
Polyethylenimine promoted electrocatalytic reduction of CO2 to CO in aqueous medium by graphene-supported amorphous molybdenum sulphide†
Fengwang
Li
ab,
Shu-Feng
Zhao
ac,
Lu
Chen
a,
Azam
Khan
ad,
Douglas R.
MacFarlane
*ab and
Jie
Zhang
*ab
aSchool of Chemistry, Monash University, Clayton 3800, Victoria, Australia. E-mail: jie.zhang@monash.edu; douglas.macfarlane@monash.edu
bARC Centre of Excellence for Electromaterials Science, School of Chemistry, Monash University, Clayton 3800, Victoria, Australia
cCSIRO Process Science and Engineering, Box 312, Clayton South 3168, Victoria, Australia
dDepartment of Chemistry, Quaid-i-Azam University, Islamabad 45320, Pakistan
First published on 13th November 2015
Abstract
Efficiently and selectively converting CO2 to value-added carbon compounds remains a major challenge in sustainable energy research. In this paper, we report the synthesis of a cost-effective catalyst, i.e. amorphous molybdenum sulphide on a polyethylenimine modified reduced graphene oxide substrate, for electrocatalytically reducing CO2 into CO in CO2 saturated aqueous NaHCO3 medium with high efficiency and selectivity. The catalyst is capable of producing CO at overpotentials as low as 140 mV and reaches a maximum faradaic efficiency of 85.1% at an overpotential of 540 mV. At an overpotential of 290 mV with respect to the formation of CO, it catalyzes the formation of syngas with high stability. Detailed investigations reveal that PEI works as a co-catalyst by providing a synergetic effect with MoSx.
Broader contextElectrochemical CO2 reduction using renewable energy as a power source could potentially solve the energy and environmental problems arising from excessive use of non-renewable fossil fuels. Currently, electrocatalysts for this reaction generally suffer from high cost, low energetic efficiency and poor product selectivity. Nevertheless, molybdenum or tungsten containing formate dehydrogenases (FDH) can efficiently and selectively catalyze reduction of CO2 to formate in Nature. Inspired by the active sites of FDH, amorphous molybdenum sulphide (MoSx) on a polyethylenimine (PEI) modified reduced graphene oxide (rGO) substrate has been synthesized and used as a heterogeneous electrocatalyst for reduction of CO2 to CO in an aqueous medium. The high efficiency and selectivity is accomplished by the synergetic effect of MoSx and PEI while rGO improves electrical conductivity and stability of the catalyst. |
Introduction
Energy and environmental problems are among the most challenging issues facing mankind in this century.1–3 Despite tremendous efforts to develop renewable energy sources, the majority of energy used today is still from non-renewable, fossil fuel sources, which leads to an increase in greenhouse CO2 levels in the atmosphere and may cause catastrophic climate changes. Electrocatalytic reduction of CO2, which recycles CO2 back to fuels and commodity chemicals by using renewable energy as a power source, could provide an attractive solution to this issue.4 However, CO2 is very stable under environmental conditions and the hydrogen evolution reaction (HER) often prevails over CO2 reduction in aqueous electrolytes under cathodic polarization,5,6 making it essential to find a suitable catalyst to achieve cost-effective CO2 reduction with high efficiency and selectivity.The past several decades have witnessed reports of a number of homogeneous and heterogeneous catalysts for electrocatalytic CO2 reduction; these mainly include metals,5 metal oxides,7–9 metal complexes10–13 and nitrogen-containing carbon materials.14–17 Among them, noble metals such as Au, Ag and Pd have attracted particular interest for they can selectively convert CO2 to CO at relatively low overpotentials.5,18 Moreover, nanostructured noble metals have been developed recently to enhance their performance.19–27 For example, oxide-derived Au nanoparticles exhibit highly selective CO2 reduction to CO in aqueous solution at a lower overpotential than a polycrystalline bulk Au electrode.20 However, precious metals are hampered by their low abundance and high cost, which restrict their practical, large-scale application. Thus, searching for abundant and efficient electrocatalysts with selectivity towards CO, or capable of producing syngas that could be readily utilized in a Fischer–Tropsch process, is vital for commercializing CO2 electroreduction technologies.28 Although less expensive materials such as Cu, Sn, Bi and their oxides have demonstrated promise for reduction of CO2 to hydrocarbons, formic acid and CO,29–32 these systems generally show low activities and/or selectivity for CO production or need non-aqueous solvents which may limit their practical applications.
In Nature, formate dehydrogenases (FDH), which contain molybdenum or tungsten centers coordinated by a selenocysteine residue and amino acid residues, can catalyze the oxidation of formate to CO2.33,34 Hirst and co-workers35,36 showed that they could efficiently and specifically catalyze the reversible transformation between CO2 and formate under mild conditions. This inspires us to explore new catalysts resembling the FDH active site for electroreduction of CO2. Molybdenum disulphide (MoS2), a two-dimensional transition metal dichalcogenide, has sparked tremendous research interest as a substitute for Pt for effectively catalysing the HER in acidic media.37–40 MoS2 consists of a planar triangular lattice of Mo atoms sandwiched between two planes of S atoms and each Mo(IV) is coordinated to six S ligands, while each S center is connected to three Mo atoms. From a biomimic point of view, MoS2 shares a similar structure with the active center of FDH, making it potentially an electrocatalyst for CO2 reduction. Recently, Nørskov et al.41 demonstrated theoretically that MoS2 could possibly be an electrocatalyst for CO2 reduction and this was subsequently proven experimentally in ionic liquid (IL) medium with water as a proton source.42 In the present work we demonstrate that amorphous molybdenum sulphide (MoSx) on a polyethylenimine (PEI) modified reduced graphene oxide (rGO) substrate can be a highly efficient and selective electrocatalyst for reduction of CO2 to CO in simple aqueous media at low overpotentials. We show that the PEI layer considerably enhances the catalytic activity of the MoSx, presumably by suppressing HER and stabilising the CO2˙− intermediate during CO2 reduction, as has been suggested previously for PEI on carbon nanotube materials.43
Experimental
Chemicals and apparatus
NaHCO3 (ACS grade), NaOH and NaH2PO4 (AR grade) were purchased from Merck. Natural graphite (crystalline, 300 mesh) was from Alfa Aesar. Polyethylenimine (PEI, Mn = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, branched), polyethylene glycol (PEG, Mn = 400), polydiallyldimethylammonium chloride (PDDA, 20 wt% in water, Mw = 100000–200000) and (NH4)2MoS4 (99.97% trace metals basis) were purchased from Sigma-Aldrich. All the chemicals were used without further purification. Graphene oxide (GO) was synthesized from natural graphite using the method described by Hummers et al.44 Phosphate buffer used for electrodeposition of MoSx was prepared by adding 5.0 M NaOH solution to 0.1 M NaH2PO4 solution to adjust the pH to 6.81. All the aqueous solutions were prepared with Milli-Q water.
000, branched), polyethylene glycol (PEG, Mn = 400), polydiallyldimethylammonium chloride (PDDA, 20 wt% in water, Mw = 100000–200000) and (NH4)2MoS4 (99.97% trace metals basis) were purchased from Sigma-Aldrich. All the chemicals were used without further purification. Graphene oxide (GO) was synthesized from natural graphite using the method described by Hummers et al.44 Phosphate buffer used for electrodeposition of MoSx was prepared by adding 5.0 M NaOH solution to 0.1 M NaH2PO4 solution to adjust the pH to 6.81. All the aqueous solutions were prepared with Milli-Q water.
Transmission electron microscopic (TEM) images were collected on a FEI Tecnai G2 T20 TWIN TEM Instrument. Scanning electron microscopic (SEM) images and energy dispersive spectrum (EDS) were recorded on a FEI Nova NanoSEM 450 FEG SEM Instrument equipped with Bruker Quantax 400 X-ray analysis system. Raman spectra were obtained using a Renishaw inVia Microscope with a 532 nm laser source and IR spectra with a Digilab (7000) Stingray Imaging FT-IR Spectrometer. X-ray diffraction (XRD) data were collected with a Philip X-ray powder diffractometer (Cu Kα radiation). Gas chromatography (GC) was performed with an Agilent 7820 A gas chromatography system equipped with a HP-plot molesieve (5A) column and a thermal conductivity detector (TCD). The carrier gas was helium (99.99%) for CO analysis while nitrogen (99.99%) was used as carrier gas for H2 analysis. The retention times were compared with known compounds. All the electrochemical experiments were conducted on a CHI 760E electrochemical workstation (CH Instruments, Austin, Texas, USA) at room temperature (22 ± 2 °C).
Fabrication of rGO–PEI–MoSx electrodes
10 mg graphene oxide (GO) powder was dispersed in 10 mL water and pH was adjusted to around 9.0 by adding an appropriate amount of aqueous NaOH solution (5.0 M) dropwise. After sonication for at least 1 hour, 500 μL of a 5 wt% PEI aqueous solution was then added into the GO dispersion quickly and sonicated for another 30 minutes. Afterwards, it was stirred at 60 °C for 12 hours to form the GO–PEI solution. In order to obtain a well-dispersed solution, centrifugation at 3000 rpm was applied for 20 minutes to remove any large aggregates. The procedure for fabricating the rGO–PEI–MoSx electrode is schematically described in Fig. 1. Firstly, 4 μL of GO–PEI solution was casted onto a glassy carbon electrode (GCE, 3 mm in diameter, CH Instruments, USA) to form the GO–PEI modified GCE and dried under an infrared lamp. The as-prepared electrode was then immersed into a solution composed of 2 mM (NH4)2MoS4 and 0.1 M phosphate buffer (pH 6.81) for electrodeposition of MoSx by cycling the potential from 0.3 V to −1.3 V (vs. Ag/AgCl (3 M KCl)) at a scan rate of 50 mV s−1 for 10 times according to the procedure developed by Merki et al.45 During this process, GO is electroreduced to reduced graphene oxide (rGO), while MoSx is electrodeposited onto the rGO. MoSx, rGO–PEI, rGO–MoSx, rGO–PEG–MoSx and rGO–PDDA–MoSx electrodes were prepared by a similar procedure. To prepare TEM samples, catalyst materials were carefully scraped off from the electrode surface and dispersed in water by sonication for 30 minutes. Afterwards, 2 μL of the dispersion was drop cast on a copper grid and dried in air.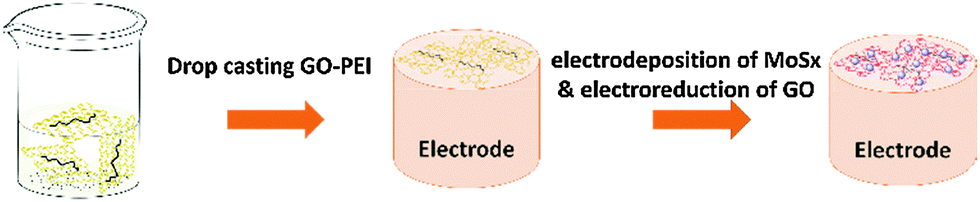 | ||
| Fig. 1 Schematic illustration of the fabrication of rGO–PEI–MoSx modified electrodes. | ||
Electrochemical measurements
Cyclic voltammetric (CV) measurements were carried out in 0.5 M aqueous NaHCO3 solution saturated with N2 or CO2 by bubbling N2 or CO2 for at least 30 minutes in a conventional three-electrode cell comprising a platinum wire counter electrode, an Ag/AgCl (3 M KCl) reference electrode and a modified or unmodified GCE as the working electrode. In some experiments, a piece of high surface area carbon cloth (1 cm × 0.5 cm) modified with rGO–PEI–MoSx was used as working electrode. For hydrogen evolution reaction (HER) catalysis, CV measurements were conducted in 0.5 M H2SO4, 0.5 M NaHCO3 and 0.5 M phosphate buffer (pH 7.2) solutions, respectively, at a scan rate of 5 mV s−1 with iR compensation. Electrolysis at different applied potentials was performed in a gas-tight two-compartment electrochemical cell with a glass frit as the separator. Each compartment contained 12.5 mL electrolyte and approximately 20 mL headspace. A piece of carbon cloth (1 cm × 1.5 cm) was used as counter electrode, an Ag/AgCl (3 M KCl) as reference electrode and a modified glassy carbon plate (0.4 cm × 1.5 cm) as working electrode. Before electrolysis, the cell was degassed by bubbling CO2 gas for at least 30 min. The solutions in both compartment were stirred during electrolysis. After the electrolysis, a small fraction of the cell's head space products (200 μL) was sampled by gas-tight syringe and analysed by GC. For each electrolysis, the products were sampled and analysed twice in a very short time interval and two electrolysis experiments were run at each applied potential. The durability test was conducted by chronoamperometry at a constant potential of −0.65 V using a rGO–PEI–MoSx modified 3 mm diameter GCE. A slightly positive CO2 pressure was applied to prevent air from entering the electrolysis cell and to maintain the concentration of CO2.All potentials were converted to the reversible hydrogen electrode (RHE) reference scale using the formula: E(vs. RHE) = E(vs. Ag/AgCl) + 0.210 V + 0.059 V × pH. All the working electrodes were polished using a 0.3 μm aqueous alumina slurry on a polishing cloth, sonicated in water and acetone, rinsed with acetone, and then dried under a flow of nitrogen before use.
Results and discussion
Fourier transform infrared spectroscopic characterization of GO–PEI
Fourier transform infrared (FT-IR) spectroscopic characterization of GO and GO–PEI was undertaken to reveal the nature of the interaction between PEI and GO. As shown in Fig. 2a, these two materials exhibit distinctly different FT-IR spectra. Two additional peaks at 2841 and 2932 cm−1 were found with GO–PEI, which were assigned to symmetric and asymmetric stretching modes of the CH2 of the PEI chains.46 Furthermore, a significant decrease of the peak at 1724 cm−1 (attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O from the carboxyl group in pristine GO) and the appearance of a new band at 1632 cm−1 (attributed to the CO stretching of the primary amide) suggest that the carbonyl groups are converted to amides on reaction with the PEI.47 This is further confirmed by the new bands observed at 1450 cm−1 (C–N stretching vibration) and 1572 cm−1 (N–H bending vibration) with GO–PEI.48 This covalent interaction between PEI and GO is beneficial in maintaining the high stability of the modified electrode under long term electrolysis conditions. It is important to point out that the band at 1592 cm−1 ascribed to the CC vibration of the aromatic rings was observed in both GO and GO–PEI, implying that the sp2 character in GO–PEI is preserved after the chemical conjugation,49 which is crucial to preserve the conductivity of the fabricated electrode.
O from the carboxyl group in pristine GO) and the appearance of a new band at 1632 cm−1 (attributed to the CO stretching of the primary amide) suggest that the carbonyl groups are converted to amides on reaction with the PEI.47 This is further confirmed by the new bands observed at 1450 cm−1 (C–N stretching vibration) and 1572 cm−1 (N–H bending vibration) with GO–PEI.48 This covalent interaction between PEI and GO is beneficial in maintaining the high stability of the modified electrode under long term electrolysis conditions. It is important to point out that the band at 1592 cm−1 ascribed to the CC vibration of the aromatic rings was observed in both GO and GO–PEI, implying that the sp2 character in GO–PEI is preserved after the chemical conjugation,49 which is crucial to preserve the conductivity of the fabricated electrode.
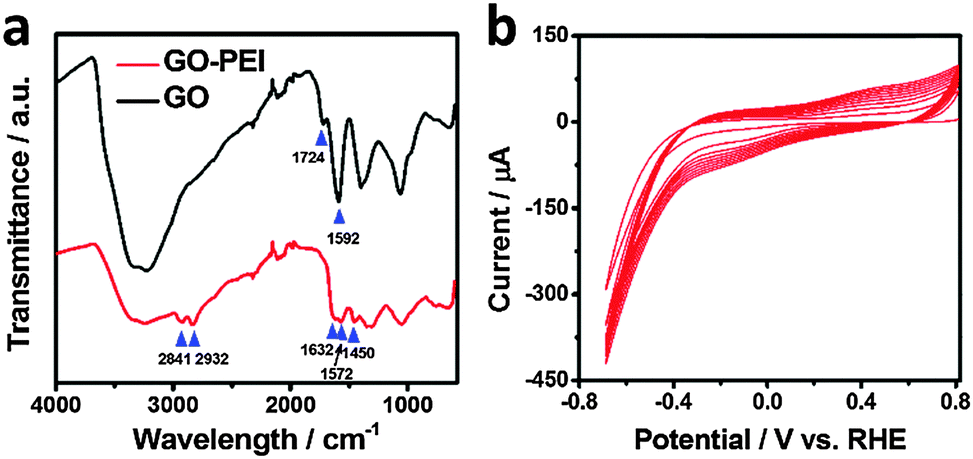 | ||
| Fig. 2 (a) FT-IR spectra of GO (black line) and GO–PEI (red line). (b) Electrodeposition of rGO–PEI–MoSx on GCE by repeating CVs for 10 cycles. | ||
Fabrication and characterization of rGO–PEI–MoSx
The rGO–PEI–MoSx electrode was fabricated by electrodeposition of MoSx onto a rGO–PEI modified electrode using the detailed procedure described in the Experimental section. The voltammogram (Fig. 2b) clearly demonstrates that a new reduction process at −0.1 V (vs. RHE, all potentials hereafter are with respect to this reference) and a new oxidation process at 0.4 V emerge and their current magnitude increases with cycling. The oxidation peak is attributed to the oxidation of [MoS4]2− to form MoS3 and S8 according to the following equation:50,51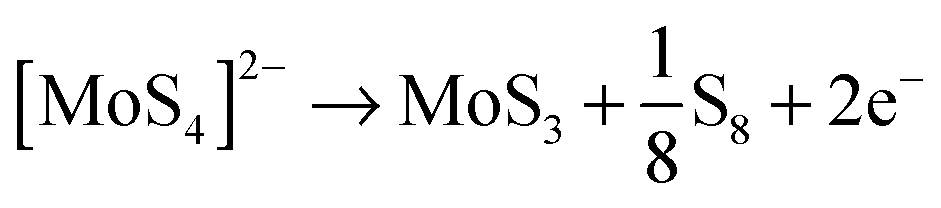
The reduction peak is assigned to the reduction of the electrogenerated S8. In the potential region more negative than −0.25 V, reductive deposition of amorphous MoIVS2 (i.e. MoSx) takes place according to the previous electrochemical quartz crystal microbalance studies.50
| [MoS4]2− + 2H2O + 2e− → MoS2 + 2HS− + 2OH− |
In the potential range of −0.4 V to −0.69 V, GO is electro-reduced to rGO.52 Thus, the deposition of MoSx and reduction of GO occurred simultaneously in this potential region, which was confirmed by energy dispersive spectroscopy (EDS) and Raman spectroscopy (Fig. S1, ESI†). The presence of hydrophilic PEI on GO could increase the space between each GO layer and make it more easily accessible for [MoS4]2− to achieve efficient deposition of MoSx.53 Moreover, both rGO–PEI and rGO–PEI–MoSx modified GCE show a large increase in capacitance compared to bare GCE (Fig. S2, ESI†) due to a significantly enlarged conductive surface area introduced by rGO.54 The further enhanced capacitance of rGO–PEI–MoSx arises from pseudo-capacitance processes associated with MoSx.55 To confirm the interaction between rGO and MoSx, transmission electron microscopic (TEM) and scanning electron microscopic (SEM) images were used to characterize the morphology. As revealed in Fig. S3 (ESI†), rGO–PEI–MoSx shows a translucent and wrinkled texture associated with flexible graphene sheets. Fig. 3a shows the magnified image and MoSx nanoparticles of diameter 17 ± 3 nm (Fig. S4, ESI†) can be clearly seen. The fast Fourier transform (FFT) pattern indicates the amorphous nature of MoSx nanoparticles (Fig. 3a inset), which is further confirmed by X-ray diffraction (XRD, Fig. S5, ESI†) with no characteristic peaks of any crystalline structures observed. Moreover, in the absence of GO–PEI, the MoSx tended to aggregate into much larger particles (Fig. S6, ESI†). By contrast, as can be seen from Fig. 3b, the MoSx nanoparticles grew uniformly on the surface of rGO–PEI allowing the exposure of more active sites of MoSx to CO2.
 | ||
| Fig. 3 TEM (a) and SEM (b) images of rGO–PEI–MoSx. Inset of (a): Corresponding FFT pattern. Scale bar: (a) 100 nm, (b) 200 nm. | ||
CO2 reduction activities of rGO–PEI–MoSx
To assess the possibility of using rGO–PEI–MoSx for electrocatalytic reduction of CO2, CV studies were firstly undertaken in N2-saturated and CO2-saturated 0.5 M aqueous NaHCO3 solutions (pH values 8.5 and 7.2, respectively) at a scan rate of 50 mV s−1 (Fig. 4a). Under both atmospheres, rapid increases of current were observed at around −0.22 V. In the more negative potential region, a broad process centred at −0.65 V appears under the CO2 atmosphere. Similar processes were also reported in the previous studies with nitrogen-doped carbon nanofibers16 or Ag23,56 as the catalysts and were assigned to the reduction of CO2. Since CV cannot provide conclusive evidence about the nature of this reduction process, potentiostatic electrolysis was conducted by applying a potential in the range of −0.25 V to −0.95 V (corresponding i–t curve is depicted in Fig. S7, ESI†) and the faradaic efficiency (FE) is plotted as a function of the applied potential in Fig. 4b. The results reveal that H2 and CO are the dominant products with a combined FE of around 100% over the whole potential range. Therefore, the liquid products, if any, were not analysed. The selectivity towards H2 and CO is strongly dependent on the applied potential. At an applied potential of −0.25 V (corresponding to an overpotential of 140 mV for the formation of CO, since the equilibrium potential is −0.11 V vs. RHE;5 all the overpotentials hereafter are reported with respect to this equilibrium potential), CO with a FE of ∼5% was detected while H2 is the dominant product. This overpotential for CO formation is significantly lower than that of polycrystalline Au or Ag5 and as low as that achieved in oxide-derived Au nanoparticles under the same conditions.20 FE of CO formation overtakes H2 evolution when the potential becomes more negative and it reaches a maximum of 85.1% at an applied potential of −0.65 V (overpotential of 540 mV), which exceeds the highest FE of the polycrystalline Ag electrode reported in the same electrolyte medium.5 H2 regains dominance in the more negative potential region. This phenomenon was also observed in the case of Ag and Au catalysts in previous reports5,27,56,57 and has been explained as follows: in the potential region of low CO2/CO overpotentials, HER is dominant since it is thermodynamically and kinetically more favourable.5,58 In the potential region of intermediate CO2/CO overpotentials (i.e. −0.35 to −0.75 V vs. RHE), CO2 reduction occurs to form CO, and HER is hindered due to the slow desorption of CO from the active sites.59 In the potential region of high CO2/CO overpotentials, kinetics of both CO2 reduction and HER increase. However, HER regains dominance since H2 evolution is the kinetically favoured process and H+ is much more readily available in the aqueous HCO3− medium. Therefore, CO2 reduction process becomes relatively slower due to either kinetic or mass transport limitations.21,42,56,57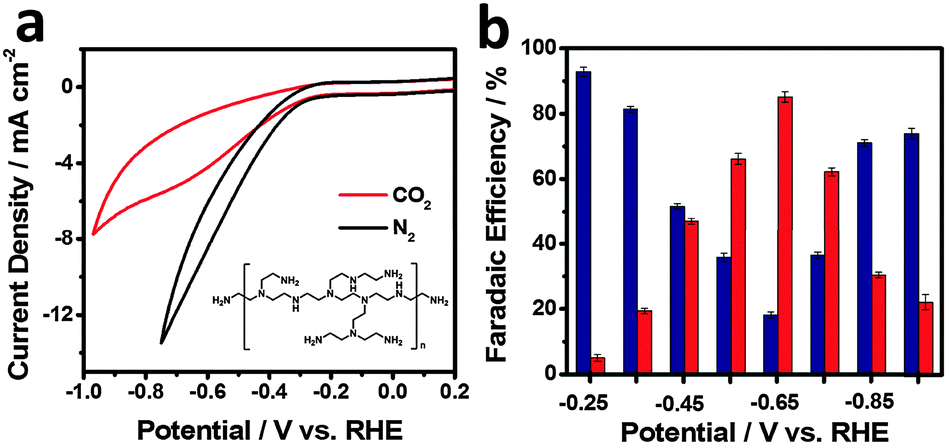 | ||
| Fig. 4 (a) CVs of rGO–PEI–MoSx modified GCE in N2-saturated (black curve) and CO2-saturated (red curve) 0.5 M aqueous NaHCO3 solution. Scan rate was 50 mV s−1. Inset: Structure of PEI. (b) Faradaic efficiency for CO (red bars) and H2 (blue bars) as a function of potential. Error bar associated with each value is also provided. | ||
The source of CO is an important issue that needed to be confirmed. In our study, no CO was detected when the electrolysis was undertaken in a N2 atmosphere, which confirms that the direct source of CO is not HCO3−. Another possible source could be the catalyst materials, which include support electrode, rGO and PEI. 13C labelled CO2 and NaHCO3 could be used to provide direct evidence. However, the formal potential is 0.52 V vs. RHE for the reaction CO(g) + 2H+ + 2e− ↔ C(s) + H2O, which is much more positive than the applied potential (−0.25 to −0.95 V vs. RHE) used in our studies. Therefore, direct oxidation of C to generate CO is not a thermodynamically favorable process. Under the mildly reducing electrochemical conditions involved in our work, extensive oxidation of rGO and PEI to produce the amount of CO detected is also very unlikely. Moreover, we also measured the weight difference of the rGO–PEI–MoSx modified glassy carbon plate electrode before and after electrolysis (see Note S1 in the ESI† for detail) and found that the weight loss is far less than the amount needed to supply the CO produced during electrolysis. Even carbon with the same mass as all of the modified catalyst materials cannot produce sufficient CO at 100% conversion yield. Therefore, neither rGO nor PEI can be the major source of CO.
The intrinsic catalytic activity toward CO2 reduction for each MoSx active site is indicated by turnover frequency (TOF), calculated using the following equation:
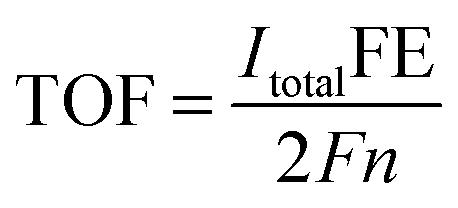
It is also worth noting that the molar ratio of H2 and CO is about 2:1, which is the composition of syngas, when the applied potential is −0.4 V (equivalent to 290 mV overpotential for CO formation) (Fig. 5). Thus, this produced gas mixture could readily serve as the source gas for the Fischer–Tropsch process, an important gas to liquids technology for generating methanol, synthetic petroleum and other liquid fuels.28 More importantly, the FE of H2 and CO remains constant at this potential over at least 3 hours of electrolysis (Fig. 5), showing great promise as a practical catalyst for this process.
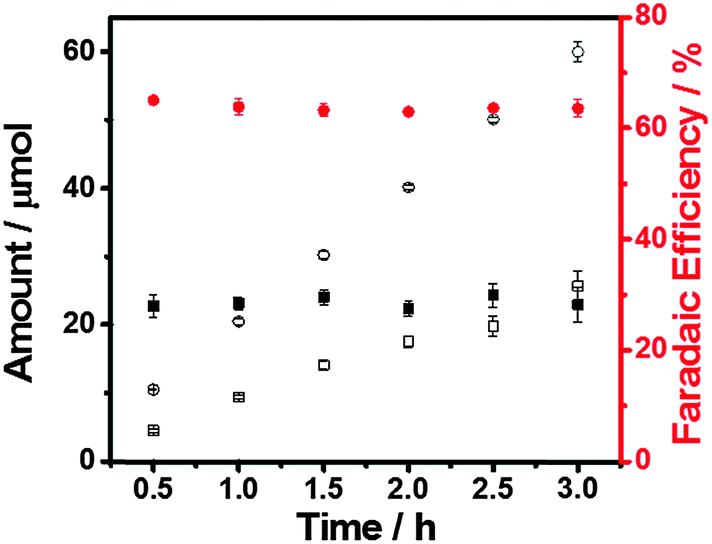 | ||
| Fig. 5 Amount and faradaic efficiency of H2 (circles) and CO (squares). Potentiostatic electrolysis at −0.4 V in CO2-saturated 0.5 M aqueous NaHCO3 solution. Measurement interval: 0.5 h. | ||
In addition, the durability of rGO–PEI–MoSx was also studied by chronoamperometry at −0.65 V, which is the optimum potential with respect to maximum CO selectivity. As shown in Fig. S10 (ESI†), the current remains stable in the time scale of more than 16 hours (60000 s). Initial decay of current, which cannot be completely attributed to the double layer charging process, is mainly due to the adsorption of gaseous products. This current was largely recovered when the electrode was left under open circuit condition for 500 seconds before applying the same potential for electrolysis, suggesting long-term durability of the rGO–PEI–MoSx. SEM images and EDS of the electrode surface after 60000 s are shown in Fig. S11 (ESI†). Compared with the SEM image taken before the electrolysis (Fig. 3b), little difference in morphology was observed after the long-term electrolysis experiment. The elemental analysis reveals the preservation of C, N, Mo and S elements, which also indicates the stability of the material. In HER catalysis, amorphous MoSx often suffers from activity decay due to mechanical loss of catalyst materials or poisoning by surface adsorbates from the electrolyte or reference electrode rather than inherent property changes.60,61 Chorkendorff et al. showed that catalyst support, such as carbon fibers, could play an important role in improving the mechanical stability of the amorphous MoSx catalyst.62 Therefore, in our case, the presence of the rGO support enhances the mechanical stability of the electrodes under catalytic turnover conditions, as suggested in regard to other types of support.55,62–64
Possible origins of the activity of rGO–PEI–MoSx for CO2 reduction
To reveal the possible origins of the activity of rGO–PEI–MoSx for CO2 reduction, the CV measurements were undertaken using rGO–PEI, rGO–MoSx and rGO–PEI–MoSx modified GCEs, respectively. As shown in Fig. 6a, in the absence of MoSx, rGO–PEI modified GCE demonstrates a reduction onset potential that is about 500 mV more negative than the other two electrodes in both N2-saturated and CO2-saturated 0.5 M aqueous NaHCO3 solutions which indicates that MoSx is the effective catalyst towards the reduction reactions observed here. Electrolysis results (Fig. 6b) show only H2 was detected with a FE of ∼93% at −0.85 V. The rGO–MoSx modified GCE presents similar CV curves in N2 and CO2 atmospheres and potentiostatic electrolysis at −0.65 V reveals that H2 is the only product with a FE of ∼95%. Interestingly, although the incorporation of PEI into rGO–MoSx system results in a decrease in current density under either N2 or CO2 atmospheres, which will be discussed later, there is a significant difference in voltammetric characteristics and a considerably higher FE for CO at −0.65 V. Hence, a synergetic effect of PEI and MoSx is responsible for the catalysis of CO2 reduction.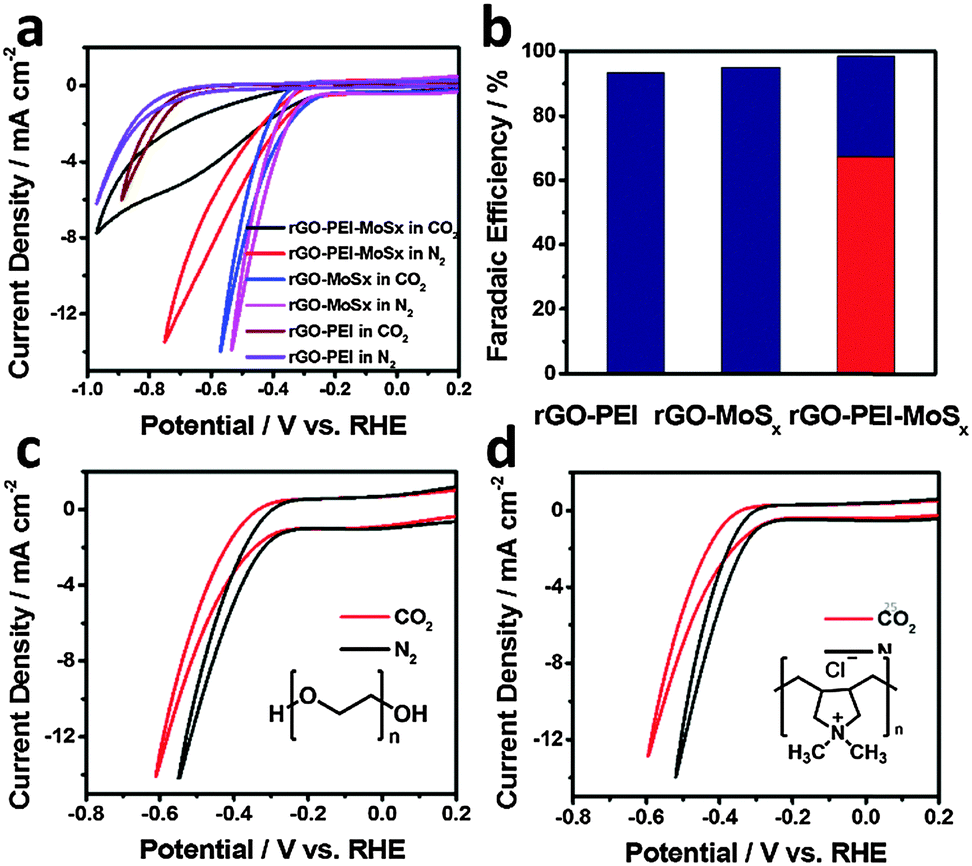 | ||
| Fig. 6 (a) CVs of rGO–PEI, rGO–MoSx, rGO–PEI–MoSx modified GCE in N2-saturated and CO2-saturated 0.5 M aqueous NaHCO3 solution. Scan rate was 50 mV s−1. (b) Faradaic efficiency for CO (red bar) and H2 (blue bars) of rGO–PEI, rGO–MoSx, rGO–PEI–MoSx, potentiostatic electrolysis at −0.65 V in CO2-saturated 0.5 M aqueous NaHCO3 solution. CVs of rGO–PDDA–MoSx (c) and rGO–PEG–MoSx (d) modified GCE in N2-saturated and CO2-saturated 0.5 M aqueous NaHCO3 solution. Scan rate was 50 mV s−1. Inset of (c) and (d) shows the structure of PDDA and PEG, respectively. | ||
Crystalline MoS2 has demonstrated its capability for effectively electroreducing CO2 in an imidazolium based IL mixed with water as proton source.42 Similar observations have also been made previously by the same group in their study of electrocatalytic reduction of CO2 at an Ag electrode in this medium.65 They found that the imidazolium cation could act as a cocatalyst via the formation of an IL–CO2 complex to reduce the activation energy barrier. Nørskov et al. recently predicted theoretically by DFT calculations that the edge sites of crystalline MoS2 could be the catalysing sites for CO2 reduction.41 They claimed that the bridging S atom at the edges of MoS2 could selectively bind the intermediate HCOO over the product CO, which resulted in a deviation from the transition-metal scaling relationship of intermediates in the CO2 reduction process and therefore significantly improved CO2 reduction activities over transition-metal catalysts. Although there are no well-defined edges in amorphous MoSx, there are many structurally and coordinatively unsaturated Mo and S atoms,45,61,66 which are equivalent to the edge sites in the crystal from a catalysis point of view. On this basis, in principle, the amorphous MoSx used in our study may be expected to be a better electrocatalyst for CO2 reduction due to its abundance of such active sites.
However, in the absence of PEI, rGO–MoSx modified GCE only showed activity towards HER, which infers the crucial role of PEI for CO2 reduction. To investigate its role, PEI was replaced with other polymers, poly diallyldimethylammonium chloride (PDDA) or polyethylene glycol (PEG) for the fabrication of rGO–polymer–MoSx modified electrodes. Fig. 6c and d present the CVs of rGO–PDDA–MoSx and rGO–PEG–MoSx modified GCE in N2-saturated and CO2-saturated 0.5 M aqueous NaHCO3 solution at the scan rate of 50 mV s−1, respectively. Again, both electrodes show similar CV curves in N2 and CO2 atmospheres with the same onset potential and only a slight decrease of current density in the present of CO2 and further potentiostatic electrolysis demonstrated H2 as the only product. Thus it is clear that the PEI possesses unique properties that produce synergetic effects with MoSx for CO2 reduction. Two factors may account for this effect:
(1) PEI can suppress HER and thus enhance the competing CO2 reduction. To probe this further, the HER catalysis properties of rGO–MoSx and rGO–PEI–MoSx were investigated in aqueous media, containing 0.5 M H2SO4, 0.5 M bicarbonate (pH 8.5) or 0.5 M phosphate buffer (pH 7.2, at which catalytic CO2 reduction was observed in our study). Results obtained are shown in Fig. S12 (ESI†). The kinetics of HER reaction were retarded in the presence of PEI in all media, as suggested by a larger overpotential. This effect of PEI on the HER is most likely due its amine groups. Rezaei et al. have shown that amine groups in ionic liquid electrolytes can inhibit hydrogen formation in aqueous H2SO4 solution.67 Rosen et al. also found this effect when they used an ionic liquid as a co-catalyst for CO2 reduction in mixed solvents containing water and [EMIM][BF4].68
(2) PEI may stabilize the intermediate species in the CO2 reduction process. Tafel slopes of ∼120 mV dec−1 have often been reported in the previous studies in aqueous media at metallic electrodes since the one-electron reduction of CO2 to form CO2˙− is the rate determining step.5 By contrast, a much smaller value of 74 mV dec−1 was obtained with our rGO–PEI–MoSx modified electrode in the present work (Fig. S13, ESI†). Considering the presence of a competing HER, this value is close to the theoretical value of 59 mV dec−1 predicted for the case where a reversible CO2/CO2˙− process occurs prior to a rate-determining chemical step.7,20,21 This enhanced kinetics of the CO2/CO2˙− process was attributed in studies of N-doped carbon nanotubes, to the stabilization of CO2˙− by the amines on PEI, through hydrogen bonding and electrostatic interactions.43
Conclusions
In summary, we have synthesized amorphous molybdenum sulphide (MoSx) on polyethylenimine (PEI) modified reduced graphene oxide (rGO–PEI–MoSx) and used it as a heterogeneous catalyst for electrocatalytic reduction of CO2 to CO in CO2 saturated aqueous NaHCO3 solution with high efficiency and selectivity. The catalyst is capable of reducing CO2 to CO at an overpotential as low as 140 mV. It reaches a maximum faradaic efficiency of 85.1% at an overpotential of 540 mV, under which this catalyst exhibits a high TOF value of 2.4 s−1 for the formation of CO. Consequently, when the catalyst was deposited on the porous carbon cloth electrode with a large active surface area, a current density of about 50 mA cm−2 can be reached at the same overpotential. At an overpotential of only 290 mV with respect to CO formation, this catalyst is also capable of producing syngas. The catalyst is highly stable in long-term electrolysis. The efficiency and selectivity towards CO2 reduction rather than hydrogen evolution at the optimal applied potential are attributed to the synergetic combination of the properties of MoSx and PEI. Our promising rGO–PEI–MoSx composite catalyst may pave a new way for efficient CO2 reduction catalysis based on inexpensive noble-metal-free materials. Catalytic performance could be further improved by fine tuning the properties of MoS2 using the well-established doping methods.39,51,69Acknowledgements
This research was supported by the Australian Research Council (ARC) through the ARC Centre of Excellence for Electromaterials Science. DRM is also grateful to the ARC for his Australian Laureate Fellowship. The authors acknowledge the Monash Centre for Electron Microscopy for use of electron microscopes.Notes and references
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- H. A. Gasteiger and N. M. Markovic, Science, 2009, 324, 48–49 CrossRef CAS PubMed.
- T. A. Faunce, W. Lubitz, A. W. Rutherford, D. R. MacFarlane, G. F. Moore, P. Yang, D. G. Nocera, T. A. Moore, D. H. Gregory, S. Fukuzumi, K. B. Yoon, F. A. Armstrong, M. R. Wasielewski and S. Styring, Energy Environ. Sci., 2013, 6, 695–698 Search PubMed.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- Y. Hori, Modern aspects of electrochemistry, Springer, 2008, pp. 89–189 Search PubMed.
- J. Schneider, H. Jia, J. T. Muckerman and E. Fujita, Chem. Soc. Rev., 2012, 41, 2036–2051 RSC.
- Y. Chen and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 1986–1989 CrossRef CAS PubMed.
- Y. Oh, H. Vrubel, S. Guidoux and X. Hu, Chem. Commun., 2014, 50, 3878–3881 RSC.
- C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 7231–7234 CrossRef CAS PubMed.
- M. R. Dubois and D. L. Dubois, Acc. Chem. Res., 2009, 42, 1974–1982 CrossRef PubMed.
- C. Costentin, M. Robert and J.-M. Savéant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- I. Bhugun, D. Lexa and J.-M. Savéant, J. Am. Chem. Soc., 1996, 118, 1769–1776 CrossRef CAS.
- S. Lin, C. S. Diercks, Y. B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang and O. M. Yaghi, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- Y. Oh and X. Hu, Chem. Soc. Rev., 2013, 42, 2253–2261 RSC.
- E. Barton Cole, P. S. Lakkaraju, D. M. Rampulla, A. J. Morris, E. Abelev and A. B. Bocarsly, J. Am. Chem. Soc., 2010, 132, 11539–11551 CrossRef CAS PubMed.
- B. Kumar, M. Asadi, D. Pisasale, S. Sinha-Ray, B. A. Rosen, R. Haasch, J. Abiade, A. L. Yarin and A. Salehi-Khojin, Nat. Commun., 2013, 4, 2819–2826 Search PubMed.
- Y. Liu, S. Chen, X. Quan and H. Yu, J. Am. Chem. Soc., 2015, 137, 11631–11636 CrossRef CAS PubMed.
- K. P. Kuhl, T. Hatsukade, E. R. Cave, D. N. Abram, J. Kibsgaard and T. F. Jaramillo, J. Am. Chem. Soc., 2014, 136, 14107–14113 CrossRef CAS PubMed.
- X. Min and M. W. Kanan, J. Am. Chem. Soc., 2015, 137, 4701–4708 CrossRef CAS PubMed.
- Y. Chen, C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 19969–19972 CrossRef CAS PubMed.
- Q. Lu, J. Rosen, Y. Zhou, G. S. Hutchings, Y. C. Kimmel, J. G. Chen and F. Jiao, Nat. Commun., 2014, 5, 3242–3247 Search PubMed.
- Q. Lu, J. Rosen and F. Jiao, ChemCatChem, 2014, 7, 38–47 CrossRef.
- S.-K. Amin, J. Huei-Ru, A. R. Brian, Z. Wei, M. Sichao, J. A. K. Paul and I. M. Richard, J. Phys. Chem. C, 2013, 117, 1627–1632 Search PubMed.
- D. Gao, H. Zhou, J. Wang, S. Miao, F. Yang, G. Wang, J. Wang and X. Bao, J. Am. Chem. Soc., 2015, 137, 4288–4291 CrossRef CAS PubMed.
- W. Zhu, R. Michalsky, O. Metin, H. Lv, S. Guo, C. J. Wright, X. Sun, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2013, 135, 16833–16836 CrossRef CAS PubMed.
- W. Zhu, Y. J. Zhang, H. Zhang, H. Lv, Q. Li, R. Michalsky, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2014, 136, 16132–16135 CrossRef CAS PubMed.
- C. Kim, H. S. Jeon, T. Eom, M. S. Jee, H. Kim, C. M. Friend, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2015, 137, 13844–13850 CrossRef CAS PubMed.
- Y. Zhang, G. Jacobs, D. E. Sparks, M. E. Dry and B. H. Davis, Catal. Today, 2002, 71, 411–418 CrossRef CAS.
- C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 7231–7234 CrossRef CAS PubMed.
- S. Zhang, P. Kang and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 1734–1737 CrossRef CAS PubMed.
- J. L. DiMeglio and J. Rosenthal, J. Am. Chem. Soc., 2013, 135, 8798–8801 CrossRef CAS PubMed.
- J. Medina-Ramos, J. L. DiMeglio and J. Rosenthal, J. Am. Chem. Soc., 2014, 136, 8361–8367 CrossRef CAS PubMed.
- J. C. Boyington, V. N. Gladyshev, S. V. Khangulov, T. C. Stadtman and P. D. Sun, Science, 1997, 275, 1305–1308 CrossRef CAS PubMed.
- K. Schuchmann and V. Müller, Science, 2013, 342, 1382–1385 CrossRef CAS PubMed.
- T. Reda, C. M. Plugge, N. J. Abram and J. Hirst, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10654–10658 CrossRef CAS PubMed.
- A. Bassegoda, C. Madden, D. W. Wakerley, E. Reisner and J. Hirst, J. Am. Chem. Soc., 2014, 136, 15473–15476 CrossRef CAS PubMed.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS PubMed.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS PubMed.
- M. Chhowalla, H. S. Shin, G. Eda, L.-J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef PubMed.
- J. D. Benck, T. R. Hellstern, J. Kibsgaard, P. Chakthranont and T. F. Jaramillo, ACS Catal., 2014, 4, 3957–3971 CrossRef CAS.
- K. Chan, C. Tsai, H. A. Hansen and J. K. Nørskov, ChemCatChem, 2014, 6, 1899–1905 CrossRef CAS.
- M. Asadi, B. Kumar, A. Behranginia, B. A. Rosen, A. Baskin, N. Repnin, D. Pisasale, P. Phillips, W. Zhu, R. Haasch, R. F. Klie, P. Kral, J. Abiade and A. Salehi-Khojin, Nat. Commun., 2014, 5, 4470–4477 CAS.
- S. Zhang, P. Kang, S. Ubnoske, M. K. Brennaman, N. Song, R. L. House, J. T. Glass and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 7845–7848 CrossRef CAS PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- D. Merki, S. Fierro, H. Vrubel and X. Hu, Chem. Sci., 2011, 2, 1262–1267 RSC.
- Y. Zhang, B. Chen, L. Zhang, J. Huang, F. Chen, Z. Yang, J. Yao and Z. Zhang, Nanoscale, 2011, 3, 1446–1450 RSC.
- X. Zhou, Z. Chen, D. Yan and H. Lu, J. Mater. Chem., 2012, 22, 13506–13516 RSC.
- Z. Y. Sui, Y. Cui, J. H. Zhu and B. H. Han, ACS Appl. Mater. Interfaces, 2013, 5, 9172–9179 CAS.
- H. Kim, R. Namgung, K. Singha, I. K. Oh and W. J. Kim, Bioconjugate Chem., 2011, 22, 2558–2567 CrossRef CAS PubMed.
- H. Vrubel and X. Hu, ACS Catal., 2013, 3, 2002–2011 CrossRef CAS.
- C. G. Morales-Guio and X. Hu, Acc. Chem. Res., 2014, 47, 2671–2681 CrossRef CAS PubMed.
- S.-X. Guo, S.-F. Zhao, A. M. Bond and J. Zhang, Langmuir, 2012, 28, 5275–5285 CrossRef CAS PubMed.
- X. Yang, C. Cheng, Y. Wang, L. Qiu and D. Li, Science, 2013, 341, 534–537 CrossRef CAS PubMed.
- F. Li, J. Chen, X. Wang, M. Xue and G. Chen, Adv. Funct. Mater., 2015, 25, 4601–4606 CrossRef CAS.
- Z. Pu, Q. Liu, A. M. Asiri, A. Y. Obaid and X. Sun, J. Power Sources, 2014, 263, 181–185 CrossRef CAS.
- T. Hatsukade, K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Phys. Chem. Chem. Phys., 2014, 16, 13814–13819 RSC.
- Y. Hori, A. Murata, K. Kikuchi and S. Suzuki, J. Chem. Soc., Chem. Commun., 1987, 10, 728–729 RSC.
- C. Delacourt, P. L. Ridgway and J. Newman, J. Electrochem. Soc., 2010, 157, B1902–B1910 CrossRef CAS.
- P. Kedzierzawski and J. Augustynski, J. Electrochem. Soc., 1994, 141, L58–L60 CrossRef CAS.
- D. Merki, H. Vrubel, L. Rovelli, S. Fierro and X. Hu, Chem. Sci., 2012, 3, 2515–2525 RSC.
- J. D. Benck, Z. Chen, L. Y. Kuritzky, A. J. Forman and T. F. Jaramillo, ACS Catal., 2012, 2, 1916–1923 CrossRef CAS.
- A. B. Laursen, P. C. Vesborg and I. Chorkendorff, Chem. Commun., 2013, 49, 4965–4967 RSC.
- D. J. Li, U. N. Maiti, J. Lim, D. S. Choi, W. J. Lee, Y. Oh, G. Y. Lee and S. O. Kim, Nano Lett., 2014, 14, 1228–1233 CrossRef CAS PubMed.
- S. Chen, J. Duan, Y. Tang, B. Jin and S. Z. Qiao, Nano Energy, 2015, 11, 11–18 CrossRef CAS.
- B. A. Rosen, A. Salehi-Khojin, M. R. Thorson, W. Zhu, D. T. Whipple, P. J. Kenis and R. I. Masel, Science, 2011, 334, 643–644 Search PubMed.
- H. Vrubel, D. Merki and X. Hu, Energy Environ. Sci., 2012, 5, 6136–6144 CAS.
- B. Rezaei, S. Mallakpour and M. Taki, J. Power Sources, 2009, 187, 605–612 CrossRef CAS.
- B. A. Rosen, W. Zhu, G. Kaul, A. Salehi-Khojin and R. I. Masel, J. Electrochem. Soc., 2012, 160, H138–H141 CrossRef.
- Y. Sun, S. Gao, F. Lei and Y. Xie, Chem. Soc. Rev., 2015, 44, 623–636 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ee02879e |
| This journal is © The Royal Society of Chemistry 2016 |