
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transition metal carbides as novel materials for CO2 capture, storage, and activation†
Christian
Kunkel
,
Francesc
Viñes
and
Francesc
Illas
*
Departament de Química Fisica & Institut de Química Teòrica i Computacional (IQTCUB), Universitat de Barcelona, c/Martí i Franqués 1, 08028 Barcelona, Spain. E-mail: francesc.illas@ub.edu
First published on 11th December 2015
Abstract
The capture and activation of the greenhouse gas carbon dioxide (CO2) is a prerequisite to its catalytic reforming or breakdown. Here we report, by means of density functional theory calculations including dispersive forces, that transition metal carbides (TMC; TM = Ti, Zr, Hf, Nb, Ta, Mo) are able to uptake and activate CO2 on their most-stable (001) surfaces with considerable adsorption strength. Estimations of adsorption and desorption rates predict a capture of CO2 at ambient temperature and even low partial pressures, suggesting TMCs as potential materials for CO2 abatement.
Carbon dioxide (CO2) is an active greenhouse gas with a rising atmospheric concentration currently approaching a value of 400 ppm. This raises concerns about serious consequences such as climate change and ocean acidification.1 As economies and energy demand are projected to grow in the future,2 a reduction of CO2 emissions nowadays only seems possible with efficient technologies for carbon capture and storage (CCS),3–6 employing absorbents or solvents.7 CO2 re-use into a saleable product has been suggested to be an appealing route to contribute to climate change mitigation, though the field is still in its infancy and questions over overall system efficiency have been raised.7–9 Active catalytic systems for such processes are known, yet most of them are made of scarce and precious metals, normally supported on high surface area porous oxides or sulfides.10,11 The effective large-scale implementation on this basis then still remains a great challenge.11–13
In the search for new active catalysts, numerous endeavours address the adsorption, activation, and subsequent catalytic conversion of CO2 on different systems. Note that a relatively high adsorption energy is needed for CO2 to stick on a catalyst surface, which is made difficult by the high stability of CO2 molecules. Moreover, when possible, CO2 adsorption normally goes without significant activation, a process that is known to require charge transfer from the substrate resulting in a concomitant bending.14 The possible capture/activation of CO2 has been theoretically tackled via first principles calculations on metals,15–17 metal oxides,18,19 graphene-based materials,20 sulfides,21 zeolites,22 and metal–organic frameworks,23 to name a few.
Since the surface chemistry of transition metal carbides (TMCs) was described as comparable to that of Pt-group metals by Levy and Boudart,24,25 these materials have received considerable attention in catalysis. TMC based catalysts have been used in a wide range of reactions like methane dry reforming,26 conversion of methane to synthesis gas,27 desulfurization,28 hydrogenation,29 the water-gas-shift reaction,30 and CO oxidation,31 demonstrating chemical robustness and oftentimes catalytic properties similar to precious Pt-group metals.
The interaction of CO2 and TMC surfaces has been recently reviewed,10,32 although one must acknowledge that most of the studies so far focused on Mo2C, MoC, WC, and TiC. From these studies it is now known that some surfaces of Mo2C, δ-MoC, and TiC are highly active for CO2 hydrogenation, producing methanol, methane, and carbon monoxide (CO) in different ratios.33–35 The catalytic activity of these TMCs has been found to be further enhanced by using them as supports for transition metal nanoclusters.34,36 Regarding CO2 adsorption and activation, density functional theory (DFT) calculations have shown that upon interaction with these surfaces a surface-bound anionic CO2δ− species with bended geometry is formed. Many CO2 surface chemistry studies37,38 evidence the key role of this activated intermediate in further reactions.
Apart from the above-mentioned extensively studied systems, information on the CO2 surface chemistry involving other TMCs is rather limited. A recent study carried out by Porosoff and coworkers35 found CO2 hydrogenation activities for TiC, ZrC, NbC, TaC, WC, and Mo2C. These results largely motivated the present theoretical systematic investigation of CO2 adsorption and activation on TMC surfaces. To facilitate a logical comparison of the results and to circumvent structural aspects and related effects, we restrained our study to TMCs with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 TM:C ratio and a face-centered cubic (fcc) crystallographic structure and, among possible surfaces, we chose the (001) one, known to be most stable one for fcc TMCs.39 In particular, we studied TiC, ZrC, and HfC (group 4), VC, NbC, and TaC (group 5), and δ-MoC (group 6). Note that for MoC fcc packing is only present in the high temperature δ-phase.
:1 TM:C ratio and a face-centered cubic (fcc) crystallographic structure and, among possible surfaces, we chose the (001) one, known to be most stable one for fcc TMCs.39 In particular, we studied TiC, ZrC, and HfC (group 4), VC, NbC, and TaC (group 5), and δ-MoC (group 6). Note that for MoC fcc packing is only present in the high temperature δ-phase.
Periodic DFT calculations aimed at studying the interaction of CO2 with the TMC(001) surfaces have been carried out within the generalized gradient approximation (GGA) using the Vienna Ab-Initio Simulation Package – VASP 5.3.5 code.40 In order to account for exchange correlation effects the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional was used,41 alone or adding the D3 dispersion correction (PBE-D3) developed by Grimme.42 Further computational details are given in the ESI.† For a better understanding of the oncoming discussion it is worth to restate that favourable adsorption energies, Eads, are defined negative, and that interactions are more favourable for lower values of Eads.
In a first step sites that might strongly bind and activate CO2 have been identified. There are MCC, MMC, TopM, and TopC sites as defined by Posada-Pérez et al.33 and all were explicitly considered for CO2 on TiC, VC, and δ-MoC(001) surfaces, thus including cases from group 4 to group 6. In no case Eads values smaller than −0.05 eV were found on the VC(001) surface, and therefore this carbide has been excluded from further discussion. The tested MCC and TopM sites yielded non-activated physisorbed situations, although on δ-MoC(001) with sensible adsorption energies around −0.5 eV.
Regardless of the previous, the adsorption energies on MMC and TopC sites are significantly high, −0.89 and −0.71 eV on δ-MoC(001), and −0.55 and −0.57 eV on TiC(001), as obtained at the PBE level, comparing well with the previously reported values33,43 and comparable to the most favourable cases involving metal surfaces. For instance, for CO2 on Ni(110), Co(110), and Fe(100) Eads values of −0.39 eV, −0.61 eV, and −0.72 eV, respectively, have been reported at the GGA level.15–17 Note, however, that these Eads values are obtained for the most open and, consequently, least stable and most reactive low Miller indices surfaces, at variance with current TMCs, where CO2 is studied on the most stable surface.
Having identified MMC and TopC sites as the most active ones for δ-MoC and TiC we further explored these sites in the rest of TMC(001) surfaces. In order to also investigate the role of dispersive (van der Waals) forces, we contemplated the study also at the PBE-D3 level. In all cases we found a favourable adsorption of CO2, see PBE-D3 Eads values in Table 1; for PBE values we refer to the ESI.† Note from these values that the competition among MMC and TopC sites is especially acute for group 4 TMCs, where the adsorption energy difference ΔEads = Eads(MMC) − Eads(TopC) is at most of 0.04 eV. The difference becomes larger for NbC and δ-MoC(001) surfaces, with ΔEads = −0.17 eV, and reaches a maximum for TaC, where ΔEads = −0.27 eV. It should be mentioned that ΔEads are equal within 0.01 eV for both PBE and PBE-D3 methods.
| E ads | TiC | ZrC | HfC | NbC | TaC | δ-MoC |
|---|---|---|---|---|---|---|
| MMC | −0.81 | −1.56 | −1.62 | −0.87 | −1.21 | −1.20 |
| TopC | −0.83 | −1.60 | −1.65 | −0.70 | −0.94 | −1.03 |
| ΔEads | 0.02 | 0.04 | 0.03 | −0.17 | −0.27 | −0.17 |
From the tabulated values, trends for adsorption energies can be withdrawn. For instance, on 3d, 4d, and 5d TMC(001) Eads rise in magnitude when going down a group. Aside, the adsorption strength decreases when moving along a d series. Indeed, the almost vanishing interaction of CO2 on the VC(001) surface can be understood from the decrease of more than 0.7 eV when going ZrC → NbC, applying the same trend in going from TiC to VC would result in an Eads value of at most 0.1 eV at the PBE level, in full concordance with the obtained results. One must warn, however, that these trends may not hold for other stable surfaces, as the surface structure can strongly alter the adsorptive properties.15,17
Last but not the least, it is mandatory to highlight that, compared to PBE Eads values, PBE-D3 predicts more stable minima by 0.21–0.32 eV. Still, the main adsorption driving force comes from non-dispersive interactions between the surface and the adsorbate, as already found for other TMCs as well.10 In the δ-MoC(001) surface a fair comparison between D3 and D2 dispersion corrections reveals that, compared to PBE,33 the latter yields a slightly larger stabilization (0.4–0.6 eV).
Note that on both MMC and TopC sites, and regardless of the TMC, the adsorbed CO2 molecule exhibits a similar geometry, see examples of the adsorbate structures on the TiC(001) surface in Fig. 1. For each system, a complete list of structural descriptors, including bond lengths and molecular angles, is reported in Table S1 of the ESI.† Briefly, and in agreement with earlier results, C ↔ C and metal ↔ O interactions between CO2 and surface site atoms imply bond lengths on the order of 1.46–1.50 and 2.09–2.37 Å, respectively. Compared to MMC sites, the latter are circa 0.2 Å longer on TopC. The adsorbed CO2 species always bend with angles α(OCO) between 120.1 and 128.8° which is a strong evidence of CO2 activation by charge transfer from the underlying TMC surface.10
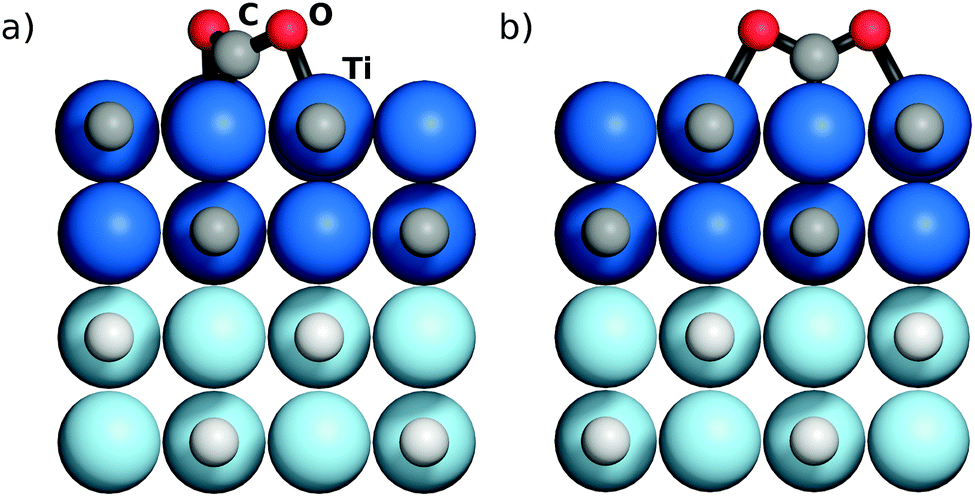 | ||
| Fig. 1 Side sketches of CO2 adsorbed on (a) MMC and (b) TopC sites of the TiC(001) surface. Atom labels indicate the sphere colouring. Lighter colour layers were fixed during optimization. For respective top views see Fig. S1 of the ESI.† | ||
This is also consistent with C–O bond lengths between 1.29 and 1.32 Å, significantly longer than the 1.176 Å value of CO2 in a vacuum.
Given the relatively high adsorption energies, there is a clear interest in estimating the temperature fringe below which these TMCs could capture and accumulate CO2. This can be easily assessed from adsorption and desorption rates, rads and rdes, respectively, which can be estimated in the framework of the transition state theory (TST). Hence, rads and rdes values have been calculated over a temperature range up to 1000 K with necessary quantities obtained from the above commented DFT calculations. The utilized equations and approaches are duly explained in the ESI.† In a nutshell, the adsorption rate depends on the impingement of CO2 to the surface and therefore, on the CO2 partial pressure (pCO2). In this sense, and for each system, rads has been calculated for (i) the current atmospheric partial pressure of CO2, pCO2 = 40 Pa,44 (ii) for pCO2 = 15000 Pa—0.15 bar—, a benchmark value for postcombustion exhaust gases,5 and (iii) pCO2 = 105 Pa—1 bar—a partial pressure regime of interest for pure CO2 stream generation from a CCS system.7
The desorption rates largely depend on the adsorption strength, and so we obtained four curves for each TMC, two for MMC and two for TopC sites, each of them as evaluated using PBE or PBE-D3 calculations respectively. DFT calculations are widespread used for evaluating adsorption strengths on a plethora of substrates, whilst TST is frequently used for simulating desorption processes as in temperature programmed desorption; a recent study for CO adsorption/desorption on a similar system (TiC nanopowders) provides a paradigmatic example.45
The estimated rads and rdes rates are graphically shown for the two fringe cases TiC and ZrC, see Fig. 2a. These are representative TMCs with low and high Eads values. Note that intersection points of rads and rdes define temperatures below which adsorption prevails and, consequently, CO2 accumulates on the TMC surface. Thus, for TiC(001) at 40 Pa CO2 partial pressure, the lowest and highest intersection temperatures are 211 K for the MMC site as calculated using PBE and 329 K for the TopC site (PBE-D3), marked as T1 and T2, respectively. At a CO2 partial pressure of 0.15 bar the intersection temperatures are shifted to higher values of 262 to 414 K (T3 and T4), and so compare to the desorption temperature of 323 K reported for the zeolite 13X benchmark material.5 At a CO2 partial pressure of 1 bar, the desorption process will likely occur in the 285–452 K range (T5 and T6), temperatures above which a surface regenerate for further use becomes feasible.
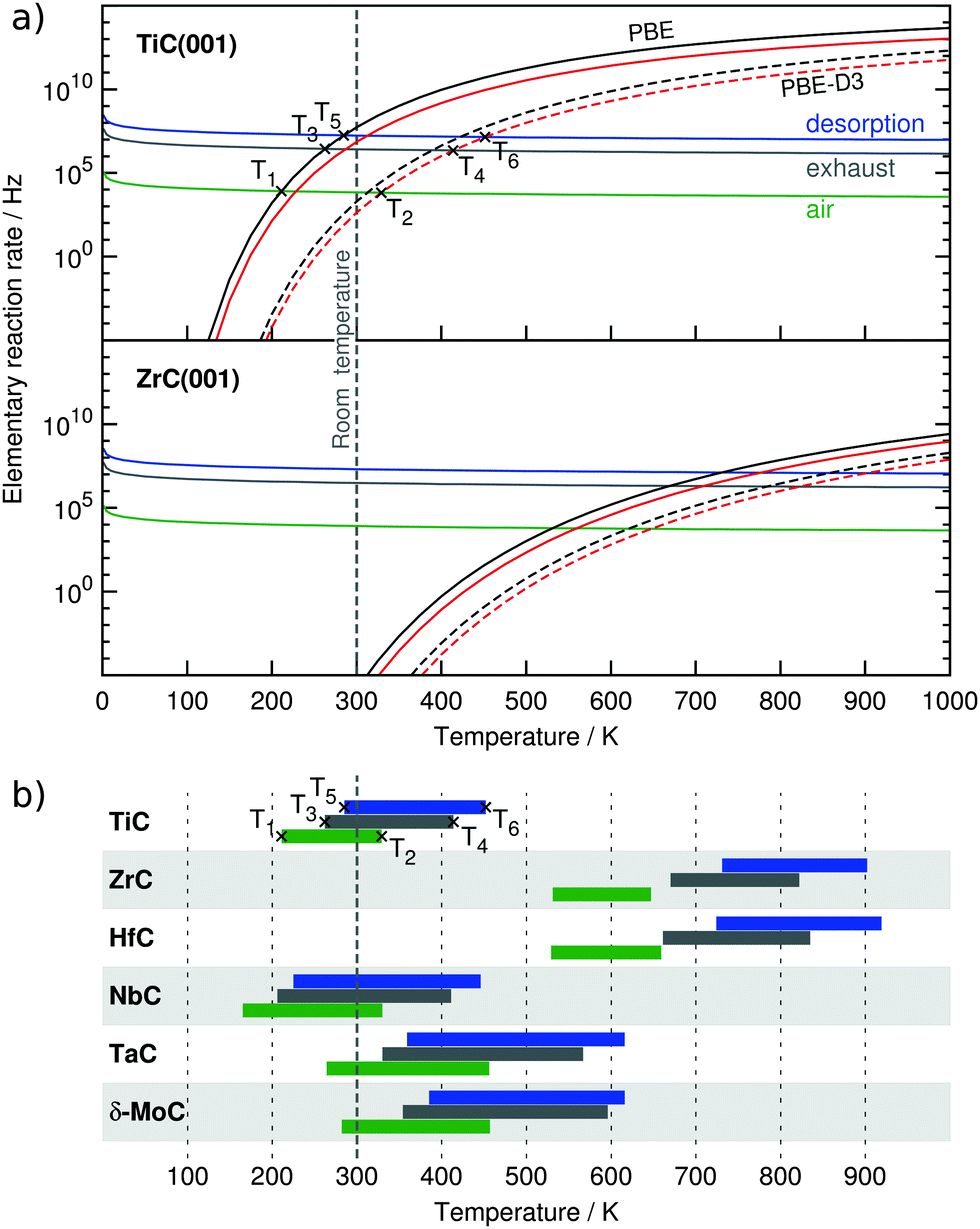 | ||
| Fig. 2 (a) Calculated rates for desorption and adsorption of CO2 on TiC and ZrC(001) surfaces. On TiC, marked points with T1–T6 labels in (a) and (b) show how desorption temperature ranges in (b) have been obtained. Legend for (a): green, grey, and blue; adsorption rates on a single site per time unit for a CO2 partial pressure of 40, 15 × 103, and 105 Pa, respectively. Black and red; desorption rates per site and time unit from MMC and TopC sites. PBE (solid) and PBE-D3 (dashed) values are provided. Legend for (b): green, grey, and blue bars belong to desorption temperature ranges for CO2 partial pressures of 40, 15 × 103, and 105 Pa, respectively. | ||
Thus T1 → T2, T3 → T4, and T5 → T6 can be interpreted as temperature ranges in which the TMC(001) surface looses its ability to initially capture and accumulate CO2 when annealing. This seems reasonable as dispersive force contributions to the adsorption energies might be overestimated by PBE-D3 and we expect the real desorption rate to lie somewhere in between. The analysis for the rest of investigated TMCs is summarized in Fig. 2b allowing for an easy comparison. Thus, ZrC- and HfC(001) surfaces display very elevated temperature ranges, in accordance with their high adsorption energies. The temperature ranges for TiC, NbC, TaC, and δ-MoC are lower but still in the range or even well above room temperature. Even though no CCS experiments on TMCs are available, it has been experimentally proven, as mentioned above, that some TMCs catalyse the CO2 hydrogenation to methanol at 500–600 K,33–35 implying that CO2 must become an adsorbed surface activated moiety.
Note that, as stated above, in most of these cases, MMC sites show a stronger adsorption compared to TopC sites, with a concomitant widening of the calculated temperature range. One must keep in mind that upon annealing, these systems may well first partially lose their adsorption capability, i.e. those TopC sites would be firstly depopulated, yet here lateral interactions are being neglected, and so the obtained rates better correlate with the initial stages of CO2 capture. Nevertheless, at low coverages on Ni surfaces,17 lateral interactions for adsorbed CO2 have been found not to be repulsive and so initial desorption rates are likely not underestimated. All in all, most of the TMC(001) surfaces are envisaged to capture and accumulate CO2 at ambient and even elevated temperatures.
In summary, periodic DFT calculations carried out at the PBE level and also including dispersion (PBE-D3) showed that CO2 molecules adsorb and get activated on a range of stable (001) surfaces of transition metal carbides (TMC–TM = Ti, Zr, Hf, Nb, Ta, Mo). Two competitive adsorption sites are identified (MMC and TopC) where CO2 chemisorbs in a bended (activated) geometry. Dispersion corrected adsorption energies are found to be favourable and lie in a range of −0.70 to −1.65 eV, depending on the TMC and surface site. These adsorption strengths are therefore considerably high, and despite dispersive forces are responsible of 0.21–0.32 eV, the major attachment force has a chemical nature. Adsorption and desorption rates, as predicted from DFT data, show that these materials theoretically can adsorb CO2 up to elevated temperatures at even low partial pressures. Thus TMCs appear to be ideal materials for CO2 capture and abatement, and actually given its activated adsorption appealing for using them as catalysts for CO2 conversion as well.
This work was supported by Spanish Ministerio grants (CTQ2012-30751) and Generalitat de Catalunya grants (2014SGR97 and XRQTC). F. V. thanks the Spanish Ministerio de Economía y Competitividad for the Ramón y Cajal postdoctoral grant (RYC-2012-10129).
Notes and references
- Intergovernmental Panel on Climate Change, Climate Change 2013 – The Physical Science Basis, Cambridge University Press, 1st edn, 2014 Search PubMed.
- International Energy Agency, World Energy Outlook 2014, Paris, 2014 Search PubMed.
- A. Sayari, Y. Belmabkhout and R. Serna-Guerrero, Chem. Eng. J., 2011, 171, 760 CrossRef CAS.
- L. Espinal, D. L. Poster, W. Wong-Ng, A. J. Allen and M. L. Green, Environ. Sci. Technol., 2013, 47, 11960 CrossRef CAS PubMed.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058 CrossRef.
- S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796 CrossRef CAS PubMed.
- M. E. Boot-Handford, J. C. Abanades, E. J. Anthony, M. J. Blunt, S. Brandani, N. Mac Dowell, J. R. Fernández, M.-C. Ferrari, R. Gross, J. P. Hallett, R. S. Haszeldine, P. Heptonstall, A. Lyngfelt, Z. Makuch, E. Mangano, R. T. J. Porter, M. Pourkashanian, G. T. Rochelle, N. Shah, J. G. Yao and P. S. Fennell, Energy Environ. Sci., 2014, 7, 130 CAS.
- M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975 RSC.
- G. Centi and S. Perathoner, Top. Catal., 2009, 52, 948 CrossRef CAS.
- S. Posada-Pérez, F. Viñes, J. Rodriguez and F. Illas, Top. Catal., 2015, 58, 159 CrossRef.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazabal and J. Perez-Ramirez, Energy Environ. Sci., 2013, 6, 3112 CAS.
- V. Havran, M. P. Duduković and C. S. Lo, Ind. Eng. Chem. Res., 2011, 50, 7089 CrossRef CAS.
- A. Otto, T. Grube, S. Schiebahn and D. Stolten, Energy Environ. Sci., 2015, 8, 3283 CAS.
- F. Viñes, A. Borodin, O. Höfft, V. Kempter and F. Illas, Phys. Chem. Chem. Phys., 2005, 7, 3866 RSC.
- V. A. de la Peña O’Shea, S. González, F. Illas and J. L. G. Fierro, Chem. Phys. Lett., 2008, 454, 262 CrossRef.
- V.-A. Glezakou, L. X. Dang and B. P. McGrail, J. Phys. Chem. C, 2009, 113, 3691 CAS.
- S.-G. Wang, D.-B. Cao, Y.-W. Li, J. Wang and H. Jiao, J. Phys. Chem. B, 2005, 109, 18956 CrossRef CAS.
- D. C. Sorescu, J. Lee, W. A. Al-Saidi and K. D. Jordan, J. Chem. Phys., 2011, 134, 104707 CrossRef.
- Q.-L. Tang and Q.-H. Luo, J. Phys. Chem. C, 2013, 117, 22954 CAS.
- Y. Jiao, A. Du, Z. Zhu, V. Rudolph, G. Q. Lu and S. C. Smith, Catal. Today, 2011, 175, 271 CrossRef CAS.
- N. Y. Dzade, A. Roldan and N. H. de Leeuw, J. Chem. Phys., 2015, 143, 094703 CrossRef CAS PubMed.
- D. Smykowski, B. Szyja and J. Szczygieł, J. Mol. Graphics Modell., 2013, 41, 89 CrossRef CAS PubMed.
- L. Valenzano, B. Civalleri, S. Chavan, G. T. Palomino, C. O. Areán and S. Bordiga, J. Phys. Chem. C, 2010, 114, 11185 CAS.
- H. H. Hwu and J. G. Chen, Chem. Rev., 2005, 105, 185 CrossRef CAS PubMed.
- R. B. Levy and M. Boudart, Science, 1973, 181, 547 CAS.
- A. Brungs, A. E. York and M. H. Green, Catal. Lett., 1999, 57, 65 CrossRef CAS.
- J. B. Claridge, A. P. E. York, A. J. Brungs, C. Marquez-Alvarez, J. Sloan, S. C. Tsang and M. L. H. Green, J. Catal., 1998, 180, 85 CrossRef CAS.
- J. A. Rodriguez, P. Liu, J. Dvorak, T. Jirsak, J. Gomes, Y. Takahashi and K. Nakamura, Surf. Sci., 2003, 543, L675 CrossRef CAS.
- P. M. Patterson, T. K. Das and B. H. Davis, Appl. Catal., A, 2003, 251, 449 CrossRef CAS.
- F. Viñes, J. A. Rodriguez, P. Liu and F. Illas, J. Catal., 2008, 260, 103 CrossRef.
- L. Ono and B. Roldán-Cuenya, Catal. Lett., 2007, 113, 86 CrossRef CAS.
- J. A. Rodriguez, P. Liu, D. J. Stacchiola, S. D. Senanayake, M. G. White and J. G. Chen, ACS Catal., 2015, 5, 6696 CrossRef CAS.
- S. Posada-Pérez, F. Viñes, P. J. Ramirez, A. B. Vidal, J. A. Rodriguez and F. Illas, Phys. Chem. Chem. Phys., 2014, 16, 14912 RSC.
- M. D. Porosoff, X. Yang, J. A. Boscoboinik and J. G. Chen, Angew. Chem., 2014, 126, 6823 CrossRef.
- M. D. Porosoff, S. Kattel, W. Li, P. Liu and J. G. Chen, Chem. Commun., 2015, 51, 6988 RSC.
- J. A. Rodriguez, J. Evans, L. Feria, A. B. Vidal, P. Liu, K. Nakamura and F. Illas, J. Catal., 2013, 307, 162 CrossRef CAS.
- U. Burghaus, Prog. Surf. Sci., 2014, 89, 161 CrossRef CAS.
- H. J. Freund and M. W. Roberts, Surf. Sci. Rep., 1996, 25, 225 CrossRef.
- F. Viñes, C. Sousa, P. Liu, J. A. Rodriguez and F. Illas, J. Chem. Phys., 2005, 122, 174709 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS.
- A. B. Vidal, L. Feria, J. Evans, Y. Takahashi, P. Liu, K. Nakamura, F. Illas and J. A. Rodriguez, J. Phys. Chem. Lett., 2012, 3, 2275 CrossRef CAS.
- T. Takahashi, S. Sutherland and A. Kozyr, Global Ocean Surface Water Partial Pressure of CO2 Database: Measurements Performed During 1957–2014 (Version 2014), Environmental Sciences Division, Oak Ridge National Laboratory, 2015 Search PubMed.
- B. P. Mant, G. G. Asara, J. A. Anderson, N. Homs, P. Ramírez de la Piscina, S. Rodríguez, J. M. Ricart and F. Illas, Surf. Sci., 2013, 613, 63 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details, list of adsorption energies and geometric descriptors, top views of adsorbate structures, brief explanation of the used rate model. See DOI: 10.1039/c5ee03649f |
| This journal is © The Royal Society of Chemistry 2016 |