
Porphyrins bearing a consolidated anthryl donor with dual functions for efficient dye-sensitized solar cells†
Chin-Li
Wang
a,
Min
Zhang
b,
Yu-Hsin
Hsiao
a,
Chuan-Kai
Tseng
a,
Chia-Lin
Liu
a,
Mingfei
Xu
b,
Peng
Wang
*b and
Ching-Yao
Lin
*a
aDepartment of Applied Chemistry, National Chi Nan University, Puli, Nantou Hsien 54561, Taiwan. E-mail: cyl@ncnu.edu.tw; Fax: +886-49-2917956; Tel: +886-49-2910960 ext. 4152
bState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022, China. E-mail: peng.wang@ciac.ac.cn
First published on 9th November 2015
Abstract
Three porphyrin dyes with different electron-donating groups were prepared to compare their photovoltaic performance in dye-sensitized solar cells. LWP12 is based on LD31 which has a complicated, piece-together electron-donating group, whereas LWP13 and LWP14 have merged substituents with electron-donating and absorption-broadening abilities. The results in this work suggest that consolidated anthryl donors may be used to design highly efficient photosensitizers.
Broader contextWith a great variety of colorful photosensitizers to choose from, a dye-sensitized solar cell (DSSC) has been considered as one of the possible means for solar energy conversion. In order to improve the photovoltaic performance of DSSCs, dyes should have broad light-harvesting ability to absorb a wide spectrum of solar energy and a push–pull structure to facilitate charge transfer processes efficiently. To accomplish both, however, chemical structures of many efficient porphyrin dyes have become very complicated. In this work, we aim to simplify the structures of porphyrin dyes by using consolidated anthryl donors to provide both absorption-broadening as well as electron-donating effects. Simplicity is not simple. Only one of the porphyrin dyes under investigation achieves overall efficiency greater than 10%. |
Introduction
The dye-sensitized solar cell (DSSC)1,2 has been of interest in recent decades because of low manufacturing costs and a great variety of dyes to choose from. Porphyrins and their derivatives have been under investigation not only because of their roles in photosynthetic processes to convert solar energy into free energy or to transfer electrons, but also due to their strong absorption in the visible/near-IR region and the ease of structural modification.3–7 In 2007, Officer, Grätzel, and co-workers achieved a power conversion efficiency (PCE) of 7.1% by using a porphyrin dye which had a fully conjugated structure for a more efficient charge transfer and a side-adsorption anchor for a higher dye-loading.8 Since then, great efforts have been made to improve photovoltaic performance of porphyrin-based DSSCs following a similar design.8–29 PCEs greater than 10% for porphyrin-based DSSCs with a conventional iodide/triiodide electrolyte12,14,24–29 or noncorrosive cobalt electrolyte9–11,13 have been reported in the literature. More recently, Mathew et al. reported a DSSC attaining 13% PCE with an SM315 porphyrin dye in conjunction with a cobalt electrolyte.9In order to achieve higher photovoltaic performance, dyes should have broad light-harvesting ability and a push–pull (or donor/acceptor) structure. To accomplish both, however, many of the efficient porphyrin dyes have complicated chemical structures. For example, porphyrin SM315 uses a bulky bis(2′,4′-bis(hexyloxy)-[1′-biphenyl]-4-yl)amine donor,9 WW-6 employs an N-annulated perylene substituent,13 LD31 attaches a dioctylaminophenyl-ethynyl-anthryl group,27 and LDD1 and YDD6 consist of porphyrin dimers.28,29 In this work, we aim to explore the possibility of using substituents with simpler chemical structures to provide both electron-donating and absorption-broadening abilities.
Chart 1 depicts chemical structures of the porphyrins under investigation (denoted as LWP12, LWP13, and LWP14). The more complicated LWP12 is based on LD31 with an additional benzothiadiazole group. In contrast, LWP13 and LWP14 utilize consolidated donors. A dioctylamino group is directly attached to anthracene for LWP13 whereas a diarylamino group is put onto the anthryl group for LWP14. All three donor groups are linked to a meso-position of the porphyrin core via an ethynyl bridge. For the anchoring groups, a benzothiadiazole is placed between the benzoic acid and the ethynyl bridge for a stronger push–pull effect.9,10 For diarylamino-anthracene, it is worth mentioning that a similar donor bearing two methoxy groups was used in a metal-free co-sensitizer recently.18 However, it has been shown that attaching longer alkyl chains at the phenyl groups of diarylamine would be a better choice than methoxy groups (i.e. YD2 vs. YD3).15
 | ||
| Chart 1 Structures of porphyrins LWP12, LWP13 and LWP14. | ||
Although the chemical structures of LWP13 and LWP14 are simpler than that of LWP12, they should still offer both electron-donating as well as absorption-broadening effects owing to the dialkylamino/diarylamino and the anthryl groups, respectively. By comparing these three porphyrins, we hope to achieve high PCE of the DSSC with a simpler dye.
Results and discussion
UV-visible absorption and fluorescence spectra
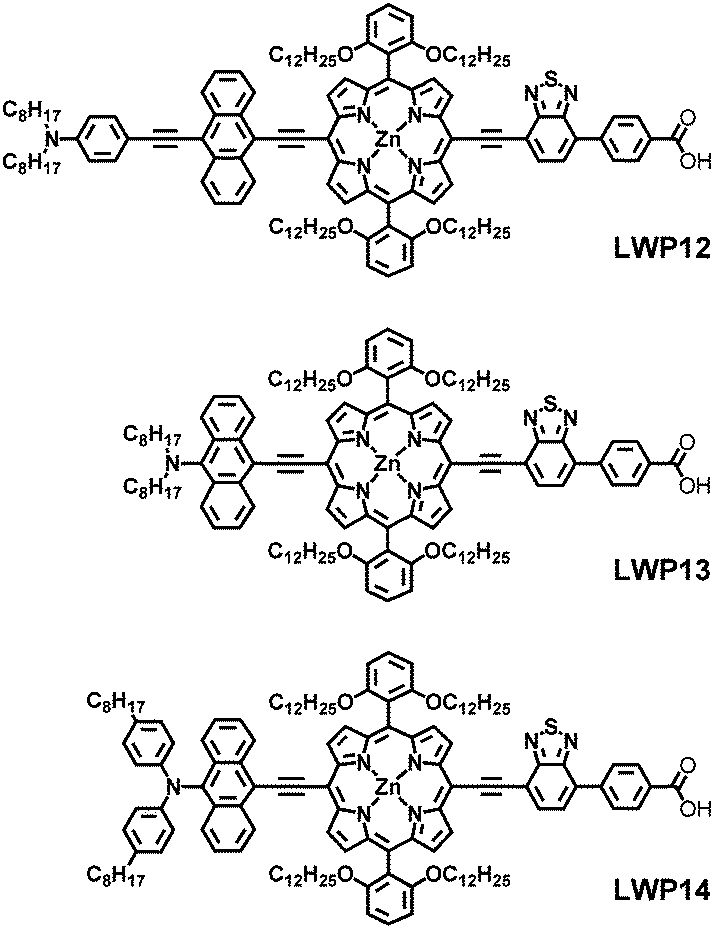
The UV–visible absorption spectra of LWP12–14 porphyrins in THF are shown in Fig. 1. The absorption maxima and extinction coefficients are listed in Table 1 (along with the data of LD14 and LD31 for comparison). As shown in the figure and table, LWP12–14 exhibit typical porphyrin absorption spectra.30 B (or Soret) bands appear in the higher energy region around 500 nm whereas Q bands locate in the lower energy region about 700 nm. As compared in Table 1, the absorption bands of the LWP12–14 porphyrins are significantly red-shifted from those of the LD1425 porphyrin. The red-shifts may be related to the fact that there are more conjugated double bonds in the chemical structures of LWP12–14, decreasing the energy gaps between the HOMOs and LUMOs, thereby those between the ground states and excited states. In addition, the very broadened absorption bands of LWP suggest that the anthryl donors and the benzothiodiazole accepter have huge impact on the absorption bands as in the cases of LD31,27 GY50,10 and SM315.9 Remarkably, the Q band absorptions of LWP12–14 are all red-shifted in comparison to those of LD31.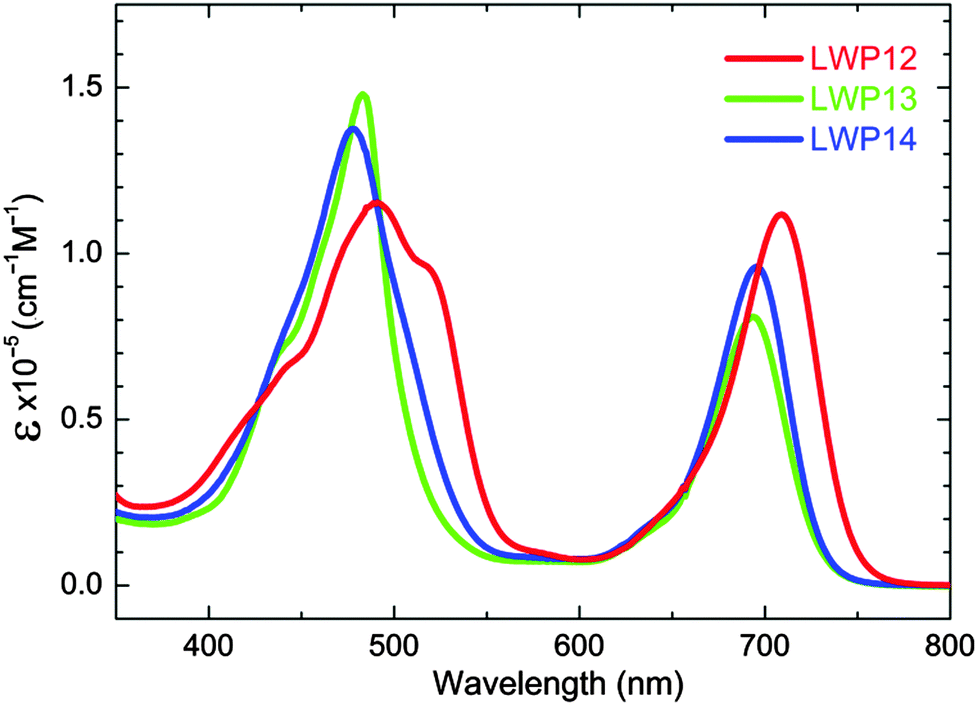 | ||
| Fig. 1 Absorption spectra of LWP12, LWP13, and LWP14 in THF. | ||
| Dye | Absorption/nm (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε, M−1 cm−1) ε, M−1 cm−1) |
Emission/nm | E 1/2/V vs. SCE | S0/S+/eV | S*/S+/eV | |
|---|---|---|---|---|---|---|
| Ox(1) | Red(1) | |||||
| a Potential determined by differential pulse voltammetry due to overlapped oxidative waves. b Taken from ref. 25. c Taken from ref. 27. | ||||||
| LWP12 | 490(5.06), 518(4.98), 709(5.05) | 736 | +0.79a | −1.14 | −5.53 | −3.81 |
| LWP13 | 484(5.17), 694(4.91) | 720 | +0.64a | −1.19 | −5.38 | −3.63 |
| LWP14 | 477(5.14), 696(4.98) | 722 | +0.83a | −1.17 | −5.57 | −3.82 |
| LD14b | 459(5.40), 667(4.82) | 682 | +0.74 | −1.32 | −5.48 | −3.65 |
| LD31c | 449(5.10), 521(4.92), 691(4.94) | 713 | +0.86 | −1.15 | −5.60 | −3.84 |
Fig. 2 compares the fluorescence spectra of the LWP12–14 porphyrins. The emission maxima are listed in Table 1. The fluorescence bands of LWP12–14 strongly resemble the mirror images of the corresponding Q bands, implying that the spacing between vibrational energy levels is similar for the ground and excited states, and the same transitions are favourable for both absorption and emission. Although the emission maximum wavelengths are similar, the trend is the same as that of the Q bands: LWP12 > LWP14 > LWP13. The Stokes shifts of LWP12, LWP13, and LWP14 are calculated to be 482, 522, and 483 cm−1, respectively. In comparison with a known dye (ESI†), fluorescence quantum yields are estimated to be 3.3%, 3.4% and 3.9% for LWP12, LWP13, and LWP14, respectively. These values are comparable to a very common porphyrin, 5,10,15,20-tetraphenylporphinato zinc(II) or ZnTPP (3.3%). Similar quantum yields and Stokes shifts among the LWP porphyrins may be related to the similarity in their chemical structures (from benzoic acid to anthracene).
 | ||
| Fig. 2 Fluorescence spectra of LWP12–14 (2.0 × 10−6 M of each porphyrin in THF, excited wavelengths per nm: LWP12, 490; LWP13, 484; LWP14, 477). | ||
Electrochemistry, molecular orbitals and energy levels
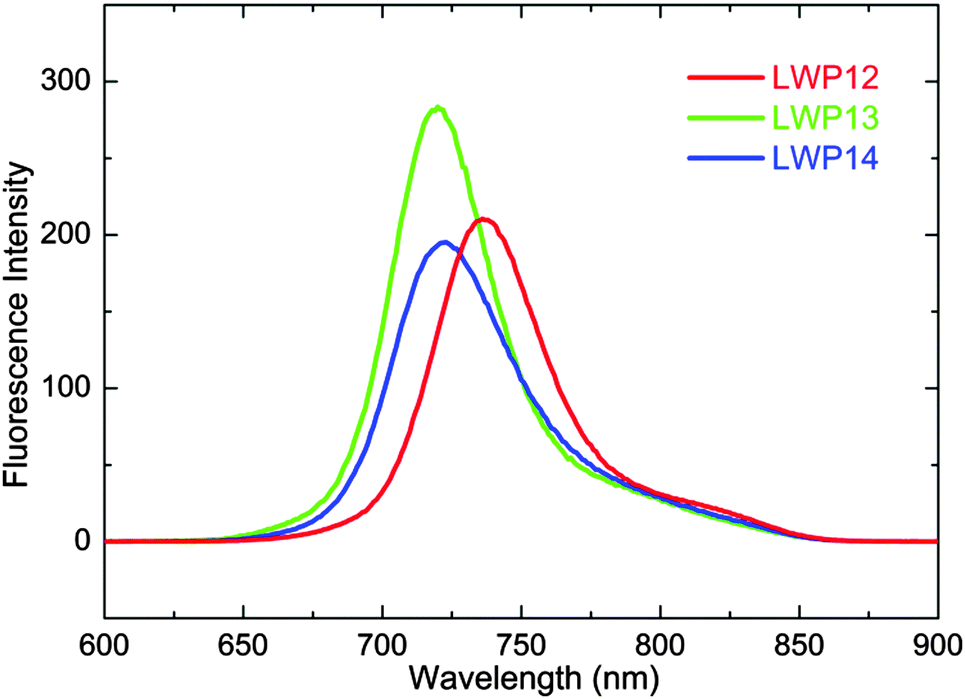
Fig. 3 overlaps the cyclic voltammograms (CV) of LWP12–14 in THF/0.1 M TBAP under nitrogen. The selected redox potentials are shown in Table 1. For the first reductions, the ill–shaped waves found at around −0.80 V vs. SCE are consistent with the reduction of the anchoring group.31 Two additional reductions are observed at more negative potentials. These quasi–reversible processes are consistent with porphyrin ring-reductions, forming porphyrin anion radicals and di-anions, respectively.32 | ||
| Fig. 3 Cyclic voltammograms (bold lines) of LWP12, LWP13 and LWP14 in THF/TBAP. For the oxidation, differential pulse voltammograms were also measured (thin lines) to resolve the overlapped waves. | ||
In general, the first porphyrin-ring reduction potentials of LWP12–14 are similar to that of LD31, but all positively shifted from that of LD14. This can be attributed to LWPs having more conjugated double bonds than LD14. On the other hand, the oxidation waves exhibit overlapped waves and stronger currents. Differential pulse voltammetry (DPV) was therefore employed to resolve the overlapped signals. As shown in Table 1 and Fig. 3, the first oxidation potentials of LWP12, 13, and 14 were found to be +0.79, +0.64, and +0.83 V vs. SCE, respectively. These potentials were used to estimate the S0/S+ energy levels (vide infra). The oxidation potentials of the porphyrins show a trend of LWP14 > LWP12 > LWP13. LWP13 being easier to oxidize than LWP12 implies that the consolidated donor of LWP13 is more effective than the complicated donor of LWP12. Also bearing a consolidated donor group, however, LWP14 is more difficult to oxidize than LWP12. This might be related to the di-arylamine group. Due to its bulkiness and the sp3-nitrogen atom, it would be difficult for the di-arylamine to be co-planar with the anthracene. As a result, π-conjugation might be hampered. This suggestion is consistent with the DFT-optimized structure of LWP14 (Fig. 4).
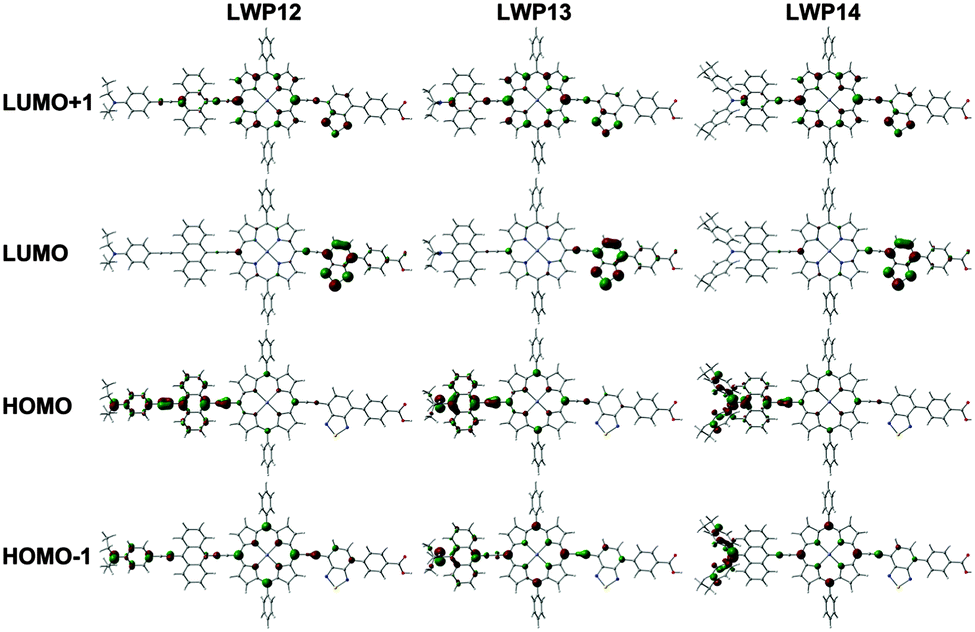 | ||
| Fig. 4 Frontier molecular orbitals of LWP12, LWP13 and LWP14 (DFT, B3LYP/LanL2DZ). | ||
Interestingly, the reduction potentials of the LWP porphyrins show a different trend of LWP13 > LWP14 > LWP12. This phenomenon may be attributed to the different patterns of the HOMOs and the LUMOs.30
Fig. 4 shows the frontier orbital patterns of LWP12–14 calculated by density-functional theory (DFT)33 at the B3LYP/LanL2DZ level. The MOs of LWP12–14 are consistent with the Gouterman's four-orbital model with deviation.30 For example, the HOMO-1 and LUMO+1 patterns resemble those of the a2u orbital and one of the eg orbitals in Gouterman's model. Because the donor and acceptor groups are attached to the porphyrin core structure via ethynyl bridges, delocalization of the MO patterns from the porphyrin centre to the substituents can be expected. In contrast, the patterns of the HOMOs and LUMOs concentrate at the electron-donating and electron-withdrawing groups, respectively, with a small contribution from the porphyrin core. Since the acceptor and donor groups dominate the LUMOs and HOMOs, respectively, it is not surprising to observe different trends in the reduction and oxidation potentials. In addition, the concentration of the HOMO and LUMO patterns at the donor and acceptor sides, respectively, suggests a stronger push–pull tendency. This is a welcome merit for n-type DSSCs. This suggestion is consistent with the calculated dipole moment of LWP12 (with benzothiadiazole, 11.90 Debye) being greater than that of LD31 (without benzothiadiazole, 10.85 Debye).
Fig. 5 illustrates the energy-level diagram of LWP12–14, upon comparing the ground-to-oxidized states (S0/S+), the first singlet excited-to-oxidized states (S*/S+) of each dye, the conduction bands (CB) of TiO2, and the redox energy of the electrolyte. The first oxidation potentials were used to estimate the S0/S+ levels. The zero–zero excitation energies (E0–0) obtained from the intersection of the corresponding normalized absorption and emission spectra were used to estimate the energy gaps between the S*/S+ and the S0/S+ levels.1 As suggested in the figure, the S*/S+ levels of LWP12–14 are considerably higher than the conduction bands of TiO2 and the S0/S+ levels are noticeably lower than the redox energy of the electrolyte. Therefore, LWP12–14 should all be capable of injecting electrons into the CB of TiO2 upon excitation and the resulting cations should be efficiently regenerated by the electrolyte.
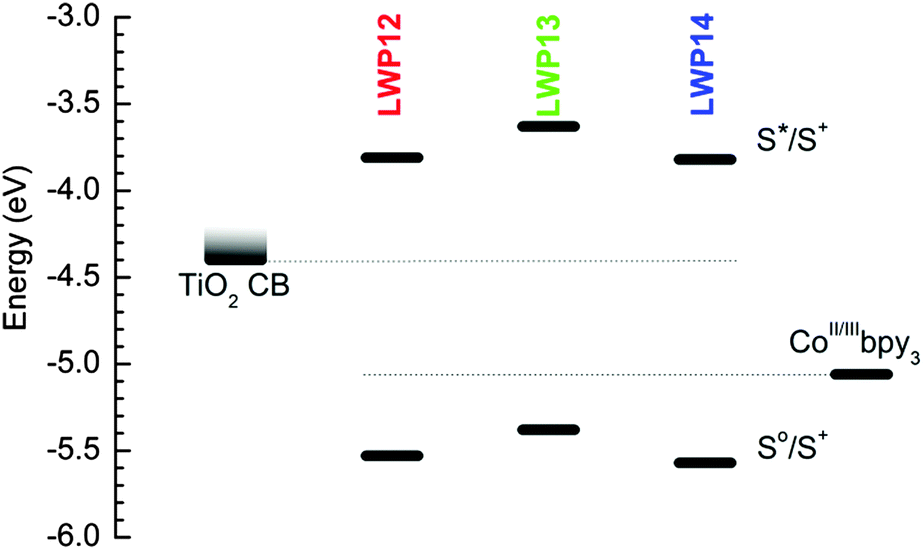 | ||
| Fig. 5 Comparison of TiO2, LWP12–14, and Co(II/III)bpy3 energy levels. | ||
Photovoltaic properties
The external quantum efficiencies (EQEs) of LWP12, LWP13, and LWP14 dye-grafted bilayer titania films (4.0 + 5.0 μm) were first inspected in conjunction with a tris(2,2′-bipyridine)cobalt (Co-bpy) electrolyte. The details of electrolyte composition and cell fabrication are described in the Experimental section. As shown in Fig. 6a, the LWP14 dye has a remarkably higher EQE summit of 77% with respect to that of 57% and 51% for LWP12 and LWP13, respectively. By recording the light-harvesting efficiency (ϕLH) (Fig. 6b) of 8 μm-thick, dye-grated translucent layer TiO2 films, we can exclude the influence of light absorption on the EQE maximum.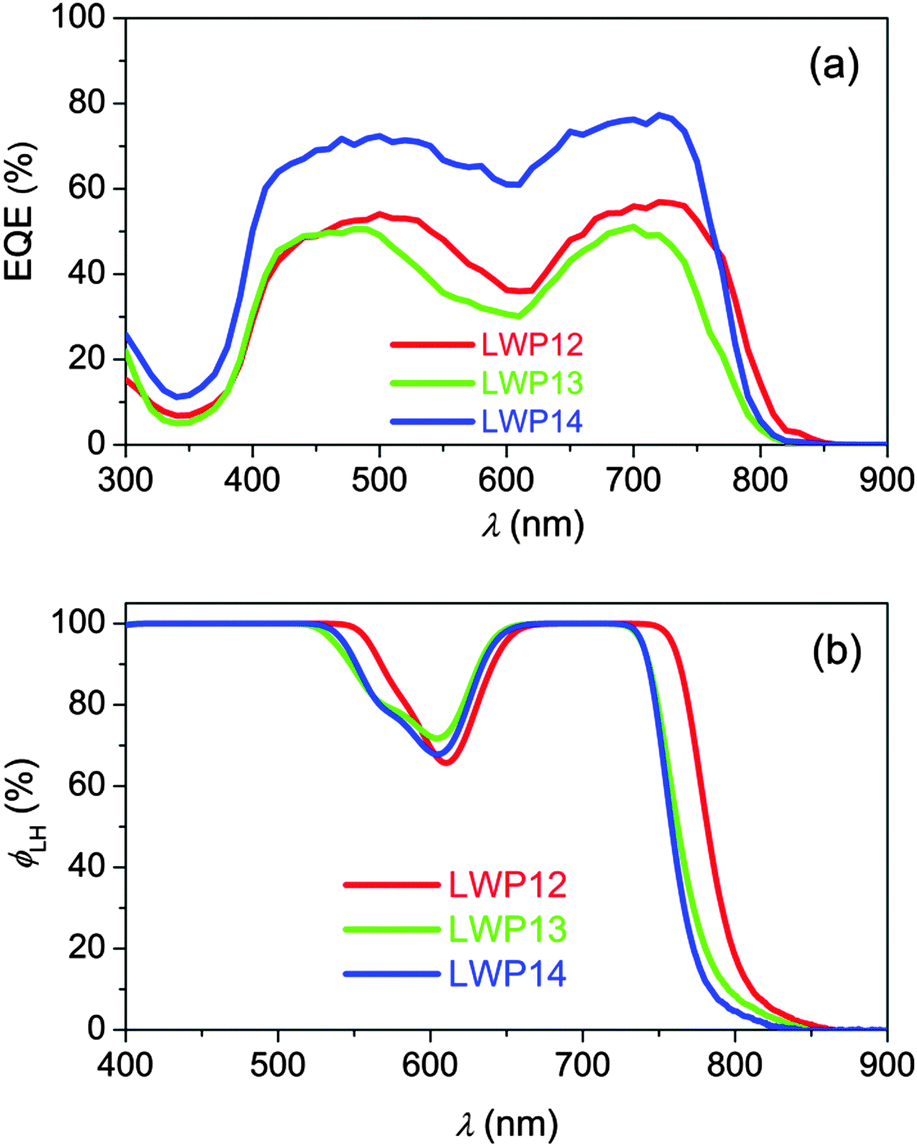 | ||
| Fig. 6 (a) Plots of external quantum efficiency (EQE) as a function of wavelength (λ) of incident monochromatic light for cells with LWP12 (red), LWP13 (green), and LWP14 (blue) dye-coated titania films. (b) Light-harvesting efficiency (ϕLH) for the 8.0 μm-thick, dye-loaded mesoporous translucent titania films in combination with a cobalt electrolyte for DSSCs. | ||
To unveil the obvious difference in the maxima of EQEs, the yields of electron injection (ϕei) from the excited states of dye molecules into the conduction band of TiO2 were estimated by resorting to the time correlated single photon counting (TCSPC) technique34 and employing the corresponding dye-grafted alumina films as control. As depicted in Fig. S10 (ESI†), due to the absence of favorable energy offsets for dye-grafted alumina films, the photoluminescence (PL) decays (blue lines) arise from the radiative and radiationless deactivations of excited-state dye molecules. Upon switching to the titania samples, considerable PL quenching is monitored owing to the occurrence of expeditious electron injection. By integrating the areas of PL traces of dye-coated titania and alumina films, the ϕei value for LWP12, LWP13, and LWP14 can be derived, being 81%, 79%, and 84%, respectively. Thereby we can conclude that the ϕei value is not the key factor controlling EQE summits.
Moreover, a tris(4,4′-dimethyl-2,2′-bipyridine)cobalt (Co-Me2bpy) electrolyte with a more negative redox potential was used to inspect the influence of hole injection efficiency (ϕhi) on the EQE summit. As shown in Fig. S11 (ESI†), the employment of the Co-Me2bpy electrolyte just slightly enhances the EQE summits of cells with LWP12 and LWP13, suggesting that the ϕhi value is also not the controlling factor of the EQE summits. Thereby, we speculate that the low EQE summits of LWP12 and LWP13 cells should be ascribed to the inferior charge collection yields, which can be originated from the swift interfacial charge recombination as observed in the following electrical analysis.
The current density–voltage (J–V) characteristics (Fig. 7) were further examined under the irradiance of 100 mW cm−2, simulated AM1.5 sunlight. The detailed device parameters are compiled in Table 2. The cell with LWP14 exhibits a considerably larger short-circuit photocurrent density (JSC) of 17.22 mA cm−2 than that of 12.07 and 10.06 mA cm−2 for LWP12 and LWP13, respectively, which is in good agreement with the preceding EQE measurements. Moreover, with respect to the low open-circuit photovoltages (VOC) of LWP12 (0.731 V) and LWP13 (0.706 V), an obviously higher VOC of 805 mV was achieved with the LWP14 dye, contributing to a higher η of 10.3%. The J–V curves under various metal-mesh attenuated lights were also measured and plotted VOC against JSC (Fig. 8). It is easy to perceive that LWP14 dye features an evidently improved VOC at a given JSC with respect to that of LWP12 and LWP13.
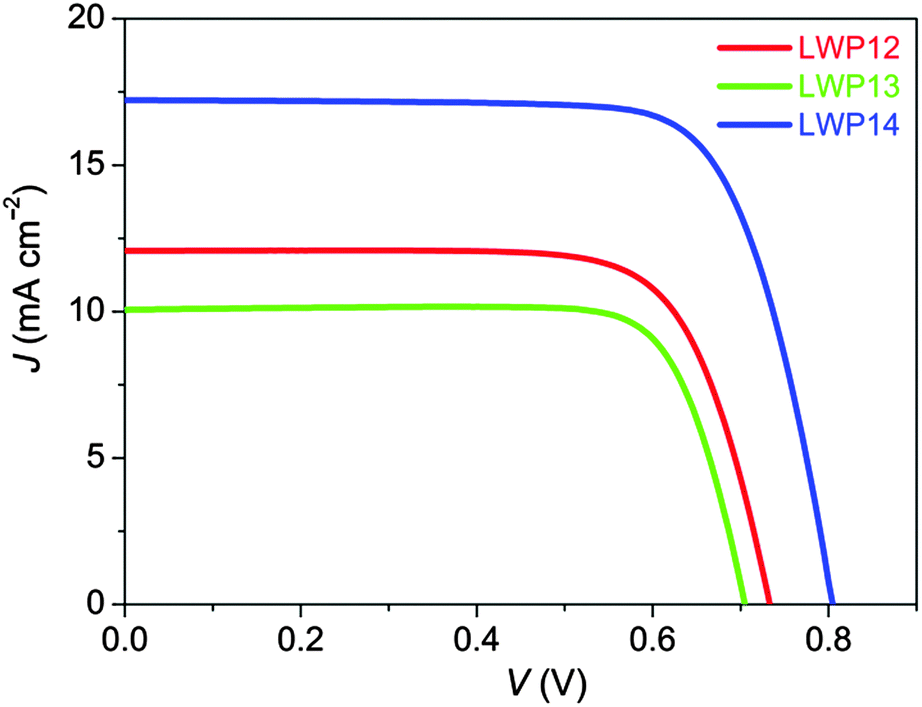 | ||
| Fig. 7 The J–V curves of cells with LWP12 (red), LWP13 (green), and LWP14 (blue) measured under an irradiance of 100 mW cm−2 simulated AM1.5G sunlight. The aperture area of the employed metal mask was 0.160 cm2. | ||
| Dye |
J
EQESCa (mA cm−2) |
J SC (mA cm−2) | V OC (mV) | FF (%) | η (%) |
|---|---|---|---|---|---|
| a J EQESC is derived via wavelength integration of the product of the standard AM1.5 emission spectrum (ASTM G173-03) and the EQEs measured at the short-circuit. The validity of measured photovoltaic parameters is evaluated by comparing the calculated JEQESC with the experimentally measured JSC. | |||||
| LWP12 | 12.16 ± 0.09 | 12.07 ± 0.08 | 731 ± 3 | 73.8 ± 0.5 | 6.5 ± 0.08 |
| LWP13 | 10.44 ± 0.07 | 10.06 ± 0.06 | 706 ± 5 | 78.0 ± 0.4 | 5.5 ± 0.10 |
| LWP14 | 16.79 ± 0.06 | 17.22 ± 0.06 | 805 ± 4 | 74.1 ± 0.5 | 10.3 ± 0.07 |
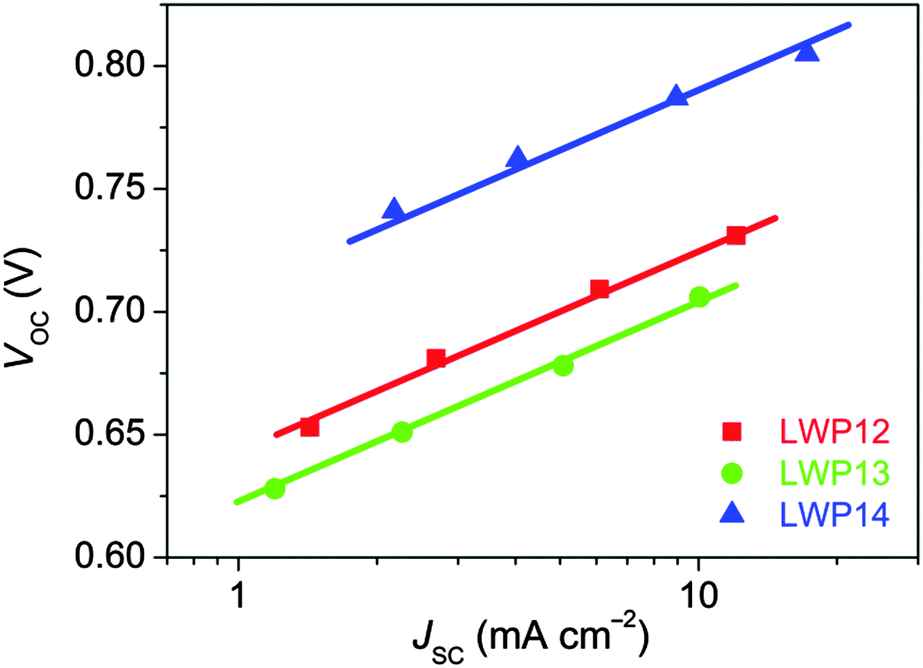 | ||
| Fig. 8 Plots of open-circuit photovoltage (VOC) vs. short-circuit photocurrent density (JSC). The solid lines are a guide to the eye. | ||
Note that for a fixed electrolyte, the enhancement of VOC stems from the up-shift of the electron quasi-Fermi-level (EF,n) of TiO2, which is positively correlated with the energy level of the conduction-band edge (Ec) of TiO2 and/or the electron density of TiO2. Therefore, to dissect the origins of the dye structure correlated VOC difference, the charge extraction (CE)35 and transient photovoltage decay (TPD)36 measurements were further carried out. As shown in Fig. 9a, more charges stored in the mesoporous titania film (QCE) can be observed for the LWP14 cell with respect to the LWP12 and LWP13 cells at a certain VOC, indicating a relatively lower Ec position of titania for LWP14. However, the cell with LWP14 dye exhibits over two orders of magnitude longer half lifetime (tTPD1/2) for photo-injected electrons at a given QCE compared with the LWP12 and LWP13 cells as presented in Fig. 9b, which outreaches the adverse effect of a lower Ec, explaining its superior photovoltage at a given Jsc (Fig. 8). The loading amount (cm) of dye molecules on TiO2 was measured by recording the small but reliable light-absorption change of a dyeing solution at a certain volume, being 0.48 × 10−8 mol cm−2 μm−1 for LWP12, 0.99 × 10−8 mol cm−2 μm−1 for LWP13, and 0.92 × 10−8 mol cm−2 μm−1 for LWP14. The slower charge recombination for LWP14 can be ascribed to a higher cm and an improved steric hindrance of the bis(4-octylphenyl)amino group to prevent the cobalt(III) ions to be in close proximity to titania. Moreover, it is valuable to note that the remarkably fast charge recombination could also result in inferior charge collection yields, providing a clue for the lower EQE maxima of LWP12 and LWP13, which have been proved by impedance spectroscopy (IS) measurements.37 As shown in Fig. S12 (ESI†), the LWP14 cell features an obviously longer electron diffusion length at a given potential bias with respect to the LWP12 and LWP13 cells.
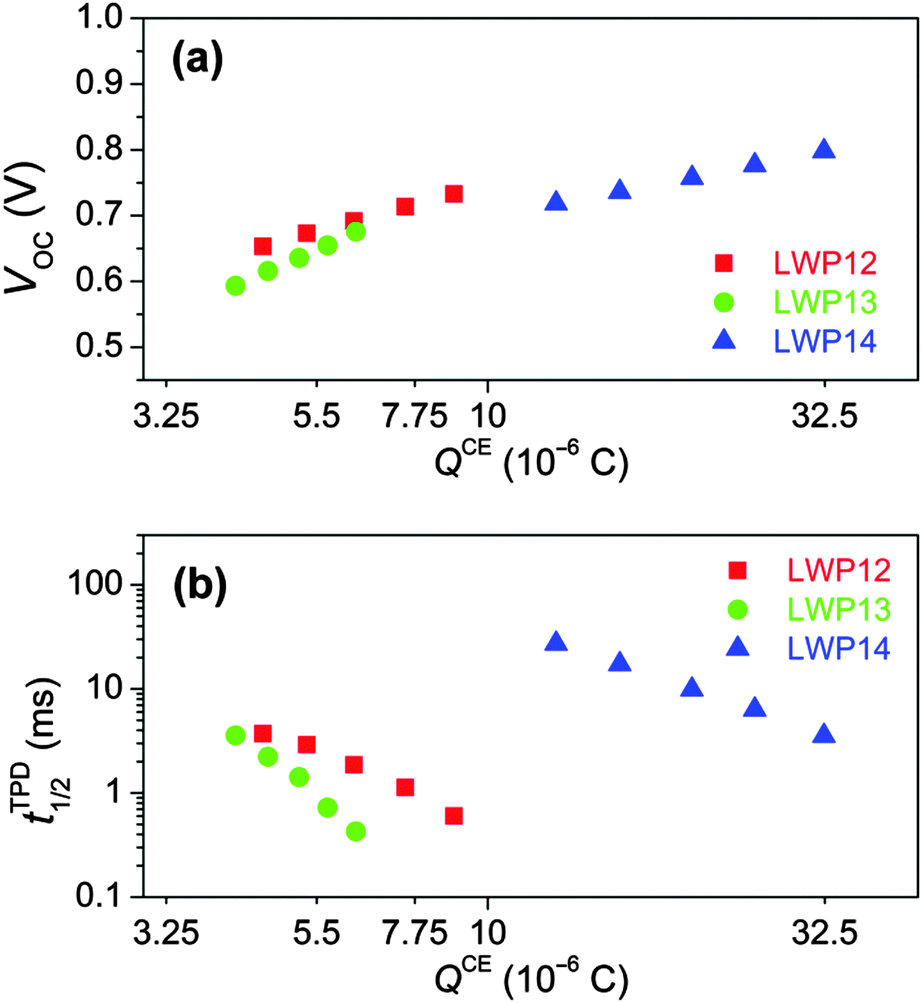 | ||
| Fig. 9 (a) Plots of charge stored in a dye-grafted titania film (QCE) measured by the charge extraction (CE) method as a function of open-circuit photovoltage (VOC). (b) Comparison of the electron half-lifetime (tTPD1/2) measured by the small-pulse transient photovoltage decay (TPD) method against QCE. | ||
Conclusions
We successfully prepared three new porphyrin dyes (LWP12, LWP13, and LWP14) bearing various donor groups. To provide both electron-donating and absorption-broadening effects, the electron-donating group of LWP12 is very complicated whereas those of LWP13 and LWP14 are considerably simplified. Significantly, LWP14-sensitized solar cells outperform others with a PCE of 10.3%. The results in this work have a significant implication that, with suitable structural design, consolidated donor groups with dual properties could be used to prepare efficient photosensitizers.Experimental
Instruments
A glove box (MBraun Uni-lab), a vacuum line and standard Schlenk glassware were employed to process all materials sensitive to air. NMR (Bruker Avance II 300 MHz), UV-visible (Agilent 8453), fluorescence (Varian Cary Eclipse), and Mass (Microfilex MALDI-TOF MS, Bruker Daltonics) spectra were obtained on the indicated instruments. Elemental analyses were carried out by the MOST Instrumentation Center at National Taiwan University (Elementar Vario EL III). Electrochemistry was carried out using a standard three-electrode cell (a Pt working electrode, a Pt auxiliary electrode, and an SCE reference electrode) on a CH Instruments Electrochemical Workstation 611A. Details of the EQE, J–V, IS, CE and TPD measurements can be found in our previous publication.38Materials and methods
Solvents for the synthesis (ACS Grade) were CH2Cl2 and CHCl3 (Mallinckrodt Baker, KE USA), hexanes (Haltermann, Hamburg Germany) and THF (Merck, Darmstadt Germany). These solvents were used as received unless otherwise stated. Other chemicals were obtained from Acros Organics, NJ, USA. THF for cross-coupling reactions was purified and dried using a solvent purification system (Asiawong SD-500, Taipei, Taiwan); about 50 ppm H2O was found in the resulting fluid. For electrochemical measurements, THF was distilled over sodium under N2. The Pd(PPh3)4 catalyst (Strem, MA, USA) and Pd2(dba)3 (Acros Organics, NJ, USA) were used as received. For chromatographic purification, we used silica gel 60 (230–400 mesh, Merck, Germany). 4-tert-butylpyridine (TBP) and lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) were purchased from Sigma-Aldrich. The light-scattering paste was purchased from Dyesol and the translucent layer paste was prepared according to the published procedure.39Dye synthesis
Syntheses of the LWP12-14 porphyrins are based on the Sonogashira cross-coupling methods.25,40,41 The detailed synthetic procedure and characterization methods are given in the ESI.†Solar cell preparation
A bilayer TiO2 film screen-printing on pre-cleaned fluorine doped tin oxide (FTO) conducting glass (NSG, TECA9X, 4.0 mm) was employed as the negative electrode of DSSCs. The semiconducting bilayer film is composed of a 4.0 μm-thick translucent layer of small particles (25 nm) and a 5.0 μm-thick light-scattering layer of large particles (350–450 nm). A titania film was further immersed into a solution of 150 μM dye and 10 mM Cheno in the binary solvent of THF/ethanol (v/v, 2/8) for 12 h for dye-loading. The dye-adsorbed TiO2 electrode was assembled with an Au/Cr coated FTO (NSG, TEC7, 2.2 mm) electrode by a 25-μm-thick Surlyn ring. A cobalt electrolyte which consists of 0.5 M TBP, 0.25 M tris(2,2′-bipyridiene)cobalt(II) di[bis(trifluoromethanesulfonyl)-imide], 0.05 M tris(2,2′-bipyridiene)cobalt(III) tris[bis(trifluoromethanesulfonyl)imide], and 0.1 M LiTFSI in acetonitrile was utilized for device fabrication.Acknowledgements
For financial support, CYL acknowledges the Ministry of Science and Technology of Taiwan (NSC 102-2113-M-260-003-MY3). MZ, MX, and PW acknowledge the National Science Foundation of China (No. 51203150, 51125015, and 91233206) and the Natural Science Foundation of Jilin Province of China (No. 20130101013JC).Notes and references
- M. Grätzel, Nature, 2001, 414, 338–344 CrossRef PubMed.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- W. M. Campbell, A. K. Burrell, D. L. Officer and K. W. Jolley, Coord. Chem. Rev., 2004, 248, 1363–1379 CrossRef CAS.
- H. Imahori, T. Umeyama and S. Ito, Acc. Chem. Res., 2009, 42, 1809–1818 CrossRef CAS PubMed.
- L.-L. Li and E. W.-G. Diau, Chem. Soc. Rev., 2013, 42, 291–304 RSC.
- M. Urbani, M. Grätzel, M. K. Nazeeruddin and T. Torres, Chem. Rev., 2014, 114, 12330–12396 CrossRef CAS PubMed.
- T. Higashino and H. Imahori, Dalton Trans., 2015, 44, 448–463 RSC.
- W. M. Campbell, K. W. Jolley, P. Wagner, K. Wagner, P. J. Walsh, K. C. Gordon, L. Schmidt-Mende, M. K. Nazeeruddin, Q. Wang, M. Grätzel and D. L. Officer, J. Phys. Chem. C, 2007, 111, 11760–11762 CAS.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242–247 CrossRef CAS PubMed.
- A. Yella, C.-L. Mai, S. M. Zakeeruddin, S.-N. Chang, C.-H. Hsieh, C.-Y. Yeh and M. Grätzel, Angew. Chem., Int. Ed., 2014, 53, 2973–2977 CrossRef CAS PubMed.
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS PubMed.
- K. Kurotobi, Y. Toude, K. Kawamoto, Y. Fujimori, S. Ito, P. Chabera, V. Sundström and H. Imahori, Chem. – Eur. J., 2013, 19, 17075–17081 CrossRef CAS PubMed.
- J. Luo, M. Xu, R. Li, K.-W. Huang, C. Jiang, Q. Qi, W. Zeng, J. Zhang, C. Chi, P. Wang and J. Wu, J. Am. Chem. Soc., 2014, 136, 265–272 CrossRef CAS PubMed.
- L. Cabau, C. V. Kumar, A. Moncho, J. N. Clifford, N. López and E. Palomares, Energy Environ. Sci., 2015, 8, 1368–1375 CAS.
- C.-P. Hsieh, H.-P. Lu, C.-L. Chiu, C.-W. Lee, C.-L. Mai, W.-N. Yen, S.-J. Hsu, E. W.-G. Diau and C.-Y. Yeh, J. Mater. Chem., 2010, 20, 1127–1134 RSC.
- M. Ishida, D. Hwang, Z. Zhang, Y. J. Choi, J. Oh, V. M. Lynch, D. Y. Kim, J. L. Sessler and D. Kim, ChemSusChem, 2015 DOI:10.1002/cssc.201500085.
- S. Rangan, S. Coh, R. A. Bartynski, K. P. Chitre, E. Galoppini, C. Jaye and D. Fischer, J. Phys. Chem. C, 2012, 116, 23921–23930 CAS.
- Y. Wang, B. Chen, W. Wu, X. Li, W. Zhu, H. Tian and Y. Xie, Angew. Chem., Int. Ed., 2014, 53, 10779–10783 CrossRef CAS PubMed.
- S. H. Kang, I. T. Choi, M. S. Kang, Y. K. Eom, M. J. Ju, J. Y. Hong, H. S. Kang and H. K. Kim, J. Mater. Chem. A, 2013, 1, 3977–3982 CAS.
- W. Li, Z. Liu, H. Wu, Y.-B. Cheng, Z. Zhao and H. He, J. Phys. Chem. C, 2015, 119, 5265–5273 CAS.
- T. Sakurada, Y. Arai and H. Segawa, RSC Adv., 2014, 4, 13201–13204 RSC.
- S. B. Mane and C.-H. Hung, Chem. – Eur. J., 2015, 21, 4825–4841 CrossRef CAS PubMed.
- J. Lu, S. Liu, H. Li, Y. Shen, J. Xu, Y. Chenga and M. Wang, J. Mater. Chem. A, 2014, 2, 17495–17501 CAS.
- C.-L. Wang, Y.-C. Chang, C.-M. Lan, C.-F. Lo, E. W.-G. Diau and C.-Y. Lin, Energy Environ. Sci., 2011, 4, 1788–1795 CAS.
- Y.-C. Chang, C.-L. Wang, T.-Y. Pan, S.-H. Hong, C.-M. Lan, H.-H. Kuo, C.-F. Lo, H.-Y. Hsu, C.-Y. Lin and E. W.-G. Diau, Chem. Commun., 2011, 47, 8910–8912 RSC.
- C.-L. Wang, C.-M. Lan, S.-H. Hong, Y.-F. Wang, T.-Y. Pan, C.-W. Chang, H.-H. Kuo, M.-Y. Kuo, E. W.-G. Diau and C.-Y. Lin, Energy Environ. Sci., 2012, 5, 6933–6940 CAS.
- C.-L. Wang, J.-Y. Hu, C.-H. Wu, H.-H. Kuo, Y.-C. Chang, Z.-J. Lan, H.-P. Wu, E. W.-G. Diau and C.-Y. Lin, Energy Environ. Sci., 2014, 7, 1392–1396 CAS.
- J.-W. Shiu, Y.-C. Chang, C.-Y. Chan, H.-P. Wu, C.-L. Wang, C.-Y. Lin and E. W.-G. Diau, J. Mater. Chem. A, 2015, 3, 1417–1420 CAS.
- H.-P. Wu, Z.-W. Ou, T.-Y. Pan, C.-M. Lan, W.-K. Huang, H.-W. Lee, N. M. Reddy, C.-T. Chen, W.-S. Chao, C.-Y. Yeh and E. W.-G. Diau, Energy Environ. Sci., 2012, 5, 9843–9848 CAS.
- M. Gouterman, J. Mol. Spectrosc., 1961, 6, 138–163 CrossRef CAS.
- P.-S. Chao, M.-Y. Kuo, C.-F. Lo, M.-H. Hsieh, Y.-H. Cheng, C.-L. Wang, H.-Y. Lu, H.-H. Kuo, Y.-N. Hsiao, C.-M. Wang and C.-Y. Lin, J. Porphyrins Phthalocyanines, 2013, 17, 92–98 CrossRef CAS.
- K. M. Kadish, E. V. Caemelbecke and G. Royal, in The Porphyrin Handbook, K. M. Kadish, K. M. Smith and G. Guilard, ed. Academic Press, New York, 2000, vol. 8, pp. 1–97, and vol. 9 Search PubMed.
- M. J. Frisch, et. al., Gaussian 03, Revision D.01, Gaussian, Inc., Pittsburgh PA, 2003 Search PubMed.
- P. Qu and G. J. Meyer, Langmuir, 2001, 17, 6720–6728 CrossRef CAS.
- N. W. Duffy, L. M. Peter, R. M. G. Rajapakse and K. G. U. Wijayant, Electrochem. Commun., 2000, 2, 658–662 CrossRef CAS.
- B. C. O'Regan, S. Scully, A. C. Mayer, E. Palomares and J. R. Durrant, J. Phys. Chem. B, 2005, 109, 4616–4623 CrossRef PubMed.
- J. Bisquert, Phys. Chem. Chem. Phys., 2003, 5, 5360–5364 RSC.
- N. Cai, R. Li, Y. Wang, M. Zhang and P. Wang, Energy Environ. Sci., 2013, 6, 139–147 CAS.
- P. Wang, S. M. Zakeeruddin, P. Comte, R. Charvet, R. Humphry-Baker and M. Grätzel, J. Phys. Chem. B, 2003, 107, 14336–14341 CrossRef CAS.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 4467–4470 CrossRef CAS.
- S. Takahashi, Y. Kuroyama and K. Sonogashira, Synthesis, 1980, 627–630 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Dye synthesis and characterization, PL transient experiments of various semi-conductor films, and photovoltaic measurements using different Co-electrolytes. See DOI: 10.1039/c5ee02505b |
| This journal is © The Royal Society of Chemistry 2016 |