
A soft, multilayered lithium–electrolyte interface†
Claudiu B.
Bucur
,
Adrian
Lita
,
Naoki
Osada
and
John
Muldoon
*
Toyota Research Institute of North America, 1555 Woodridge Ave, Ann Arbor, MI 48105, USA. E-mail: john.muldoon@tema.toyota.com
First published on 26th November 2015
Abstract
It is desirable that a thin film lithium–electrolyte interface is flexible and self-healing to accommodate the large volume expansion during lithium deposition without rupturing and impede electrolyte decomposition. The most influential area of polymer thin films is the alternating adsorption of oppositely charged polyelectrolytes popularized by the introduction of the layer-by-layer (LBL) self-assembly technique. We demonstrate that the introduction of a pH sensitive component into a nanomembrane assembled by the LBL method can have a dramatic effect on the interface governing the coulombic efficiency and morphology of lithium depositing.
Broader contextConsumer demand for cell phones that last longer and lower cost EVs that drive further results in an on-going thirst for higher energy batteries. This quest attracts interest towards finding a post lithium ion battery with 2–3 times the energy density of what lithium ion can offer. An insertion graphite anode with a specific capacity of 339 mA h g−1 could be upgraded to a metallic lithium anode with a specific capacity of 3861 mA h g−1. Challenges associated with the use of a lithium metal anode are rooted in its highly electronegative potential which causes degradation of battery electrolytes and dendritic growth. Our work shows that interfacial nanoscale engineering by nanomembranes can improve lithium metal cycling and we believe that this is crucial towards the development of high energy batteries such as lithium–sulfur or lithium–air. |
The most far-reaching electrochemical success has been the development and commercialization of the lithium ion battery which has changed the world in numerous ways. In addition to portable electronics and power tools, lithium ion batteries now power electric vehicles and are starting to replace gas backup generators with the introduction of the Powerwall. However, consumer demand for cell phones that last longer and lower cost EVs drive further results in a never ending thirst for higher energy batteries. This quest attracts interest to finding a post lithium ion battery with 2–3 times the energy density of what lithium ion can offer. The banner of post lithium ion battery research is carried by lithium/sulfur, lithium/air and multivalent metal batteries which operate with metallic anodes.1–10 One of the ways in which commercial lithium ion batteries offer high stability upon cycling is at the expense of their capacity storage, by utilizing spectator host materials such as graphitic carbon in the anode. In order to increase the capacity of the anode, innovative methods must be adopted without sacrificing stability and safety. For example, a graphite anode with a specific capacity of 339 mA h g−1 could be upgraded to a lithium metal anode with a specific capacity of 3861 mA h g−1. Challenges associated with the use of lithium metal anodes are rooted in its highly electronegative potential which causes high reactivity with battery electrolytes.11 The reduction of electrolytes on the surface of the lithium results in the formation of a solid electrolyte interface (SEI).12 This SEI is unstable and non-uniform due to rupturing and reformation throughout the reversible plating of lithium. The rigid SEI cannot withstand the mechanical stress caused by the uneven nucleation and dendrite growth or mossy deposition of lithium. Growth of dendrites causes a rapid and large increase in the surface area of the anode and can lead to thermal runaway or shorting of the battery. With each regeneration of the SEI, additional electrolyte is decomposed by reduction which severely limits the coulombic efficiency between the deposition and dissolution of lithium to below 50% after a few cycles at high rates. The low coulombic efficiencies impact cycle life negatively. For example, a 50% coulombic efficiency renders half the deposited lithium unrecoverable on each cycle and quickly depletes the lithium in the battery. In addition to this electrochemically inactive lithium, rapid degradation of the electrolyte which occurs during SEI regeneration also limits cycle life. Therefore, for practical batteries with metallic anodes, a coulombic efficiency above 99% is desirable (less than 1% of lithium “lost” on each cycle). Since to our knowledge there have been no reports of electrolytes which are reductively stable on lithium metal it is vital to have a uniform and flexible SEI on the lithium anode which allows the use of currently unstable electrolytes. Recent approaches to stabilize the lithium metal anode and avoid dendrite formation include development of new electrolyte additives (such as cesium salts or nitrates) which promote more uniform lithium electrodeposition.13,14 A related approach has been the ex situ formation of the SEI by pretreatment with chlorosilanes, chlorophosphines, and chloroboranes, which appear to form protective layers on the lithium anode surface through reaction with Li.15,16 Solid electrolytes, polymers, ceramics and interconnected carbon have also been explored to mitigate dendrite nucleation by blocking their growth.17–29 These solid state approaches suffer from interface issues which remain largely unresolved.30–40
It is desirable that a thin film lithium–electrolyte interface is flexible and self-healing to accommodate the large volume expansion during lithium deposition without rupturing and impede electrolyte decomposition therefore enhancing coulombic efficiencies. It also should be in close contact and provide gentle tension against the nucleating lithium to discourage dendritic growth. In addition, it should promote a uniform flux of lithium ions, which is required for the uniform plating of lithium.
The most influential area of polymer thin films is the alternating adsorption of oppositely charged polyelectrolytes popularized by the introduction of the layer-by-layer self-assembly technique. The LBL method permits the fabrication of multilayered thin films on solid supports by the self-limiting adsorption of cationic and anionic species with more than the stoichiometric number of charges (relative to the substrate), from dilute aqueous solutions.41–43 Each adsorbed layer reverses the surface charge. The driving force for the multilayer film buildup is primarily ion exchange between polyelectrolyte counterions and oppositely charged sites on the “partner” polymer backbone. Precise measurements have concluded that these compensation exchanges responsible for assembly are entropic in nature.44 While the “electrostatic” model is the more popular LBL method of thin film formation, other methods such as based on the hydrogen bond as a driving force were also reported where polymers bearing hydrogen bond donors and acceptors are employed as building blocks.45 We have previously demonstrated a flexible (self-healing46–48) LBL nanomembrane assembled based on ion exchange which allows the rapid diffusion of lithium ions while accommodating volume expansion and impeding the dissolution of polysulfides in a lithium–sulfur battery.49 Diffusion of a charged species through a LBL nanomembrane assembled based on the principle of ion-exchange is enabled by ion hopping as previously reported50 as well as by migration through pockets of electrolyte which might form inside such membrane.
Here we envision analogs of the nanomembrane which may have the capability to impede dendritic growth of lithium and enhance coulombic efficiencies. For example, the adhesion strength between a pH sensitive nanomembrane and its copper substrate may be tuned51,52 during assembly to promote nucleation of lithium between the copper substrate and the conformal membrane coating it. (Typical anode current collectors are thin copper foils.) Since the nanomembrane is conformal to the substrate, uniform and permeable to lithium ions,53 weakening of its pH sensitive contacts to the copper substrate may permit its controlled elevation during lithium plating without excessive stress on the integrity of its interpenetrated structure. As a result, membrane areas with a low flux of lithium ions would experience less elevation than areas with a higher flux. Since LBL membranes assembled based on ion exchange tend to be intrinsically flexible and elastic,54,55 it is envisioned they may recover their original state once lithium is stripped off. The process can then restart on the subsequent battery cycle. The nanomembrane morphology may reversibly change in response to the stimulus of lithium deposition. As a result, it may provide continuous, gentle tension against the growing lithium deposits which may limit uncontrolled lithium offshoots and may stop the formation of dendrites. Due to intimate affinity, it may also stabilize newly deposited lithium thus hindering decomposition of the electrolyte and improving cycle life and coulombic efficiency.
Initial efforts consisted of a multilayer nanomembrane assembled by the sequential adsorption of a positive polyelectrolyte such as polydiallyldimethylammonium chloride or PDAD and a negative polyelectrolyte such as poly(3,4-ethylenedioxythiophene)–poly(styrenesulfonate) or PEDOT:PSS on a copper disk substrate. Layer by layer assembly of such polymer multilayer films is presented in ESI,† Fig. S1. The reversible lithium deposition originating from a lithium metal counter electrode source was characterized electrochemically in a EL-CELL. A coulombic efficiency of ∼70% over the first 15 cycles is observed for the membrane modified anode, which offers no significant improvement over the unprotected anode (ESI,† Fig. S2). Unfortunately, the addition of this nanomembrane did not stabilize the SEI which forms during the reversible lithium deposition. We hypothesized that if the nanomembrane adheres too strongly to the copper substrate, lithium nucleation can only occur in concert with fragmentation of the membrane. It is desirable to create space for lithium nucleation beneath the lithium permeable nanomembrane. We envisioned a pH sensitive interface between the copper substrate and the multilayer nanomembrane which would permit the creation of “open space” for lithium nucleation via pH modification of this interface after fabrication. The positively charged polymer polyallylamine hydrochloride or PAH contains –NH3(+) functionalities. PAH can be deprotonated to its neutral form (PA) in basic media. If used as a first layer PAH will allow for the assembly of the multilayer due to its charged (–NH3(+)) functionality. Post assembly modification can be achieved by exposure to pH's higher than 8.8 which will partially (or completely) deprotonate PAH to form PA. Converting the charged PAH of the initial membrane layer to neutral PA will only cause a partial decoupling of the nanomembrane from the substrate52 which is in part due to the interpenetration of the membrane layers.56 Typically, the first four to five layers are often needed to obtain full coverage of a substrate (due to filling in of uncoated “gaps” see Fig. 1a) after which there is a substantial growth of the multilayer (linear or exponential, depending on the type of polymers used).56 As a result, since direct anchorage between the third positively charged polymer layer (PDAD) and the substrate is still expected, deprotonation of the first PAH layer will not fully decouple the nanomembrane from the copper substrate. This post assembly modification of the interface is corroborated by the following experiment. A multilayer consisting of a first layer of PAH and 14 layers of PDAD/PEDOT:PSS on a polished silicon wafer substrate becomes 50% thicker upon exposure to a pH 10 solution of sodium hydroxide. This is presumably due to the “open space” created at the interface between the substrate and the nanomembrane as PAH converts to PA (see Fig. 1b). It is noteworthy that a control multilayer composed of 14 layers of PDAD/PEDOT:PSS undergoes only a 5% change in thickness following exposure to a pH 10 solution. A distinct enhancement in coulombic efficiency is provided by the modification of the interface between the nanomembrane and the copper current collector.
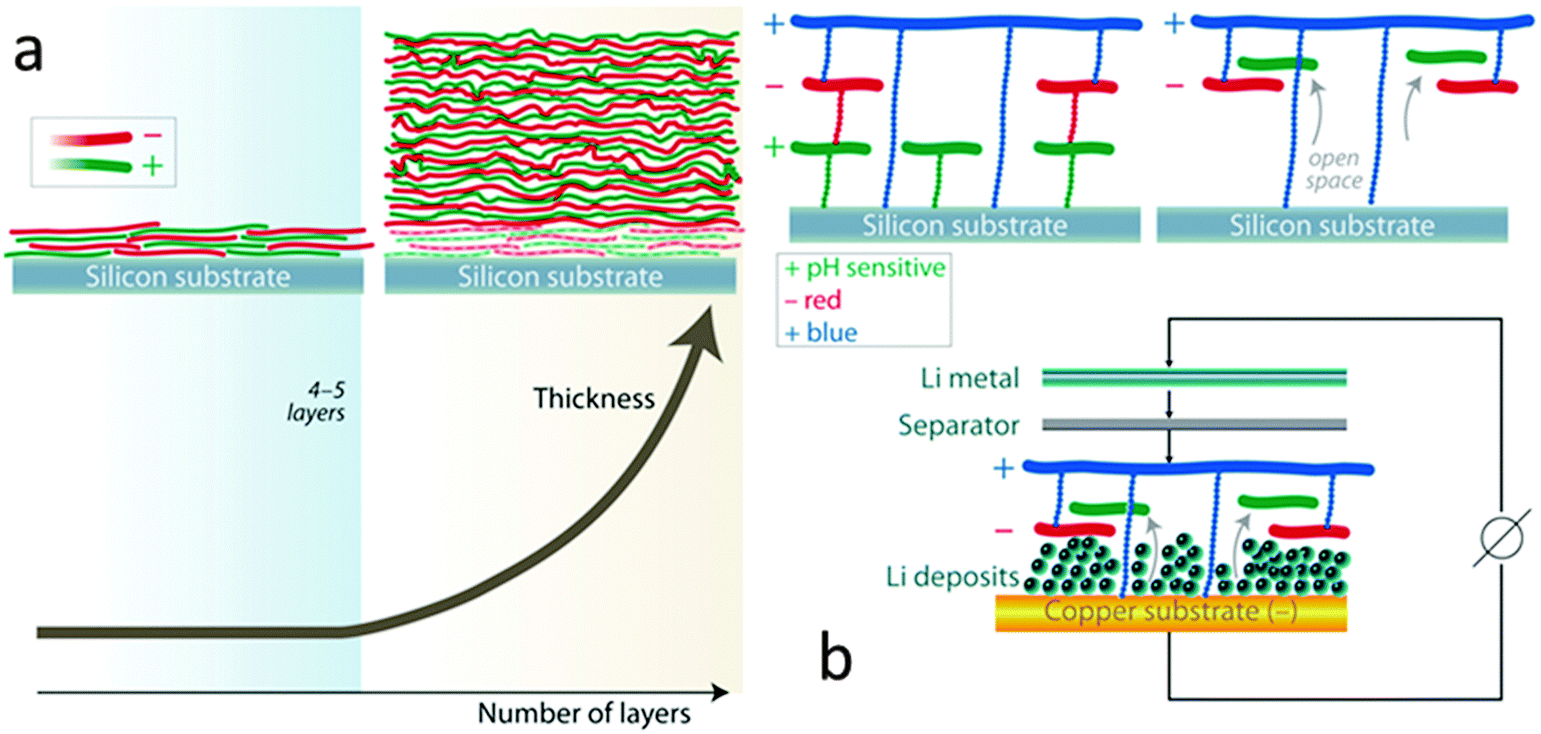 | ||
| Fig. 1 (a) Schematic depicting the layer by layer (LBL) deposition of oppositely charged polymers on a planar surface. The role of the first 4–5 layers is to fully cover the substrate by filling in any “gaps”. Subsequent layers contribute directly to thickness in a linear or exponential fashion, depending on the polymer pairs used; (b) in order to accommodate the non-dendritic deposition of lithium, “open spaces” can be cleared on the substrate by modifying the ionic character of a pH sensitive polymer (post assembly!). When in ionic form, the polymer layer (PAH) adheres tightly to the substrate due to the positively charged –NH3(+) functionality but when deprotonated (PA) (neutral –NH2 terminal group), it detaches leaving behind voids in the structure of the LBL film. | ||
On average, 95% of the lithium deposited is recovered for 50 cycles when a 14 layers of PDAD/PEDOT:PSS, PPy/PEDOT:PSS or PDAD/Nafion are interfaced with the copper substrate by the pH sensitive PAH layer (Fig. 2a) (85% of the deposited lithium could be recovered after 200 cycles – Fig. 2b). In contrast, the coulombic efficiency for the reversible deposition of lithium on a copper electrode without a nanomembrane protection drops below 60% by cycle 20. 0.5 mA h cm−2 of lithium was deposited in all instances. The rate of deposition/dissolution was 0.5 mA cm−2. Voltage profiles can be seen in Fig. 2c. In tandem with the high coulombic efficiency achieved for the deposition/dissolution process, the nanomembrane treatment of the copper anode also provides regular, non-dendritic lithium deposits. The morphology of the lithium deposited on the untreated copper electrode is dendritic with visible needle-like structures (ESI,† Fig. S3). In contrast, the deposits on the nanomembrane coated copper are granular, dense and needle-like structures are absent (Fig. 2d).
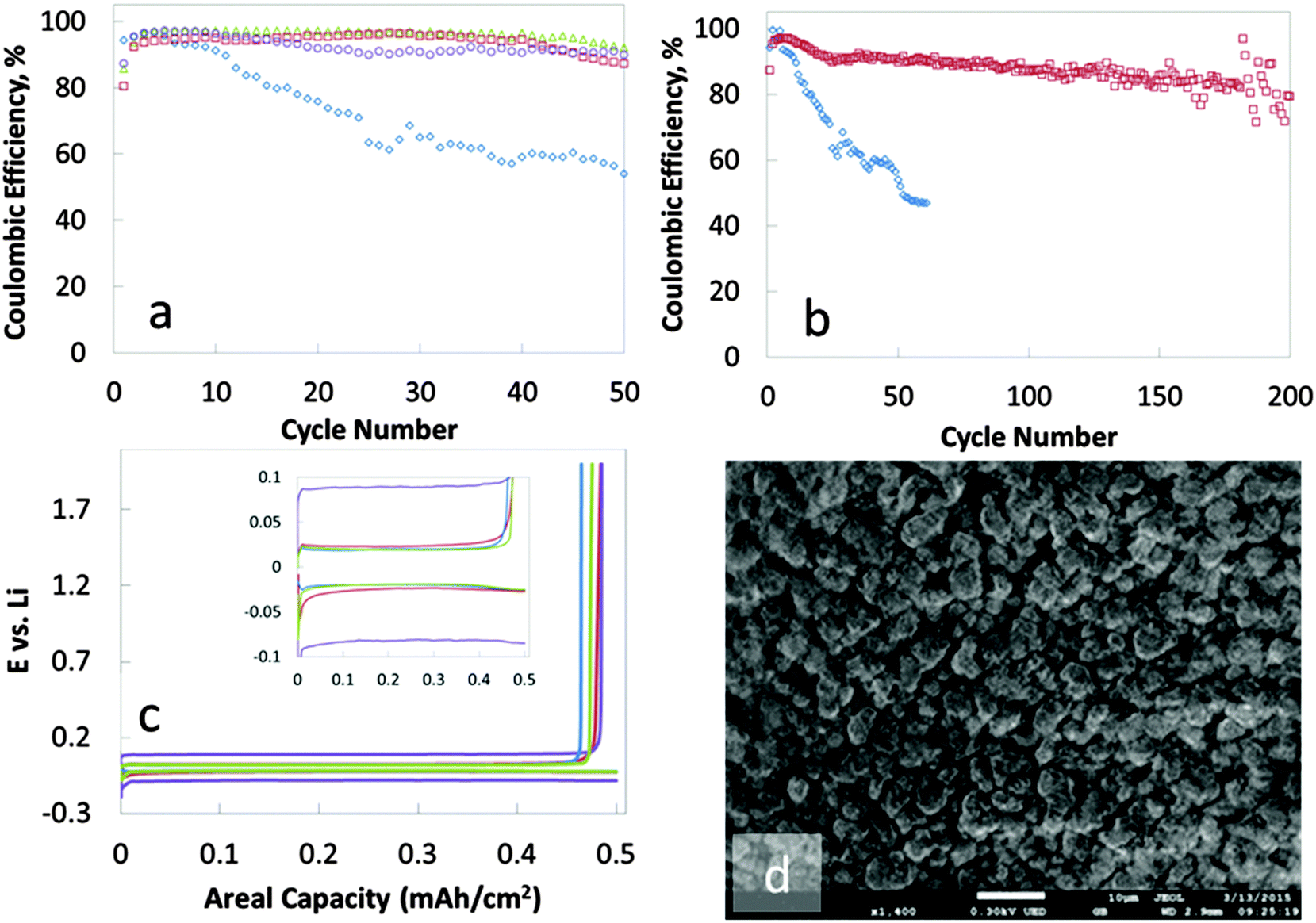 | ||
| Fig. 2 (a and b) Comparison of cycling performances of untreated copper current collector (diamond, ◊) to copper current collectors coated by nanomembranes composed of PAH + (PEDOT:PSS/PDAD)14 (square, □), PAH + (PEDOT:PSS/PPy)14 (circles, ○) and PAH + (Nafion/PDAD)14 (triangles, △); (c) voltage profiles for the 7th cycle of the lithium deposition/dissolution process with lithium metal as the reference electrode. Comparison of reversible deposition on a bare copper electrode (blue) to the following nanomembrane coatings: PAH + (PEDOT:PSS/PPy)14 (purple), PAH + (PEDOT:PSS/PDAD)14 (garnet) and PAH + (Nafion/PDAD)14 (green). Inset shows similar overpotential for the bare and nanomembrane coated copper electrodes. The higher overpotential of PAH + (PEDOT:PSS)14 (garnet) may suggest a smaller diffusion coefficient for the lithium ions through this multilayer which requires a higher overpotential to initiate and maintain the reduction of lithium; (d) SEM image of lithium deposits on a nanomembrane coated copper current collector. No visible dendrites. Deposits are regular, dense and granular. 0.5 mA h cm−2 lithium is deposited each time. Both the deposition and the dissolution rates are 0.5 mA cm−2. Dissolution is carried out to 2.0 V. | ||
Here we have shown that an SEI polymer membrane layer containing a pH sensitive interface with the substrate can have a dramatic effect on both coulombic efficiency and the morphology of lithium deposition. Further research is ongoing to investigate the effect of several pH sensitive layers in the nanomembrane and electrolytes additives, in hopes of increasing the free volume available for lithium nucleation and enhance coulombic efficiency and quantity of lithium deposited without rupturing of the membrane. This approach may lead towards commercial applicability where high quantities of lithium (10 mA h lithium or higher per cm2) is routinely cycled.
References
- A. C. Luntz and B. D. McCloskey, Chem. Rev., 2014, 114, 11721–11750 CrossRef CAS PubMed.
- J. Muldoon, C. B. Bucur and T. Gregory, Chem. Rev., 2014, 114, 11683–11720 CrossRef CAS PubMed.
- J. Muldoon, C. B. Bucur, N. Boaretto, T. Gregory and V. di Noto, Polym. Rev., 2015, 55, 208–246 CrossRef CAS.
- A. Manthiram, Y. Fu, S. H. Chung, C. Zu and Y. S. Su, Chem. Rev., 2014, 114, 11751–11787 CrossRef CAS PubMed.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed.
- J.-J. Xu, Z.-L. Wang, D. Xu, L.-L. Zhang and X.-B. Zhang, Nat. Commun., 2013, 4, 2438 Search PubMed.
- J.-J. Xu, D. Xu, Z.-L. Wang, H.-G. Wang, L.-L. Zhang and X.-B. Zhang, Angew. Chem., Int. Ed., 2013, 52, 3887–3890 CrossRef CAS PubMed.
- J.-J. Xu, Z.-L. Wang, D. Xu, F.-Z. Meng and X.-B. Zhang, Energy Environ. Sci., 2014, 7, 2213–2219 CAS.
- Z.-L. Wang, D. Xu, J.-J. Xu and X.-B. Zhang, Chem. Soc. Rev., 2014, 43, 7746–7786 RSC.
- C. B. Bucur, J. Muldoon and A. Lita, Energy Environ. Sci., 2015 10.1039/c5ee02367j.
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 CrossRef CAS PubMed.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- F. Ding, W. Xu, G. L. Graff, J. Zhang, M. L. Sushko, X. Chen, Y. Shao, M. H. Engelhard, Z. Nie, J. Xiao, X. Liu, P. V. Sushko, J. Liu and J. G. Zhang, J. Am. Chem. Soc., 2013, 135, 4450–4456 CrossRef CAS PubMed.
- J. Zhang, W. Xu, G. L. Graff, X. Chen, F. Ding and Y. Shao, US20130199936 A1, 2013.
- F. Marchioni, K. Star, E. Menke, T. Buffeteau, L. Servant, B. Dunn and F. Wudl, Langmuir, 2007, 23, 11597–11602 CrossRef CAS PubMed.
- G. A. Umeda, E. Menke, M. Richard, K. L. Stamm, F. Wudl and B. Dunn, J. Mater. Chem., 2011, 21, 1593–1599 RSC.
- G. Zheng, S. W. Lee, Z. Liang, H. W. Lee, K. Yan, H. Yao, H. Wang, W. Li, S. Chu and Y. Cui, Nat. Nanotechnol., 2014, 9, 618–623 CrossRef CAS PubMed.
- L. DeJonghe, S. J. Visco, Y. S. Niman and A. M. Sukeshimi, US6911280 B1, 2005.
- M. G. Laramie, Y. V. Mikhaylik, J. A. Phipps and V. G. Viner, WO2015003123 A1, 2015.
- Y. S. Nimon, M. Y. Chu and S. J. Visco, US6537701 B1, 2003.
- S. Fleischmann, T. Petsh, A. Misske, R. Schmidt, V. G. Viner and B. Sankaran, WO2014068036 A1, 2014.
- Y. Chan and T. B. Atwater, US7598000 B1, 2009.
- H. B. Eitonni, M. Singh, N. Balsara, W. Hudson and I. R. Gur, US20110033755 A1, 2011.
- T. A. Skotheim, C. J. Sheehan, Y. V. Mikhaylik and J. D. Affinito, US8728661 B2, 2014.
- D. J. Lee, D. M. Im, V. Roev, S. B. Ma, M. S. Park, O. Yamamoto, N. Imanishi, W. S. Choi and Y. Takeda, US20140178777 A1, 2014.
- K. Starr, J. Muldoon, F. Marchioni, F. Wudl, B. Dunn and K. L. Stamm, WO2008008130 A3, 2008.
- J. Muldoon, E. Nicolas, M. Richard and K. L. Stamm, WO2008036742 A3, 2008.
- Q.-C. Liu, J.-J. Xu, S. Yuan, Z.-W. Chang, D. Xu, Y.-B. Yin, L. Li, H.-X. Zhong, Y.-S. Jiang, J.-M. Yan and X.-B. Zhang, Adv. Mater., 2015, 27, 5241–5247 CrossRef CAS PubMed.
- Q.-C. Liu, J.-J. Xu, D. Xu and X.-B. Zhang, Nat. Commun., 2015, 6, 7892 CrossRef CAS PubMed.
- J. N. Chazalviel, Phys. Rev. A: At., Mol., Opt. Phys., 1990, 42, 7355–7367 CrossRef CAS.
- G. Ma, Z. Wen, Q. Wang, C. Shen, J. Jin and X. Wu, J. Mater. Chem. A, 2014, 2, 19355–19359 CAS.
- M. Mori, Y. Naruoka, K. Naoi and D. Fauteux, J. Electrochem. Soc., 1998, 145, 2340–2348 CrossRef CAS.
- X. Yu, J. B. Bates, G. E. Jellison and F. X. Hart, J. Electrochem. Soc., 1997, 144, 524–532 CrossRef CAS.
- I. S. Kang, Y. S. Lee and D. W. Kim, J. Electrochem. Soc., 2014, 161, A53–A57 CrossRef CAS.
- Y. M. Lee, N. S. Choi, J. H. Park and J. K. Park, J. Power Sources, 2003, 119–121, 964–972 CrossRef CAS.
- S. M. Choi, I. S. Kang, Y. K. Sun, J. H. Song, S. M. Chung and D. W. Kim, J. Power Sources, 2013, 244, 363–368 CrossRef CAS.
- M. Rosso, T. Gobron, C. Brissot, J. N. Chazalviel and S. Lascaud, J. Power Sources, 2001, 97–98, 804–806 CrossRef CAS.
- N. S. Choi, Y. M. Lee, W. Seol, J. A. Lee and J. K. Park, Solid State Ionics, 2004, 172, 19–24 CrossRef CAS.
- A. A. Arie and J. K. Lee, Diamond Relat. Mater., 2011, 20, 403–408 CrossRef CAS.
- D. G. Belov, O. V. Yarmolenko, A. Peng and O. N. Efimov, Synth. Met., 2006, 156, 745–751 CrossRef CAS.
- G. Decher, Science, 1997, 277, 1232–1237 CrossRef CAS.
- F. Caruso, R. A. Caruso and H. Möhwald, Science, 1998, 282, 1111–1114 CrossRef CAS PubMed.
- H. Ejima, J. J. Richardson, K. Liang, J. P. Best, M. P. van Koeverden, G. K. Such, J. Cui and F. Caruso, Science, 2013, 341, 154–157 CrossRef CAS PubMed.
- C. B. Bucur, Z. Sui and J. B. Schlenoff, J. Am. Chem. Soc., 2006, 128, 13690–13691 CrossRef CAS PubMed.
- G. Wu and X. Zhang, in Multilayer Thin Films, ed. G. Decher and J. B. Schlenoff, Wiley-VCH Verlag GmbH & Co. KGaA, 2012, pp. 43–67 Search PubMed.
- X. Wang, F. Liu, X. Zheng and J. Sun, Angew. Chem., Int. Ed., 2011, 50, 11378–11381 CrossRef CAS PubMed.
- D. O. Grigoriev, K. Köhler, E. Skorb, D. G. Shchukin and H. Möhwald, Soft Matter, 2009, 5, 1426–1432 RSC.
- C. W. Park, A. B. South, X. Hu, C. Verdes, J.-D. Kim and L. A. Lyon, Colloid Polym. Sci., 2010, 289, 583–590 Search PubMed.
- C. B. Bucur, J. Muldoon, A. Lita, J. B. Schlenoff, R. A. Ghostine, S. Dietz and G. Allred, Energy Environ. Sci., 2013, 6, 3286–3290 CAS.
- T. R. Farhat and J. B. Schlenoff, J. Am. Chem. Soc., 2003, 125, 4627–4636 CrossRef CAS PubMed.
- S. Sukhishvili, in Multilayer Thin Films, ed. G. Decher and J. B. Schlenoff, Wiley-VCH Verlag GmbH & Co. KGaA, 2012, pp. 337–362 Search PubMed.
- J. Choi, PhD thesis, Massachusetts Institute of Technology, 2004.
- J. B. Schlenoff, in Multilayer Thin Films, ed. G. Decher and J. B. Schlenoff, Wiley-VCH Verlag GmbH & Co. KGaA, 2012, pp. 281–320 Search PubMed.
- J. L. Lutkenhaus, K. D. Hrabak, K. McEnnis and P. T. Hammond, J. Am. Chem. Soc., 2005, 127, 17228–17234 CrossRef CAS PubMed.
- Y. Li, S. Chen, M. Wu and J. Sun, Adv. Mater., 2012, 24, 4578–4582 CrossRef CAS PubMed.
- S. T. Dubas and J. B. Schlenoff, Macromolecules, 1999, 32, 8153–8160 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ee03056k |
| This journal is © The Royal Society of Chemistry 2016 |