specific inhibitor KU-55933 reversed curcumin-induced phosphorylation of H2A.X. These results collectively show that curcumin treatment induced the DNA damage response via triggering an ATM-activated Chk2-Cdc25C-Cdc2 signaling pathway. These observations provide novel mechanisms and potential targets for the better understanding of the anti-cancer mechanisms of curcumin.
Introduction
Thyroid cancer is the most common endocrine tumor. The worldwide incidence of thyroid cancer has been steadily increasing over the past 30 years.1 The most frequent type of thyroid malignancy is papillary thyroid carcinoma (PTC).Nowadays, an alternative dimension in cancer management is turning its attention to the prevention and therapeutic intervention by dietary phytochemicals. Curcumin [bis(4-hydroxy-3-methoxy-phenyl)-1,6-heptadiene-3,5-dione] is a type of natural dietary polyphenolic compound isolated as a yellow pigment from turmeric (Curcuma longa), which is extensively used in Indian and Chinese medicine for the management of various diseases. Curcumin has been reported to have various pharmacological properties, including immunomodulation,2 anti-hyperalgesic,3 anti-oxidative4 and anti-cancer effects.5 In our previous study, we reported that curcumin induces apoptosis-dependent cell death in K1 papillary thyroid cancer cells.6 Recent studies from our group have also proved that curcumin inhibits the invasion and metastasis of K1 cells under both normoxic7,8 and hypoxic conditions9via modulating E-cadherin and matrix metalloproteinase-9 expressions. However, the detailed mechanisms underlying the anti-cancer effects of curcumin, especially against thyroid cancers, are still unclear.
Recently, several studies showed that curcumin or its derivatives induces DNA damage in certain human cancer cell lines.10,11 DNA lesions, constantly produced by environmental agents such as the ultraviolet (UV) component of sunlight, ionizing radiation and endogenous DNA damage-inducing agents, activate the DNA damage response (DDR), which involves detection, signaling and repair of the damage to safeguard the integrity of the genome. The kinases, ATM (Ataxia telangiectasia mutated) and ATR (Ataxia telangiectasia and Rad3 related), play key roles in sensing DNA breaks. ATM and ATR which phosphorylate a variety of downstream signaling proteins such as checkpoint kinases 1 and 2 (Chk1, Chk2), and p53, lead to further transmission of the checkpoint signals.12 Chk2 phosphorylates Cdc25C at Ser216, which acts as a negative regulator of Cdc25C. Cdc25C is an activator of Cdc2, therefore this cascade involves the inhibition of Cdc2. During the G2-M transition, Cdc2 is rapidly converted into the active form by Tyr15 dephosphorylation catalyzed by the Cdc25 phosphatase and drives cells into mitosis.
In the present study, we show that curcumin induces significant DNA damage, resulting in G2/M cell cycle arrest and following apoptosis in BCPAP cells. Our results provide evidence for the activation of an ATM-Chk2-Cdc25C-Cdc2 cascade as a central mechanism of curcumin-induced G2/M phase arrest and growth inhibition in human papillary thyroid carcinoma BCPAP cells.
Materials and methods
Chemicals, reagents and antibodies
Curcumin (Cur, Biochemical, product no. C7727), propidium iodide (PI), cisplatin, catalase and butylated hydroxyanisole (BHA) were purchased from Sigma (St. Louis, MO, USA). Crystal violet staining solution, N-acetyl-L-cysteine (NAC) and zVAD.fmk were purchased from Beyotime Institute of Biotechnology (Nantong, China). Hoechst 33342 was purchased from Invitrogen. Dihydroethidium was purchased from Molecular Probes. Mito-TEMPO was purchased from Alexis Biochemicals. Low-melting agarose was purchased from Takara. KU-55933 was purchased from Abcam. The mitochondria isolation kit for cultured cells was purchased from Pierce Biotechnology. All other chemicals were of analytical reagent grade and purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). The antibodies used were as follows: anti-ATM, anti-ATR, anti-Chk1, anti-Chk2 and anti-Cyclin B1, purchased from Santa Cruz Biotechnology. Anti-Bax, anti-Bcl-XL, anti-Bcl-2, anti-phospho-Cdc2 (tyr15) and Alexa Fluor 555 goat anti-rabbit IgG were purchased from Beyotime Institute of Biotechnology. Anti-caspase-3, anti-caspase-8, anti-caspase-9, anti-phospho-histone H2A.X (Ser139), anti-phospho-ATM (Ser1981), anti-phospho-ATR (Ser248), anti-phospho-Chk1 (Ser345), anti-phospho-Chk2 (Thr68), anti-Cdc25C and anti-Cdc25C (Ser216) were purchased from Cell Signaling Technology.Cell culture and treatments
The BCPAP cell line, derived from a human papillary thyroid carcinoma, was obtained from the German Collection of Micro-organisms and Cell Cultures (Braunschweig, Germany). BCPAP cells were cultured in RPMI 1640 medium supplemented with 10% new born bovine serum, 100 units per ml penicillin and 100 μg per ml streptomycin sulfate in a standard incubator with humidified air containing 5% CO2 at 37 °C. Curcumin was dissolved in DMSO at 50 mM as a stock solution and stored at −20 °C until diluted before use. Unless otherwise indicated cells were treated with various concentrations (12.5, 25, 50 μM) of curcumin for 24 h. The solvent control contains an equivalent amount of DMSO corresponding to the highest used concentration of curcumin (final concentration, 0.1% DMSO).Colony formation assay
BCPAP cells were seeded into 6-well plates at a density of 100 cells per well. After pretreatment with various concentrations of curcumin for 6 h, the medium was replaced with complete growth medium. After 14 d, cells were fixed using 4% paraformaldehyde and stained with crystal violet, and colonies consisting of more than 50 cells were counted for each dose point. The visible colonies were photographed by using a common Canon camera. Plating efficiency (PE) was calculated according to the formula PE = (number of colonies formed/number of cells seeded) × 100%. Plating efficiencies were routinely about 60–65%. The surviving fraction was calculated as the plating efficiency of the treated cells divided by the plating efficiency of untreated control cells.Western blotting
Western blots were performed as previously reported.13Immunofluorescence cytochemistry
BCPAP cells adhered to coverslips were treated with or without curcumin for 24 h. Then cells were fixed with 4% paraformaldehyde (15 min, room temperature), stained with an anti-γH2A.X antibody (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100), and detected with secondary antibodies (Alexa Fluor 555 goat anti-rabbit IgG, 1:1000). The coverslips were counterstained with 10 μg per ml of Hoechst 33342 and imaged with a fluorescence microscope (X51, Olympus).
:100), and detected with secondary antibodies (Alexa Fluor 555 goat anti-rabbit IgG, 1:1000). The coverslips were counterstained with 10 μg per ml of Hoechst 33342 and imaged with a fluorescence microscope (X51, Olympus).
Comet assay
The comet assay, or single cell gel electrophoresis assay (SCGE), is a common technique for measuring cellular DNA damage as single-strand breaks.14 Briefly, BCPAP cells plated at a density of 5 × 105 cells per well in 6-well plates were treated with or without various concentrations of curcumin for 24 h. Then the cells were washed and resuspended at 1 × 105 cells per ml in ice-cold PBS (without Mg2+ and Ca2+). Twenty microliters of the cell suspension was mixed with 160 μl of 0.6% low-melting agarose and placed on frosted slides pre-layered with 1% regular agarose. The slides were maintained horizontally and kept in the dark for 15 min. After solidification, the slides were immersed in a lysing solution (2.5 M NaCl, 100 mM EDTA, 10 mM Tris, pH 10, and 10% DMSO with 1% Triton X-100) at 4 °C in the dark for 1 h. After the lysis, the slides were placed in alkaline solution (1 mM Na2EDTA and 300 mM NaOH, pH 13) for 30 min to allow DNA unwinding, and then they were electrophoresed under alkaline (pH 13) conditions for 30 min with a current setting of 300 mA. Following electrophoresis, slides were neutralized in 0.4 M Tris (pH 7.5). Slides were dehydrated in 70% ethanol and allowed to air dry. Then slides were stained with 50 μl ethidium bromide (20 μg ml−1) and incubated at room temperature for 15 minutes. After the slides were viewed by a fluorescence microscope, the DNA damage was quantified by measuring the displacement between the genetic material of the nucleus (‘comet head’) and the resulting ‘tail’. Tail moment was measured using the following method: Extent tail moment = 100 × Tail DNA intensity/Cell DNA intensity × Length of tail. Each treatment was carried out in duplicate, and at least 100 randomly selected cells from two microscope slides were analyzed.Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay
TUNEL was used to assess the internucleosomal DNA fragmentation. Cells were fixed with 4% paraformaldehyde at room temperature and permeabilized with 0.1% Triton X-100 in PBS on ice for 2 min. The treated cells were incubated with the one step TUNEL reaction mixture in the presence of cy3-dUTP for 60 min at 37 °C. Then the coverslips were co-stained with DAPI and observed under a fluorescence microscope.DNA staining
Cell cycle and the sub-G1 (subdiploid) cell population were analyzed by PI staining. Cell cycle analysis was determined as described previously.15 The synchronization was performed by incubating cells in RPMI 1640 supplemented with 0.5% NBS for 12 h. Then cells were incubated in fresh RPMI 1640 supplemented with 10% FBS and treated with or without curcumin for 24 h. After incubation, cells were harvested and fixed with 70% ethanol at 4 °C overnight. Then the cells were washed and resuspended in 1 ml of the DNA staining reagent containing 50 μg per ml RNase, 0.1% Triton X-100, 0.1 mM EDTA (pH 7.4), and 50 μg per ml PI in the dark for 30 min, and the DNA content was analyzed using the FACS flow cytometry. For sub-G1 DNA content detection, ten thousand cells in each sample were analyzed and the percentage of apoptotic cells accumulating in the sub-G1 peak was calculated by CellQuest software.Measurement of cytochrome c release
The proteins in the curcumin-treated cells were separated into cytosolic and mitochondrial fractions using the Mitochondria Isolation Kit according to the procedures provided by the manufacturer. To check the selectivity of proteins from subcellular fractionation, β-actin and CoxIV were used as marker proteins representing the cytosolic and mitochondrial fractions, respectively.Cell viability assay
Cell viability was assessed by the MTT assay which was performed as previously reported.16Measurement of intracellular superoxide levels
Dihydroethidium (DHE), a nonfluorescent membrane-permeable probe, interacts with superoxide, causing the liberation of membrane-impermeable ethidium cations. After drug treatment, cells were washed once with PBS and stained on culture plates with DHE (10 μM) at 37 °C for 30 min. Then culture plates were placed on ice to stop the labeling, trypsinized, and resuspended in ice-cold PBS. Samples were analyzed using a FACScan flow cytometer (Becton Dickinson).Statistical analysis
All experiments were performed with at least 3 independent biological replicates. For western blots, one representative blot is shown in figures; quantifications were performed from 3 independent experiments and expressed as mean ± SD. Statistical evaluations were performed with the Student t test when 2 value sets were compared. P < 0.05 was considered to be statistically significant.Results
Curcumin inhibits clonogenic survival in human papillary thyroid carcinoma BCPAP cells
Firstly, to examine the potential cell growth inhibition of curcumin in thyroid cancer cells, the effects of curcumin on BCPAP cell morphology were investigated. As illustrated in Fig. 1A, BCPAP cells exposed to different dosages of curcumin (12.5 to 50 μM) underwent significant morphological changes compared with untreated control cells. Curcumin treatment resulted in an obvious cell loss and shrinkage, rounding and partial detachment, demonstrating the cytotoxic effects of curcumin on BCPAP cells. In addition, in vitro clonogenic assays were performed to determine the antitumor activities of curcumin. As shown in Fig. 1B, the clonogenicity of BCPAP cells was reduced in a concentration-dependent manner after curcumin treatment. These results indicate that curcumin can inhibit the growth of BCPAP cells.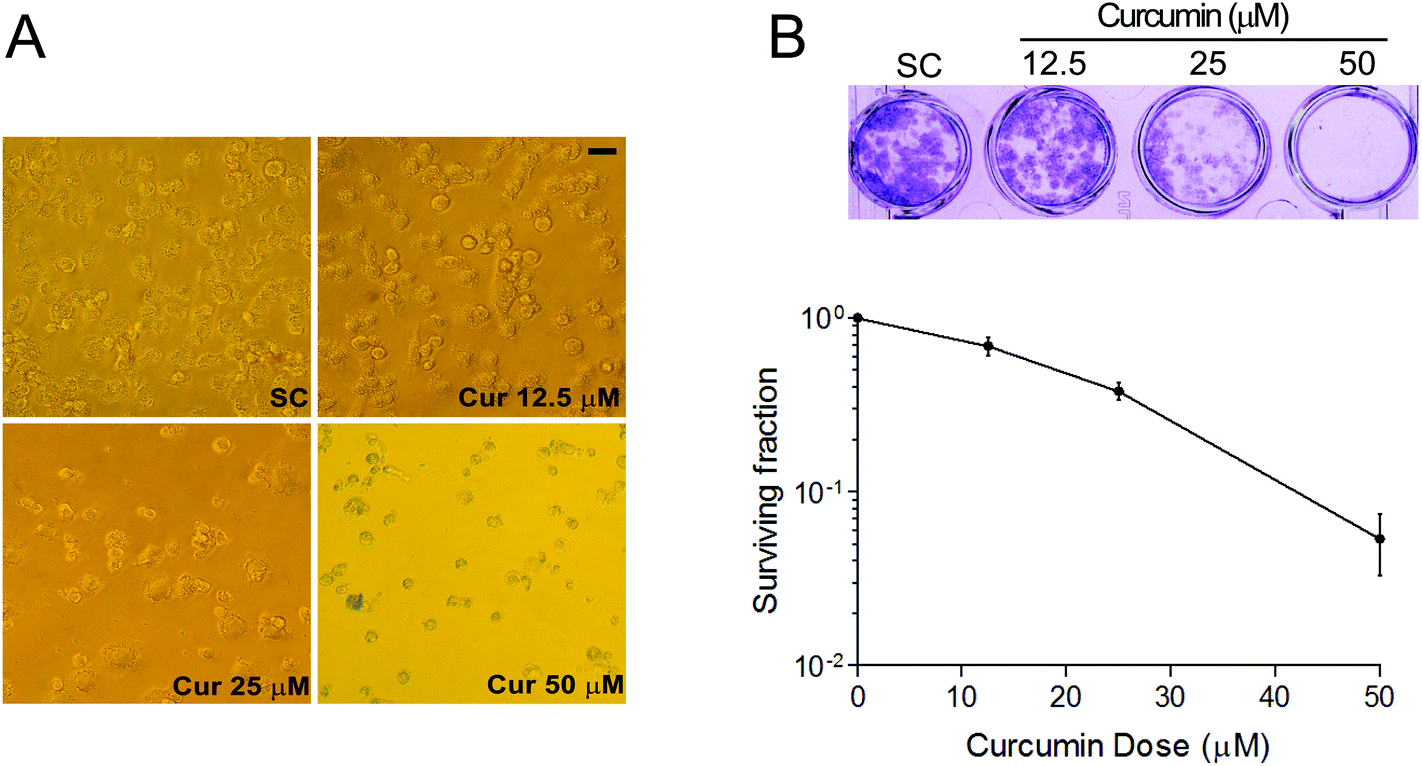 | ||
| Fig. 1 Effects of curcumin on cell morphology and colony formation ability of BCPAP cells. (A) Effects of curcumin on the morphological alterations in BCPAP cells. Cells were exposed to different concentrations of curcumin (0–50 μM) for 24 h and cell morphological changes were assessed by microscopic examination. Scale bar, 20 μm. (B) Curcumin inhibits the clonogenic ability of BCPAP cells. To determine surviving fractions, counts were normalized using the plating efficiency of the corresponding control group. SC, solvent control. | ||
Curcumin induces DNA double-strand breaks (DSBs) in BCPAP cells
The phosphorylation of H2A.X on Ser-139 (γH2A.X) is one of the major and early cellular responses to the induction of nuclear foci, the sites of DNA damage to which various DNA damage response regulators are recruited.17 As shown in Fig. 2A, H2A.X phosphorylation was increased in a dose-dependent manner upon curcumin exposure as analyzed by western blotting. Corroborating this observation, immunofluorescence analysis of curcumin-treated cells also confirmed the expression of γ-H2A.X. Excess DNA damage after curcumin treatment is documented in terms of dose-dependent increased number of γH2A.X foci counted in the nuclei of BCPAP cells (Fig. 2B). This indicates the most serious forms of genetic damage because each focus within the nucleus represents a DNA double strand break. The comet assay is a gel electrophoresis-based method that can be used to measure DNA damage in individual cells.18 Next, curcumin-induced DNA damage was measured by using the comet assay. As shown in Fig. 2C, after 24 h curcumin treatment, BCPAP cells showed differently sized and fragmented dead cells and comet structures with a longer tail length. In contrast, the untreated control cells exhibited almost condensed nuclei. Within the concentrations ranging from 12.5, 25 to 50 μM, there was a dose-dependent increase in the comet extent tail moment values from 6 to 395, 520 and 906 respectively, compared to the untreated cells. DNA fragmentation was similarly calculated by counting positive TUNEL stained cells per microscopic field. Compared with the control cells, the number of TUNEL-positive cells of curcumin treated groups displayed a dramatic and dose-dependent increase (Fig. 2D). Altogether, these results suggest that curcumin induces significant DNA damage in BCPAP cells, as evidenced by an increased number of DSBs.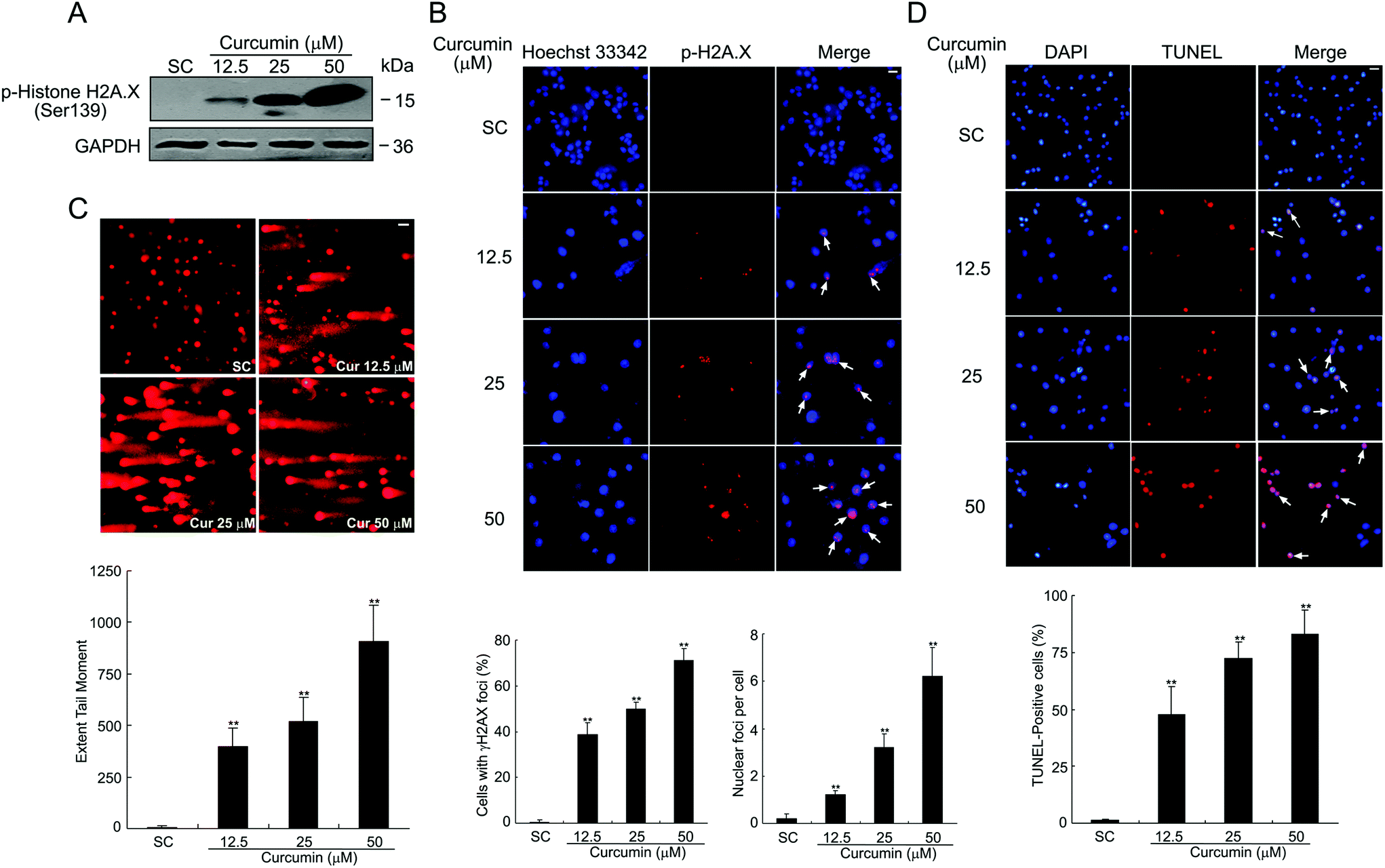 | ||
| Fig. 2 Curcumin induces DNA damage in BCPAP cells. (A) Cells were treated with 12.5, 25 and 50 μM of curcumin for 24 h and the level of γH2A.X was detected using western blot analysis. (B) Effects of curcumin on induction of γH2A.X foci measured by immunocytochemistry. γ-H2A.X foci formation (red) in BCPAP cells was observed under immunofluorescence microscopy. Nuclei were stained with Hoechst 33342 (blue). Scale bar, 10 μm. Bottom graphs quantified the percentage of cells with γ-H2A.X foci and the mean number of foci per cell after curcumin treatment. (C) Cells were treated with different dosages of curcumin for 24 h and DNA damage was detected by the comet assay. Nuclei with damaged DNA have a comet feature with a bright head and a tail, whereas nuclei with undamaged DNA appear round with no tail. Each figure represents a typical comet tail of the observed cells (at least 100 cells) from two slides in each experiment. Typical micrographs of comet assays are shown. Scale bar, 20 μm. (D) Representative images of TUNEL staining of BCPAP cells after treatment with the indicated concentration of curcumin for 24 h. The number of TUNEL-positive nuclei was expressed as a percentage of total nuclei detected by DAPI staining. Scale bar, 20 μm. **P < 0.01 vs. the SC group. | ||
Curcumin induces G2/M cell cycle arrest in BCPAP cells
To assess whether curcumin-induced cell growth inhibition is mediated by the alterations lying in the cell cycle, we evaluated the effects of curcumin on cell cycle distribution by propidium iodide (PI) staining and the DNA content was examined by flow cytometry. As shown in Fig. 3, compared to the solvent treated controls which had 20.78% cells in the G2/M phase, curcumin treatment caused an appreciable G2/M phase arrest accompanied by a decrease in the G0/G1 populations in BCPAP cells. The treatment caused an arrest of 28.98% of the cells in the G2/M phase of the cell cycle at a concentration of 12.5 μM; the percentage of cells arrested in this phase further increased to 30.54% at 25 μM and 43.68% at 50 μM. These data indicate that curcumin at doses from 12.5 μM to 50 μM causes cell cycle arrest at the G2/M phase in BCPAP cells.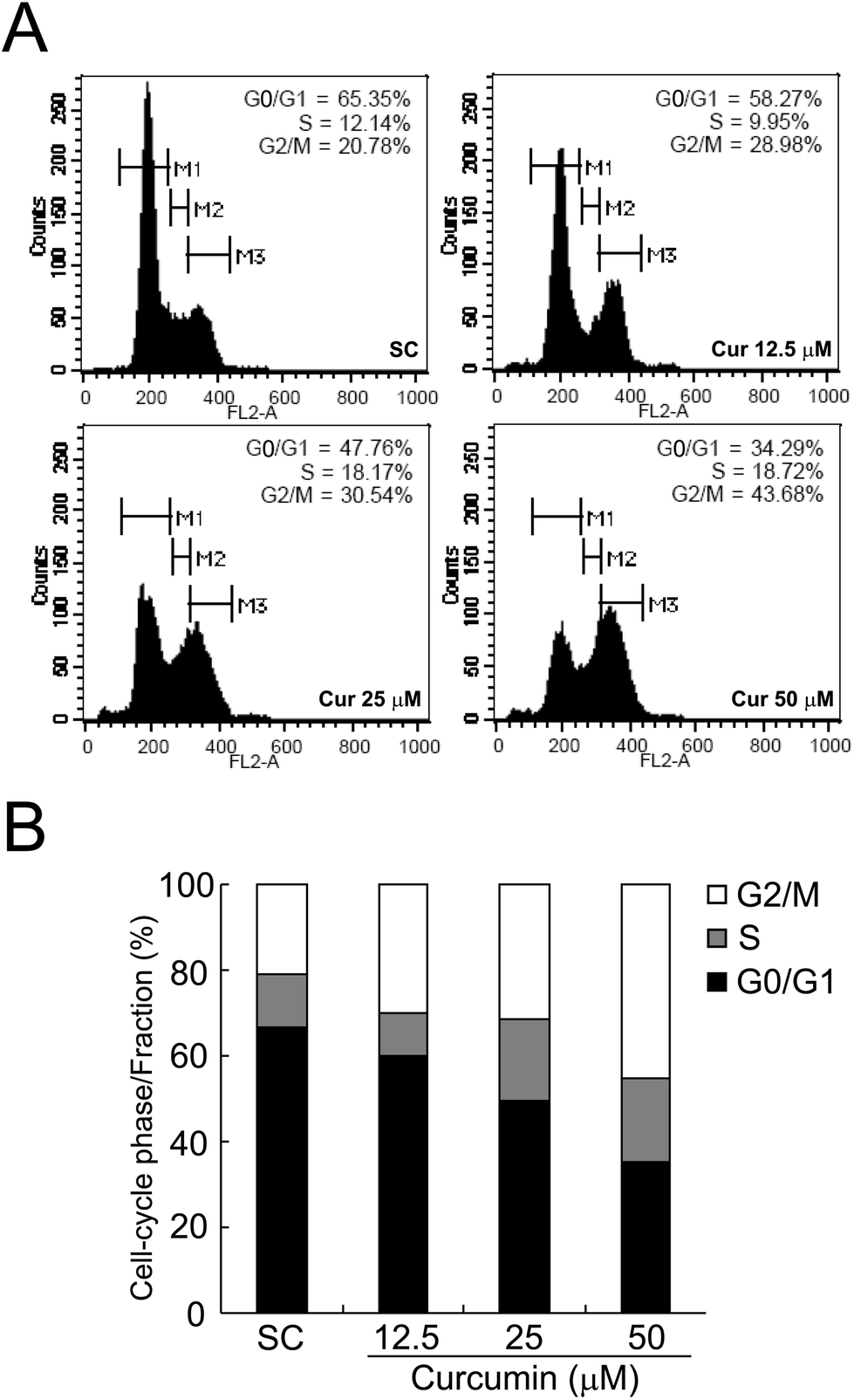 | ||
| Fig. 3 Curcumin treatment causes G2/M cell cycle arrest in BCPAP cells. (A) Synchronized BCPAP cells were treated with or without different concentrations of curcumin for 24 h. Cells were stained with PI solution followed by the analyses of cell cycle distribution using flow cytometry. Histograms were made by using CellQuest software. Gates M1, M2 and M3 indicated the G0/G1 phase, S phase and G2/M phase, respectively. (B) The cell cycle distribution after exposure to curcumin in BCPAP cells was determined and the data was plotted in the bar chart. | ||
Curcumin elicits a caspase-dependent apoptosis in BCPAP cells
Excessive DNA damage may trigger a series of cellular signaling pathways, such as apoptosis. As shown in Fig. 4A, curcumin increased the sub-G1 cell fraction in a dose-dependent manner. Hence, to characterize the apoptotic process, we examined the expression of apoptosis-related proteins by western blotting. As illustrated in Fig. 4B, curcumin treatment led to an increased expression of pro-apoptotic Bax with a concomitant decrease in the level of anti-apoptotic proteins Bcl-XL and Bcl-2. In addition, curcumin treatment induced the cleavage of caspase-3, -8 and -9, as well as the proteolysis of PARP. By curcumin treatment at 25 or 50 μM, the release of cytochrome c (cyt c) from mitochondria into cytosol, a critical early event leading to apoptosis, was also observed (Fig. 4C). Moreover, the recovery of cell viability was achieved by treating the cells with a pan-caspase inhibitor, Z-VAD-FMKfmk, which provides another strong piece of evidence for a caspase-dependent cell death in curcumin-treated cells (Fig. 4D).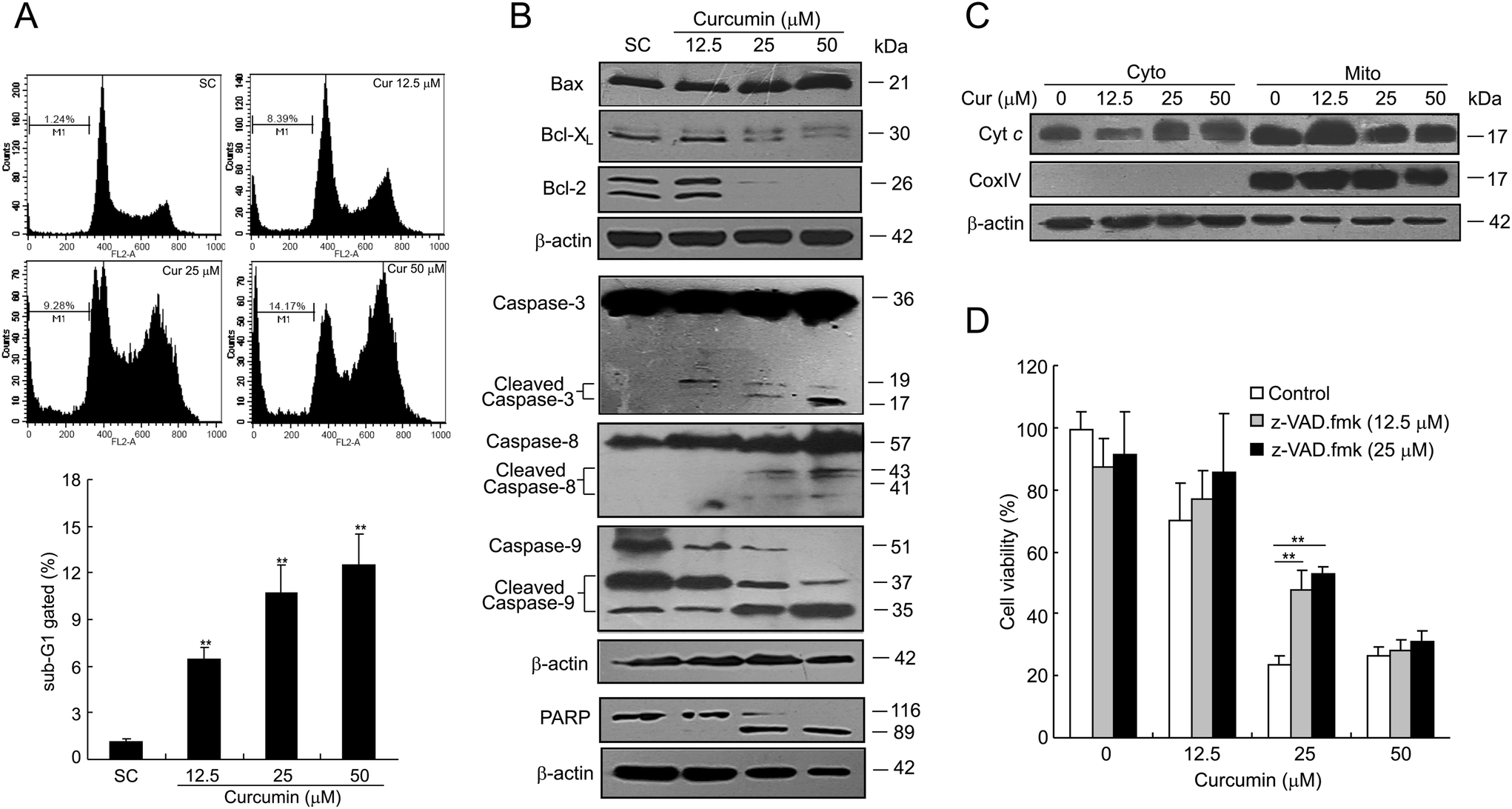 | ||
| Fig. 4 Curcumin induces caspase-dependent apoptosis in BCPAP cells. (A) The sub-G1 peaks were determined by flow cytometry. BCPAP cells were treated with 12.5 to 50 μM of curcumin for 24 h, then washed and stained with propidium iodide. Analysis of sub-G1 populations were performed using CellQuest software. The data were expressed as the mean ± SD of the three experiments. **P < 0.01 vs. the SC group. (B) Effects of curcumin on the expression of apoptosis-related proteins Bax, Bcl-XL, Bcl-2, caspase-3, -8, -9, and PARP cleavage in BCPAP cells measured by western blot analysis. (C) Curcumin induces the release of cytochrome c from mitochondria. At the end of the treatment period, cells were harvested and separated into cytosolic and mitochondrial fractions using the commercial fractionation kit. The expressions of cytochrome c in cytosol and mitochondria were analyzed by western blotting. (D) Curcumin-induced cell death is caspase-dependent. BCPAP cells were pretreated with different concentrations of z-VAD.fmk for 6 h, and then incubated with different dosages of curcumin at 12.5, 25 or 50 μM for 24 h. Cell viability was measured by the MTT assay. *P < 0.05. | ||
Curcumin fails to induce DNA conformational changes
Due to the fact that curcumin itself could emit bright green fluorescence, the dynamic process of curcumin entering cells could be observed. As shown in Fig. 5A, curcumin readily entered BCPAP cells and stayed in the cytoplasm for 6 h. Then it dispersed in both cytoplasm and nucleus at 12 h, while it was mostly confined to the nucleus at 24 h. It increased the possibility that curcumin may cause DNA damage by directly changing DNA conformation as most chemotherapeutic agents execute. To verify this hypothesis, the DNA binding assay was performed. The supercoiled DNA pUC19 was incubated with different concentrations of either curcumin or cisplatin for 12 h at 37 °C followed by DNA agarose gel analysis. As a positive control, cisplatin was able to unwind pUC19 DNA starting at a low concentration of 1 μM, while there was no observable band shifting for those DNA bound with curcumin even at a concentration of 50 μM, suggesting that curcumin can't cause conformational change of pUC19 DNA (Fig. 5B, upper). Similar results were obtained using another DNA plasmid, pSuper (Fig. 5B, lower).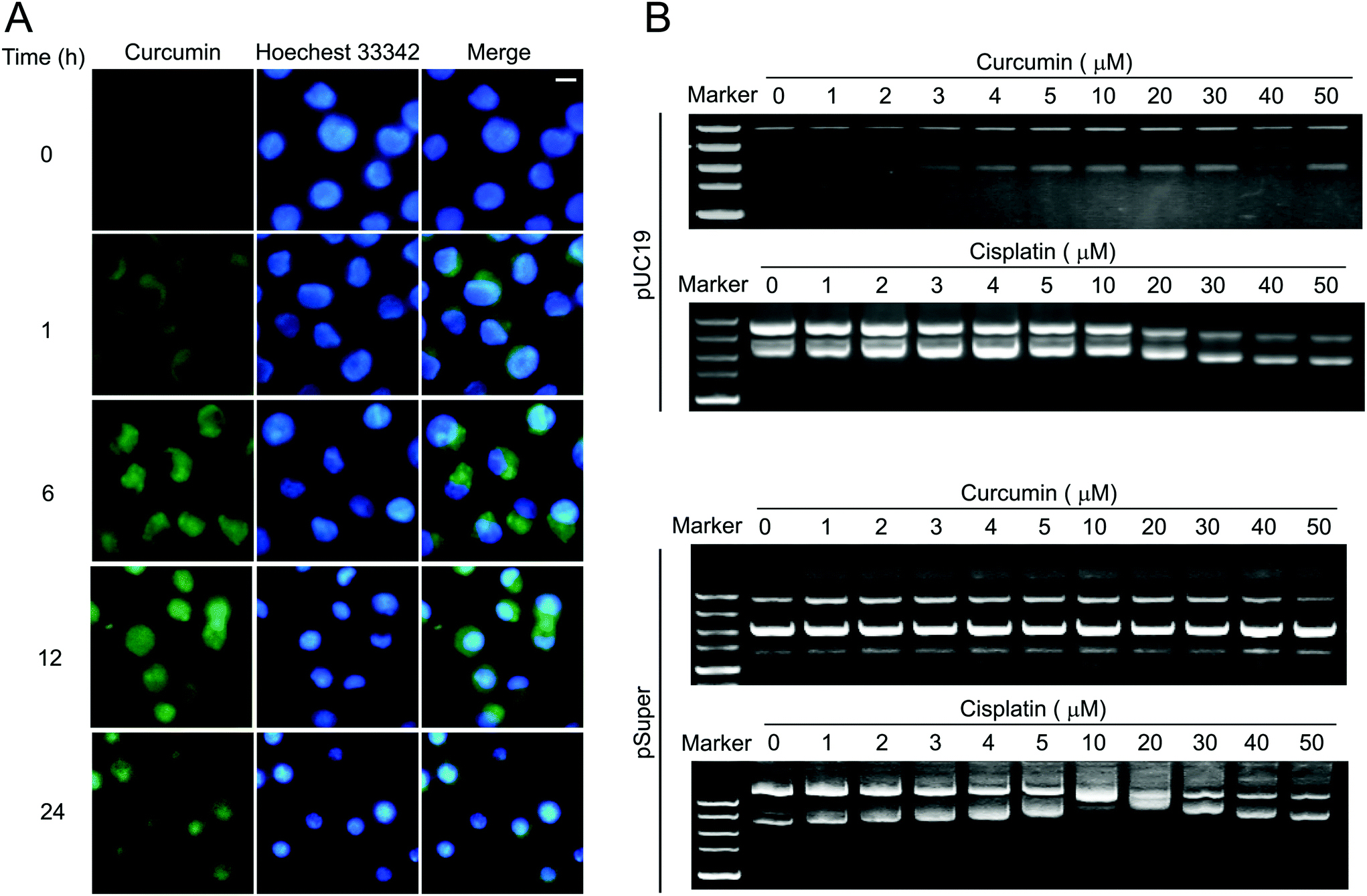 | ||
| Fig. 5 Effects of curcumin on DNA conformation. (A) Dynamic process of curcumin entering cells. Curcumin was added into BCPAP cells which were grown on coverslips at a final concentration of 50 μM. Curcumin emits green fluorescence. Nuclei were stained with Hoechst 33342 (blue). Fluorescence images were taken at indicated times. Scale bar, 5 μm. (B) DNA binding assay. pUC19 DNA or pSuper (200 ng) was used in each sample and incubated with different concentrations of curcumin or cisplatin at 37 °C for 12 h. The conformational changes of the supercoiled DNA caused by the binding of the complexes were displayed as the backward shifting of the band on the 1% agarose gel. | ||
Curcumin induces DNA DSBs in a ROS-independent way
Oxidative damage caused by excess ROS has been involved in DNA based modifications like single- and double-strand breaks.19 Based on our previous observations that curcumin-induced cell apoptosis in human papillary thyroid cancer K1 cells was ROS dependent,6 we next investigated whether curcumin-induced ROS caused DNA damage in BCPAP cells. Firstly, we measured ROS with dihydroethidium which was used to specifically detect superoxides. As shown in Fig. 6A, curcumin treatment dose-dependently induced ROS production, as demonstrated by a significant increase of superoxide anion (O2−) positive cell numbers. To determine whether ROS was responsible for DNA damage by curcumin treatment, cells were incubated with an ROS scavenger, N-acetylcysteine (NAC). As shown in Fig. 6B, an obvious elevated H2A.X phosphorylation level was detected at 24 h after curcumin treatment, however such an increase was not attenuated by pretreatment with the antioxidant NAC although it was at 10 mM or even higher dosage at 20 mM. This result was further confirmed by other antioxidants such as butylated hydroxyanisole (BHA) and catalase. Unexpectedly, both BHA (100 μM) and catalase (5000 U ml−1) not only failed to prevent curcumin-induced phosphorylation of H2A.X, but rather caused a slight induction of phosphorylated H2A.X (Fig. 6C). Considering that ROS is mainly generated inside the mitochondria, Mito-TEMPO, a specific mitochondria-targeting antioxidant was used. Similar results were obtained which indicated that Mito-TEMPO could not block curcumin-induced phosphorylation of H2A.X (Fig. 6D). Altogether, these results demonstrate that curcumin is capable of inducing ROS, while ROS is not required for DNA damage induced by curcumin.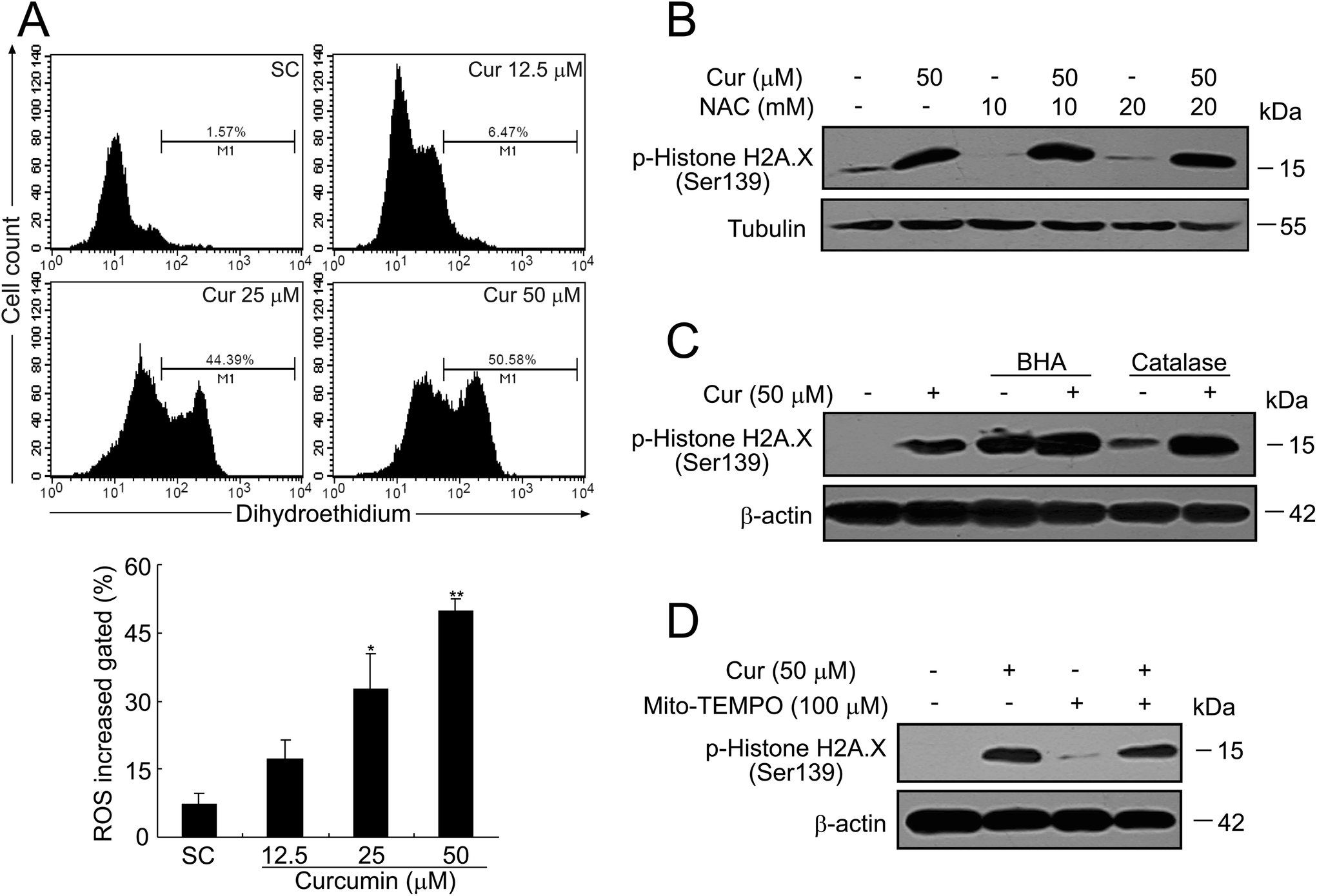 | ||
| Fig. 6 Effects of curcumin on intracellular superoxide generation in BCPAP cells. (A) BCPAP cells were treated with different dosages of curcumin for 24 h, stained with dihydroethidium (10 μM) and analyzed by flow cytometry. Data were representative of three independent experiments. Superoxide accumulation in curcumin-treated BCPAP cells was quantitatively analyzed. *P < 0.05, **P < 0.01 vs. the SC group. (B) BCPAP cells were pretreated with 10 or 20 mM NAC for 30 min, followed by exposure to 50 μM of curcumin for another 24 h. Then equal amounts of proteins were subjected to western blot detection of γH2A.X. (C and D) BCPAP cells were treated with curcumin (50 μM) in the absence or presence of various antioxidants (BHA, 100 mM; catalase, 5000 U ml−1; Mito-TEMPO, 100 μM) for 24 h. Equal amounts of proteins were subjected to western blot detection of γH2A.X. | ||
Curcumin modulates cell-cycle regulatory proteins in BCPAP cells
To further elucidate the molecular mechanism leading to curcumin mediated G2/M phase arrest, the signaling pathway responsible for DNA damage induced cell cycle checkpoint was detected. Key DNA-damage response (DDR) signaling components in mammalian cells are the protein kinases, ATM and ATR.20 The ATM and ATR kinases are activated by the presence of DNA double strand breaks or DNA replication stress, respectively.21 Therefore, the effects of curcumin on the expression and phosphorylation of ATM and ATR were firstly investigated. Compared with untreated control, the phosphorylation levels of ATM on Ser1981 and ATR on Ser248 were markedly increased in a dose-dependent manner after curcumin treatment while the total ATM only slightly decreased and total ATR remained unaffected (Fig. 7, upper). The checkpoint functions of ATR and ATM are mediated, in part, by a pair of checkpoint effector kinases known as Chk1 and Chk2. Next, the phosphorylation status of Chk1 and Chk2 were determined. The phosphorylation of Chk2 at Thr68 was dose-dependently up-regulated by curcumin. Strikingly, the BCPAP cells did not express phospho-Chk1-Ser345 at basal levels (Fig. 7, middle), indicating that Chk2 but not Chk1 might play a dominant role in the response to curcumin induced DNA DSBs. In response to DNA damage and DNA replication stress, both Chk1 and Chk2 can phosphorylate Cdc25C at Ser216, a site known to be involved in the negative regulation of Cdc25C.22 Consistent with the observed activation of Chk2 induced by curcumin, the level of Ser216-phosphorylated Cdc25C was dramatically increased in curcumin treated cells while there was no apparent effect on the total Cdc25C protein levels (Fig. 7, lower). During the G2-M transition, Cdc2 is rapidly converted into the active form by Thr14 and Tyr15 dephosphorylation which is catalyzed by a member of the Cdc25 family of phosphatases. The activation of Cdc2/Cyclin B1 is the trigger for entering into mitosis.23 As shown in Fig. 7 (lower), the inactive form of Cdc2 (phosphorylation on Tyr15) was also up-regulated by curcumin treatment. Moreover, there was a strong accumulation of the Cdc2 regulatory subunit cyclin B1 as well. Collectively, these observations support that the ATM-Chk2-Cdc25C-Cdc2 signal participates in the G2/M checkpoint in curcumin-induced DNA damage in BCPAP cells.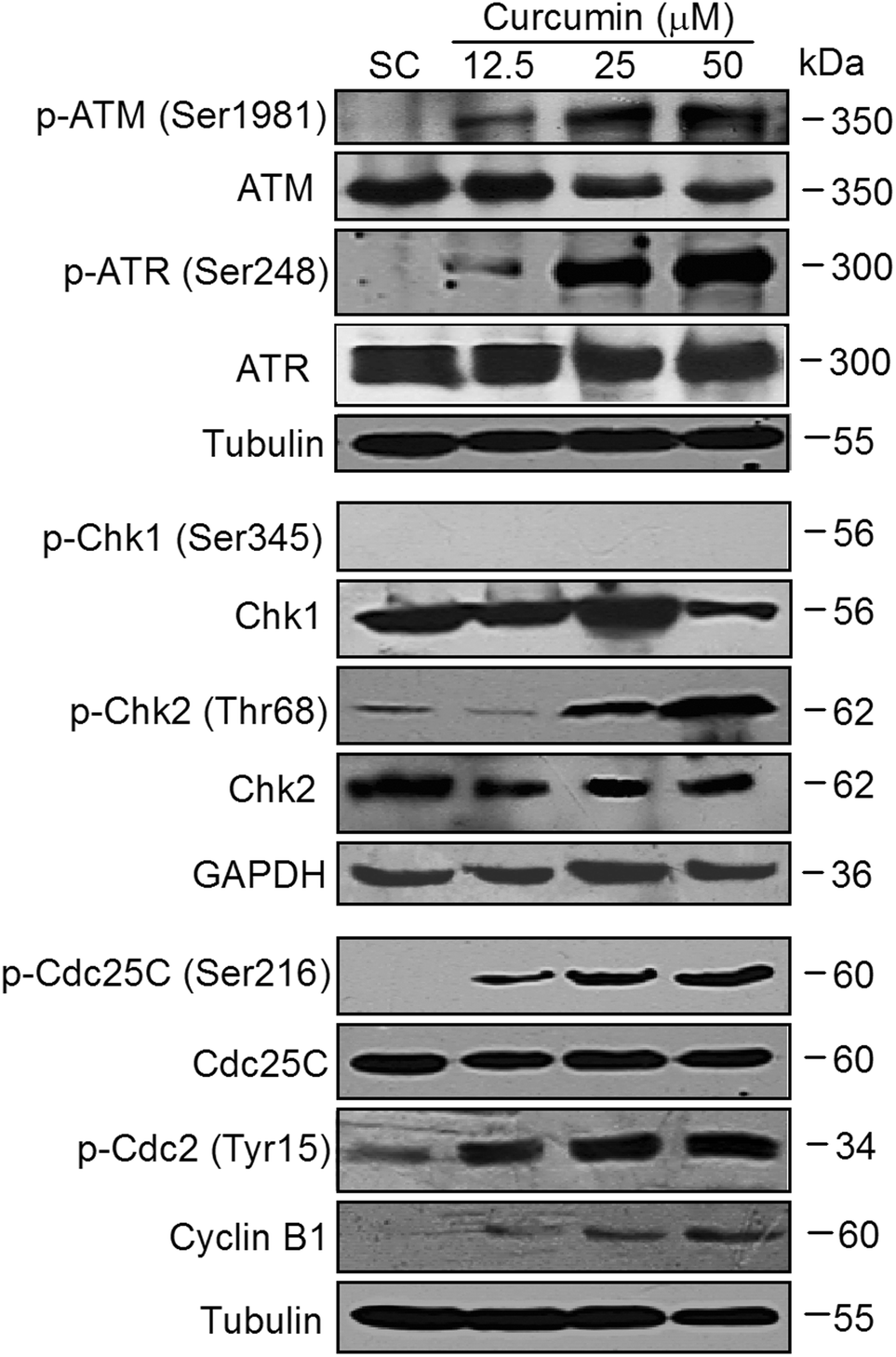 | ||
| Fig. 7 Curcumin modulates cell-cycle regulatory proteins in BCPAP cells. BCPAP cells were harvested after exposure to various concentrations of curcumin for 24 h. Equal amounts of proteins were subjected to western blot detection of indicated proteins. | ||
ATM kinase inhibitor blocks curcumin-induced DDR
As shown in Fig. 8A, curcumin treatment (50 μM) of cells for 24 h resulted in a strong increase in the phosphorylation levels of H2A.X, which was abrogated to a considerable extent when cells were treated in the presence of 5 or 10 μM of KU-55933, a potent and selective inhibitor of the ATM kinase.24 Next, we found by the MTT assay that pretreatment with KU-55933 significantly prevented curcumin-induced cell death in a dose-dependent manner in BCPAP cells (Fig. 8B). Moreover, KU-55933 at 10 μM could inhibit the expression of phospho-ATM successfully without killing cells (Fig. 8C and D). These data show that ATM kinase activation plays an essential role in curcumin-induced DDR.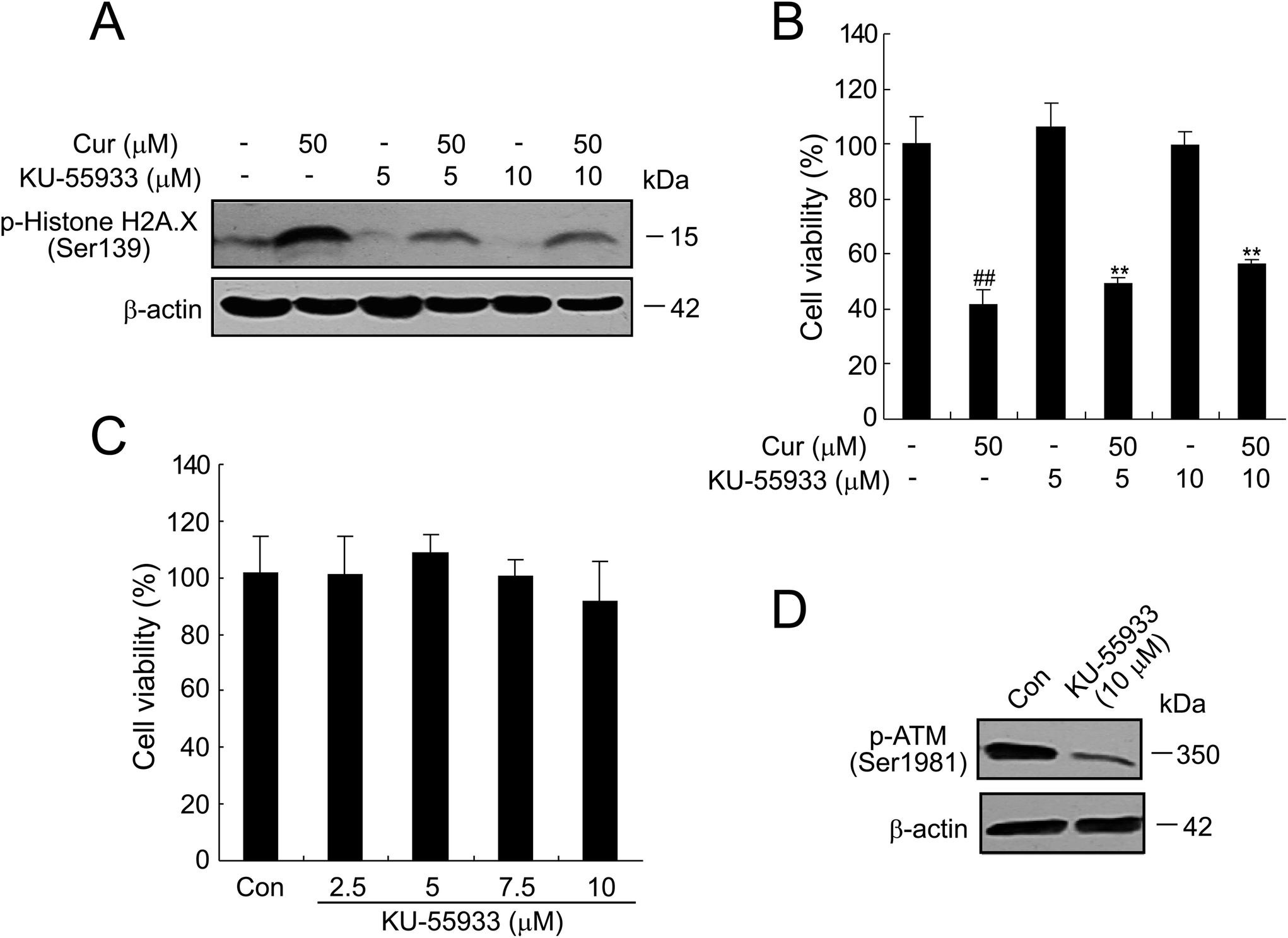 | ||
| Fig. 8 Inhibitor of ATM kinase, KU-55933, attenuates γH2A.X formation in BCPAP cells. (A and B) BCPAP cells treated with 50 μM curcumin and/or 5 or 10 μM KU-55933 for 24 h. The levels of γH2A.X were determined by western blot in (A) and cell viability was measured by the MTT assay in (B). (C) Effects of KU-55933 on the viability of BCPAP cells. Cells were exposed to the indicated concentration of KU-55933 and cell viability was analyzed by the MTT assay. DMSO was used as control. (D) Western blot analysis of phosphorylation of ATM in BCPAP cells treated with KU55933 at 10 μM. | ||
Discussion
Papillary thyroid cancer (PTC; account for about 80%), the most frequently occurring form of thyroid cancer usually has a good prognosis. However, there are still more than 6% thyroid cancers with aggressive invasion and metastasis.25Curcumin, a yellow pigment obtained from turmeric, is commonly used as a spice and a food coloring agent. Curcumin is also considered as a cancer chemopreventive agent. A mass of accumulated evidence reinforces that curcumin exerts potent anti-proliferative effects on a wide range of tumor cell types. Consistent with the published reports by Jutooru et al. on other cancer cells26 and our previous results in K1 papillary thyroid cancer cells,6 curcumin could also inhibit the proliferation and colony formation of BCPAP, another papillary thyroid cancer cell line, in a dose-dependent manner (Fig. 1). Furthermore, we found that curcumin induced DNA damage in BCPAP cells in a dose-dependent manner as evidenced by the upregulated phosphorylation of H2A.X at Ser139, which was further confirmed by the long tails in the comet assay and the increase in the number of TUNEL-positive cells (Fig. 2).
The induction of DNA damage in dividing cells results in the activation of cell cycle checkpoints which halt the proliferating cell in its cell cycle progression in order to give time to the DNA damage repair machinery to repair the DNA damage. Eventually, when repair is complete, the cell may proceed into its cell cycle. Alternatively, if the repair process fails, the cell cycle can be blocked permanently, leading to cell senescence or apoptosis.27 Cell cycle dysregulation resulting in uncontrolled cell proliferation is one of the most frequent alterations in tumor development. Thus, in recent years, cell cycle inhibition has become a promising target for cancer management.28 It has been reported that curcumin and its derivatives cause different phase cell cycle arrest in different types of cancer cells. For instance, T63, a 4-arylidene curcumin analogue, was reported to induce G0/G1 cell cycle arrest and apoptosis in A549 and H460 human lung cell lines.29 In our present study, we showed that curcumin induced G2/M phase arrest which was followed by the induction of apoptosis in BCPAP cells (Fig. 3 and 4), and that one of the plausible mechanisms accounting for the growth inhibition activity of curcumin in thyroid cancer cells occurs through DNA damage.
In most cases, DNA damage caused by anti-cancer agents, such as platinum, are attributed to the binding of the complexes to DNA, which leads to DNA conformational changes that block the normal replication and transcription processes.30 We noticed that after incubation for 24 h, curcumin localized to nucleus indicating a possibility for curcumin to access DNA (Fig. 5A). However, distinct from cisplatin, a DNA-damaging agent, curcumin had no effects on DNA conformation (Fig. 5B). It appears, therefore, that curcumin induces DNA damage without directly affecting DNA conformation. Our results were consistent with previous work by others which showed that curcumin bound to the major and minor grooves of the DNA duplex and to RNA bases, yet no conformational changes were observed upon curcumin interaction with these biopolymers.31
The widely held view is that reactive oxygen species (ROS; i.e., superoxide anions, hydroxyl radicals, and hydrogen peroxide) derived from oxidative respiration and products of lipid peroxidation, can cause severe damage to cellular macromolecules, especially the DNA.27 Indeed, a significant ROS production was observed by curcumin treatment in BCPAP cells (Fig. 6A). Pretreatment with NAC at 20 mM for 30 min successfully inhibited curcumin-induced ROS (data not shown). In contrast, we found that various ROS scavengers, including NAC, BHA, catalase and Mito-TEMPO failed to abrogate curcumin-induced H2A.X phosphorylation in BCPAP cells (Fig. 6B–D). Thus, our results indicate that curcumin induces DNA damage in a ROS-independent manner. Consistent with our results, luteolin induced ROS-independent DNA damage in HL-60 cells.32 In contrast, cucurbitacin B-induced DNA damage was ROS dependent in A549 cells.15 The reasons for these differences are yet to be uncovered but may reflect that maybe ROS is not necessary for all DNA damage events induced by different chemicals. Furthermore, the possibility of the existence of cell type specific signaling cascades could not be ruled out.
Once DNA damage response (DDR) signaling is activated, the key mediators ATM and ATR act via its downstream targets to promote DNA repair to maintain chromosome stability and restart stalled replication forks and transient cell cycle arrest. Consistent with these findings, we also showed that ATM/ATR activation by curcumin induced phosphorylation of Chk2 (Thr68) followed by that of Cdc25C (Ser216) and Cdc2 (Tyr15), and Cyclin B1 accumulation, which was the basis for the curcumin-induced G2/M block (Fig. 7). Moreover, the ATM-specific inhibitor KU-55933 reversed curcumin-induced phosphorylation of H2A.X, further confirming that ATM did participate in the curcumin-induced DNA damage in BCPAP cells (Fig. 8).
Conventional therapy of thyroid cancers relied on surgery, radioiodine ablation and thyroid-stimulating hormone suppression. Moreover, 131I is a major weapon in the fight against metastatic thyroid carcinoma.33 Whether curcumin treatment by inducing DNA damage in tumor cells will lead to enhance the sensitivity to 131I and have certain therapeutic implications in thyroid cancer therapy needs to be further investigated.
Conclusions
Collectively, our study provides evidence that one of the mechanisms by which curcumin triggers G2/M cell cycle arrest is by inducing DNA damage, which in turn results in the activation of the apoptotic cascade. Furthermore, exploring the detailed anti-cancer mechanisms of curcumin has direct implications in our standpoint of these important intra-cellular mechanisms as well as for developing therapies for treatment of thyroid cancers.Abbreviations
| ATM | Ataxia telangiectasia mutated |
| ATR | Ataxia telangiectasia and Rad3 related |
| BHA | Catalase and butylated hydroxyanisole |
| Chk1,2 | Checkpoint kinases 1 and 2 |
| cur | Curcumin |
| DDR | DNA damage response |
| DHE | Dihydroethidium |
| DSBs | Double-strand breaks |
| NAC | N-Acetyl-L-cysteine |
| Cyt c | Cytochrome c |
| PTC | Papillary thyroid carcinoma |
| ROS | Reactive oxygen species. |
Conflict of interest statement
The authors declare that they have no conflicts of interest.Acknowledgements
This study was supported by the grants from the National Natural Science Foundation of China (no. 81402214, 81422050 and 91313303), Jiangsu Province Clinical Science and Technology Project (Clinical Research Center, BL2012008), the Ministry of Health Foundation of China (W201304), and the Public Service Platform for Science and Technology Infrastructure Construction Project of Jiangsu Province (BM2012066).References
- Y. E. Nikiforov and M. N. Nikiforova, Nat. Rev. Endocrinol., 2011, 7, 569–580 CrossRef CAS PubMed.
- G. J. Zhao, Z. Q. Lu, L. M. Tang, Z. S. Wu, D. W. Wang, J. Y. Zheng and Q. M. Qiu, Int. Immunopharmacol., 2012, 14, 99–106 CrossRef CAS PubMed.
- A. K. Singh and M. Vinayak, Neurochem. Res., 2015, 40, 463–472 CrossRef CAS PubMed.
- X.-C. Zhao, L. Zhang, H.-X. Yu, Z. Sun, X.-F. Lin, C. Tan and R.-R. Lu, Food Chem., 2011, 129, 387–394 CrossRef CAS.
- K. G. Troselj and R. N. Kujundzic, Curr. Pharm. Des., 2014, 20, 6682–6696 CrossRef CAS PubMed.
- F. Song, L. Zhang, H.-X. Yu, R.-R. Lu, J.-D. Bao, C. Tan and Z. Sun, Food Chem., 2012, 132, 43–50 CrossRef CAS PubMed.
- C. Y. Zhang, L. Zhang, H. X. Yu, J. D. Bao and R. R. Lu, Biotechnol. Lett., 2013, 35, 995–1000 CrossRef CAS PubMed.
- C.-Y. Zhang, L. Zhang, H.-X. Yu, J.-D. Bao, Z. Sun and R.-R. Lu, Food Chem., 2013, 139, 1021–1028 CrossRef CAS PubMed.
- C. Tan, L. Zhang, X. Cheng, X. F. Lin, R. R. Lu, J. D. Bao and H. X. Yu, Exp. Biol. Med., 2015, 240, 925–935 CrossRef CAS PubMed.
- H. F. Lu, J. S. Yang, K. C. Lai, S. C. Hsu, S. C. Hsueh, Y. L. Chen, J. H. Chiang, C. C. Lu, C. Lo, M. D. Yang and J. G. Chung, Neurochem. Res., 2009, 34, 1491–1497 CrossRef CAS PubMed.
- J. Cao, L. Jia, H. M. Zhou, Y. Liu and L. F. Zhong, Toxicol. Sci., 2006, 91, 476–483 CrossRef CAS PubMed.
- J. Smith, L. M. Tho, N. Xu and D. A. Gillespie, Adv. Cancer Res., 2010, 108, 73–112 CrossRef CAS PubMed.
- W. J. Guo, Y. M. Zhang, L. Zhang, B. Huang, F. F. Tao, W. Chen, Z. J. Guo, Q. Xu and Y. Sun, Autophagy, 2013, 9, 996–1008 CrossRef CAS PubMed.
- N. P. Singh and R. E. Stephens, Mutat. Res., 1997, 383, 167–175 CAS.
- J. Guo, G. Wu, J. Bao, W. Hao, J. Lu and X. Chen, PLoS One, 2014, 9, e88140 Search PubMed.
- L. Zhang, H. Yu, Y. Sun, X. Lin, B. Chen, C. Tan, G. Cao and Z. Wang, Eur. J. Pharmacol., 2007, 564, 18–25 CrossRef CAS PubMed.
- L. J. Mah, A. El-Osta and T. C. Karagiannis, Leukemia, 2010, 24, 679–686 CrossRef CAS PubMed.
- C. B. Bennett, A. L. Lewis, K. K. Baldwin and M. A. Resnick, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5613–5617 CrossRef CAS.
- M. S. Cooke, M. D. Evans, M. Dizdaroglu and J. Lunec, FASEB J., 2003, 17, 1195–1214 CrossRef CAS PubMed.
- A. M. Weber and A. J. Ryan, Pharmacol. Ther., 2015, 149, 124–138 CrossRef CAS PubMed.
- S. P. Jackson and J. Bartek, Nature, 2009, 461, 1071–1078 CrossRef CAS PubMed.
- A. Tyagi, R. P. Singh, C. Agarwal, S. Siriwardana, R. A. Sclafani and R. Agarwal, Carcinogenesis, 2005, 26, 1978–1987 CrossRef CAS PubMed.
- C. P. De Souza, K. A. Ellem and B. G. Gabrielli, Exp. Cell Res., 2000, 257, 11–21 CrossRef CAS PubMed.
- I. Hickson, Y. Zhao, C. J. Richardson, S. J. Green, N. M. Martin, A. I. Orr, P. M. Reaper, S. P. Jackson, N. J. Curtin and G. C. Smith, Cancer Res., 2004, 64, 9152–9159 CrossRef CAS PubMed.
- L. Davies and H. G. Welch, Arch. Otolaryngol. Head Neck Surg., 2010, 136, 440–444 CrossRef PubMed.
- I. Jutooru, G. Chadalapaka, P. Lei and S. Safe, J. Biol. Chem., 2010, 285, 25332–25344 CrossRef CAS PubMed.
- J. H. Houtgraaf, J. Versmissen and W. J. van der Giessen, Cardiovasc. Revasc. Med., 2006, 7, 165–172 CrossRef PubMed.
- G. Bretones, M. D. Delgado and J. Leon, Biochim. Biophys. Acta, 2015, 1849, 506–516 CrossRef CAS PubMed.
- H. Liu, B. H. Zhou, X. Qiu, H. S. Wang, F. Zhang, R. Fang, X. F. Wang, S. H. Cai, J. Du and X. Z. Bu, Free Radicals Biol. Med., 2012, 53, 2204–2217 CrossRef CAS PubMed.
- S. Ramachandran, B. R. Temple, S. G. Chaney and N. V. Dokholyan, Nucleic Acids Res., 2009, 37, 2434–2448 CrossRef CAS PubMed.
- S. Nafisi, M. Adelzadeh, Z. Norouzi and M. N. Sarbolouki, DNA Cell Biol., 2009, 28, 201–208 CrossRef CAS PubMed.
- N. Yamashita and S. Kawanishi, Free Radicals Res., 2000, 33, 623–633 CrossRef CAS PubMed.
- R. J. Robbins and M. J. Schlumberger, J. Nucl. Med., 2005, 46(Suppl 1), 28S–37S CAS.
| This journal is © The Royal Society of Chemistry 2016 |