
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of interlayer anions on [NiFe]-LDH nanosheet water oxidation activity†
B. M.
Hunter
a,
W.
Hieringer
b,
J. R.
Winkler
a,
H. B.
Gray
a and
A. M.
Müller
*a
aBeckman Institute and Division of Chemistry and Chemical Engineering, California Institute of Technology, M/C 139-74, Pasadena, California 91125, USA. E-mail: astridm@caltech.edu
bTheoretical Chemistry and Interdisciplinary Center for Molecular Materials, Department of Chemistry and Pharmacy, Friedrich-Alexander Universität Erlangen-Nürnberg, Egerlandstrasse 3, 91058 Erlangen, Germany
First published on 17th March 2016
Abstract
We synthesized nickel–iron layered double hydroxide ([NiFe]-LDH) nanosheets with different interlayer anions to probe their role in water oxidation catalysis. In alkaline electrolyte in ambient air, carbonate rapidly replaced other interlayer anions and catalytic activity was highest. Electrocatalytic water oxidation in virtually carbonate-free alkaline electrolyte revealed that activity was a function of anion basicity. Our [NiFe]-LDH nanosheets, prepared by pulsed laser ablation in liquids, were regenerated in carbonate-containing aqueous KOH. Anion binding motifs were assessed by X-ray photoelectron spectroscopy in combination with density functional theory calculations, suggesting that nitrite species bound to edge-site Fe in the precatalyst correlated with higher water oxidation activity.
Broader contextThere is an urgent need to develop carbon-neutral technologies to accelerate the transition to renewable energy for use on our planet. Much work has been done on solar-driven water splitting to produce hydrogen fuel from water, but successful development of a scalable technology will depend critically on the availability of highly active catalysts made from abundant elements. At the least, it will be absolutely essential to have structural and mechanistic understanding of heterogeneous materials to facilitate the construction of robust catalytic photoanodes. In our work, we have discovered that interlayer anions play key roles in the catalytic performance of nickel–iron layered double hydroxide nanosheets, as incorporating anions with different basicities tunes the water oxidation activities of these materials. By probing anion binding sites by X-ray photoelectron spectroscopy along with DFT calculations, we have found that water oxidation occurs mainly on nanosheet edges. Importantly, our work opens the way for rational design of a new generation of highly active water splitting catalysts. |
1. Introduction
Global supply of sustainable fuels affects every aspect of human life. Electrocatalytic water splitting (2H2O → 2H2 + O2) is a promising approach towards transportable, carbon-neutral hydrogen fuel. The water oxidation half reaction is more demanding because it involves four electron and proton transfer steps,1,2 for which highly active, earth-abundant catalysts are needed.Layered double hydroxides (LDHs), materials based on mineral structures readily found in nature, have been shown to be active for water oxidation.3–22 We recently reported that a [NiFe]-LDH nanomaterial synthesized by pulsed laser ablation in liquids (PLAL) is among the best water oxidation catalysts made of earth abundant elements.20
The structures of these nanosheet precatalysts were analyzed by X-ray diffraction (XRD) and Raman spectroscopy.20 The [NiFe]-LDH materials consist of sheets of edge-shared nickel oxide octahedra, with varying amounts of ferric iron substituting at nickel sites. Hydroxide ligands extend into the interlayer space, which also contains water. The excess positive charges of Fe3+ substituting for Ni2+ are balanced by interlayer anions23 (Fig. 1).
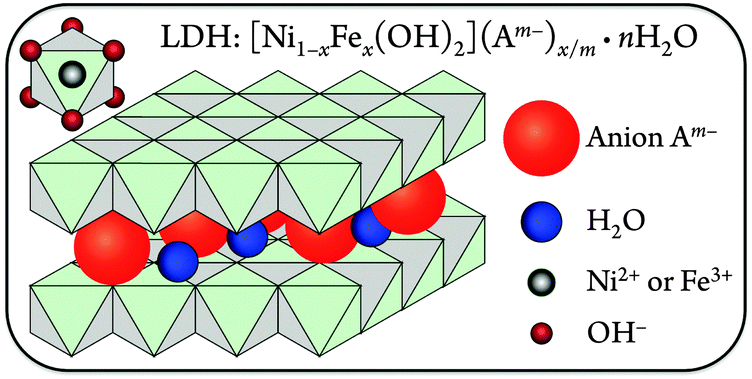 | ||
| Fig. 1 Schematic illustration of the [NiFe]-LDH structure. Anions and water are present in the interlayer space, and Ni2+ or Fe3+ ions are surrounded by six hydroxides in distorted octahedral coordination (upper left). | ||
The affinities for various mono- and divalent anions were established for [MgAl]-LDH hydrotalcite materials, which are structurally similar to our [NiFe]-LDHs. Data derived from anion-exchange isotherms revealed that divalent anions generally had higher ion selectivity than monovalent ones. The ion-exchange equilibrium constants followed the sequence CO32− > SO42− > OH− > F− > Cl− > I−.24 Some hydroxide ions are likely always present in the interlayer galleries because OH− partakes in strong interlamellar hydrogen bonding networks with the interlayer water, the ionic radius of hydroxide is very similar to that of water, and hydroxide is required to form the basic layers.25 Moreover, given the large slab hydroxide and intercalated water content of LDHs, the experimental determination of minute amounts of interlayer OH− ions is not practical. Nevertheless, we were interested in the identity of non-hydroxide interlayer anions in [NiFe]-LDHs during turnover in aqueous base in ambient air and their effects on water oxidation activity.
We exploited the benefits of PLAL as a synthetic method to investigate the role of interlayer anions in [NiFe]-LDH water oxidation catalysts. PLAL produces small, mono-dispersed, surfactant-free nanoparticles.26 The small size (<25 nm) allowed for the complete metathesis of interlayer anions simply by soaking in aqueous solutions. In addition, by modifying the anion content of the ablation liquid, we were able to synthesize species with different interlayer ions.
2. Experimental section
Materials and methods
Nanomaterial synthesis by pulsed laser ablation in liquids was performed in the Beckman Institute Laser Resource Center at California Institute of Technology. X-ray photoelectron spectroscopy was carried out at the Molecular Materials Research Center (Beckman Institute at California Institute of Technology).All chemicals were used as received. Deionized water was obtained from a Barnstead Diamond Nanopure system and had a resistivity of ≥16 MΩ cm−1. Data analysis and graphing was performed with Igor Pro 6.37 (Wavemetrics).
Synthesis
PLAL26 was used to synthesize [NiFe]-LDH nanosheets.20 Suspensions of 0.5 g iron (Alfa, −200 mesh) powder were stirred in 10 mL aqueous nickel salt solutions, using a magnetic stirrer in a 30 mL glass beaker at room temperature in ambient air. Metal salt solutions consisted of 3.0 M nickel nitrate (Ni(NO3)2·6H2O, Alfa), or 2.5 M nickel chloride (NiCl2·6H2O, JT Baker) and 0.5 M nickel nitrate, or 1.5 M each nickel sulfate (NiSO4·6H2O, EMD) and nickel nitrate. The limited solubilities of nickel chloride and sulfate required addition of nickel nitrate to keep the total nickel concentration of all three solutions at 3.0 M. The material resulting from use of 3.0 M aqueous nickel nitrate solution is henceforward called [NiFe]–(NO3−)-LDH. Beakers and stir bars were thoroughly cleaned with aqua regia before use. A 355 nm, 8 ns pulse length, 90 mJ per pulse laser beam, provided by the third harmonic of a 10 Hz Q-switched Nd:YAG laser (Spectra-Physics Quanta-Ray PRO-Series), was focused 0.5 mm below the surface of the liquid with a 100 mm focal length plano-convex quartz lens. Each sample was irradiated for 60 min. After synthesis, unreacted iron ablation target powder was separated from the nanoparticle suspensions using a rare-earth magnet. Solid nanopowders were obtained by centrifugation and washing with water until the supernatant did not show any nickel salt absorption. The nanoparticles were then washed twice with 3 mL acetone (EMD, OmniSolv®) and dried under vacuum.Anion exchange was effected by suspending 10 mg [NiFe]–(NO3−)-LDH nanosheets in 2 mL 1.0 M aqueous alkali salt solutions and letting the rigorously vortexed nanopowder soak for 45 min; the soaking time was 10 min for aqueous KOH solutions. The salts were K2CO3, KOH, KCl, NaF (all Mallinckrodt), Na2SO4 (EMD), NaClO4·1H2O (Fisher Scientific), KI, KBF4, K3PO4, and K2C2O4·1H2O (all Sigma-Aldrich). For exchange as a function of solution pH, 1.5 mg [NiFe]–(NO3)-LDH were dispersed in 1.0 mL 1.0 M (pH 14.0), 1.0 × 10−3 M (pH 11.0), or 1 × 10−6 M (pH 8.0) aqueous KOH solutions for 5 min. Anion-exchanged [NiFe]-LDH powders were obtained by centrifugation and washing first three times with 5 mL water and then twice with 3 mL acetone; the powders were dried under vacuum.
Physical characterisation
X-ray photoelectron spectra (XPS) were collected using a Surface Science Instruments M-probe surface spectrometer. Monochromatic Al Kα radiation (1486.6 eV) was used to excite electrons from the samples, which had either been deposited as dry powders on double-sided adhesive carbon tape (EM Sciences) or drop-cast from aqueous suspension on clean Cu foil and dried in ambient air at room temperature; we did not find different results for the two sample mounting methods. The sample chamber was maintained at <5 × 10−9 Torr. Survey scans from 0 to 1000 eV were performed to identify the elements present in the nanoparticles. Binding energies were referenced to the C 1s peak arising from adventitious carbon, taken to have a binding energy of 284.8 eV.27 High-resolution spectra were collected for all materials for the Fe 2p, Ni 2p, C 1s, N 1s, and O 1s regions. In addition, depending on the elemental composition of the material, high-resolution spectra were taken in the Cl 2s, S 2s, I 3d, B 1s, F 1s, and P 2s regions. Quantitative peak areas were derived after Shirley background subtraction28 and using relative sensitivity factors. Binding energies were obtained from the same peak fits. Quantitative XPS analysis was performed with CasaXPS (Version 2.3.16 PR 1.6).XRD data were collected with a Bruker D2 PHASER diffractometer. Monochromatic Cu Kα radiation (1.5418 Å; tube power 30 kV, 10 mA) was used; the instrument was equipped with 0.1° divergence, 1.5° Soller, and 0.6 mm detector slits, and had a 3 mm secondary anti-scatter screen. Diffracted radiation was collected with a Lynxeye detector. The instrument resolution was 0.030° in 2θ, and the counting time was 4.5 seconds per step, resulting in a total scan time of about 3.5 hours for each sample. Solid samples were deposited with vaseline (X-Alliance GmbH) on a zero-diffraction silicon plate (MTI Corporation). XRD background subtraction and Scherrer analysis29 were performed with the Bruker DIFFRAC.SUITE software. Reflections were analyzed using a Scherrer constant K of 0.89 and integral breadth, defined as the total area under the diffraction maximum divided by the peak intensity.30 Basal spacings d were derived using Braggs law,31nλ = 2d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sin(θ), where the integer n equals one, λ is the wavelength of the incident light, and θ is the angle of incidence. The (003) peaks of background-subtracted XRD data were fitted with Gaussians (ESI†), whose centers at 2θ were divided by two to obtain the angle of incidence required to calculate basal spacings.
sin(θ), where the integer n equals one, λ is the wavelength of the incident light, and θ is the angle of incidence. The (003) peaks of background-subtracted XRD data were fitted with Gaussians (ESI†), whose centers at 2θ were divided by two to obtain the angle of incidence required to calculate basal spacings.
Attenuated total reflectance infrared (IR) spectra of dry nanoparticulate powders were collected with a Thermo Nicolet iS50 FT-IR spectrometer, equipped with a Pike Technologies GladiATR accessory plate, an uncooled pyroelectric deuterated triglycine sulfate (DTGS) detector, and a KBr beamsplitter. Spectra of the solid nanoparticulate powders were collected at room temperature in ambient air, and 132 scans were averaged for each sample.
Electrochemical characterisation
Nanosheet powders were weighed with a high precision balance (Sartorius CPA225D), and aqueous 2 mg mL−1 suspensions were prepared. A catalyst loading of 40 μg was used throughout this work and was obtained by drop-casting 20 μL of these suspensions on highly-ordered pyrolytic graphite (HOPG) disks, which were dried in ambient air under a heat lamp at 50 °C. The electrode disk had stabilizing epoxy around its side and a surface area of 0.20 cm2. HOPG electrodes were cleaned by soaking for 5 min in concentrated hydrochloric acid, washed with water, and their surfaces were polished using 400 and 600 grit sandpaper, after which the graphite was cleaved with adhesive tape to obtain a fresh HOPG surface for each catalyst.Measurements were carried out in ambient air or argon atmosphere in 100 mL three-neck round-bottom flasks, filled with 25 mL electrolyte. An Hg/HgO reference electrode (CH Instruments) and a Ni gauze (Alfa) counter electrode were used. All data were collected at room temperature, using a rotating disk electrode (RDE) setup at 1500 rpm. Experiments in Ar atmosphere were carried out in a glove box, whose catalyst tolerates water; during oxygen evolution the catalyst was shut off, and after experiments the box was thoroughly flushed with Ar. In ambient air, a Pine MSR variable speed rotator and a Gamry Reference 600 potentiostat were used. In Ar atmosphere, a Pine WaveNow potentiostat and a (smaller) home-built RDE apparatus were employed; the rotation speed was determined to be 1500 rpm with a laser tachometer.
In ambient air, the electrolyte was aqueous 1.0 M (pH 14.0) KOH. The electrolyte took up CO2 from ambient air, which was dissolved as carbonate at this high pH. Even highest purity reagent grade alkali hydroxide solutions contain a minimum of 1% (∼0.1 M) alkali carbonate.32 Measurements in virtually carbonate-free electrolyte were performed under Ar atmosphere in a glove box. The electrolyte was prepared in the box from thoroughly degassed water, 1.0 M high purity KOH (Alfa, 99.98%, packaged under Ar), and 0.43 M barium hydroxide (Ba(OH)2·1H2O, Sigma-Aldrich). The barium hydroxide was added in excess to precipitate all dissolved carbonate as BaCO3, rendering the supernatant electrolyte virtually free of carbonate (see IR data below). Before use, an Hg/HgO reference electrode was equilibrated for one week in this solution. The pH of the virtually carbonate-free electrolyte was measured to be 14.6. Electrolyte from the same stock solution was used for all experiments requiring carbonate-free electrolyte.
Chronopotentiometry was performed for 3.5 hours at 1 mA cm−2. Observed fluctuations in the data were due to formation and release of oxygen bubbles from the electrode surface. Cyclic voltammograms were measured at 0.1 V s−1 scan rate and after the working electrode had been held for 10 min at a current density of 1.0 mA cm−2. The current density versus potential data were post-measurement corrected for uncompensated resistance losses (details are described elsewhere).20 All polarization potentials reported here are relative to the normal hydrogen electrode (NHE), and current densities are per geometric area. Overpotentials η were calculated from polarization potentials Ep as η = Ep − (1.23 V − 0.059 V pH), taking into account the different pH values of electrolytes with and without carbonate.
Computational methods
Density-functional theory calculations were performed with the Turbomole program package.33 The PBE0 hybrid functional34–36 was used throughout this work. The SV(P) basis set37 was employed for geometry optimizations as well as for N 1s core level energy calculations. Solvent screening effects were approximately included via the conductor-like screening model (COSMO)38,39 with a solvent permittivity of 80. All calculations were spin-unrestricted. In this work, we imposed the lowest total spin projection Sz = 0.5 possible for the cluster models used in this work (one excess spin-up spin orbital in the Slater determinant; see ESI† for further details). No point group symmetries were assumed. The model clusters (Fig. 12 and 13; ESI†) were subjected to constrained geometry optimizations, where the atomic Ni, O, H positions of the parent LDH structure were kept fixed at the experimental positions, whereas the positions of all other atoms (Fe, additional OH groups, nitrate/nitrite anions) were optimized (more details and Cartesian atomic positions are in the ESI†). Standard convergence criteria (Turbomole defaults) were used in general. Only for the calculation of the core-level binding energies, SCF convergence criteria were tightened to 10−7 a.u. for the energy and density matrix.The calculation of N 1s core-level binding energies (BEs) was performed using N 1s orbital energies and with the so-called Slater transition state (STS) method.40 Initial state contributions to core level shifts (is-CLS) were estimated as orbital energy differences of the N 1s orbital energies ε in the chosen SCF solutions of the clusters without any core hole. A limited overview of the performance of the approach for nitrate and nitrite is given in Table S2 (ESI†). The adequacy of the SV(P) basis set for N 1s core level calculations was verified (Table S3, ESI†), and the influence of additional explicit water molecules on N 1s binding energy shifts was discussed (Table S4, ESI†). Only shifts in core-level binding energies (core-level shifts, CLS) were relevant in this work, absolute binding energies should not be compared to experimental XPS data. Further detail on the calculations and additional results can be found in the ESI.†
3. Results and discussion
We synthesized twelve materials with different interlayer anions, either by anion exchange from [NiFe]-LDH nanosheets made by PLAL from nitrate-containing ablation liquid,20 denoted [NiFe]–(NO3−)-LDH, or with Cl− or SO42− in the PLAL ablation liquid. Aqueous solutions for anion exchange contained 1.0 M BF4−, Cl−, ClO4−, CO32−, C2O42−, F−, I−, PO43−, or SO42−.We discovered that all interlayer anions quickly exchanged for carbonate in 1.0 M aqueous KOH in ambient air, as evidenced by IR, XPS, and XRD data (see below). Strongly alkaline aqueous electrolyte exposed to ambient air is self-buffered regarding carbonate content, as dissolved carbonate is in steady-state equilibrium with gaseous CO2.41 The active catalyst was therefore the carbonate-containing species, regardless of the precatalyst composition.
Physical characterisation of materials
The Ni 2p core-level binding energies of all catalysts were consistent with assignment to hydrated Ni(OH)2,43 with Ni 2p3/2 binding energies close to 855.5 eV. After exposure to 1.0 M aqueous KOH in ambient air, all [NiFe]-LDH materials featured an additional, lower binding-energy peak in the Ni 2p3/2 core level region, consistent with NiO.44,45 The Fe 2p core level spectra of all catalysts showed peaks attributable to iron oxides and oxyhydroxides,13,46 with Fe 2p3/2 binding energies close to 711.9 eV. Various iron oxides and oxyhydroxides, such as FeO, Fe2O3, Fe3O4, and FeOOH, have similar Fe core-level binding energies and spectral shapes.46 Therefore, it is impossible to distinguish different Fe phases in our materials from Fe 2p XPS data. All materials contained 22% Fe relative to the total metal content.
The N 1s core level spectra of [NiFe]–(NO3−)-LDH, [NiFe]–(Cl−)-LDH made by PLAL and [NiFe]–(SO42−)-LDH made by PLAL showed peaks with binding energies around 407.3 eV, consistent with nitrate.47 In addition, [NiFe]–(SO42−)-LDH made by PLAL exhibited an N 1s peak at 403.5 eV, attributable to nitrite.48 As reported by us before, PLAL-synthesized [NiFe]–(NO3−)-LDH featured in addition to the nitrate signal an N 1s peak centered at 405.1 eV.20 After exposure to 1.0 M aqueous KOH in ambient air, we could no longer detect any peaks in the N 1s core level region, indicating that all nitrogen species were readily exchanged by other anions. The O 1s spectra were consistent with Fe or Ni oxide and hydroxide species.44 Contributions attributable to oxygen-containing anions44 and from adventitious sources were also present. The C 1s spectra of as-synthesized [NiFe]-LDH materials with different interlayer anions showed mostly the presence of adventitious carbon, whereas those of nanosheets that had been exposed to 1.0 M aqueous KOH in ambient air exhibited additional peaks, which were consistent with more highly oxidized carbon.44 High-resolution XP spectra of anionic hetero-atoms (other than Ni, Fe, C, N, or O) showed that the initially present anions were lost after exposure to pH 14 aqueous KOH solution in ambient air (ESI†). Quantification of non-metal atom content in the as-synthesized [NiFe]-LDH materials is summarized in Table 1. Additional hydroxide ions may have been present to balance the overall charge in the materials.
| Material | XPS line | % Non-metal atoms |
|---|---|---|
| [NiFe]–(NO3−)-LDH (PLAL) | N 1s | 10 |
| [NiFe]–(BF4−)-LDH (exch.) | B 1s | 18 |
| F 1s | 72 | |
| [NiFe]–(Cl−)-LDH (PLAL) | Cl 2s | 16 |
| N 1s | 4.7 | |
| [NiFe]–(Cl−)-LDH (exch.) | Cl 2s | 21 |
| [NiFe]–(ClO4−)-LDH (exch.) | Cl 2s | 10 |
| [NiFe]–(CO32−)-LDH (exch.) | C 1s | 20 |
| [NiFe]–(C2O42−)-LDH (exch.) | C 1s | 20 |
| [NiFe]–(F−)-LDH (exch.) | F 1s | 18 |
| [NiFe]–(I−)-LDH (exch.) | I 3d | 19 |
| [NiFe]–(PO43−)-LDH (exch.) | P 2s | 8.0 |
| [NiFe]–(SO42−)-LDH (PLAL) | S 2s | 10 |
| N 1s | 4.2 | |
| [NiFe]–(SO42−)-LDH (exch.) | S 2s | 17 |
Knowing that all nitrogen species of the as-synthesized [NiFe]–(NO3−)-LDH nanosheets were replaced by carbonate in strong aqueous base in ambient air, we exposed [NiFe]–(NO3−)-LDH to aqueous KOH solutions with pH values of 8.0, 11.0, and 14.0 and took XP spectra (Fig. 2). For comparison, we also collected XP spectra of commercial Ni(NO3)2·6H2O, whose N 1s region featured a single peak centered at 407.4 eV, consistent with previous reports for metal nitrate.48 The observed C 1s signal for Ni(NO3)2·6H2O was attributable to adventitious carbon, and the O 1s peak was consistent with a transition metal nitrate.44 Its Ni 2p3/2 peak was centered at 857.0 eV as expected,44 and no peaks in the Fe 2p core level region were observed.
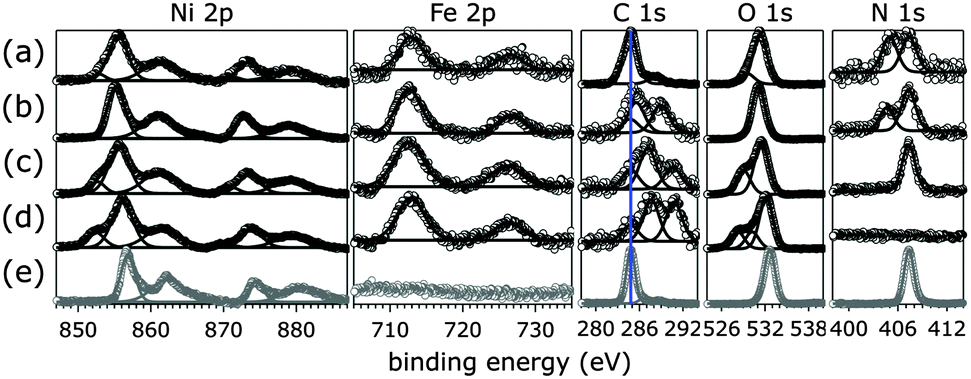 | ||
| Fig. 2 XPS data of [NiFe]–(NO3−)-LDH (as synthesized, a) and exchanged in different pH aqueous KOH solutions (b, pH 8.0; c, pH 11.0; d, pH 14.0). Depicted in grey are data of commercial Ni(NO3)2·6H2O (e). The blue line in the C 1s panel indicates the binding energy of adventitious carbon. Open circles, data; lines, peak fits. | ||
We found that all nitrogen species of the original material were gradually exchanged into carbon species with higher C 1s binding energy than adventitious carbon. As the solution pH rose, the N 1s peak centered at 405.1 eV disappeared before that at 407.3 eV. After exposure to pH 14.0 solution in ambient air, no nitrogen species were detectable (Fig. 2). Instead the material took up carbonate from the aqueous base (see also IR spectra below). Quantification of XPS signals as a function of solution pH showed a linear decline of N 1s and a concomitant increase of C 1s signals (ESI†). In aqueous KOH solutions at room temperature with pH values above 10, the predominant species resulting from dissolution of ambient CO2 is carbonate, whereas at pH 8.0 dissolved inorganic carbon speciation favors HCO3− over CO32− ions.41 The observed C 1s signals of [NiFe]-LDH materials after exposure to different pH aqueous KOH solutions were consistent with transition metal bicarbonates and carbonates.44
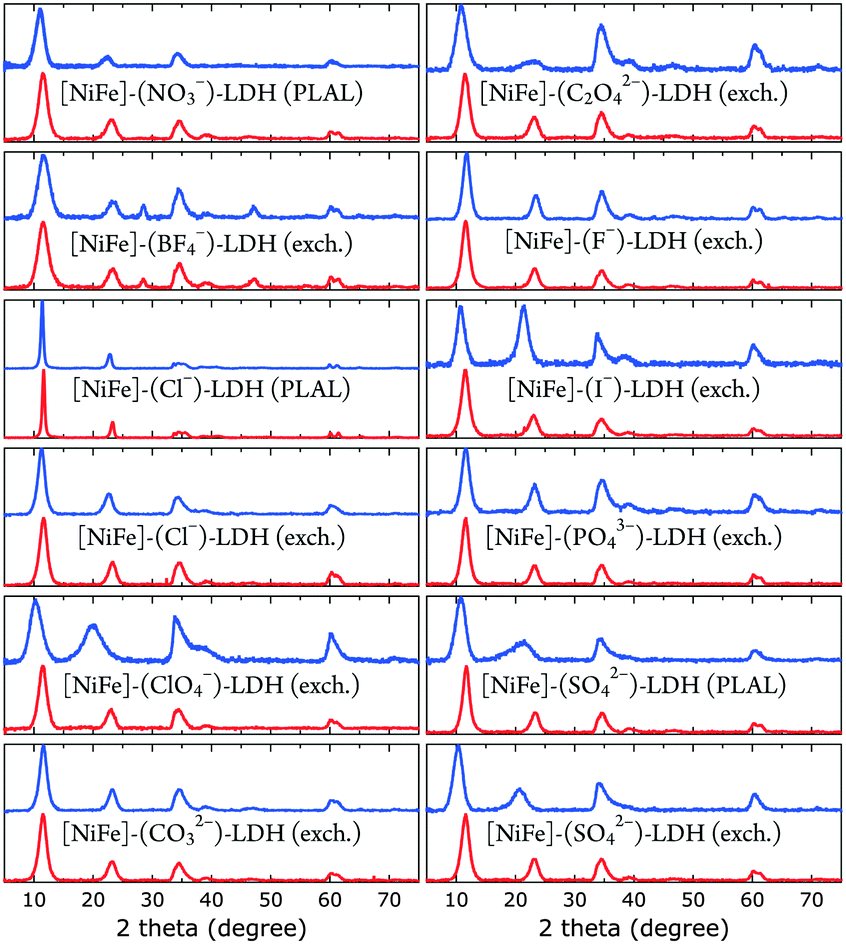 | ||
| Fig. 3 XRD data of [NiFe]-LDH nanosheets with different interlayer anions as synthesized (blue) and after suspension in 1.0 M aqueous KOH in ambient air (red). | ||
All materials showed XRD patterns characteristic for layered double hydroxides.23 The intensities of the basal (00l) reflections decrease as l increases. The intensity ratio of the (006) and (003) reflections is a measure of the interlamellar electron density. Loss of water in the interlayer galleries, associated with reduction in interlamellar electron density, has been reported to lead to less intense (006) reflections with respect to (003) diffractions.49
All XRD peaks were significantly broadened, owing to small crystallite size and stacking faults. The XRD data of [NiFe]–(ClO4−)-LDH (exch., as synthesized) and [NiFe]–(SO42−)-LDH (PLAL, as synthesized) materials showed non-uniform (“saw-tooth”) broadening of (h0l) reflections, which are indicative of turbostratic disorder.25 The observed reflections were indexed in a three-layer 3R polytype with rhombohedral symmetry, such as in synthetic hydrotalcite50 (ESI†).
We obtained crystallite sizes from Scherrer analysis29 of our XRD data (Table 2). Crystalline domain sizes in the a and c unit cell directions were derived from broadening of the (110) and (00l) (l = 3, 6) reflections, respectively.25 In case of [NiFe]–(ClO4−)-LDH (exch., as synthesized) and [NiFe]–(SO42−)-LDH (PLAL, as synthesized) materials, only (003) reflections were used to determine nanosheet thicknesses, as the asymmetric line shape of the (006) reflections hampered proper analysis. The unit cell directions a and c correspond to lateral size (diameter) and nanosheet thickness, respectively. Most materials had very similar size. However, the [NiFe]–(Cl−)-LDH (PLAL) materials, both as synthesized and after suspension in 1.0 M aqueous KOH in ambient air, exhibited significantly narrower peaks, indicating larger crystallite sizes.
| Am− (prep.) | Thickness (nm) as synth. | Thickness (nm) after base | Diameter (nm) as synth. | Diameter (nm) after base |
|---|---|---|---|---|
| NO3− (PLAL) | 4 | 4.4 | 13 | 13 |
| BF4− (exch.) | 3.9 | 3.9 | 14 | 12 |
| Cl− (PLAL) | 12 | 13 | 21 | 23 |
| Cl− (exch.) | 4.8 | 4.7 | 13 | 14 |
| ClO4− (exch.) | 3.2 | 4.5 | 12 | 12 |
| CO32− (exch.) | 4.8 | 4.6 | 12 | 13 |
| C2O42− (exch.) | 3.6 | 4.6 | 10 | 13 |
| F− (exch.) | 3.9 | 4.7 | 11 | 14 |
| I− (exch.) | 4.7 | 4.5 | 12 | 13 |
| PO43− (exch.) | 4.5 | 4.8 | 11 | 14 |
| SO42− (PLAL) | 3.8 | 5.2 | 12 | 14 |
| SO42− (exch.) | 3.8 | 4.7 | 10 | 13 |
Gaussian fits of the (003) peaks (ESI†) were performed to obtain the angle of incidence required to calculate basal spacings. The fits had an average error in 2θ of ±0.05°, resulting in a basal spacing error of ±0.03 Å. The derived basal spacings of [NiFe]–(Am−)-LDH materials with different intercalated anions Am−, prepared by PLAL or by anion exchange from [NiFe]–(NO3−)-LDH (exch.), are depicted in Fig. 4.
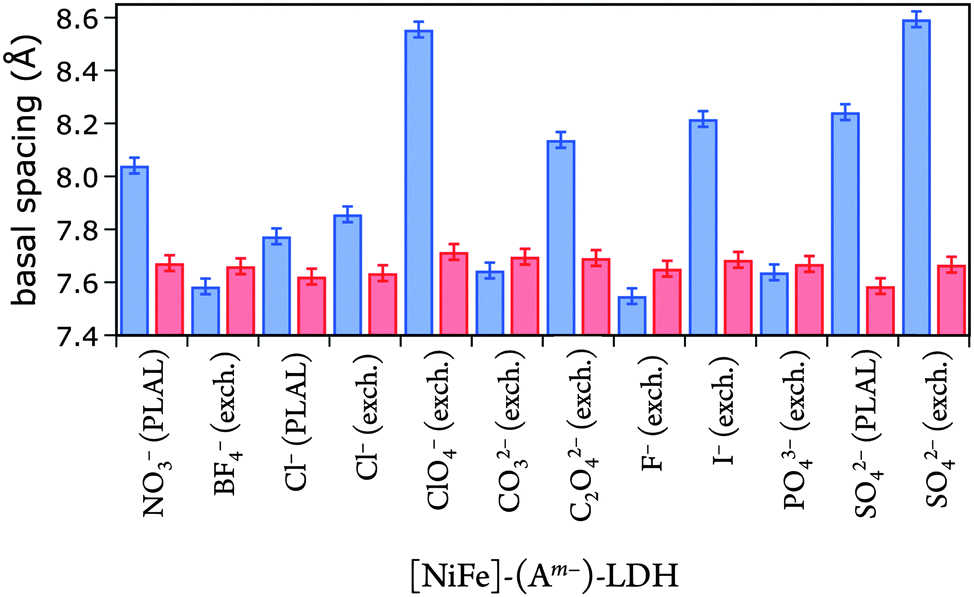 | ||
| Fig. 4 Basal spacings of [NiFe]-LDH nanosheets with different interlayer anions as synthesized (blue) and after suspension in 1.0 M aqueous KOH in ambient air (red). | ||
We correlated the basal spacings of [NiFe]-LDH materials with the ionic radii of the anions. Non-spherical anions occupy interlamellar galleries of LDH materials at various angles, and their arrangement depends on many factors.24,25,51 Therefore, we limited our analysis to the spherical halogen anions. We found a linear correlation of measured basal spacing with anionic radii. Our observation indicates that the anions did indeed reside in the interlayer galleries of our LDH materials (Fig. 5).
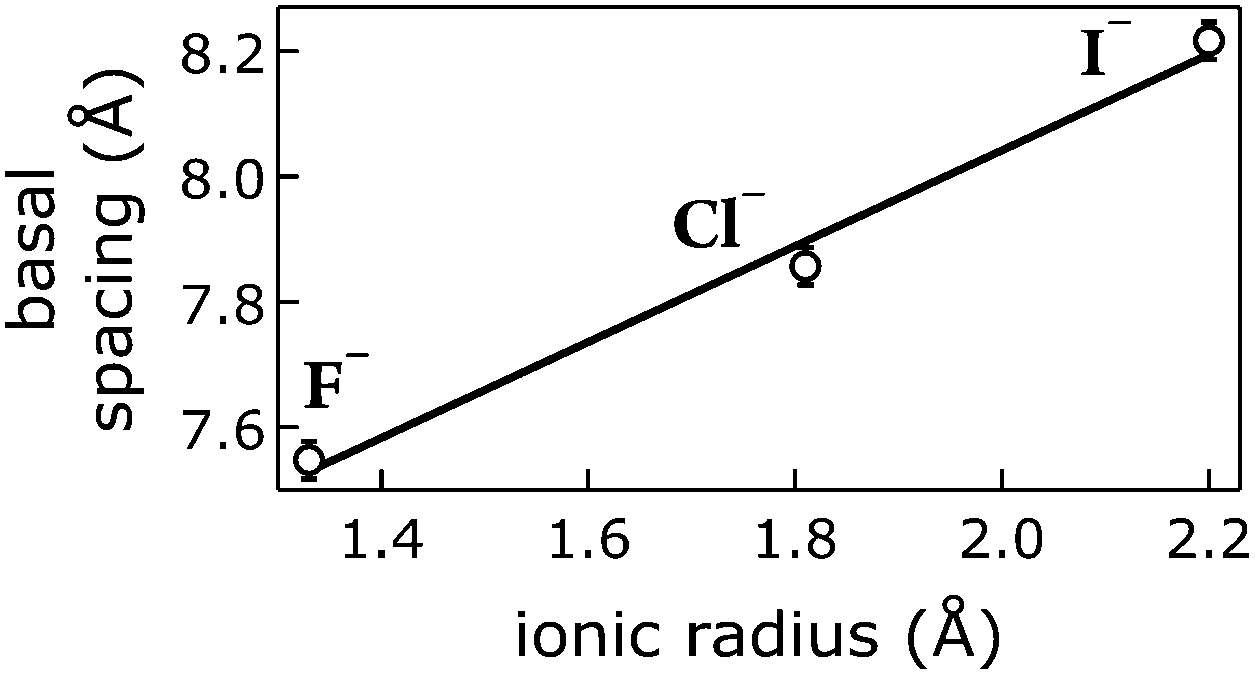 | ||
| Fig. 5 Correlation of ionic radii52 of intercalated halogen anions Am− with basal spacings of [NiFe]–(Am−)-LDH materials. Open circles, data; line, linear fit. | ||
Our [NiFe]–(NO3−)-LDH nanosheets turned into [NiFe]–(CO32−)-LDH materials in strong aqueous base in ambient air (Fig. 6). We confirmed the identity of interlayer carbonate by collecting IR data after soaking [NiFe]–(NO3−)-LDH in 0.1 M aqueous K2CO3 solution.
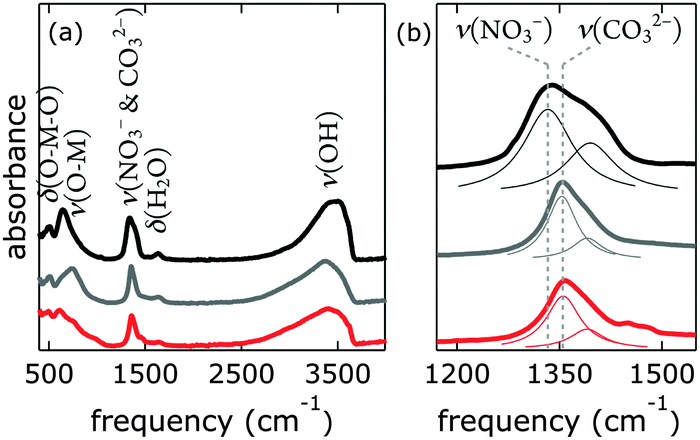 | ||
| Fig. 6 Infrared spectra of [NiFe]–(NO3−)-LDH nanosheets as synthesized (black), after being suspended in 0.1 M aqueous K2CO3 solution (grey), and after being suspended in 0.1 M aqueous KOH (red); (a) full spectra, (b) magnification of the region characteristic for NO3− and CO32−, Lorentzian fits are depicted as thin lines. | ||
One way of de-carbonation is precipitation of dissolved carbonate with Ba2+ as BaCO3.56 We used this method in inert atmosphere (Fig. 7), using Ba(OH)2, as to not introduce anions other than hydroxide. All attempts to achieve carbonate-free electrolyte in ambient air failed (ESI†). Virtually carbonate-free electrolyte was prepared under Ar as described above. In the glove box, we dispersed [NiFe]–(NO3−)-LDH in this electrolyte, let it settle overnight, and removed the supernatant. Still in Ar atmosphere, we dispersed the resulting material in water, let it settle overnight, removed the supernatant, and repeated this procedure twice. After the last supernatant removal, we let the material dry in the glove box. The IR spectrum (Fig. 7) of this material clearly shows the characteristic redshift and broadening of the nitrate with respect to the carbonate band, indicating that we succeeded to exclude carbonate from our [NiFe]–(NO3−)-LDH and electrolyte in the glove box.
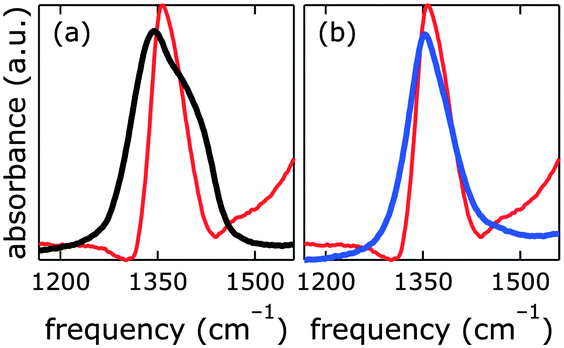 | ||
| Fig. 7 Infrared spectra of [NiFe]–(NO3−)-LDH nanosheets as synthesized (black), after being suspended in carbonate-free strong base electrolyte in Ar atmosphere as described above (blue), and after being suspended in 0.1 M aqueous KOH in ambient air (red). | ||
The nitrate peak in the infrared spectrum of the as-synthesized [NiFe]–(NO3−)-LDH nanosheets was broader than that observed for the [NiFe]–(NO3−)-LDH nanosheets which had been soaked in carbonate-free aqueous base. We attributed this to a change in the morphology of the material at high pH. This is in agreement with the dramatic changes observed in the OH and Ni–O lattice regions upon exposure to highly alkaline solutions (see Fig. S12, ESI†).
Electrochemical characterisation
We prepared 1.0 M aqueous KOH electrolyte in a virtually CO2-free atmosphere (glove box) and scrubbed the solution of any residual dissolved carbonate by saturating with barium hydroxide. IR data confirmed that the [NiFe]-LDH catalysts did not take up carbonate under these conditions (see above). Constant current electrolysis was used to assess long-term water oxidation activity; measured potentials were converted to overpotentials η to facilitate comparison between electrolytes with and virtually without carbonate. In Fig. 8, the overpotentials as a function of time are shown for [NiFe]–(NO3−)-LDH in alkaline electrolytes with and without carbonate. The [NiFe]–(CO32−)-LDH catalyst performed better than the [NiFe]–(NO3−)-LDH, indicating that interlayer anions matter during water oxidation catalysis.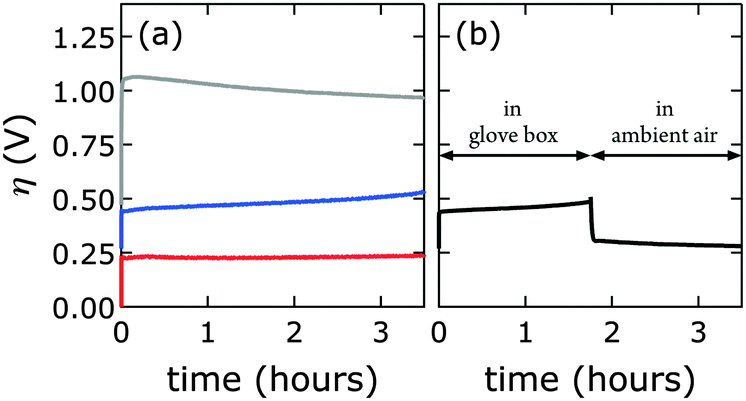 | ||
| Fig. 8 Constant current electrolysis of [NiFe]–(NO3−)-LDH (PLAL) at 1 mA cm−2. Measured applied potentials were converted to overpotentials η to account for the different pH values of alkaline electrolytes with and virtually without carbonate. (a) Synthesized nanosheets were exposed to only one electrolyte: virtually carbonate-free (glove box, blue), with carbonate (ambient air, red). The grey curve corresponds to the bare graphite electrode. (b) A [NiFe]–(NO3−)-LDH electrode was first anodically polarized in virtually carbonate-free electrolyte (glove box), then brought into ambient air and further polarized in electrolyte with carbonate. | ||
Interestingly, after water oxidation catalysis with [NiFe]–(NO3−)-LDH catalyst in virtually carbonate-free alkaline electrolyte, most of the activity of the material recovered when the electrode was removed from the glove box, and the electrolysis was continued in aqueous base in ambient air (Fig. 8b). Enhanced catalytic activity was observed instantaneously as [NiFe]–(CO32−)-LDH was formed. The [NiFe]-LDH nanosheets were regenerated in aqueous base in ambient air, and high activity was detected even after initial catalytic turnover without any carbonate (Fig. 8). Differences in electrochemical response of the bare graphite electrode in carbonate-free and carbonate-containing electrolytes were insignificant (Fig. S8, ESI†).
We probed all twelve materials in a virtually carbonate-free alkaline electrolyte to assess the effects of different interlayer anions (other than hydroxide) in the precatalysts on water oxidation catalysis. We obtained overpotential values at 1 mA cm−2 current density after 10 min of catalytic turnover. At the chosen current density most catalysts exhibited appreciable stability under anodic polarization. We compared activities in carbonate-free and carbonate-containing alkaline electrolytes. In all cases, catalytic activity in aqueous base in ambient air was superior (Fig. 9).
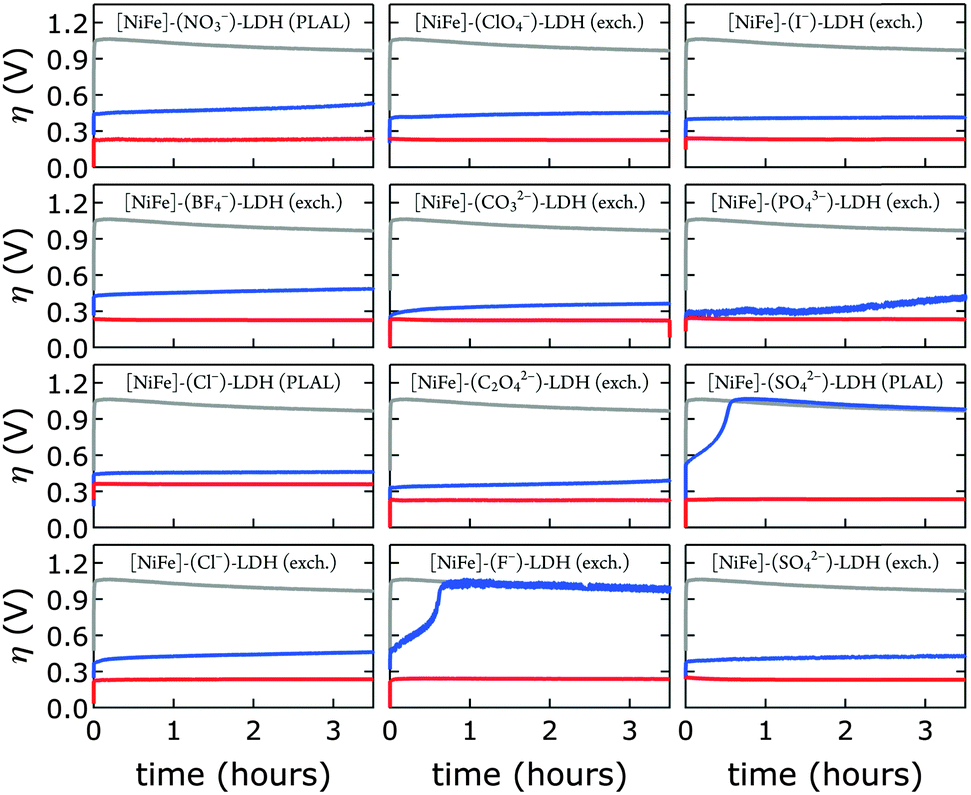 | ||
| Fig. 9 Constant current electrolysis of [NiFe]-LDH materials with different interlayer anions in virtually carbonate-free electrolyte in a glove box (blue) and in 1.0 M aqueous KOH in ambient air (red). Chronopotentiometry data of bare HOPG are shown in grey. All data were collected at a constant current density of 1 mA cm−2. | ||
After exposure to carbonate-containing electrolyte (1.0 M aqueous KOH in ambient air), all materials immediately featured low, very similar overpotentials, consistent with formation of [NiFe]–(CO32−)-LDH materials; only the [NiFe]–(Cl−)-LDH material synthesized by PLAL, which consisted of larger nanosheets (see Table 2), exhibited slightly inferior performance. Likewise, the cyclic voltammograms of all materials after exposure to 1.0 M aqueous KOH in ambient air were very similar; again, the [NiFe]–(Cl−)-LDH synthesized by PLAL showed slightly less current density (ESI†). All materials exhibited remarkable stability over several hours in carbonate-containing (self-buffered) alkaline electrolyte.
We discovered a strong correlation between water oxidation activity and the pKa values of the conjugate acids of the interlayer anions (ESI†); there is sigmoidal behavior with a midpoint of 3.4 ± 0.7 (Fig. 10).
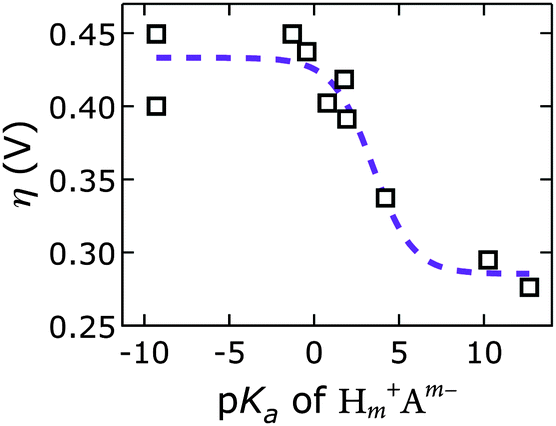 | ||
| Fig. 10 Overpotentials η of [NiFe]-LDH materials with different interlayer anions Am− derived from constant current electrolysis at 1 mA cm−2 in virtually carbonate-free electrolyte as a function of anion basicity. Open squares, data; dashed line, sigmoidal fit. | ||
Mechanistically, this behavior suggests a base-assisted deprotonation step during turnover. The apparent pKa, determined electrochemically, may be related to the pKa of a higher-valent metal–oxo species, which is typically much lower than 7. This effect manifests as an “oxo wall” between the Fe and Co triads of the periodic table.57 Transition metals to the left of the wall make stable oxo complexes, whereas those to the right are too basic and are easily attacked by electrophiles. Different Lewis basicities, which would lower the potential required to reach the necessary Fe and/or Ni oxidation states, could also be at play.
Our discovery of the sigmoidal dependence of water oxidation activity on anion basicity is perhaps surprising, given that there is 1 M OH− in the electrolyte. Hydroxide is of course also a good base. It is unclear, however, how much hydroxide resides in the interlayer galleries. Our results imply that the di- and tri-valent anions outcompeted hydroxide presence in the interlayer space. This is consistent with previous reports on anion affinities in LDHs (see above).24 Our observation that the [NiFe]–(CO32−)-LDH catalyst regenerated in carbonate-containing aqueous base and showed exceptional robustness supports our conclusion that this higher-valent anion was strongly bound and played an active role during turnover. We cannot exclude that in the regime of lower activity, where we have monovalent interlayer anions (to the left in Fig. 10), the catalysis is governed by hydroxide-mediated deprotonation. It is important to note, however, that catalytic activity can be enhanced by di- and tri-valent anions. The higher charge of multivalent anions makes them stronger proton acceptors and electron donors than monovalent anions. It is possible that a strongly bound proton acceptor is required to reduce the activation barrier for water oxidation.
Data correlations
We emphasize that we did not find that catalytic activity was a function of Ni3+/2+ prewave position (ESI†),58 indicating that the Ni3+/2+ couple is not the rate-determining step for water oxidation. Likewise, measured overpotentials did not depend on the slab [NiFe](OH)2 structure, as evidenced by analysis of far-IR spectra (ESI†); intensities of characteristic OH-deformation modes as a function of soaking solution pH showed sigmoidal behavior with a midpoint of approximately 12, whereas overpotentials vs. pKa values from anion-conjugate-acid data had a midpoint of 3.4 ± 0.7, indicating that intrasheet structural changes were unrelated to observed activities.Perhaps surprisingly, measured overpotentials also did not depend on the basal spacings. In Fig. 11, overpotentials of [NiFe]-LDH materials with different interlayer anions, collected in virtually carbonate-free electrolyte (glove box), are plotted as a function of the materials' basal spacings. Layer-to-layer spacings would only matter if reactants and products had to diffuse though the interlamellar galleries for redox reactions mediated by (central) intrasheet species. Clearly, this is not the case for our materials.
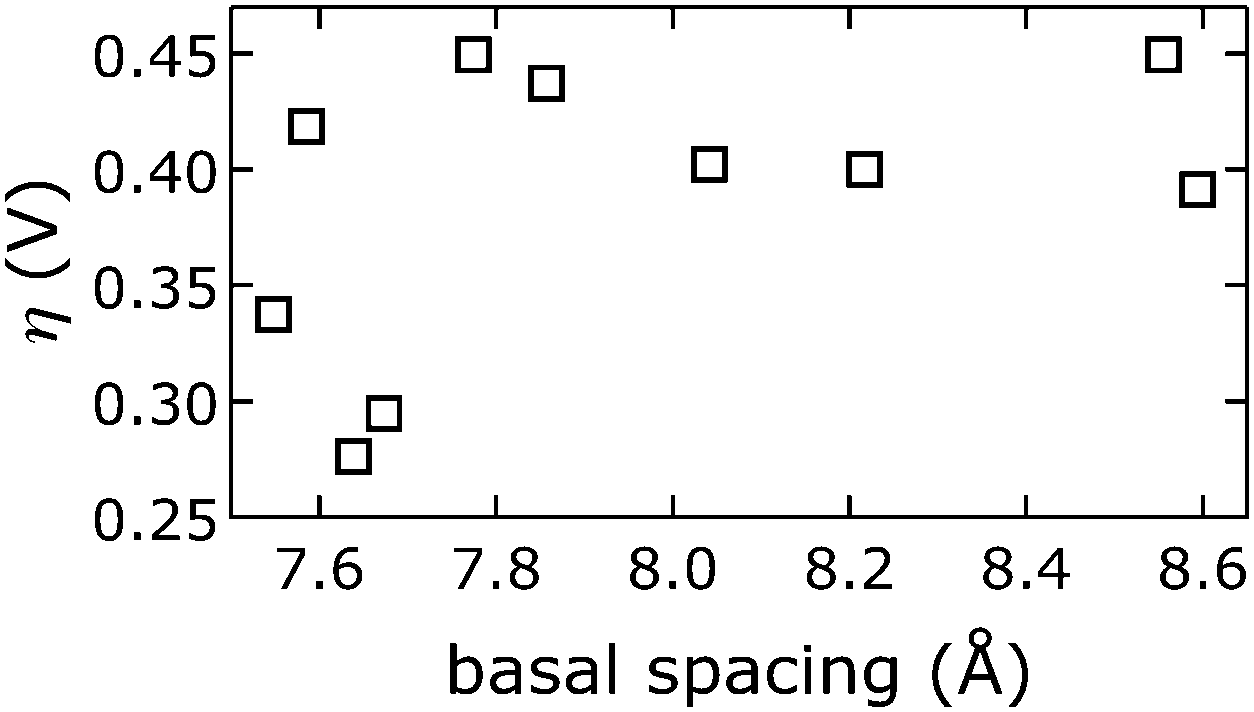 | ||
| Fig. 11 Observed overpotentials η at 10 min from chronopotentiometry data collected in virtually carbonate-free electrolyte (glove box) as a function of the basal spacing of [NiFe]-LDH materials with different interlayer anions. | ||
Density functional theory calculations
We recently reported two distinct N 1s signals in the XPS data of PLAL-synthesized [NiFe]–(NO3−)-LDH precatalysts.20 One peak was centered at 407.3 eV, attributable to nitrate,47 and the other at 405.1 eV, which we tentatively ascribed to nitrate or nitrite in an unusual chemical environment. Curiously, the turnover frequency of [NiFe]–(NO3−)-LDH precatalysts with different Ni/Fe ratios depended on the relative proportion of the two XPS peaks.20We performed DFT calculations on model LDH clusters, [Ni9Fe1(OH)20(NO2or3)], to obtain information about the previously unassigned N 1s XPS signal at 405.1 eV. We wanted to find out if binding nitrate or nitrite to the [NiFe]-LDH nanosheets in various positions and binding motifs could explain the observed N 1s binding energy (BE) of 405.1 eV. This BE would require either (i) a lowering of the N 1s BE of nitrate by 2.2 eV or (ii) an increase of the N 1s BE of nitrite by 1.6 eV. We calculated N 1s core level shifts relative to a nitrate anion, which was bound by a hydrogen bridge to the top-side of the cluster (ESI,† Fig. S15). Technical details concerning the calculation of the N 1s core level binding energies and the choice of the cluster models are described above and in the ESI.†
We assessed the binding of nitrate and nitrite anions to different Fe sites within our prolate model clusters (central, edge at the rim along the long axis, and corner at the short-axis rim, Fig. 12). All geometries can be inspected in detail using the Cartesian coordinates given in the ESI.†
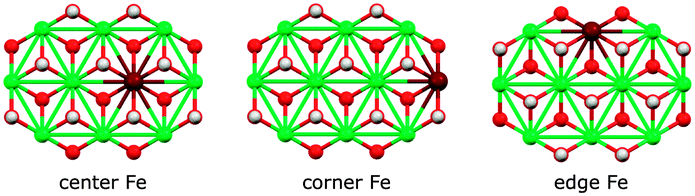 | ||
| Fig. 12 Fe sites in the basic [Ni9Fe1(OH)18]3+ cluster fragment. Colors: Ni green, Fe maroon, O red, H white. | ||
We also investigated different anion binding motifs: nitrate binding via one of its O atoms, nitrite binding by both O and N atom coordination (Fig. 13).
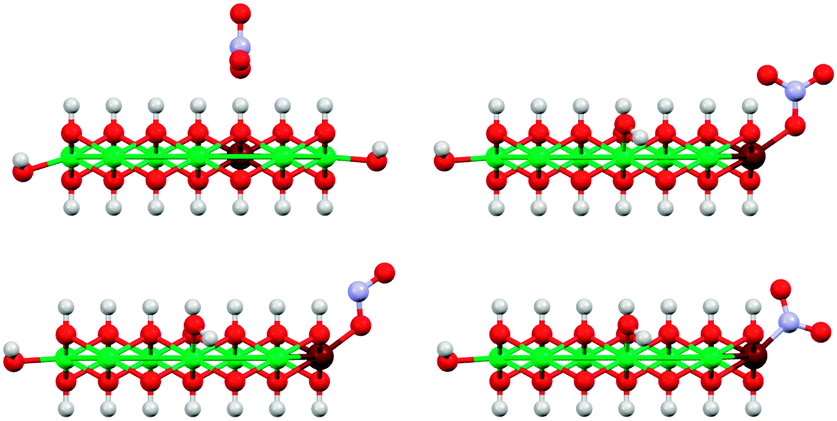 | ||
| Fig. 13 Illustration of the basic anion coordination motifs of NO3− (top) and NO2− (bottom) to [Ni9Fe1(OH)20] clusters. Only coordination to corner sites of the clusters are shown here, other sites were also investigated (Fig. 12). The top left structure shows the reference for the calculation of core level shift from core level binding energies (see also ESI†). Colors: Ni green, Fe maroon, N blue, O red, H white. | ||
The experimentally observed N 1s XPS peak at 405.1 eV corresponds to a core level shift of −2.2 eV with respect to the electron binding energy of pristine nitrate at 407.4 eV.48 Calculations of nitrate binding to the clusters yielded core level shifts that were inconsistent with the XPS signal at 405.1 eV (Fig. 14 and ESI,† Table S6). However, N-coordination of nitrite to edge-site Fe resulted in a core level shift of −2.1 eV (initial state contribution, see ESI† for details), which is in very close agreement with the experimentally observed shift. Other nitrite binding motifs were not in accord with the experimental XPS data (ESI†). We note that formation of nitrite was possible during our PLAL from nitrate-containing ablation solutions, as evidenced by the fact that PLAL-made [NiFe]–(SO42−)-LDH exhibited an N 1s peak at 403.5 eV, attributable to nitrite (see above).48
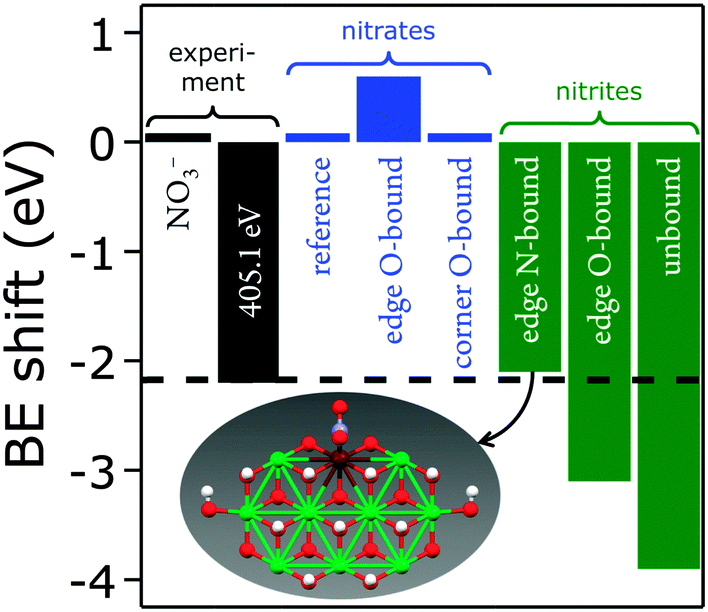 | ||
| Fig. 14 Measured (black) and calculated XPS binding energy (BE) shifts (is-CLS) for differently bound nitrates (blue) and nitrites (green) with a calculated structure that is consistent with the feature at 405.1 eV. Atom colors: Ni green, Fe maroon, N blue, O red, H white. | ||
Assignment of the experimentally observed 405.1 eV N 1s peak of the [NiFe]–(NO3−)-LDH nanosheet precatalyst to nitrite bound by its N-atom to edge-site Fe suggested that this XPS signal provided a means to quantify Fe edge sites. It also rationalized why we had previously observed higher catalytic turnover frequencies as the relative proportion of the 405.1 eV peak with respect to that at 407.3 eV increased.20 Therefore, we conclude that edge-site Fe plays a major role in water oxidation catalyzed by our PLAL-synthesized [NiFe]-LDH nanosheets. Our calculations are consistent with our observation that the overpotentials of [NiFe]-LDH materials with different interlayer anions did not correlate with basal spacings.
Conclusions
We synthesized a series of [NiFe]-LDH nanosheets with various intercalated anions, both directly by PLAL and by anion metathesis. We found that their water oxidation activity correlated with the pKa of the conjugate acid of the interlayer anions and propose that the anions' Brønsted or Lewis basicity plays a role in the water oxidation mechanism. Our nanocatalysts were regenerated and most active in alkaline electrolyte in ambient air, as carbonate rapidly replaced other interlayer anions. Finally, our DFT calculations suggest that a previously unassigned N 1s species in the precatalyst, which correlated with higher water oxidation activity,20 is consistent with nitrite bound by its N-atom to edge-site iron. This finding implies that iron sites at the edges of our [NiFe]-LDH nanosheets are active in water oxidation catalysis.Acknowledgements
We thank George Rossman for help with solid-state IR spectroscopy. Research was carried out in the Laser Resource Center and the Molecular Materials Research Center of the Beckman Institute of the California Institute of Technology. This work was supported by the NSF CCI Solar Fuels Program (CHE-1305124) and the Arnold and Mabel Beckman Foundation. B. M. H. is a Fellow of the Resnick Sustainability Institute at Caltech. W. H. thanks the Deutsche Forschungsgemeinschaft and the Cluster of Excellence “Engineering of Advanced Materials” at the University of Erlangen-Nürnberg for support.Notes and references
- H. B. Gray, Nature Chem., 2009, 1, 7 CrossRef CAS PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- Y. Lee, J. H. Choi, H. J. Jeon, K. M. Choi, J. W. Lee and J. K. Kang, Energy Environ. Sci., 2011, 4, 914–920 CAS.
- M. Gong, Y. Li, H. Wang, Y. Liang, J. Z. Wu, J. Zhou, J. Wang, T. Regier, F. Wei and H. Dai, J. Am. Chem. Soc., 2013, 135, 8452–8455 CrossRef CAS PubMed.
- B. Li, Y. Zhao, S. Zhang, W. Gao and M. Wei, ACS Appl. Mater. Interfaces, 2013, 5, 10233–10239 CAS.
- Y. Zhang, B. Cui, C. Zhao, H. Lin and J. Li, Phys. Chem. Chem. Phys., 2013, 15, 7363–7369 RSC.
- X. Zou, A. Goswami and T. Asefa, J. Am. Chem. Soc., 2013, 135, 17242–17245 CrossRef CAS PubMed.
- S. J. Kim, Y. Lee, D. K. Lee, J. W. Lee and J. K. Kang, J. Mater. Chem. A, 2014, 2, 4136–4139 CAS.
- Y. Li, L. Zhang, X. Xiang, D. Yan and F. Li, J. Mater. Chem. A, 2014, 2, 13250–13258 CAS.
- D. Tang, Y. Han, W. Ji, S. Qiao, X. Zhou, R. Liu, X. Han, H. Huang, Y. Liu and Z. Kang, Dalton Trans., 2014, 43, 15119–15125 RSC.
- O. Diaz-Morales, I. Ledezma-Yanez, M. T. M. Koper and F. Calle-Vallejo, ACS Catal., 2015, 5, 5380–5387 CrossRef CAS.
- I. J. Godwin and M. E. G. Lyons, Electrochem. Commun., 2013, 32, 39–42 CrossRef CAS.
- M. Gong, Y. Li, H. Wang, Y. Liang, J. Z. Wu, J. Zhou, J. Wang, T. Regier, F. Wei and H. Dai, J. Am. Chem. Soc., 2013, 135, 8452–8455 CrossRef CAS PubMed.
- M. Gao, W. Sheng, Z. Zhuang, Q. Fang, S. Gu, J. Jiang and Y. Yan, J. Am. Chem. Soc., 2014, 136, 7077–7084 CrossRef CAS PubMed.
- F. Song and X. Hu, Nat. Commun., 2014, 5, 4477–4486 CAS.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- Y. Zhao, B. Li, Q. Wang, W. Gao, C. J. Wang, M. Wei, D. G. Evans, X. Duan and D. O'Hare, Chem. Sci., 2014, 5, 951–958 RSC.
- C. G. Morales-Guio, M. T. Mayer, A. Yella, S. D. Tilley, M. Grätzel and X. Hu, J. Am. Chem. Soc., 2015, 137, 9927–9936 CrossRef CAS PubMed.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef CAS PubMed.
- B. M. Hunter, J. D. Blakemore, M. Deimund, H. B. Gray, J. R. Winkler and A. M. Müller, J. Am. Chem. Soc., 2014, 136, 13118–13121 CrossRef CAS PubMed.
- M. S. Burke, L. J. Enman, A. S. Batchellor, S. Zou and S. W. Boettcher, Chem. Mater., 2015, 27, 7549–7558 CrossRef CAS.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CrossRef CAS PubMed.
- X. Duan and D. G. Evans, Layered double hydroxides, Springer Science & Business Media, 2006 Search PubMed.
- S. Miyata, Clays Clay Miner., 1983, 31, 305–311 CAS.
- D. G. Evans and R. C. Slade, Struct. Bonding, 2006, 119, 1–87 CAS.
- J. D. Blakemore, H. B. Gray, J. R. Winkler and A. M. Müller, ACS Catal., 2013, 3, 2497–2500 CrossRef CAS.
- T. L. Barr and S. Seal, J. Vac. Sci. Technol., A, 1995, 13, 1239–1246 CAS.
- D. A. Shirley, Phys. Rev. B: Solid State, 1972, 5, 4709–4714 CrossRef.
- P. Scherrer, Nachr. Ges. Wiss. Goettingen, Math.-Phys. Kl., 1918, 26, 98–100 Search PubMed.
- J. I. Langford and A. J. C. Wilson, J. Appl. Crystallogr., 1978, 11, 102–113 CrossRef CAS.
- W. H. Bragg and W. L. Bragg, Proc. R. Soc. London, Ser. A, 1913, 88, 428–438 CrossRef CAS.
- J. E. S. Han and T. Y. Chao, Ind. Eng. Chem., Anal. Ed., 1932, 4, 229–232 CrossRef CAS.
- R. Ahlrichs, F. Furche, C. Hättig, W. M. Klopper, M. Sierka and F. Weigend, Turbomole program package, version 6.6, Karlsruhe, Germany, since 1989 (http://www.turbomole.com).
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029–5036 CrossRef CAS.
- J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799 RSC.
- A. Schäfer, A. Klamt, D. Sattel, J. C. Lohrenz and F. Eckert, Phys. Chem. Chem. Phys., 2000, 2, 2187–2193 RSC.
- J. C. Slater, Adv. Quantum Chem., 1972, 6, 92 CrossRef.
- N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, 1984 Search PubMed.
- G. P. Williams, X-ray Data Booklet, 2001 Search PubMed.
- A. N. Mansour and C. A. Melendres, Surf. Sci. Spectra, 1994, 3, 247–254 CrossRef CAS.
- NIST X-ray Photoelectron Spectroscopy Database, X-ray Photoelectron Spectroscopy Database 20, Version 4.1 (National Institute of Standards and Technology, Gaithersburg, 2012); http://srdata.nist.gov/xps/.
- M. C. Biesinger, B. P. Payne, L. W. M. Lau, A. Gerson and R. S. C. Smart, Surf. Interface Anal., 2009, 41, 324–332 CrossRef CAS.
- W. Temesghen and P. Sherwood, Anal. Bioanal. Chem., 2002, 373, 601–608 CrossRef CAS PubMed.
- C. E. Nanayakkara, P. M. Jayaweera, G. Rubasinghege, J. Baltrusaitis and V. H. Grassian, J. Phys. Chem. A, 2013, 118, 158–166 CrossRef PubMed.
- J. Baltrusaitis, P. M. Jayaweera and V. H. Grassian, Phys. Chem. Chem. Phys., 2009, 11, 8295–8305 RSC.
- C. Vaysse, L. Guerlou-Demourgues and C. Delmas, Inorg. Chem., 2002, 41, 6905–6913 CrossRef CAS PubMed.
- T. S. Stanimirova, G. Kirov and E. Dinolova, J. Mater. Sci. Lett., 2001, 20, 453–455 CrossRef CAS.
- S. Carlino, Solid State Ionics, 1997, 98, 73–84 CrossRef CAS.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, FL, USA, 72nd edn, 1991 Search PubMed.
- V. Rives, Mater. Chem. Phys., 2002, 75, 19–25 CrossRef CAS.
- T. Stanimirova, N. Piperov, N. Petrova and G. Kirov, Clay Miner., 2004, 39, 177–191 CrossRef CAS.
- F. A. Miller and C. H. Wilkins, Anal. Chem., 1952, 24, 1253–1294 CrossRef CAS.
- W. L. F. Armarego and D. D. Perrin, Purification of Laboratory Chemicals, Butterworth-Heinemann, Oxford, UK, 4th edn, 1987 Search PubMed.
- J. R. Winkler and H. B. Gray, Struct. Bonding, 2012, 142, 17–28 CrossRef CAS.
- The prewaves of nickel–iron double hydroxide cyclic voltammograms have previously been assigned to the Ni3+/2+ redox couple ( L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed ).
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ee00377j |
| This journal is © The Royal Society of Chemistry 2016 |