
In vivo immunomodulatory effect of the lectin from edible mushroom Agaricus bisporus
Yanina
Ditamo
,
Lucia L.
Rupil†
,
Victor G.
Sendra‡
,
Gustavo A.
Nores
,
German A.
Roth
and
Fernando J.
Irazoqui
*
Centro de Investigaciones en Química Biológica de Córdoba (CIQUIBIC, UNC-CONICET), Departamento de Química Biológica, Facultad de Ciencias Químicas, Universidad Nacional de Córdoba, Argentina. E-mail: irazoqui@fcq.unc.edu.ar; Fax: +54 351 5353855; Tel: +54 351 5353855
First published on 16th September 2015
Abstract
Lectins are glycan-binding proteins that are resistant to digestion in the gastrointestinal tract and enter intact to blood circulation. The aim of this study was to evaluate the influence of edible mushroom Agaricus bisporus lectin (ABL) on innate and adaptive immune responses as well as its effect in two different experimental pathologies that involve the immune system. ABL inhibited in vitro nitric oxide (NO) production by mouse peritoneal macrophages in response to the pro-inflammatory stimuli lipopolysaccharides (LPS). However, it did not modify the activity of arginase, showing that while ABL downregulates M1 activation, it does not affect M2 activation. ABL also inhibited mononuclear cell proliferation in response to mitogen Con A, or in a mixed lymphocyte reaction. During the in vivo studies, oral administration of ABL to BALB/c mice induced a marked inhibition of NO production by peritoneal macrophages after LPS stimuli. The influence of ABL on tumor growth was studied in BALB/c mice receiving daily oral doses of ABL and implanted with CT26 tumor cells. ABL treatment induced significantly higher rate of tumor growth when compared with control mice. On the other hand, oral ABL administration in Wistar rats induced a marked diminution of the incidence of the disease and the severity of the clinical signs of experimental autoimmune encephalomyelitis. We can conclude that ABL has an in vivo immunomodulatory effect reducing the innate and adaptive responses. This food lectin shows potential therapeutic application on control of inflammatory autoimmune pathologies.
Lectins are proteins that recognize glycans in a specific and reversible manner. They are implicated in many molecular recognition and essential cellular processes, such as rolling adhesion of circulating leukocytes to activated endothelium, via interaction between selectins and their glycan ligands.1 Endogenous lectins such as mannose-binding lectin (MBL) recognize terminal mannose, fucose, and N-acetylglucosamine, which are common in microbial glycans, providing a specific mechanism for identification and neutralization of pathogens of the innate immune system.2 Galectins are β-galactosyl-binding lectins that can function as a homeostatic agent by modulating innate and adaptive immune responses. Galectin-1 induces cell growth inhibition, inhibits T cell activation, and promotes apoptosis of activated T cells.3
The current trend of consuming minimally processed plant-based foods, results in a higher intake of compounds such as lectins. Lectins are typically globular proteins that are resistant to digestion in the gastrointestinal tract. Dietary lectins are able to cross the gastrointestinal barrier rapidly and enter intact into the circulation4 and it has been shown that orally administered lectins can have diverse effects. Dietary lectins have the ability to interact with components of the immune system: most of them are mitogenic and show proinflammatory effects as the amplification of HLA class II expression in intestinal epithelial cell lines, stimulation of T cell proliferation, stimulation of IFN-γ causing HLA class II expression and stimulation of the production of inflammatory cytokines (IL-1, TNFα).5,6 High dietary intake of lectins such as wheat germ agglutinin, may affect the allergic response to oral antigens in the gut-associated lymphoid tissue.7 Lectins present in banana and garlic can induce adipogenic differentiation of mesenchymal cells via active signaling mechanism, suggesting they can affect the functions of stem cells.8
The lectin from the edible mushroom Agaricus bisporus (ABL) has been studied for its capacity to bind with high affinity to Galβ3GalNAcαSer/Thr, core 1 mucin-type O-glycan residue, known as Thomsen-Friedenreich antigen (T antigen).9 With a lower affinity constant, ABL also binds sialyl-T antigen and Tn antigen (GalNAcαSer/Thr). In addition, it has a second carbohydrate-recognition domain with GlcNAc specificity.10 ABL has proved to have a marked antiproliferative effect in in vitro assays. Yu et al. (1999) described that ABL blocks the nuclear sequence-dependent protein import which is associated with its inhibitory effect on HT29 cell line.11 Proliferation of several colorectal cancer cell lines was also inhibited by ABL12 as well as normal cells such as primary human subconjunctival fibroblasts, retinal pigment epithelium cells, and human keratinocytes.13–16 On the other hand, ABL increases the pancreatic β-cell proliferation through regulation of cell cycle proteins. In vivo assays showed that ABL administration expands β-cell mass with significant decrease in blood glucose concentration in animals with partial pancreatectomy.17
Therefore, since lectins incorporated to organisms could have important consequences on immunological homeostasis, the aim of this study was to evaluate the in vitro action of ABL on immune cells as well as in vivo modulation of immune responses of animals which received oral administration of purified ABL, analyzing the physiological repercussion of this food protein.
Materials and methods
ABL purification
ABL was purified from the edible mushroom Agaricus bisporus (white type) fruiting bodies by affinity chromatography in a column of human erythrocyte stroma incorporated into a polyacrylamide gel as previously described.18 Briefly, mushrooms were homogenized with 300 ml of phosphate buffer saline (PBS) and centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 20 min at 4 °C. The supernatant was heated to 60 °C with constant stirring for 2 h, and then centrifuged again. The supernatant was loaded onto a column of human erythrocyte stroma incorporated into polyacrylamide gel. Elution of the lectin was carried out with 0.1 M NH4OH, pH 11.0. Protein fractions were pooled, PBS dialyzed, concentrated with sucrose and stored at −20 °C until use. Purity of ABL was evaluated by using sodium dodecyl sulfate–polyacrylamide gel electrophoresis with Coomassie Brilliant Blue staining. Bicinchoninic acid assay was performed to measure total proteins using bovine serum albumin as standard protein (Pierce Protein Biology Products, Thermo Scientific, Rockford. Ill., USA).
000g for 20 min at 4 °C. The supernatant was heated to 60 °C with constant stirring for 2 h, and then centrifuged again. The supernatant was loaded onto a column of human erythrocyte stroma incorporated into polyacrylamide gel. Elution of the lectin was carried out with 0.1 M NH4OH, pH 11.0. Protein fractions were pooled, PBS dialyzed, concentrated with sucrose and stored at −20 °C until use. Purity of ABL was evaluated by using sodium dodecyl sulfate–polyacrylamide gel electrophoresis with Coomassie Brilliant Blue staining. Bicinchoninic acid assay was performed to measure total proteins using bovine serum albumin as standard protein (Pierce Protein Biology Products, Thermo Scientific, Rockford. Ill., USA).
Animals
Female BALB/c and C57/b mice (7 to 10 week old) were purchased from the School of Veterinary, La Plata National University (La Plata, Argentina) and housed in our animal facility. Male rats from a Wistar strain inbred in our laboratory for 40 years were used for experimental autoimmune encephalomyelitis (EAE) induction. Animals were provided with food and water ad libitum. Handling and animal procedures were performed in compliance with the Institutional Review Board and Ethical Committee of the School of Chemical Sciences, National University of Cordoba.Isolation of peritoneal macrophages
Mouse macrophages were isolated from the peritoneal cavities of mice injected 72 h previously with 500 μl of 3% sterile peptone.19 Normal resident peritoneal cells were obtained by a peritoneal washing with 20 ml Dulbecco's PBS containing 2% FBS and 40 μg ml−1 gentamicin. After centrifugation at 350g for 5 min, red blood cells were lysed in Ammonium–Chloride–Potassium buffer and mononuclear cells were resuspended in complete medium (RPMI 1640 containing 2 mM L-glutamine, 50 μg ml−1 gentamicin and 10% FBS) (cRPMI) and incubated at 37 °C for 120 min in plastic culture plates. Then, the non-adherent cells were discarded, and the adherent cells were cultured in duplicate with 1 ml cRMPI without phenol red in the presence or absence of lipopolysaccharides (LPS) (1 μg ml−1) plus varying concentrations (0.5–2 μg ml−1) of ABL in PBS for 48 h at 37 °C, 5% CO2. The supernatants of the cultured macrophages were then collected for measurement of nitric oxide (NO) production.Quantification of nitrite in culture supernatants
Supernatants from macrophages which had been cultured for 48 h with or without the indicated stimuli or in the presence of 1 mM aminoguanidine (AG), a selective inhibitor of the iNOS enzyme as a positive control, were analysed. The concentration of nitrites was assayed in duplicate by a standard Griess reaction adapted to microplate as an indirect measurement of NO synthesis. 50 μl of Griess reagent (1% sulphanilamide + 0.1% N-[1-naphthy]ethylenediamine in 5% phosphoric acid) was added to 50 μl of the harvested supernatants. The absorbance at 540 nm was obtained with a microplate reader model 680 (Bio-Rad Laboratories, Hercules, CA, USA). The data were referred to a standard curve of sodium nitrite.20Arginase activity
To determine arginase activity, peritoneal macrophages were cultured as described above, for 18 h with different concentrations of ABL. The cells were then harvested and lysed with 100 μl 0.2% Triton X-100. The arginase activity was assessed by incubating 50 μl of the lysate with 50 μl of 10 nM MnCl2 and 50 μl 50 mM Tris-HCl (pH 7.5) for 10 min at 56 °C to induce the activation of the enzyme. The hydrolysis of L-arginine was conducted by incubating the samples with 100 μl 0.5 M L-arginine, pH 9.7 at 37 °C for 60 min. The enzymatic reaction was stopped by adding 900 μl of a mixture of H2SO4 (96%)/H3PO4 (85%)/H2O (1:3:7 v/v/v). To determine the content of urea produced, 40 μl of 9% isonitrosopropiophenone (dissolved in 100% ethanol) were added and the samples were heated at 95 °C for 45 min. The absorbance at 550 nm was measured with a microplate reader. A standard curve of urea was used to calculate urea content and the results are expressed as μg urea per μg proteins.21 Cell viability was evaluated using the vital dye trypan blue.
Mixed lymphocyte culture
Total splenocytes from BALB/c mice (2.5 × 105 per well) were preincubated for 3 h with cRPMI in the presence or absence of ABL (0.25–1 μg ml−1). Subsequently, 2.5 × 105 per well splenocytes from C57/b mice were added and cultured for 4 days. T cell proliferation was measured by [3H]thymidine ([3H]TdR) incorporation.Proliferation measurement of mononuclear cells (MNC)
MNC from naïve mice spleen were cultured by triplicate in 96-well flat bottom plates essentially as previously indicated.22 Briefly, 1.25 × 106 cells per ml were cultured in a total volume of 200 μl per well of cRPMI with 0.5 μg ml−1 concanavalin A (Con A) in the presence or absence of ABL (0.25–1 μg ml−1) for 4 days at 37 °C, 5% CO2. Each well was pulsed with 20 μl of medium containing 1 μCi of [3H]TdR during the last 18 h of culture, then the cells were harvested onto fiberglass filters and the radioactivity incorporated counted using standard liquid scintillation techniques. Each treatment was performed in triplicate, and the results expressed as stimulation index (SI), defined as the ratio between mean counts per minute (cpm) of antigen-stimulated culture/mean cpm of the unstimulated culture, considering a SI ≥2 as positive response.ABL oral administration and analysis of NO production by macrophages
For the study of ABL in vivo effects, groups of mice received daily oral doses of ABL (10 μg per animal per day) in 20% sucrose-PBS or vehicle alone. Mouse macrophages were isolated from the peritoneal cavities and NO determination was performed as indicated above.In vivo tumor growth
Groups of 4 mice (BALB/c female, age 8 weeks) were implanted subcutaneously on the right side with mouse colon carcinoma CT26 cells (1.5 × 105 cells per mouse). Animals received daily oral doses of ABL (10 μg per animal per day) or vehicle alone seven days before tumor implantation and through the end of the experiment. Tumor volume was measured daily using an electronic digital caliper [tumor volume (mm3) = (longer diameter) × (shorter diameter)2 × 0.4]. CT26 tumor growth was monitored from 50 mm3 (5 mm diameter) to 3.200 mm3 (20 mm diameter).23 Animals were sacrificed when tumor diameter exceeded 50 mm diameter, or when there were signs of distress.Experimental autoimmune encephalomyelitis (EAE)
Male rats from a Wistar strain were anaesthetized and intradermally immunized in both hind feet with 0.5 ml of an emulsion of 0.05 ml saline solution and 0.45 ml of complete Freund's adjuvant containing 8 mg bovine myelin as previously described.24 Starting one week before immunization and through the end of the experiment, one group of animals were orally administered with 80 μg per kg per day of ABL in sucrose and the control group received mock treatment. The animals were weighed and examined daily for clinical signs of EAE. The degree of clinical signs was assessed as 0, normal; 1, flaccid tail; 2, hind limb paralysis; 3, definitive hindquarter weakness and urinary incontinence; and 4, severe quadriparesis, moribund state or death. Disease index (DI) was calculated as a sum of the daily clinical score for each animal throughout the experimental period divided by the day of onset of EAE signs × 100.Statistical analysis
Mean and standard error of the mean (s.e.m.) were calculated. Significant differences between experimental groups were calculated using the Student's t-test.Results
ABL affects the NO production of macrophages
LPS promote the differentiation of classically activated M1 macrophages. These cells produce high levels of oxidative metabolites and proinflammatory cytokines.25 Conversely, activated M2 macrophages suppress destructive effect of immunity, antagonizing M1 macrophages and suppressing the production of NO from L-arginine by inducible NO synthase. Taking into account that macrophages can actively participate in the promotion or the resolution of inflammatory processes, the effect of ABL on the L-arginine metabolism as a biochemical indicator of the type of activation of macrophages was evaluated. We observed that ABL inhibited in vitro NO production by mouse peritoneal macrophages in response to the pro-inflammatory stimuli LPS. Activated macrophages yielded lower NO production in an ABL concentration-dependent manner, showing 45% less NO in presence of 2 μg ml−1 ABL (Fig. 1A). As expected, the positive control aminoguanidine caused a marked inhibition of NO production. We also examined the ability of ABL to modulate arginase activity. We found that the concentrations of lectin used in these experiments did not modify the arginase activity in a significant proportion, demonstrating that while ABL downregulates M1 activation it does not affect M2 activation (Fig. 1B). Cell viability was not affected by ABL presence (Fig. 1C). | ||
| Fig. 1 ABL inhibits LPS-induced production of nitric oxide without altering arginase activity. (A) Peritoneal macrophages from naïve mice were cultured in the presence of 1 μg ml−1 LPS and treated with different concentrations of ABL or 1 mM aminoguanidine (AG). NO concentration (55 μM = 100%) was measured in supernatants by the Griess method and expressed as percent of inhibition in function of different ABL concentrations. Mean values were significantly different from those on the control group (*P < 0.01, **P < 0.001). (B) Arginase activity was determined in homogenates of peritoneal macrophages cultured in the absence or presence of ABL. Results are expressed in μg urea per μg protein related to different ABL concentrations. (C) Viability of cells with several ABL concentrations was evaluated using trypan blue exclusion. | ||
Influence of ABL in T cell proliferation
The capacity of ABL to affect MNC proliferation was evaluated. The addition of ABL (1 μg ml−1) to spleen cell culture in presence of the mitogen Con A caused 65% reduction of the spleen cells proliferation as compared to the culture without ABL (Fig. 2A). This result demonstrates that ABL reduced the capacity of total spleen cells to proliferate in vitro. Cell staining with trypan blue vital dye did not show significant loss of viability (Fig. 2B). | ||
| Fig. 2 ABL inhibits mitogen induced T cell activation. (A) Proliferation of T cells from naïve mice was measured by [3H] thymidine incorporation in the absence or presence of ABL. Results are expressed as counts per minute ± s.e.m. from quadruplicate wells. Mean values were significantly different in 1 μg ml−1 ABL treated cells compared with mock treated cells (*P < 0.01). (B) Cell viability of ABL treated cells was evaluated using trypan blue exclusion. | ||
To determine whether the inhibition of T cell proliferation correlated with a functional defect in antigen presentation, we analyzed the proliferation capacity of the cells in a mixed lymphocyte reaction. A concentration-dependent inhibition of T cell proliferation was observed as consequence of ABL presence (Fig. 3).
 | ||
| Fig. 3 ABL suppress mixed lymphocyte proliferation. For mixed lymphocyte reaction, total spleen cells from BALB/c mice were preincubated with ABL and cocultured with splenocytes from C57/b mice for 4 days. T cell proliferation was measured by [3H]thymidine incorporation. Data show mean cpm ± s.e.m. of quadruplicate wells from one of two independent experiments (**P < 0.001). | ||
Oral treatment with ABL impairs in vivo immune response
We next evaluated the in vivo effects of ABL on innate immune system components. Mice were supplemented with daily oral doses of ABL in sucrose and then peritoneal macrophages were obtained from ABL and mock treated animals. NO production in response to the pro-inflammatory stimuli of LPS was affected by oral ABL administration. Activated macrophages from ABL supplemented animals produced up to 53% less NO than macrophages from mock treated animals (Fig. 4). | ||
| Fig. 4 ABL oral treatment inhibits NO production by peritoneal macrophages. Macrophages were obtained from mice daily oral treated with ABL (5 or 10 μg/dose) or mock treated. NO production (50 μM = 100%) in response to LPS stimuli was measured using Griess method and expressed as percent of inhibition. Significant difference between macrophages from mock and ABL treated mice is indicated (***P < 0.02). | ||
In vivo effect of ABL on tumor growth
Fig. 5 shows the in vivo influence of ABL on tumor growth. BALB/c mice, implanted with CT26 tumor cells and receiving daily oral doses of ABL, showed faster tumor growth rate than animals treated with vehicle. 15 days after CT26 cells were implanted, the mean tumor volume in animals treated with ABL was 2020 mm3, whereas in mock treated animals the mean tumor volume was 640 mm3. In addition, similar results showing greater tumor growth were observed when C57/b mice implanted with Ta3HA cells received oral administration with ABL in relation to mock vehicle (data not shown).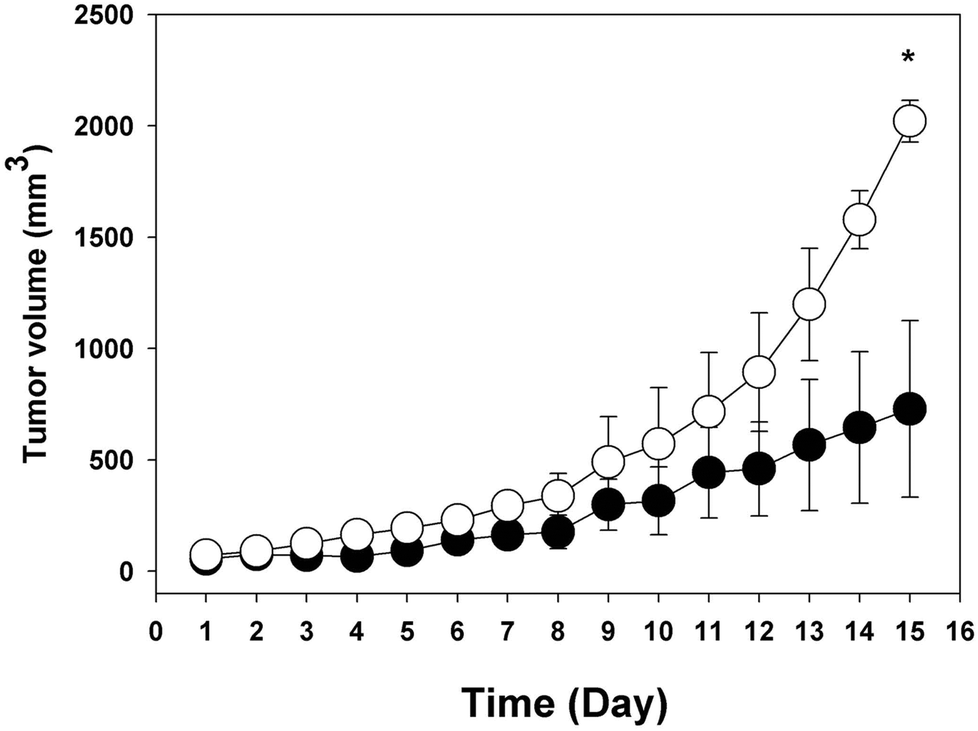 | ||
| Fig. 5 Effect of ABL oral treatment on tumor growth. CT26 tumor growth was evaluated in mice treated with oral doses of ABL (open circles) or vehicle alone (closed circles). Four mice per group in each experiment were assayed, three times each experiment was independently performed. Average tumor volume was indicated as function of time. The tumor volume at day 15 reported significant difference between ABL and mock treated animals (* P < 0.01). | ||
ABL ameliorates EAE clinical signs
To determine whether the observed in vitro and in vivo immunomodulatory effects of ABL could be extended to an autoimmune disease, we used the Multiple Sclerosis animal model EAE. In this model, clinical sings are related to the generation of autoreactive T cells that infiltrates central nervous system (CNS). It has been shown that NO contributes to the inflammation and the clinical expression of the disease.26 ABL administration induced a marked diminution of the disease incidence: only 50% of the ABL treated animals developed clinical signs while 100% of the mock treated animals developed EAE. The onset of the disease was retarded in animals treated with ABL, and the severity of the clinical signs was also diminished. Table 1 shows that maximum clinical score and disease index reached by ABL treated rats were clearly lower than the observed in mock control.| Animal group | Sick vs. total (%) | Clinical score | Day of onset | Disease index | Length of the disease (days) | ||||
|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | |||||
| *P = 0.031. | |||||||||
| ABL | 4/8 (50) | 4 | 1 | 1 | 2 | 0 | 12.5 ± 2.9 | 61 ± 19* | 6.2 ± 1.0 |
| Mock | 8/8 (100) | 0 | 0 | 0 | 6 | 2 | 11.5 ± 2.8 | 129 ± 51 | 8.0 ± 2.2 |
Discussion
Endogenous lectins such as MBL, DC-SIGN, Siglec, selectin and galectin play key roles in immune system. These proteins have the common property of a fine carbohydrate-recognition to glycans exposed in surface of cells. Glycans are important residues of plasmatic membranes that mediate the cell–cell and cell-matrix environment communication. Differential glycan expression in cell populations of immune system has been previously described.27 As consequence of the lectin–glycan interaction, extrinsic effects such as the modulation of cellular function can be observed. Because lectins incorporated to organisms could alter immunological homeostasis, the aim of the present work was to study the in vivo immunomodulatory capacity of a protein present in human food such as ABL.Macrophages are important members of innate immunity, where mucin-type O-glycans have a significant contribution to the cellular phenotype. Mucin-type O-glycans present in macrophages frequently expose T antigen and sialyl-T antigen as terminal residues.28 T antigen and sialyl-T antigen are most important ligands of ABL.9 As a result of ABL-macrophage interaction, we observed a reduction in the NO production in macrophages with a direct consequence in innate immune response. ABL presence in in vitro assays inhibited 45% NO production, whereas in in vivo assays, when mice received oral ABL, NO production was 53% lower compared with mock treated animals. Arginase activity was not affected by oral treatment with ABL. These results showed clear evidence that ABL downregulated the classical M1 activation of macrophages without affecting the M2 activation. Inhibition of NO production could be a consequence of altered Akt signaling. Akt is a serine/threonine kinase that plays critical roles in multiple cellular processes. Particularly, inhibition of Akt in macrophages leads to a decreased production of proinflammatory cytokines, and reactive oxygen and nitrogen intermediates.29,30 Cheung et al. (2012) demonstrated that ABL treatment inhibits Akt phosphorylation in RPE cells, indicating that this could be the mechanism involved in the inhibition of NO production.15 Similar property was previously described for Galectin-1.31 Galectin-1, an endogenous lectin, has an important role in control of NO production in macrophages, showing that interaction of carbohydrate-binding proteins is a way of innate immune modulation.
With the purpose to study the effect of ABL on adaptive immunity, we analyzed how this lectin affects the T lymphocyte proliferation. Using two different strategies we observed similar results: the T cell proliferation was inhibited by ABL. Treatment with 1 μg ml−1 ABL produced 65% inhibition of T cell proliferation when lymphocytes were stimulated with Con A, and 36% inhibition in a mixed allogeneic mice lymphocyte (BALB/c and C57/b) culture. CD43 and CD45 are two of the most abundant transmembrane glycoproteins on the T cell surface, and they interact with endogenous lectins to regulate several T cell functions. Throughout T cell development and activation, O- and N-glycans that decorate CD43 and CD45, are modulated and so are the interactions with endogenous lectins and potential functional outcomes. It has been shown that binding of multivalent lectins to CD45 results in clustering or oligomerization of CD45 and negatively regulate T cell receptor signaling.32,33 Taking into account that T antigen is the main ligand of ABL, and that activated CD8+, activated CD4+ and Th1 cells from periphery expose T antigen in CD43 and CD45 glycoproteins,34 the interaction between ABL and O-glycans from CD43 and CD45 could explain the inhibitory effect of ABL on T cell proliferation.
The in vivo effect of ABL in immune response was also studied in two different pathologies where the immunological system has a relevant role: a tumor growth model and an inflammatory autoimmune disease. The evolution and progression of tumors have a direct relation with the homeostasis of immune system where immunosupression facilitates the tumor cell growth.35 We observed that tumor volume was 3-times (2020/640) greater in animals treated with ABL than in mock treated animals at day 15. These results show the significance of ABL inducing downregulation of macrophage and T lymphocyte protective functions with the consequence of faster tumor cell growth. It has been described that appropriately activated M1 macrophages have cytotoxic activity toward cancer cells and mediate resistance against tumors36 hence, NO inhibition could have allowed an enhancement of tumor cell growth. ABL has been described as an important inhibitor of epithelial tumor cell proliferation in in vitro assays of colon HT29, colon Caco-2, breast MCF7,37 retinal pigment epithelium14,38 and colon CT26 cells.23 However the in vivo immunosupression generated by ABL allowed an enhancement of CT26 tumor cell growth. This result shows that downregulation by oral treatment of ABL was more relevant in immune system that in in vivo tumor cell proliferation.
EAE is an autoimmune disease mediated by autoantigen-specific T cells infiltrating the CNS,39 and it has been described that the production of NO in the CNS, in excess of normal levels, is required for clinical expression of disease.40 Oral administration of ABL to rats resulted in markedly diminished disease incidence and milder clinical signs compared with mock treated animals. Since the inflammatory Th1 immunity has a relevant role in EAE development,41 these results suggest a regulatory effect of ABL on inflammatory Th1 response.
Gastrointestinal tract and immune system are the two principal target tissues of food lectins, and most of the studied lectins are toxic.42 Consequently, it is imperative to not only differentiate between benefitial and harmful lectins, but also to examine the effect of dietary lectins in different in vitro as well as in vivo assays. This work shows that ABL has an in vivo immunomodulatory effect, reducing the innate immunity and adaptive Th1 response with potential application on control of inflammatory autoimmune pathologies.
Financial support
This study was supported by funding (to FJI) from the following Argentinean Institutions: Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Secretaría de Ciencia y Tecnología, Universidad Nacional de Córdoba (SeCyT, UNC), Ministerio de Ciencia y Tecnología, Provincia de Córdoba (MinCyT, Cba), Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT-PICT 967).Disclosure/Conflict of interest
The authors declare no conflict of interest.Authorship
The authors’ contributions to the study were as follows: Y. D. and F. J. I. designed the research, Y. D.; L. L. R and V. G. S conducted the research; Y. D., L. L. R., V. G. S., G. A. R., G. A. N. and F. J. I. analyzed the data and wrote the article. Y. D. and F. J. I. wrote the paper and had primary responsibility for final content. All authors read and approved the final manuscript. No authors have any conflict of interest to declare.Abbreviations
| ABL | Agaricus bisporus lectin |
| CFA | Complete Freund's adjuvant |
| Con A | Concanavalin A |
| CPM | Counts per minute |
| DC-SIGN | Dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin |
| DI | Disease index |
| EAE | Experimental autoimmune encephalomyelitis |
| FBS | Fetal bovine serum |
| [3H]TdR | [3H]thymidine |
| LPS | Lipopolysaccharides |
| MBL | Mannose-binding lectin |
| MNC | Mononuclear cells |
| NO | Nitric oxide |
| PBS | Phosphate buffer saline |
| SI | Stimulation index |
| Siglec | Sialic acid-binding immunoglobulin-type lectin |
| T antigen | Thomsen-Friedenreich antigen |
Acknowledgements
We thank S. Deza and G. Schachner for cell culture assistance and Dr C. Luquez for English editing.References
- S. Schmidt, M. Moser and M. Sperandio, Mol. Immunol., 2013, 55, 49–58 CrossRef CAS PubMed.
- T. R. Kjaer, S. Thiel and G. R. Andersen, Mol. Immunol., 2013, 56, 413–422 CrossRef CAS PubMed.
- G. A. Rabinovich, M. M. Iglesias, N. M. Modesti, L. F. Castagna, C. Wolfenstein-Todel, C. M. Riera and C. E. Sotomayor, J. Immunol., 1998, 160, 4831–4840 CAS.
- A. Pusztai, S. Bardocz and S. W. Ewen, Front. Biosci., 2008, 13, 1130–1140 CrossRef CAS.
- L. Cordain, L. Toohey, M. J. Smith and M. S. Hickey, Br. J. Nutr., 2000, 83, 207–217 CrossRef CAS PubMed.
- R. S. Singh, R. Bhari and H. P. Kaur, Crit. Rev. Biotechnol., 2010, 30, 99–126 CrossRef CAS PubMed.
- B. Watzl, C. Neudecker, G. M. Hansch, G. Rechkemmer and B. L. Pool-Zobel, Br. J. Nutr., 2001, 85, 483–490 CrossRef CAS PubMed.
- M. Bajaj, A. Hinge, L. S. Limaye, R. K. Gupta, A. Surolia and V. P. Kale, Glycobiology, 2011, 21, 521–529 CrossRef CAS PubMed.
- F. J. Irazoqui, M. A. Vides and G. A. Nores, Glycobiology, 1999, 9, 59–64 CrossRef CAS PubMed.
- M. E. Carrizo, S. Capaldi, M. Perduca, F. J. Irazoqui, G. A. Nores and H. L. Monaco, J. Biol. Chem., 2005, 280, 10614–10623 CrossRef CAS PubMed.
- L. G. Yu, D. G. Fernig, M. R. White, D. G. Spiller, P. Appleton, R. C. Evans, I. Grierson, J. A. Smith, H. Davies, O. V. Gerasimenko, O. H. Petersen, J. D. Milton and J. M. Rhodes, J. Biol. Chem., 1999, 274, 4890–4899 CrossRef CAS PubMed.
- M. Jordinson, I. El-Hariry, D. Calnan, J. Calam and M. Pignatelli, Gut, 1999, 44, 709–714 CrossRef CAS PubMed.
- M. Batterbury, C. A. Tebbs, J. M. Rhodes and I. Grierson, Exp. Eye Res., 2002, 74, 361–370 CrossRef CAS PubMed.
- D. Kent, C. M. Sheridan, H. A. Tomkinson, S. J. White, P. Hiscott, L. Yu and I. Grierson, Wound Repair Regen., 2003, 11, 285–291 CrossRef PubMed.
- Y. H. Cheung, C. M. Sheridan, A. C. Lo and W. W. Lai, Invest. Ophthalmol. Vis. Sci., 2012, 53, 7469–7475 CAS.
- R. Parslew, K. T. Jones, J. M. Rhodes and G. R. Sharpe, Br. J. Dermatol., 1999, 140, 56–60 CrossRef CAS PubMed.
- Y. Wang, Y. Liu, H. Wang, C. Li, P. Qi and J. Bao, Exp. Biol. Med., 2012, 237, 287–296 CrossRef CAS PubMed.
- F. J. Irazoqui, F. E. Zalazar, G. A. Chiabrando, O. Romero and M. A. Vides, J. Immunol. Methods, 1992, 156, 199–204 CrossRef CAS PubMed.
- L. L. Rupil, A. F. de Bem and G. A. Roth, Innate Immun., 2012, 18, 627–637 CrossRef CAS PubMed.
- L. R. Dulgerian, V. V. Garrido, C. C. Stempin and F. M. Cerban, Immunology, 2011, 133, 29–40 CrossRef CAS PubMed.
- A. Andersson, R. Kokkola, J. Wefer, H. Erlandsson-Harris and R. A. Harris, J. Leukocyte Biol., 2004, 76, 1118–1124 CrossRef CAS PubMed.
- A. L. Degano and G. A. Roth, J. Neurosci. Res., 2000, 59, 283–290 CrossRef CAS PubMed.
- V. G. Sendra, N. Zlocowski, Y. Ditamo, S. Copioli, M. P. Tarp, E. P. Bennett, H. Clausen, G. A. Roth, G. A. Nores and F. J. Irazoqui, Mol. Immunol., 2009, 46, 3445–3453 CrossRef CAS PubMed.
- Y. Ditamo, A. L. Degano, D. R. Maccio, M. C. Pistoresi-Palencia and G. A. Roth, Immunol. Cell Biol., 2005, 83, 75–82 CrossRef CAS PubMed.
- D. M. Mosser and J. P. Edwards, Nat. Rev. Immunol., 2008, 8, 958–969 CrossRef CAS PubMed.
- S. Ljubisavljevic, I. Stojanovic, D. Pavlovic, M. Milojkovic, S. Vojinovic, D. Sokolovic and I. Stevanovic, Acta Neurobiol. Exp., 2012, 72, 33–39 Search PubMed.
- M. A. Toscano, G. A. Bianco, J. M. Ilarregui, D. O. Croci, J. Correale, J. D. Hernandez, N. W. Zwirner, F. Poirier, E. M. Riley, L. G. Baum and G. A. Rabinovich, Nat. Immunol., 2007, 8, 825–834 CrossRef CAS PubMed.
- Y. Cao, P. Stosiek, G. F. Springer and U. Karsten, Histochem. Cell Biol., 1996, 106, 197–207 CrossRef CAS PubMed.
- M. V. Rajaram, L. P. Ganesan, K. V. Parsa, J. P. Butchar, J. S. Gunn and S. Tridandapani, J. Immunol., 2006, 177, 6317–6324 CrossRef CAS.
- A. Sica, M. Erreni, P. Allavena and C. Porta, Cell. Mol. Life Sci., 2015 DOI:10.1007/s00018-015-1995-y.
- S. G. Correa, C. E. Sotomayor, M. P. Aoki, C. A. Maldonado and G. A. Rabinovich, Glycobiology, 2003, 13, 119–128 CrossRef CAS PubMed.
- L. A. Earl, S. Bi and L. G. Baum, J. Biol. Chem., 2010, 285, 2232–2244 CrossRef CAS PubMed.
- R. Majeti, A. M. Bilwes, J. P. Noel, T. Hunter and A. Weiss, Science, 1998, 279, 88–91 CrossRef CAS PubMed.
- M. C. Clark and L. G. Baum, Ann. N. Y. Acad. Sci., 2012, 1253, 58–67 CrossRef CAS PubMed.
- A. Corthay, Front. Immunol., 2014, 5, 197 Search PubMed.
- P. J. Murray, J. E. Allen, S. K. Biswas, E. A. Fisher, D. W. Gilroy, S. Goerdt, S. Gordon, J. A. Hamilton, L. B. Ivashkiv, T. Lawrence, M. Locati, A. Mantovani, F. O. Martinez, J. L. Mege, D. M. Mosser, G. Natoli, J. P. Saeij, J. L. Schultze, K. A. Shirey, A. Sica, J. Suttles, I. Udalova, J. A. van Ginderachter, S. N. Vogel and T. A. Wynn, Immunity, 2014, 41, 14–20 CrossRef CAS PubMed.
- L. Yu, D. G. Fernig, J. A. Smith, J. D. Milton and J. M. Rhodes, Cancer Res., 1993, 53, 4627–4632 CAS.
- H. Wenkel, D. Kent, P. Hiscott, M. Batterbury, C. Groenewald, C. M. Sheridan, L. G. Yu and J. Milton, Invest. Ophthalmol. Vis. Sci., 1999, 40, 3058–3062 CAS.
- A. G. Baxter, Nat. Rev. Immunol., 2007, 7, 904–912 CrossRef CAS PubMed.
- D. O. Willenborg, M. Staykova, S. Fordham, N. O'Brien and D. Linares, J. Neuroimmunol., 2007, 191, 16–25 CrossRef CAS PubMed.
- A. P. Robinson, C. T. Harp, A. Noronha and S. D. Miller, Handb. Clin. Neurol., 2014, 122, 173–189 Search PubMed.
- I. M. Vasconcelos and J. T. Oliveira, Toxicon, 2004, 44, 385–403 CrossRef CAS PubMed.
Footnotes |
| † Present address: Centro de Investigaciones y Desarrollo en Inmunología y Enfermedades Infecciosas (CIDIE-CONICET,) Facultad de Medicina, Universidad Católica de Córdoba, Argentina. |
| ‡ Present address: Schepens Eye Research Institute/Massachusetts Eye and Ear, Department of Ophthalmology, Harvard Medical School, Boston, Massachusetts, USA. |
| This journal is © The Royal Society of Chemistry 2016 |