
Catalytic reduction of CO2 by H2 for synthesis of CO, methanol and hydrocarbons: challenges and opportunities
Marc D.
Porosoff
a,
Binhang
Yan
b and
Jingguang G.
Chen
*ab
aDepartment of Chemical Engineering, Columbia University, 500 W. 120th Street, New York, NY 10027, USA. E-mail: jgchen@columbia.edu
bChemistry Department, Brookhaven National Laboratory, 2 Center Street, Upton, NY 11973, USA
First published on 22nd October 2015
Abstract
Ocean acidification and climate change are expected to be two of the most difficult scientific challenges of the 21st century. Converting CO2 into valuable chemicals and fuels is one of the most practical routes for reducing CO2 emissions while fossil fuels continue to dominate the energy sector. Reducing CO2 by H2 using heterogeneous catalysis has been studied extensively, but there are still significant challenges in developing active, selective and stable catalysts suitable for large-scale commercialization. The catalytic reduction of CO2 by H2 can lead to the formation of three types of products: CO through the reverse water–gas shift (RWGS) reaction, methanol via selective hydrogenation, and hydrocarbons through combination of CO2 reduction with Fischer–Tropsch (FT) reactions. Investigations into these routes reveal that the stabilization of key reaction intermediates is critically important for controlling catalytic selectivity. Furthermore, viability of these processes is contingent on the development of a CO2-free H2 source on a large enough scale to significantly reduce CO2 emissions.
 Marc D. Porosoff | Dr Marc Porosoff received his BS in 2009 and MS in 2010, both in Chemical and Biomolecular Engineering from the Johns Hopkins University. In 2015, he completed his PhD in Chemical Engineering at Columbia University. Currently, he is National Research Council sponsored postdoctoral fellow at the Naval Research Laboratory. His research interests include CO2 conversion to chemicals and fuels through applications of supported catalysts and transition metal carbides. |
 Binhang Yan | Dr Binhang Yan is a visiting scholar in the Chemistry Department at Brookhaven National Laboratory and a postdoctoral fellow at Tsinghua University. He received his BS in 2008 and PhD in 2013, both in Chemical Engineering from Tsinghua University. His current research activities focus on catalytic reduction of carbon dioxide to produce low-carbon fuels and value-added chemicals over supported monometallic and bimetallic catalysts. |
 Jingguang G. Chen | Dr Jingguang Chen is the Thayer Lindsley Professor of Chemical Engineering at Columbia University. He started his career at the Exxon Corporate Research Laboratories before joining the faculty at the University of Delaware. His current research activities include experimental and theoretical studies aimed at the utilization of carbide and bimetallic materials in catalysis and electrocatalysis. |
Broader contextCatalytic conversion of CO2 into chemicals and fuels is crucial for mitigating climate change and ocean acidification. Depending on the particular catalyst and reaction conditions, the products from the reaction of CO2 and H2 can be selectively controlled. These products are divided into three reaction pathways, reverse water–gas shift (RWGS), methanol synthesis and CO2 Fischer–Tropsch (CO2-FT). Understanding the thermodynamics and kinetics of the three possible pathways is necessary to design new and improved catalysts. Significant advances have been made in catalysts for RWGS and methanol synthesis, but catalysts for CO2-FT require further development from the difficulty of understanding this complex reaction pathway. This perspective describes the challenges and opportunities for each of the three pathways and concludes with recommendations and suggestions for designing improved catalysts through a combination of density functional theory (DFT) calculations and in situ analytical techniques to identify and stabilize key reaction intermediates. Ultimately, CO2 conversion by H2 also requires a CO2-free H2 source, which must be developed together with these catalysts. |
Introduction
As atmospheric concentrations of CO2 continue to rise, efforts must be put forth to avoid negative effects of climate change and ocean acidification.1,2 Stabilization of atmospheric CO2 levels requires both significant cuts in emissions and active removal of CO2 from the atmosphere.3 Utilizing CO2 in a catalytic process to manufacture valuable chemicals and fuels is more desirable than sequestration because the net amount of CO2 mitigated by conversion with renewable energy is 20–40 times greater than sequestration over a 20 year span.4–6 Additionally, the products of CO2 conversion are value-added and can be used as fuels or precursors to produce more complex chemicals and fuels.To substantially reduce CO2 emissions by catalytic conversion, only reactions which produce fuels or commodity chemicals can be considered as viable solutions. The demand for fine chemicals is simply not large enough to effectively reduce emissions through a CO2 conversion process.7 For example, assuming all fuels and chemicals would be produced using CO2 as the feedstock, demand for organic chemicals only accounts for 4% of CO2 emissions, while fuels account for 30% of total CO2 emissions and 100% of emissions from power plants.8 Therefore, conversion to fuels represents a greater impact than to specialty chemicals for achieving a substantial CO2 reduction.
Current efforts into CO2 reduction focus on the development of highly active, selective and stable catalysts in two categories, electrochemical and thermal reduction of CO2. Electrochemical reduction of CO2 would most likely operate on a smaller scale and is more desirable for localized CO2 conversion and production of fine chemicals. There are extensive reports regarding electrochemical CO2 reduction, but they are outside the scope of this perspective and can be found elsewhere.9,10 Research into catalysts for the thermal reduction of CO2 can be further divided into the production of three classes of products, CO, methanol (MeOH) and hydrocarbons.
CO produced by reverse water–gas shift (RWGS) offers high flexibility because CO can be used in both MeOH synthesis and downstream Fischer–Tropsch (FT) for chemicals and fuels. However, RWGS is an endothermic process, which requires high temperatures and the conversion is equilibrium limited to ∼23% at 300 °C and 1 MPa.11 Because the maximum conversion of CO2 ranges from 10% to 50% from 200 °C to 500 °C with a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2:CO2 ratio, efforts must be put forth to develop active catalysts to overcome the slow kinetics and ensure CO is produced at the maximum allowable yield.
:1 H2:CO2 ratio, efforts must be put forth to develop active catalysts to overcome the slow kinetics and ensure CO is produced at the maximum allowable yield.
CO2 conversion to MeOH is the most direct route for CO2 utilization because MeOH can be used as a fuel additive, fuel substitute and precursor to many commodity chemicals.12 Although MeOH synthesis from CO2 and H2 is exothermic, CO2 conversion to MeOH is kinetically limited at low temperatures and thermodynamically limited at high temperatures, resulting in a low theoretical MeOH yield of 0.06% at 300 °C and 0.1 MPa.13 In typical industrial MeOH synthesis, CO, H2 and a small amount of CO2 are reacted over a Cu/ZnO/Al2O3 catalyst between 5–10 MPa at 220–300 °C.14 Cu/ZnO/Al2O3 has also been investigated for MeOH synthesis from CO2 and H2, but further improvements are needed to improve MeOH selectivity and yield.
Direct hydrogenation of CO2 can also lead to the production of hydrocarbons, including both alkanes and olefins. Direct hydrogenation of CO2 to –CH2– species is possible through dissociative adsorption followed by hydrogenation, but the extent to which this occurs is not well known.11 Another possible route is direct FT from CO2 and H2 (CO2-FT) by performing RWGS followed by FT in one reactor, which is thermodynamically easier than RWGS because the overall process is exothermic.15 The CO2-FT process is very attractive because it provides a route to directly produce alkanes and olefins from CO2 and H2, but designing catalysts that are water resistant with high olefin selectivity is challenging. Out of the three CO2 conversion processes mentioned, CO2 hydrogenation to long-chain hydrocarbons is the least studied and characterized process.
In this perspective, each of the three pathways of CO2 reduction by H2 will be reviewed in the order of (1) CO2 to CO via the RWGS reaction over bimetallic and carbide catalysts, (2) CO2 to MeOH over Cu-based catalysts and other materials and (3) CO2 to hydrocarbons via CO2-FT over redesigned FT catalysts. The perspective will conclude by discussing challenges and opportunities for further advancing the field of CO2 reduction by H2.
CO production through reverse water–gas shift
Typical RWGS catalysts consist of well isolated and dispersed nanoparticles supported on a metal-oxide to maximize the interfacial area between the metal and the support.16 The interfacial region is important because both the metal and support are involved in the RWGS chemistry. Two reaction pathways have been proposed for CO formation from RWGS. One is the redox mechanism, where over Cu-based catalysts, CO2 oxidizes Cu0 to generate CO and Cu+ while H2 reduces Cu+ to form H2O.17 Further evidence for this mechanism is provided by FTIR spectroscopy studies over a Cu/ZnO catalyst which indicate CO2 dissociates to CO,18 but formate has also been detected over Cu0.19The other widely accepted pathway is the formate decomposition mechanism in which CO2 is first hydrogenated into formate,20 followed by cleavage of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond. Therefore, an effective RWGS catalyst should be dual functional with high activity for both hydrogenation and CO bond scission. Metal nanoparticles supported on metal-oxides are popular materials because dispersed metal catalytic sites dissociate hydrogen relatively easily,21 which then allows reactive atomic hydrogen to spill-over onto the support and hydrogenate CO2 that is adsorbed on the oxides.22
O bond. Therefore, an effective RWGS catalyst should be dual functional with high activity for both hydrogenation and CO bond scission. Metal nanoparticles supported on metal-oxides are popular materials because dispersed metal catalytic sites dissociate hydrogen relatively easily,21 which then allows reactive atomic hydrogen to spill-over onto the support and hydrogenate CO2 that is adsorbed on the oxides.22
Based on the proposed mechanisms, an active and selective catalyst for RWGS should consist of both an active metal and metal-oxide support that participate in the reaction steps. Cu-based catalysts, noble metals and catalysts supported on CeO2 have been studied extensively.4,5 Pt-based catalysts are generally popular because of their high hydrogenation activity, with Pt–Co bimetallics showing higher CO production than their parent metals.23 A detailed study into Pt–Co supported on MCF-17 with ambient pressure X-ray photoelectron spectroscopy (AP-XPS) and environmental transmission electron microscopy (eTEM) reveals that the surface is enriched in Pt, explaining the Pt-like selectivity of Pt–Co. In comparison with the pure Co catalyst, the addition of Pt aids the reduction of Co, shifting the selectivity primarily toward CO.24 Details of the activity and selectivity with reaction conditions of several representative RWGS catalysts are compared in Table 1.
| Catalyst | H2:CO2 ratio |
Temperature (°C) | Pressure (MPa) | Conversion (%) | Selectivity (%) |
|---|---|---|---|---|---|
| NiO/CeO216 | 1:1 |
700 | 0.1 | ∼40 | ∼100 |
| Cu/Al2O320 | 1:9 |
500 | N/A | ∼60 | N/A |
| Co/MCF-1724 | 3:1 |
200–300 | 0.55 | ∼5 | ∼90 |
| Pt–Co/MCF-1724 | 3:1 |
200–300 | 0.55 | ∼5 | ∼99 |
| Cu/SiO234 | 1:1 |
600 | 0.1 | 5.3 | N/A |
| Cu/K/SiO234 | 1:1 |
600 | 0.1 | 12.8 | N/A |
| Cu–Ni/γ-Al2O335 | 1:1 |
600 | 0.1 | 28.7 | 79.7 |
| Cu–Fe/SiO236 | 1:1 |
600 | 0.1 | 15 | N/A |
| Li/RhY37 | 3:1 |
250 | 3 | 13.1 | 86.6 |
| Rh/SiO238 | 3:1 |
200 | 5 | 0.52 | 88.1 |
| Rh/TiO225 | 1:1 |
270 | 2 | 7.9 | 14.5 |
| Fe/TiO225 | 1:1 |
270 | 2 | 2.7 | 73.0 |
| Rh–Fe/TiO225 | 1:1 |
270 | 2 | 9.2 | 28.4 |
| Fe–Mo/γ-Al2O326 | 1:1 |
600 | 1 | ∼45 | ∼100 |
| Mo/γ-Al2O327 | 1:1 |
600 | 1 | 34.2 | 97 |
| Pd/Al2O330 | 1:1 |
260 | 0.1 | N/A | 78 |
| Pd/CeO2/Al2O330 | 1:1 |
260 | 0.1 | N/A | 87 |
| Pd/La2O3/Al2O330 | 1:1 |
260 | 0.1 | N/A | 70 |
| CeO2–Ga2O332 | 1:1 |
500 | 0.1 | 11.0 | N/A |
| Pt/TiO233 | 1.4:1 |
400 | N/A | ∼30 | N/A |
| Pt/Al2O333 | 1.4:1 |
400 | N/A | ∼20 | N/A |
| PtCo/CeO239 | 3:1 |
300 | 0.1 | 3.3 | 71.0 |
| Co/CeO239 | 3:1 |
300 | 0.1 | 3.8 | 39.4 |
| PtCo/γ-Al2O339 | 3:1 |
300 | 0.1 | 5.1 | 89.4 |
| Co/γ-Al2O339 | 3:1 |
300 | 0.1 | 3.8 | 67.0 |
| Mo2C39 | 3:1 |
300 | 0.1 | 8.7 | 93.9 |
| Mo2C40 | 5:1 |
250 | 2 | 17 | 34 |
| Cu–Mo2C40 | 5:1 |
250 | 2 | 13 | 40 |
| Ni–Mo2C40 | 5:1 |
250 | 2 | 21 | 29 |
| Co–Mo2C40 | 5:1 |
250 | 2 | 23 | 24 |
Although Pt-based catalysts are active and selective for RWGS, their high cost is unattractive for large scale conversion of CO2. Fe-based catalysts are promising and show high activity and selectivity for RWGS,25 while a bimetallic Fe–Mo catalyst has a decreased particle size with higher Fe dispersion and improved stability from the formation of a Fe2(MoO4)3 phase.26 Bimetallic Ni–Mo shows similar behavior to the Fe–Mo system27 and NiO supported on mesoporous CeO2 shows high CO selectivity when the NiO particles are well dispersed on the support.16
While the metallic phase is clearly important for RWGS selectivity, the reducibility of the metal-oxide support can significantly influence the activity. CeO2 is a common support for RWGS because of its reducibility and high intrinsic activity toward CO2 adsorption. DFT studies indicate that the CeO2(110) surface is more catalytically active than (100) or (111), likely because the creation of oxygen vacancies is most facile on CeO2(110).28 For Pt nanoparticles supported on CeO2, temporal analysis of products (TAP) studies with isotopically labeled CO2 indicate that the order of H2 and CO2 adsorption on the surface is critical. The presence of Pt improves oxygen exchange of CO2 with oxygen defects in CeO2.29 The addition of CeO2 to catalysts supported on irreducible oxides can also improve activity, as Pd/CeO2–γ-Al2O3 is more active than Pd/γ-Al2O3 because of the ability of CeO2 to exchange oxygen.30
CeO2 is clearly a well-studied reducible support for RWGS, but other reducible metal-oxides are also promising. CO2 binds on In2O3 in a bent configuration and has an exothermic energy of adsorption, which contributes to the high activity.31 Ga2O3 is an active support and can be further improved by the addition of CeO2, which enhances the generation of bicarbonate intermediates that readily dissociate into CO and H2O.32 TiO2 is another reducible support that is active for RWGS and it has been shown that Pt/TiO2 outperforms the irreducible Pt/γ-Al2O3 catalyst.33
The aforementioned combinations of metal and oxide phases require the presence of active and stable interfacial regions for RWGS. In principle, an ideal catalyst should consist of one phase that can perform both hydrogenation and CO bond scission to selectively produce CO from CO2. One promising class of catalysts are transition metal carbides (TMCs), which have shown desirable behavior for reactions involving CO2,41 and properties similar to precious metals for many other reactions.42 Perhaps the most interesting TMC for RWGS is Mo2C because of its low cost, dual functionality for H2 dissociation and CO bond scission, and potential to behave similarly to reducible oxides, such as CeO2.39 As compared in Fig. 1, Mo2C outperforms Pt-based bimetallic catalysts supported on CeO2 in terms of both activity for CO2 conversion and selectivity toward CO production.
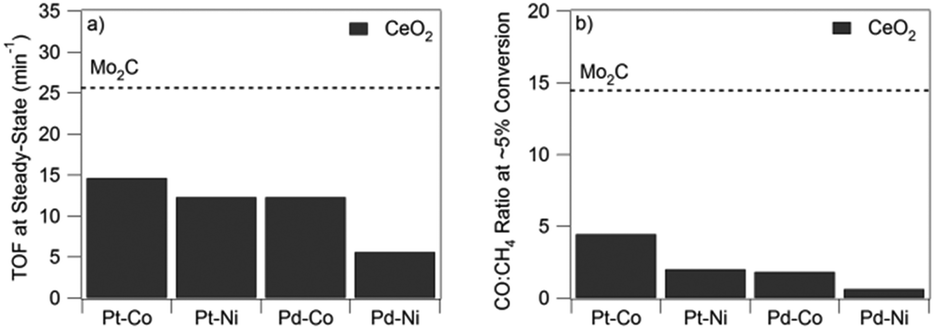 | ||
| Fig. 1 TOF (a) and selectivity (b) at 300 °C on bimetallic supported catalysts on CeO2 (black bars) and Mo2C (dashed line). (Reproduced from ref. 39 with permission from John Wiley and Sons.) | ||
Several mechanistic studies have been performed for CO2 activation over Mo2C to understand the high intrinsic activity towards CO2. The findings show that CO2 binds to Mo2C in a bent configuration and one of the CO bonds can spontaneously break,39,43 leaving adsorbed CO and O on the catalyst surface. The adsorbed CO can desorb, but the adsorbed O, in the form of an oxycarbide (Mo2C–O), must be removed by H2 to complete the catalytic cycle. Because CO2 activation over TMCs results in oxycarbide formation, the oxygen binding energy (OBE) on the TMC surface is an important descriptor for predicting high RWGS activity.44 Future studies of CO2 reduction by H2 over TMCs should investigate metal-modified carbides as it has been shown that metal can modify the electronic properties of the TMC, thus influencing the OBE and possibly product selectivity.40
Methanol synthesis
Currently the CAMERE (carbon dioxide hydrogenation to form methanol via reverse-water gas shift) process produces MeOH from CO2 and H2 at a capacity of ∼75 Mt y−1. The overall process scheme involves RWGS over ZnAl2O4 followed by water removal and MeOH synthesis over Cu/ZnO/ZrO2/Ga2O3, but the disadvantage is that it requires two different catalysts and reactors.45 An ideal process should use one catalyst in a single reactor, much like current research over Cu/ZnO/Al2O3, the commercial catalyst for MeOH synthesis from CO and H2.46,47 This catalyst has shown varying degrees of CO2 conversion, selectivity and space-time yield, as compared in Table 2 with other catalysts. Although Cu/ZnO/Al2O3 exhibits promising performance (with a space-time yield up to 7729 gMeOH kgcat−1 h−1) under certain conditions (36 MPa and 10:1 H2:CO2 ratio),48 the pressure is likely too high for economic conversion of CO2.
| Catalyst | H2:CO2 ratio |
Temperature (°C) | Pressure (MPa) | Conversion (%) | Selectivity (%) | Space-time yield (gMeOH kgcata−1 h−1) |
|---|---|---|---|---|---|---|
| Cu–ZnO/Al2O348 | 10:1 |
260 | 36 | 22.7 | 77.3 | 7729 |
| CuO–ZnO/Al2O349 | 3.89:1 |
280 | 5 | 19.5 | 37 | 311 |
| CuO–ZnO/CeO249 | 3.89:1 |
280 | 5 | 12.8 | 37 | 210 |
| Cu–Zn–Ga50 | 3:1 |
270 | 3 | 15.9 | 29.7 | 135.9 |
| Cu/ZrO2/CNF51 | 3:1 |
180 | 3 | 14 | N/A | 34 |
| Cu/plate ZnO/Al2O352 | 2.2:1 |
270 | 4.5 | 10.9 | 72.7 | N/A |
| Cu/γ-Al2O353 | 3.8:1 |
200 | 36 | 8.4 | 37.3 | 103.4 |
| Cu–K/γ-Al2O353 | 3.8:1 |
280 | 36 | 28.6 | 2.1 | 18.2 |
| Cu–Ba/γ-Al2O353 | 3.8:1 |
280 | 10 | 25.2 | 9.3 | 70.7 |
| Pd–CaO/MCM-4154 | 3:1 |
250 | 3 | 12.1 | 65.2 | N/A |
| Mo2C55 | 1:3 |
220 | 6 | 4.6 | 17.7 | ∼21.5 |
| WC55 | 1:3 |
220 | 6 | 1.4 | 22.4 | ∼8.3 |
| Cu–Mo2C55 | 1:3 |
220 | 6 | 4 | 31.5 | ∼33.3 |
| Cu–WC55 | 1:3 |
220 | 6 | 0.6 | 21.3 | ∼3.4 |
| Cu–SiO255 | 1:3 |
220 | 6 | 5.3 | 34.2 | ∼47.9 |
| Cu–ZnO/ZrO256 | 3:1 |
240 | 3 | 17.0 | 41.5 | ∼48.8 |
| Cu–ZnO/TiO2–ZrO256 | 3:1 |
240 | 3 | 17.4 | 43.8 | ∼52.7 |
| CuO–ZnO/ZrO257 | 3:1 |
240 | 3 | 18.0 | 51.2 | 305 |
| Fe–Cu/MCM-4158 | 3:1 |
200 | 1 | ∼2 | 99.97* | N/A |
| Pd–Cu/SiO259 | 3:1 |
250 | 4.1 | 6.6 | 34.0 | 35.7 |
| Pd–Cu/SBA-1559 | 3:1 |
250 | 4.1 | 6.5 | 23.0 | 23.0 |
| CoMoS60 | 3:1 |
310 | 10.4 | 28 | 31 | N/A |
| Rh–Sn/SiO261 | 3:1 |
240 | 5 | 2.8 | 43.1 | ∼23.5 |
| NiGa/SiO262 | 3:1 |
160–260 | 0.1 | N/A | N/A | 90–125 |
| Cu–ZnO/γ-Al2O363 | 3:1 |
250 | 3 | 10.1 | 78.2 | 76.8 |
| Cu/ZnO64 | 9:1 |
165 | 0.1 | N/A | 61.3 | 5.2 |
| Cu@ZnO65 (core–shell) | 3:1 |
250 | 3 | 2.3 | 100 | 147.2 |
| La–Mn–Zn–Cu–O66 | 3:1 |
270 | 5 | 13.1 | 54.5 | 100 |
| Cu–ZnO–TiO267 | 3:1 |
220 | 3 | 14.8 | 50.5 | 51.5 |
| CuO/ZnO68 | 3:1 |
240 | 3 | 16.5 | 78.2 | 550 |
| Au/ZrO269 | 3:1 |
240 | 0.5 | 9.3 | 3.4 | 21.1 |
| Cu/ZrO2/CNT70 | 3:1 |
260 | 3 | 16.3 | 43.5 | 84.0 |
| Pd–ZnO/CNT71 | 3:1 |
270 | 5 | 19.63 | 35.5 | 343 |
| Pd/Ga2O372 | 3:1 |
250 | 5 | 17.33 | 51.62 | ∼175.6 |
| La–Zr–Cu–Zn–O73 | 3:1 |
250 | 5 | 12.6 | 52.5 | 100 |
| Cu/Zn/Al/Y74 | 3:1 |
250 | 5 | 26.9 | 52.4 | 520 |
| Ga–Cu–ZnO–ZrO275 | 3:1 |
250 | 7 | 22 | 72 | 704 |
| Cu–ZnO–ZrO276 | 3:1 |
240 | 5 | 9.7 | 62 | 1200 |
| La–Cu/ZrO277 | 3:1 |
220 | 3 | 6.2 | 66 | N/A |
| Pd–Ga/CNT78 | 3:1 |
250 | 5 | 16.5 | 52.5 | 512 |
| LaCr0.5Cu0.5O379 | 3:1 |
250 | 2 | 10.4 | 90.8 | ∼278 |
| Ga2O3–Pd/SiO280 | 3:1 |
250 | 3 | 1.34 | 58.9 | 283.4 |
| Cu/ZnO–ZrO281 | 3:1 |
220 | 8 | 21 | 68 | 181 |
| Au/ZnO–ZrO281 | 3:1 |
220 | 8 | 2 | 100 | 19 |
| PdO–CuO–ZnO82 | 3:1 |
240 | 6 | 9.19 | 66.2 | 322 |
| Cu–Ga/ZnO83 | 3:1 |
270 | 2 | 6.0 | 88 | 378 |
| YBa2Cu3O784 | 3:1 |
240 | 3 | 3.4 | 50.7 | N/A |
Similar to the commercial Cu/ZnO/Al2O3 catalyst, Cu-based materials are popular choices for MeOH synthesis from CO2;49–51 however, activity over Cu-based catalysts is structure sensitive. Ultra-high vacuum (UHV) experiments indicate that Cu(110) is not intrinsically active for CO2 dissociation,85 while other studies show Cu(110) is more active toward CO2 than Cu(111) and Cu(100).19 To improve interactions with CO2, many researchers have shown that adding promoters can significantly improve the CO2 adsorption strength and MeOH selectivity. For example, potassium (K) promoters on Cu/Al2O3 stabilize surface intermediates and enhance formate dissociation, lanthanum (La) doping on Cu/ZrO2 promotes formate hydrogenation to MeOH and inhibits its dissociation into CO,77 barium (Ba) promoters inhibit formate dissociation and promote MeOH synthesis,53 and adding CaO to Pd/MCM-41 improves CO2 adsorption and leads to higher CO2 conversion and MeOH selectivity.54 A similar conclusion is obtained over transition metal carbides and those modified with Cu and Au.55 Cu and Au nanoparticles supported on TiC(001) become charge polarized, which increases CO2 binding energy, making some of these systems more active than traditional Cu/ZnO catalysts.86
The size of the Cu and ZnO crystallites in Cu–ZnO catalysts can also influence the CO2 adsorption strength on the catalyst,56 with the catalyst synthesis method playing an important role. CuO/ZrO2 prepared by deposition–precipitation has a smaller particle size and exhibits higher activity when compared to impregnation or co-precipitation.87 Catalysts synthesized by the gel-oxalate coprecipitation method show a higher interfacial surface area and MeOH yield than coprecipitation with sodium bicarbonate and complexation with citric acid.57 On the other hand, a study over Fe–Cu/MCM-41 demonstrates that larger particles with less metal–support interaction are more favorable for CO2 hydrogenation to alcohols.58
In addition to interacting strongly with CO2, catalysts should stabilize the desired intermediate for high MeOH yield. There are some conflicting studies reporting carboxyl, formic acid or formaldehyde as important intermediates.88 Other researchers hypothesize that formate is the intermediate over Zn-modified Cu(111),89,90 while infrared studies on Cu/SiO2 contradict the previous study and hypothesize that carboxyl is the intermediate with formate simply acting as a spectator.91 Furthermore, DFT calculations show that methanol synthesis on Cu(111) is more energetically favorable from hydrocarboxyl (trans-COOH) than formate in the presence of H2O.92
An extensive study combining DFT and UHV experiments on Cu-based model surfaces confirms that stabilization of formyl combined with facile hydrogenation of formate and dioxomethylene (H2COO) are critical for high MeOH yield.93 In this case, an ideal catalyst should lower the barrier for H2COO hydrogenation and exhibit an intermediate CO binding energy. Out of several metals supported on Cu(111), Ni/Cu(111) exhibits the lowest barrier for H2COO hydrogenation with an intermediate CO binding energy, leading to the highest MeOH production out of Pt, Rh, Pd, Cu and Au supported on Cu(111).94
It is well established that Cu is an important metal for promoting MeOH synthesis, but the reducibility of Cu and the nature of the support material can also have a significant effect on the catalytic performance. For example, deactivation over Cu/ZnO/Al2O3 can be caused by several factors, excess surface hydroxyls, Cu sintering, and decreasing catalyst reducibility from fixation of Cu in the monovalent oxidation state.95 To improve the catalytic activity and selectivity, Graciani et al. supported a reducible oxide, CeOx on Cu(111).96 AP-XPS and infrared reflection absorption spectroscopy (IRRAS) experiments reveal that the metal-oxide Cu–ceria interface directly activates CO2 in the form of an unstable carboxylate (CO2δ−), which is a desirable intermediate and opens a new reaction pathway for MeOH synthesis. The low stability of the CO2δ− species over CeOx/Cu(111) and Cu/CeOx/TiO2(110) leads to MeOH synthesis rates that are significantly faster than those over traditional Cu/ZnO catalysts, as seen in Fig. 2.
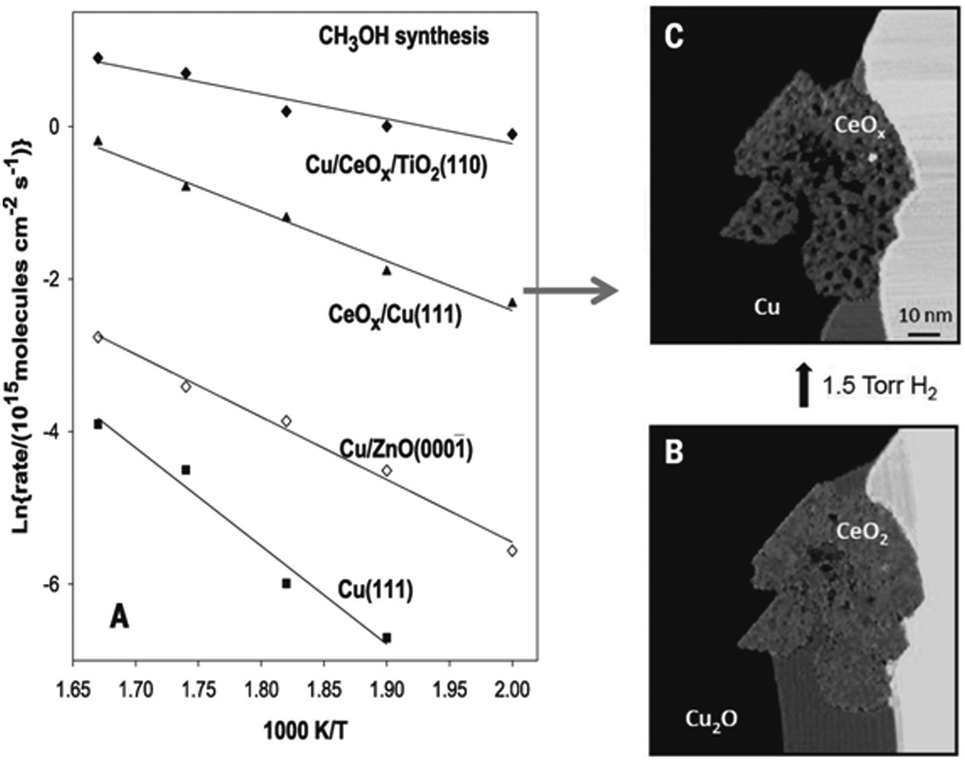 | ||
| Fig. 2 Arrhenius plot for methanol synthesis on Cu(111), a 0.2 ML of Cu on ZnO(0001), a Cu(111) surface covered 20% by ceria, and a 0.1 ML of Cu on a TiO2(110) surface pre-covered 15% with ceria (A). STM image of a CeOx/Cu(111) surface as prepared (B). In situ STM image taken during exposure to 1.5 Torr of H2 at 27 °C after 26 hours of reaction (C). (Reproduced from ref. 96 with permission from the American Association for the Advancement of Science.) | ||
The study by Behrens et al. over CeOx/Cu(111) offers a different mechanism from the majority of researchers for MeOH synthesis from CO2.97 Most studies propose that the first step of MeOH synthesis is the direct hydrogenation of CO2 through a formate intermediate, while Graciani et al. propose that the overall mechanism is RWGS followed by CO hydrogenation to MeOH. A recent study over Pd–Cu/SiO2 also shows that CO produced through RWGS contributes to MeOH synthesis.59
Similar to the study of Graciani et al., DFT calculations over Mo6S8, a structural building block of MoS2, show that MeOH synthesis proceeds through RWGS followed by CO hydrogenation to MeOH,98 which is consistent with studies over CoMoS.60 Investigations of Rh-based bimetallic catalysts indicate that CO is the intermediate,61 with XPS measurements over Rh–Co/SiO2 showing a surface enriched in Co; however, the more desirable surface is enriched in Rh, which correlates with CO stabilization and higher MeOH selectivity.99 Observations from these studies indicates that the mechanism of MeOH synthesis from CO2 is controversial, with researchers providing evidence for both formate and CO being the intermediates.
A recent study of low-pressure CH3OH synthesis over Au/CeOx/TiO2 model catalysts also indicate that charge redistribution over metal particles may play a role.100 The addition of CeOx over Au/CeOx/TiO2 leads to increases in both CO2 conversion and CH3OH selectivity. AP-XPS measurements reveal that Au is partially negatively charged and CeOx is in the Ce3+ state. The presence of adjacent negatively charged Au and Ce3+ sites enhances the adsorption strength of CO2, leading to the higher CH3OH yield.
Regardless of the exact nature of the intermediate, there is a necessity for more researchers to take advantage of DFT to identify potential descriptors that correlate with MeOH yield. By using the BEEF-vdW101 functional, it has been shown that all of the relevant energy kinetics of MeOH synthesis can be mapped using one parameter, the oxygen adsorption energy (ΔEO). Plotting TOF of CO2 hydrogenation versus ΔEO leads to a volcano relationship with Cu/ZnO and Ni–Ga at the peak. These two materials exhibit an optimal interaction with oxygen, resulting in stabilization of intermediates without poisoning the surface.62 As more experimental results become available, future studies should continue to use DFT to develop descriptors to identify other novel and active materials for MeOH synthesis from CO2 and H2.
CO2-FT for alkane and olefin production
Another promising route is the direct production of hydrocarbons, including both alkanes and olefins, from direct Fischer–Tropsch with CO2 and H2 (CO2-FT). Olefins are produced on the order of 200 Mt per year and result in 1.2–1.8 tons of CO2 emitted per ton of olefin produced.15 By manufacturing these products with a CO2 feedstock, the net CO2 emissions of the process will substantially decrease. However, designing active catalysts for CO2-FT is difficult because they should be active for both RWGS and FT. Thermodynamics suggest that CO2-FT becomes more favorable as higher chain compounds are formed because RWGS is slightly endothermic and the FT process is exothermic.102,103 Furthermore, high conversion of CO2 can only be achieved if the FT step is fast enough to overcome the thermodynamic limitation of RWGS, which is the main challenge for CO2-FT.104 Other difficulties with designing catalysts for CO2-FT are that (1) CO2 is likely a poison for CO hydrogenation catalysts15 and (2) water, an unavoidable byproduct during CO2-FT, is a known poison that influences catalyst activity and product selectivity,105 as seen in Fig. 3. | ||
| Fig. 3 Comparison of model prediction for CO2 conversion (○), C3H6 yield (□), and water (△) in catalytic tubular reactor with water removal, represented by hollow symbols and without water removal, represented by solid symbols. (Reproduced from ref. 104 with permission from Elsevier.) | ||
The most commonly used metals in typical FT with syngas (CO + H2) are Fe at higher temperatures and Co at lower temperatures. Generally, when comparing CO and CO2 FT, CO conversion (up to 87%) is much higher than CO2 conversion (up to 45%),15 indicating that current FT catalysts are not adequate for CO2-FT. Furthermore, in CO2-FT, Co catalysts lead to high methane production and a deviation from the Anderson–Schultz–Flory (ASF) distribution.106 This is further supported by a study over Co-based catalysts which shows that CO forms typical FT products, while CO2 produces CH4 over Co/SiO2 and Co–Pt/γ-Al2O3.107 Therefore, new and improved catalysts should be investigated to synthesize typical FT products with CO2 as the carbon source.
Current research into CO2-FT primarily focuses on Fe-based catalysts, which yield higher olefins than Co-based catalysts.108,109 Fe supported on γ-Al2O3 promotes C2+ hydrocarbon formation, while Ni catalysts yield CH4 as the primary product.110 The active site of these Fe-based catalysts is under intense debate. Some studies indicate that an iron carbide phase is active,111 while others show that the FeO phase is active and interacts strongly with the support.112 CO2 reduction into long-chain hydrocarbons is significantly improved with the addition of effective promoters, for example, K promoters in Fe catalysts help stabilize the iron carbide phase and adding boron (B) leads to light olefin formation.113 One hypothesis is that K promotes CO2 binding and hinders hydrogen adsorption,114 which leads to suppressed methane formation and increases the olefin to alkane ratio.103 Adding manganese (Mn) in a Fe/γ-Al2O3 catalyst also promotes long-chain olefin synthesis and suppresses methane formation as seen in Table 3.109
| Catalyst | H2:CO2 ratio |
Temperature (°C) | Pressure (MPa) | Conversion (%) | Selectivity (%) |
|---|---|---|---|---|---|
| Fe–La–Cu–K/TiO2103 | 3:1 |
300 | 1 | 27 | C5–C15 (40) |
| Fe–Ru–Zn–K/TiO2103 | 3:1 |
300 | 1 | 27 | C5–C15 (37) |
| Fe–Zr–Cu–K/TiO2103 | 3:1 |
300 | 1 | 25 | C5–C15 (30) |
| Co–Pt/Al2O3106 | 1:1 |
220 | 1.9 | 6.8 | CH4 (93.1), C2–C4 (6.8) |
| Fe/Al2O3109 | 3:1 |
290 | 1.4 | 18.2 | C2–C5 + (34.9) |
| Mn–Fe/Al2O3109 | 3:1 |
290 | 1.4 | 37.7 | C2–C5 + (55.3) |
| K–Mn–Fe/Al2O3109 | 3:1 |
290 | 1.4 | 41.4 | C2–C5 + (62.4) |
| Fe/Al2O3110 | 3:1 |
300 | 1.1 | 12.1 | C2–C7 (38) |
| Fe/K–OMS-2111 | 2:1 |
120–320 | 13.7 | 45 | C2–C6 (68.7) |
| Fe–K/Al2O3–MgO112 | 3:1 |
300 | 1.01 | 27.5 | C2–C5 + (58.5) |
| Fe–Co–K/Al2O3114 | 3:1 |
300 | 1.1 | 31 | C2 + (69) |
| Co/Al2O3115 | 6:1 |
260 | 0.1 | 2.5 | N/A |
| Co/MgO115 | 6:1 |
260 | 0.1 | 2.0 | N/A |
| Co/SiO2115 | 6:1 |
260 | 0.1 | 1.5 | N/A |
| Ni/SiO2116 | 4:1 |
350 | 0.1 | 28.4 | CH4 (86.7) |
| Ni/CexZr1−xO2116,117 | 4:1 |
350 | 0.1 | 70.6 | CH4 (98.6) |
| Ni/CeO2118 | 4:1 |
350 | 0.1 | ∼90 | CH4 (∼100) |
| Ru/γ-Al2O3119 | 4:1 |
150–325 | 0.1 | N/A | CH4 (∼100) |
| Ru/TiO2120 | 4:1 |
160 | 0.1 | 100 | CH4 (100) |
| Pd–Mg/SiO2121 | 4:1 |
450 | 0.1 | 59.2 | CH4 (95.3) |
| Pd–Ni/SiO2121 | 4:1 |
450 | 0.1 | 50.5 | CH4 (89.0) |
| Pd–Li/SiO2121 | 4:1 |
450 | 0.1 | 42.6 | CH4 (88.5) |
When comparing results from the Fe-based catalysts to those of Co and Ni, the CO2 conversion over Fe materials is generally higher due to their increased RWGS activity. However, it is possible that the active phase in Co-based materials is difficult to stabilize under reaction conditions. In situ X-ray absorption near edge spectroscopy (XANES) and XPS measurements of Co/TiO2 show that the CoO phase is more active than Co metal for CO2 hydrogenation and larger particles are more active because they are more easily oxidized.122 Traditional FT Co-based catalysts show similar intermediates during CO2 and CO hydrogenation according to FTIR measurements of a Co/γ-Al2O3 catalyst, suggesting that the hydrogenation pathway might be the same for both reactants. When CO2 and CO are introduced together as feed, CO hydrogenation is primarily observed with CO2 hydrogenation as a minor pathway because of competitive adsorption.123
The future direction of CO2-FT should be focused on synthesizing catalysts that are highly active, selective and water-resistant in the range of 100–300 °C. It has been shown that catalysts synthesized with silica improve stability in water, with examples being HZSM-5 zeolite124 and iron-based catalysts,125 while the type of support material can prevent sintering of the active metallic phase to ensure catalytic stability.109 Additionally, carbon composites synthesized through deposition of mesoporous carbon by impregnation of sugars126 are promising materials as they improve activity by increasing metal dispersion and preventing leaching into aqueous reaction media. Because there are several different promising synthesis routes and metals for CO2-FT, a facile means of rapidly screening new materials with DFT calculated descriptors should help develop a new generation of improved catalysts.
Another hydrogenation route, CO2 methanation, is appropriate in certain geographical regions. Although natural gas supplies are abundant in the U.S., CO2 methanation is an attractive energy storage route for many European nations where renewable energy is relatively abundant and CO2 emissions are regulated.127 Several catalysts have shown promise for CO2 methanation, including Ni–Fe,128 Rh/TiO2,129 Ni/CeO2,118 Ni/CeO2–ZrO2,117 and Ru/γ-Al2O3.119 Supported Ni-based catalysts are the most promising and well-studied systems for CO2 methanation, while noble metal (e.g., Ru and Rh) based catalysts show better activity and stability at low temperatures.120,130 Ru/γ-Al2O3 is particularly interesting as the catalyst can be treated with cycles of CO2 and H2 and remains active after multiple reaction cycles. Another study has shown similar behavior for reduced Ru/CeO2,131 while high methane yield (100% at 160 °C) can be achieved on highly dispersed Ru nanoparticles supported on TiO2.120 Low temperature (25–150 °C) CO2 methanation over Rh/γ-Al2O3 has been reported,130 while high temperature operation is required over Pd–Mg/SiO2 (450 °C)121 and Pd–Ni/CeO2 (300 °C).23
Dual-functional materials that can both adsorb and hydrogenate CO2 to CH4 are very promising for commercial applications. By combining Ru/γ-Al2O3 with CaO, the catalyst can adsorb CO2 from flue gas, then hydrogenate the adsorbed CO2 to CH4 when treated with pure H2.132 This type of dual-functional material shows significant promise for practical applications as it can be used directly in a flue gas stream, without the need to purify and transport CO2.
Two primary mechanisms have been proposed for CO2 methanation. In the first one, CO2 undergoes CO bond cleavage to form CO, which is subsequently converted into methane. Here, adsorbed surface carbon (Cads) is considered to be a possible key intermediate.133–135 The second mechanism proposes that CO2 is first activated into carbonates, which are then hydrogenated into formate and subsequently hydrogenated into methoxy species. This mechanism suggests that weak basic sites are required for CO2 adsorption, which is supported by the higher activity of Ni/CeO2–ZrO2 over Ni/SiO2.116 DRIFTS studies by Das et al. show that CO2 adsorbs as carbonate species on Al2O3 and MgO supports with some formate, which is stabilized by the metal–support interface.115
Challenges and opportunities for CO2 reduction
Controlling the selectivity of CO2 conversion by H2 requires thorough understanding of the thermodynamics, kinetics and key reaction intermediates of the aforementioned three pathways. CO2-FT and MeOH synthesis are both exothermic processes, but RWGS is endothermic. Therefore, the temperature regime should be carefully chosen depending on the reaction of interest, as shown in Fig. 4. Furthermore, for MeOH synthesis and CO2-FT, higher reaction pressures can help drive the reaction forward. Clearly, low temperature operation would result in significant energy and economic benefits; however, CO2-FT and MeOH synthesis are kinetically limited while RWGS is thermodynamic limited under these low-temperature conditions. | ||
| Fig. 4 Thermodynamic equilibrium composition of the product gas of RWGS reaction at 0.1 MPa for a molar H2:CO2 inlet ratio of 3:1. (Reproduced from ref. 8 with permission from John Wiley and Sons.) | ||
As outlined in detail above, the conversion of CO2 to CO, CH3OH, CH4 and other hydrocarbons can occur via several possible routes. Fig. 5 depicts some of the proposed pathways.20,88,93,97,115,116,136 Along the formate pathways, the initial hydrogen transfer to CO2 forms a formate (HCOO) species which undergoes series of hydrogenation and dissociation reactions to form CH4 and CH3OH. In contrast, along the RWGS and CO hydrogenation pathways, the initial hydrogenation of CO2 forms a carboxylate (HOCO) species which undergoes dissociation reaction to form CO and OH. The CO intermediate then either desorbs or undergoes further hydrogenation reactions to form CH3OH, CH4 or other hydrocarbons.
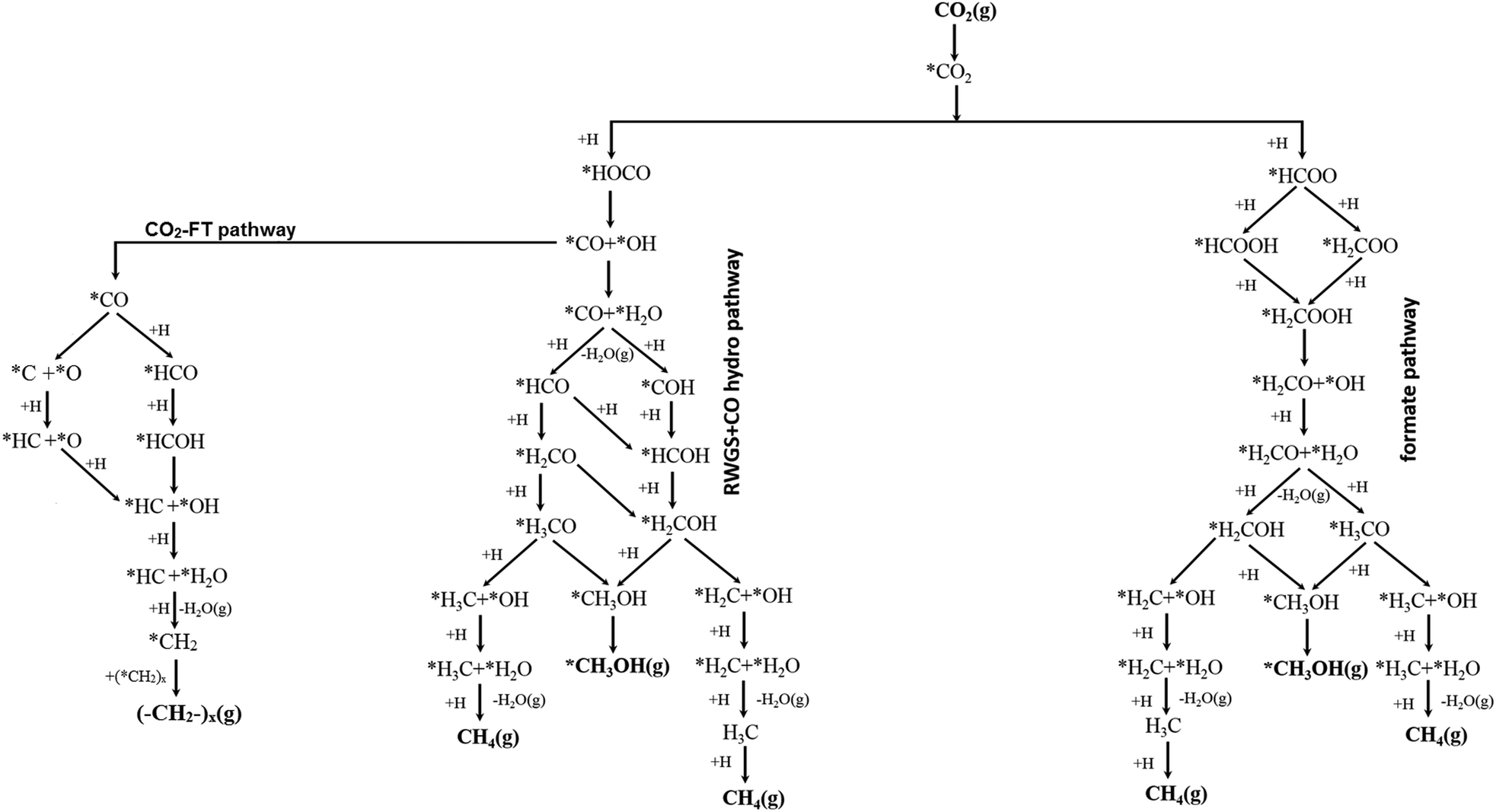 | ||
| Fig. 5 Representatives of proposed reaction schemes for the conversion of CO2 to CO, CH3OH, CH4 and other hydrocarbons. | ||
For all three pathways of CO2 reduction by H2, there are significant challenges that should be addressed when designing active, selective and stable catalysts, as described below:
Stabilization of key intermediates
As described in each section for CO2 reduction by H2, the identification and stabilization of intermediates are critical for controlling the selectivity for each pathway. CO is perhaps the most important intermediate because catalysts with a stronger CO binding energy would favor MeOH93 and hydrocarbon synthesis, while a weaker CO binding energy would favor RWGS. For MeOH synthesis, there is more work to be done in identifying the correct intermediate(s) and structure–property descriptors, but the latest research indicates that stabilization of CO is necessary for high MeOH yield. Identification of other descriptors with DFT calculations, such as oxygen adsorption energy,62 adsorption configurations of CO2 and key intermediates, and activation barriers for key reaction steps, should save a significant amount of time for catalyst screening and development.Utilization of in situ techniques
Parallel experiments on well-defined model surfaces are critical to support DFT calculations. However, most of the conventional UHV techniques are not very useful due to the weak adsorption strength of CO2. Ambient pressure techniques, such as AP-XPS, AP-Temperature Programmed Reaction (AP-TPR), and infrared spectroscopy, should be utilized to determine the adsorption strength and configurations of CO2 and key intermediates. Furthermore, in situ techniques, such as environmental TEM and synchrotron-based XRD and X-ray absorption techniques, should be employed to characterize the electronic and structural properties of supported catalysts under reaction conditions.Identification of low-cost catalysts
Significant reduction of CO2 emissions requires large-scale processes and low-cost catalysts. These catalysts should also exhibit reducible properties, which are an important feature of many catalysts for CO2 reduction by H2.137 One promising material is Mo2C, which is cost effective, reducible and has already been proven to reduce CO2 by H2.39 However, Mo2C is not ideal for CO2-FT because it binds hydrocarbon intermediates relatively strongly, resulting in coke formation. Future efforts should focus on metal-modifications to attenuate the Mo2C binding energy of intermediates, much like what is seen in a MeOH synthesis study with Cu–Mo2C.55Poisoning by water
In all cases of CO2 reduction by H2, the production of large amounts of water is unavoidable, leading to catalyst poisoning through hydroxyl formation.95 New water-tolerant catalysts should be identified that are stable under CO2 reduction by H2 conditions. Some promising materials are bimetallic particles encapsulated in porous SiO2138 and carbon shells.139 Recent results by Qiao et al. show outstanding thermal stability and good recyclability for Pd and Pt particles encased in microporous Si shells138 and PtCo has been proven to be active for CO2 hydrogenation when encased in SiO2 microspheres.140 If this SiO2 microsphere technology can be extended from precious metals to lower-cost materials, it could be possible to design highly active and stable catalysts which repel water.Development of CO2-free H2 sources
Currently, 95% of H2 is produced from hydrocarbon based feedstocks (steam reforming of CH4, coal gasification and partial oxidation of light oil residues), with CO2 as a byproduct. A large-scale reduction of CO2 requires sources of relatively inexpensive, renewable and CO2-free H2.141 If the cost of renewable H2 can be reduced to $2.75 kg−1, fuel from CO2 becomes cost competitive with gasoline,142 and the production of light olefins becomes economically viable.143 Currently biomass conversion144 and water electrolysis show promise for producing CO2-free H2. On a large-scale, the latter is likely the only suitable source of CO2-free H2 as it does not result in other byproducts except O2.137 Although recent studies have identified lower-cost electrocatalysts for hydrogen evolution in both acid145 and alkaline146 electrolytes, significant improvement in overall process cost is needed to produce enough H2 for substantially reducing CO2 emissions.CO2 reduction by alkanes
Alternatively, until CO2-free H2 can be produced on a large scale, light alkanes can be used to replace H2 for CO2 reduction. Researchers have attempted dry reforming of methane to produce synthesis gas, but high reaction temperatures (∼700 °C) along with rapid deactivation of catalysts have prevented breakthroughs.147 Dry reforming of ethane, however, becomes thermodynamically favorable about 100 °C lower than that of methane, making the process more feasible under milder conditions.12 Furthermore, by reducing CO2 with light alkanes, it might be possible to produce synthesis gas and olefins, both of which are valuable products.148Comparison with electrochemical reduction
Although the current perspective focuses on thermal catalysis, it should be pointed out that significant efforts are taking place in the electrocatalytic reduction of CO2, as summarized in recent reviews.149,150 One of the main advantages of electrocatalysis is that the hydrogen source for CO2 reduction is from water instead of from H2 in thermal catalysis. Some of the current challenges in electrocatalysis include the relatively low Faradic efficiency for CO2 conversion due to the high activity of the competing hydrogen evolution reaction (HER). Product separation might also present a challenge if low concentrations of oxygenate products, such as methanol and formic acid, are produced in water-based electrolytes. Opportunities in utilizing hybrid thermal-electrochemical approaches should be explored for CO2 reduction.Conclusions
In summary, several routes have been explored for CO2 reduction by H2. CO production through RWGS can be used in down-stream FT and MeOH synthesis, direct MeOH synthesis offers a liquid product with many industrial applications and finally, CO2-FT produces olefins and alkanes that can be used directly as fuels or in the synthesis of plastics, surfactants, and detergents. Currently there is no preferred route for CO2 reduction by H2 because the specific application ultimately dictates which route is the most attractive. In any event, mitigation of atmospheric CO2 is required on a large scale to prevent ocean acidification and climate change. Significant efforts must be put forth to both identify new catalysts and reduce the cost of CO2-free H2 to make CO2 reduction by H2 scientifically and economically viable.Acknowledgements
The work was sponsored by the United States Department of Energy under Contract No. DE-FG02-13ER16381.References
- T. R. Knutson and R. E. Tuleya, J. Clim., 2004, 17, 3477–3495 CrossRef.
- J. Hansen, M. Sato, R. Ruedy, K. Lo, D. W. Lea and M. Medina-Elizade, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14288–14293 CrossRef CAS PubMed.
- M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975–2992 RSC.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- G. Centi and S. Perathoner, Catal. Today, 2009, 148, 191–205 CrossRef CAS.
- S. Perathoner and G. Centi, ChemSusChem, 2014, 7, 1274–1282 CrossRef CAS PubMed.
- C. Song, Catal. Today, 2006, 115, 2–32 CrossRef CAS.
- P. Kaiser, R. B. Unde, C. Kern and A. Jess, Chem. Eng. Technol., 2013, 85, 489–499 CAS.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 CAS.
- Q. Lu, J. Rosen, Y. Zhou, G. S. Hutchings, Y. C. Kimmel, J. G. Chen and F. Jiao, Nat. Commun., 2014, 5, 6 Search PubMed.
- T. Riedel, G. Schaub, K.-W. Jun and K.-W. Lee, Ind. Eng. Chem. Res., 2001, 40, 1355–1363 CrossRef CAS.
- X. Xiaoding and J. A. Moulijn, Energy Fuels, 1996, 10, 305–325 CrossRef CAS.
- X.-M. Liu, G. Q. Lu, Z.-F. Yan and J. Beltramini, Ind. Eng. Chem. Res., 2003, 42, 6518–6530 CrossRef CAS.
- J. B. Hansen and P. E. Højlund Nielsen, Handbook of Heterogeneous Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, 2008 Search PubMed.
- Green Carbon Dioxide: Advances in CO2 Utilization, ed. G. Centi and S. Perathoner, John Wiley & Sons, Hoboken, NJ, 2014 Search PubMed.
- B. W. Lu and K. Kawamoto, Mater. Res. Bull., 2014, 53, 70–78 CrossRef CAS.
- M. Gines, A. J. Marchi and C. R. Apesteguia, Appl. Catal., A, 1997, 154, 155–171 CrossRef CAS.
- S. Fujita, M. Usui and N. Takezawa, J. Catal., 1992, 134, 220–225 CrossRef CAS.
- J. Yoshihara and C. T. Campbell, J. Catal., 1996, 161, 776–782 CrossRef CAS.
- C.-S. Chen, W.-H. Cheng and S.-S. Lin, Catal. Lett., 2000, 68, 45–48 CrossRef CAS.
- R. Todorovic and R. J. Meyer, Catal. Today, 2011, 160, 242–248 CrossRef CAS.
- W. C. Conner and J. L. Falconer, Chem. Rev., 1995, 95, 759–788 CrossRef CAS.
- M. D. Porosoff and J. G. Chen, J. Catal., 2013, 301, 30–37 CrossRef CAS.
- S. Alayoglu, S. K. Beaumont, F. Zheng, V. V. Pushkarev, H. M. Zheng, V. Iablokov, Z. Liu, J. H. Guo, N. Kruse and G. A. Somorjai, Top. Catal., 2011, 54, 778–785 CrossRef CAS.
- M. R. Gogate and R. J. Davis, Catal. Commun., 2010, 11, 901–906 CrossRef CAS.
- A. G. Kharaji, A. Shariati and M. A. Takassi, Chin. J. Chem. Eng., 2013, 21, 1007–1014 CrossRef CAS.
- A. G. Kharaji, A. Shariati and M. Ostadi, J. Nanosci. Nanotechnol., 2014, 14, 6841–6847 CrossRef CAS PubMed.
- Z. Cheng, B. J. Sherman and C. S. Lo, J. Chem. Phys., 2013, 138, 1–12 Search PubMed.
- A. Bueno-Lopez, K. Krishna and M. Makkee, Appl. Catal., A, 2008, 342, 144–149 CrossRef CAS.
- D. J. Pettigrew, D. L. Trimm and N. W. Cant, Catal. Lett., 1994, 28, 313–319 CrossRef CAS.
- Q. D. Sun, J. Y. Ye, C. J. Liu and Q. F. Ge, Greenhouse Gases: Sci. Technol., 2014, 4, 140–144 CrossRef CAS.
- B. Zhao, Y.-x. Pan and C.-j. Liu, Catal. Today, 2012, 194, 60–64 CrossRef CAS.
- S. S. Kim, H. H. Lee and S. C. Hong, Appl. Catal., A, 2012, 423–424, 100–107 CAS.
- C. S. Chen, W. H. Cheng and S. S. Lin, Appl. Catal., A, 2003, 238, 55–67 CrossRef CAS.
- Y. Liu and D. Liu, Int. J. Hydrogen Energy, 1999, 24, 351–354 CrossRef CAS.
- C. S. Chen, W. H. Cheng and S. S. Lin, Appl. Catal., A, 2004, 257, 97–106 CrossRef CAS.
- K. K. Bando, K. Soga, K. Kunimori and H. Arakawa, Appl. Catal., A, 1998, 175, 67–81 CrossRef.
- H. Kusama, K. K. Bando, K. Okabe and H. Arakawa, Appl. Catal., A, 2001, 205, 285–294 CrossRef CAS.
- M. D. Porosoff, X. Yang, J. A. Boscoboinik and J. G. Chen, Angew. Chem., Int. Ed., 2014, 53, 6705–6709 CrossRef CAS PubMed.
- W. Xu, P. Ramírez, D. Stacchiola, J. Brito and J. Rodriguez, Catal. Lett., 2015, 1–9 Search PubMed.
- J. A. Rodriguez, J. Evans, L. Feria, A. B. Vidal, P. Liu, K. Nakamura and F. Illas, J. Catal., 2013, 307, 162–169 CrossRef CAS.
- R. B. Levy and M. Boudart, Science, 1973, 181, 547–549 CAS.
- S. Posada-Perez, F. Vines, P. J. Ramirez, A. B. Vidal, J. A. Rodriguez and F. Illas, Phys. Chem. Chem. Phys., 2014, 16, 14912–14921 RSC.
- M. D. Porosoff, S. Kattel, W. Li, P. Liu and J. G. Chen, Chem. Commun., 2015, 51, 6988–6991 RSC.
- O.-S. Joo, K.-D. Jung, I. Moon, A. Y. Rozovskii, G. I. Lin, S.-H. Han and S.-J. Uhm, Ind. Eng. Chem. Res., 1999, 38, 1808–1812 CrossRef CAS.
- S. Kuld, C. Conradsen, P. G. Moses, I. Chorkendorff and J. Sehested, Angew. Chem., Int. Ed., 2014, 53, 5941–5945 CrossRef CAS PubMed.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B.-L. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlögl, Science, 2012, 336, 893–897 CrossRef CAS PubMed.
- A. Bansode and A. Urakawa, J. Catal., 2014, 309, 66–70 CrossRef CAS.
- L. Angelo, K. Kobl, L. M. M. Tejada, Y. Zimmermann, K. Parkhomenko and A. C. Roger, C. R. Chim., 2015, 18, 250–260 CrossRef CAS.
- W. J. Cai, P. R. de la Piscina, J. Toyir and N. Homs, Catal. Today, 2015, 242, 193–199 CrossRef CAS.
- I. U. Din, M. S. Shaharun, D. Subbarao and A. Naeem, J. Power Sources, 2015, 274, 619–628 CrossRef.
- F. L. Liao, Y. Q. Huang, J. W. Ge, W. R. Zheng, K. Tedsree, P. Collier, X. L. Hong and S. C. Tsang, Angew. Chem., Int. Ed., 2011, 50, 2162–2165 CrossRef CAS PubMed.
- A. Bansode, B. Tidona, P. R. von Rohr and A. Urakawa, Catal. Sci. Technol., 2013, 3, 767–778 CAS.
- Y. Q. Song, X. R. Liu, L. F. Xiao, W. Wu, J. W. Zhang and X. M. Song, Catal. Lett., 2015, 145, 1272–1280 CrossRef CAS.
- J.-L. Dubois, K. Sayama and H. Arakawa, Chem. Lett., 1992, 5–8 CrossRef CAS.
- J. Xiao, D. Mao, X. Guo and J. Yu, Appl. Surf. Sci., 2015, 338, 146–153 CrossRef CAS.
- G. Bonura, M. Cordaro, C. Cannilla, F. Arena and F. Frusteri, Appl. Catal., B, 2014, 152–153, 152–161 CrossRef CAS.
- S. Kiatphuengporn, M. Chareonpanich and J. Limtrakul, Chem. Eng. J., 2014, 240, 527–553 CrossRef CAS.
- X. Jiang, N. Koizumi, X. W. Guo and C. S. Song, Appl. Catal., B, 2015, 170, 173–185 CrossRef.
- D. L. S. Nieskens, D. Ferrari, Y. Liu and R. Kolonko Jr, Catal. Commun., 2011, 14, 111–113 CrossRef CAS.
- H. Kusama, K. Okabe, K. Sayama and H. Arakawa, Catal. Today, 1996, 28, 261–266 CrossRef CAS.
- F. Studt, I. Sharafutdinov, F. Abild-Pedersen, C. F. Elkjær, J. S. Hummelshøj, S. Dahl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2014, 6, 320–324 CrossRef CAS PubMed.
- H. Ahouari, A. Soualah, A. Le Valant, L. Pinard, P. Magnoux and Y. Pouilloux, React. Kinet., Mech. Catal., 2013, 110, 131–145 CrossRef CAS.
- S. Fujita, Y. Kanamori, A. M. Satriyo and N. Takezawa, Catal. Today, 1998, 45, 241–244 CrossRef CAS.
- A. Le Valant, C. Comminges, C. Tisseraud, C. Canaff, L. Pinard and Y. Pouilloux, J. Catal., 2015, 324, 41–49 CrossRef CAS.
- H. Zhan, F. Li, C. Xin, N. Zhao, F. Xiao, W. Wei and Y. Sun, Catal. Lett., 2015, 145, 1177–1185 CrossRef CAS.
- J. Xiao, D. Mao, X. Guo and J. Yu, Energ. Tech., 2015, 3, 32–39 CrossRef CAS.
- H. Lei, R. Nie, G. Wu and Z. Hou, Fuel, 2015, 154, 161–166 CrossRef CAS.
- Y. Hartadi, D. Widmann and R. J. Behm, ChemSusChem, 2015, 8, 456–465 CrossRef CAS PubMed.
- G. Wang, L. Chen, Y. Sun, J. Wu, M. Fu and D. Ye, RSC Adv., 2015, 5, 45320–45330 RSC.
- X.-L. Liang, J.-R. Xie and Z.-M. Liu, Catal. Lett., 2015, 145, 1138–1147 CrossRef CAS.
- J. Qu, X. Zhou, F. Xu, X.-Q. Gong and S. C. E. Tsang, J. Phys. Chem. C, 2014, 118, 24452–24466 CAS.
- H. Zhan, F. Li, P. Gao, N. Zhao, F. Xiao, W. Wei, L. Zhong and Y. Sun, J. Power Sources, 2014, 251, 113–121 CrossRef CAS.
- P. Gao, F. Li, N. Zhao, F. Xiao, W. Wei, L. Zhong and Y. Sun, Appl. Catal., A, 2013, 468, 442–452 CrossRef CAS.
- R. Ladera, F. J. Perez-Alonso, J. M. Gonzalez-Carballo, M. Ojeda, S. Rojas and J. L. G. Fierro, Appl. Catal., B, 2013, 142, 241–248 CrossRef.
- F. Arena, G. Mezzatesta, G. Zafarana, G. Trunfio, F. Frusteri and L. Spadaro, J. Catal., 2013, 300, 141–151 CrossRef CAS.
- X. Guo, D. Mao, G. Lu, S. Wang and G. Wu, J. Mol. Catal. A: Chem., 2011, 345, 60–68 CrossRef CAS.
- H. Kong, H.-Y. Li, G.-D. Lin and H.-B. Zhang, Catal. Lett., 2011, 141, 886–894 CrossRef CAS.
- L. Jia, J. Gao, W. Fang and Q. Li, Catal. Commun., 2009, 10, 2000–2003 CrossRef CAS.
- D. L. Chiavassa, J. Barrandeguy, A. L. Bonivardi and M. A. Baltanas, Catal. Today, 2008, 133, 780–786 CrossRef.
- J. Sloczynski, R. Grabowski, A. Kozlowska, P. Olszewski, J. Stoch, J. Skrzypek and M. Lachowska, Appl. Catal., A, 2004, 278, 11–23 CrossRef CAS.
- I. Melian-Cabrera, M. L. Granados and J. Fierro, Catal. Lett., 2002, 79, 165–170 CrossRef CAS.
- J. Toyir, P. R. de la Piscina, J. Fierro and N. Homs, Appl. Catal., B, 2001, 34, 255–266 CrossRef CAS.
- L. Z. Gao and C. T. Au, J. Catal., 2000, 189, 1–15 CrossRef CAS.
- J. Nakamura, J. A. Rodriguez and C. T. Campbell, J. Phys.: Condens. Matter, 1989, 1, SB149 CrossRef CAS.
- A. B. Vidal, L. Feria, J. Evans, Y. Takahashi, P. Liu, K. Nakamura, F. Illas and J. A. Rodriguez, J. Phys. Chem. Lett., 2012, 3, 2275–2280 CrossRef CAS PubMed.
- J. Liu, J. Shi, D. He, Q. Zhang, X. Wu, Y. Liang and Q. Zhu, Appl. Catal., A, 2001, 218, 113–119 CrossRef CAS.
- E. L. Uzunova, N. Seriani and H. Mikosch, Phys. Chem. Chem. Phys., 2015, 17, 11088–11094 Search PubMed.
- J. Nakamura, I. Nakamura, T. Uchijima, T. Watanabe and T. Fujitani, in Studies in Surface Science and Catalysis, ed. W. N. D. E. I. Joe, W. Hightower and T. B. Alexis, Elsevier, 1996, vol. 101, pp. 1389–1399 Search PubMed.
- T. Fujitani, I. Nakamura, T. Uchijima and J. Nakamura, Surf. Sci., 1997, 383, 285–298 CrossRef CAS.
- Y. Yang, C. A. Mims, D. H. Mei, C. H. F. Peden and C. T. Campbell, J. Catal., 2013, 298, 10–17 CrossRef CAS.
- Y.-F. Zhao, Y. Yang, C. Mims, C. H. F. Peden, J. Li and D. Mei, J. Catal., 2011, 281, 199–211 CrossRef CAS.
- Y. Yang, J. Evans, J. A. Rodriguez, M. G. White and P. Liu, Phys. Chem. Chem. Phys., 2010, 12, 9909–9917 RSC.
- Y. Yang, M. G. White and P. Liu, J. Phys. Chem. C, 2012, 116, 248–256 CAS.
- O. Martin and J. Perez-Ramirez, Catal. Sci. Technol., 2013, 3, 3343–3352 CAS.
- J. Graciani, K. Mudiyanselage, F. Xu, A. E. Baber, J. Evans, S. D. Senanayake, D. J. Stacchiola, P. Liu, J. Hrbek, J. F. Sanz and J. A. Rodriguez, Science, 2014, 345, 546–550 CrossRef CAS PubMed.
- M. Behrens, Angew. Chem., Int. Ed., 2014, 53, 12022–12024 CrossRef CAS PubMed.
- P. Liu, Y. Choi, Y. Yang and M. G. White, J. Phys. Chem. A, 2010, 114, 3888–3895 CrossRef CAS PubMed.
- H. Kusama, K. Okabe and H. Arakawa, Appl. Catal., A, 2001, 207, 85–94 CrossRef CAS.
- X. Yang, S. Kattel, S. D. Senanayake, J. A. Boscoboinik, X. Nie, J. Graciani, J. A. Rodriguez, P. Liu, D. J. Stacchiola and J. G. Chen, J. Am. Chem. Soc., 2015, 137, 10104–10107 CrossRef CAS PubMed.
- F. Studt, F. Abild-Pedersen, J. Varley and J. Nørskov, Catal. Lett., 2013, 143, 71–73 CrossRef CAS.
- K. Müller, L. Mokrushina and W. Arlt, Chem. Eng. Technol., 2014, 86, 497–503 Search PubMed.
- U. Rodemerck, M. Holeňa, E. Wagner, Q. Smejkal, A. Barkschat and M. Baerns, ChemCatChem, 2013, 5, 1948–1955 CrossRef CAS.
- H. D. Willauer, R. Ananth, M. T. Olsen, D. M. Drab, D. R. Hardy and F. W. Williams, J. CO2 Util., 2013, 3–4, 56–64 CrossRef CAS.
- Y.-x. Pan, C.-j. Liu and Q. Ge, J. Catal., 2010, 272, 227–234 CrossRef CAS.
- R. W. Dorner, D. R. Hardy, F. W. Williams, B. H. Davis and H. D. Willauer, Energy Fuels, 2009, 23, 4190–4195 CrossRef CAS.
- Y. Q. Zhang, G. Jacobs, D. E. Sparks, M. E. Dry and B. H. Davis, Catal. Today, 2002, 71, 411–418 CrossRef CAS.
- R. W. Dorner, D. R. Hardy, F. W. Williams and H. D. Willauer, Energy Environ. Sci., 2010, 3, 884–890 CAS.
- R. W. Dorner, D. R. Hardy, F. W. Williams and H. D. Willauer, Appl. Catal., A, 2010, 373, 112–121 CrossRef CAS.
- R. Satthawong, N. Koizumi, C. Song and P. Prasassarakich, Top. Catal., 2014, 57, 588–594 CrossRef CAS.
- B. Hu, S. Frueh, H. F. Garces, L. Zhang, M. Aindow, C. Brooks, E. Kreidler and S. L. Suib, Appl. Catal., B, 2013, 132–133, 54–61 CrossRef CAS.
- G. Kishan, M. W. Lee, S. S. Nam, M. J. Choi and K. W. Lee, Catal. Lett., 1998, 56, 215–219 CrossRef CAS.
- Z. You, W. Deng, Q. Zhang and Y. Wang, Chin. J. Catal., 2013, 34, 956–963 CrossRef CAS.
- R. Satthawong, N. Koizumi, C. S. Song and P. Prasassarakich, Catal. Today, 2015, 251, 34–40 CrossRef CAS.
- T. Das and G. Deo, J. Mol. Catal. A: Chem., 2011, 350, 75–82 CrossRef CAS.
- P. A. U. Aldana, F. Ocampo, K. Kobl, B. Louis, F. Thibault-Starzyk, M. Daturi, P. Bazin, S. Thomas and A. C. Roger, Catal. Today, 2013, 215, 201–207 CrossRef CAS.
- F. Ocampo, B. Louis, L. Kiwi-Minsker and A.-C. Roger, Appl. Catal., A, 2011, 392, 36–44 CrossRef CAS.
- S. Tada, T. Shimizu, H. Kameyama, T. Haneda and R. Kikuchi, Int. J. Hydrogen Energy, 2012, 37, 5527–5531 CrossRef CAS.
- C. Janke, M. S. Duyar, M. Hoskins and R. Farrauto, Appl. Catal., B, 2014, 152, 184–191 CrossRef.
- T. Abe, M. Tanizawa, K. Watanabe and A. Taguchi, Energy Environ. Sci., 2009, 2, 315–321 CAS.
- J.-N. Park and E. W. McFarland, J. Catal., 2009, 266, 92–97 CrossRef CAS.
- G. Melaet, W. T. Ralston, C.-S. Li, S. Alayoglu, K. An, N. Musselwhite, B. Kalkan and G. A. Somorjai, J. Am. Chem. Soc., 2014, 136, 2260–2263 CrossRef CAS PubMed.
- C. G. Visconti, L. Lietti, E. Tronconi, P. Forzatti, R. Zennaro and E. Finocchio, Appl. Catal., A, 2009, 355, 61–68 CrossRef CAS.
- Y. Zhao, H. Wu, W. Tan, M. Zhang, M. Liu, C. Song, X. Wang and X. Guo, Catal. Today, 2010, 156, 69–73 CrossRef CAS.
- T. Herranz, S. Rojas, F. J. Pérez-Alonso, M. Ojeda, P. Terreros and J. L. G. Fierro, Appl. Catal., A, 2006, 308, 19–30 CrossRef CAS.
- A. Jean-Marie, A. Griboval-Constant, A. Y. Khodakov and F. Diehl, Catal. Today, 2011, 171, 180–185 CrossRef CAS.
- W. Davis and M. Martin, J. Cleaner Prod., 2014, 80, 252–261 CrossRef CAS.
- D. Pandey and G. Deo, J. Mol. Catal. A: Chem., 2014, 382, 23–30 CrossRef CAS.
- J. C. Matsubu, V. N. Yang and P. Christopher, J. Am. Chem. Soc., 2015, 137, 3076–3084 CrossRef CAS PubMed.
- M. Jacquemin, A. Beuls and P. Ruiz, Catal. Today, 2010, 157, 462–466 CrossRef CAS.
- D. C. Upham, A. R. Derk, S. Sharma, H. Metiu and E. W. McFarland, Catal. Sci. Technol., 2015, 5, 1783–1791 CAS.
- M. S. Duyar, M. A. A. Treviño and R. J. Farrauto, Appl. Catal., B, 2015, 168–169, 370–376 CrossRef CAS.
- D. E. Peebles, D. W. Goodman and J. M. White, J. Phys. Chem., 1983, 87, 4378–4387 CrossRef CAS.
- J. L. Falconer and A. E. Zagli, J. Catal., 1980, 62, 280–285 CrossRef CAS.
- G. D. Weatherbee and C. H. Bartholomew, J. Catal., 1982, 77, 460–472 CrossRef CAS.
- M. Ojeda, R. Nabar, A. U. Nilekar, A. Ishikawa, M. Mavrikakis and E. Iglesia, J. Catal., 2010, 272, 287–297 CrossRef CAS.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazabal and J. Perez-Ramirez, Energy Environ. Sci., 2013, 6, 3112–3135 CAS.
- Z.-A. Qiao, P. Zhang, S.-H. Chai, M. Chi, G. M. Veith, N. C. Gallego, M. Kidder and S. Dai, J. Am. Chem. Soc., 2014, 136, 11260–11263 CrossRef CAS PubMed.
- G.-H. Wang, J. Hilgert, F. H. Richter, F. Wang, H.-J. Bongard, B. Spliethoff, C. Weidenthaler and F. Schüth, Nat. Mater., 2014, 13, 293–300 CrossRef CAS PubMed.
- S. Takenaka, A. Hirata, E. Tanabe, H. Matsune and M. Kishida, J. Catal., 2010, 274, 228–238 CrossRef CAS.
- G. Centi, E. A. Quadrelli and S. Perathoner, Energy Environ. Sci., 2013, 6, 1711–1731 CAS.
- Q. Smejkal, U. Rodemerck, E. Wagner and M. Baerns, Chem. Eng. Technol., 2014, 86, 679–686 CAS.
- G. Centi, G. Iaquaniello and S. Perathoner, ChemSusChem, 2011, 4, 1265–1273 CrossRef CAS PubMed.
- M. R. Stonor, T. E. Ferguson, J. G. Chen and A.-H. A. Park, Energy Environ. Sci., 2015, 8, 1702–1706 CAS.
- D. V. Esposito and J. G. Chen, Energy Environ. Sci., 2011, 4, 3900–3912 CAS.
- Q. Lu, G. S. Hutchings, W. Yu, Y. Zhou, R. V. Forest, R. Tao, J. Rosen, B. T. Yonemoto, Z. Cao, H. Zheng, J. Q. Xiao, F. Jiao and J. G. Chen, Nat. Commun., 2015, 6 DOI:10.1038/ncomms7567.
- M. S. Fan, A. Z. Abdullah and S. Bhatia, ChemCatChem, 2009, 1, 192–208 CrossRef CAS.
- M. D. Porosoff, M. Myint, S. Kattel, Z. Xie, E. Gomez, P. Liu and J. G. Chen, Angew. Chem., Int. Ed., 2015 DOI:10.1002/anie.201508128R1.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- R. J. Lim, M. Xie, M. A. Sk, J.-M. Lee, A. Fisher, X. Wang and K. H. Lim, Catal. Today, 2014, 233, 169–180 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |