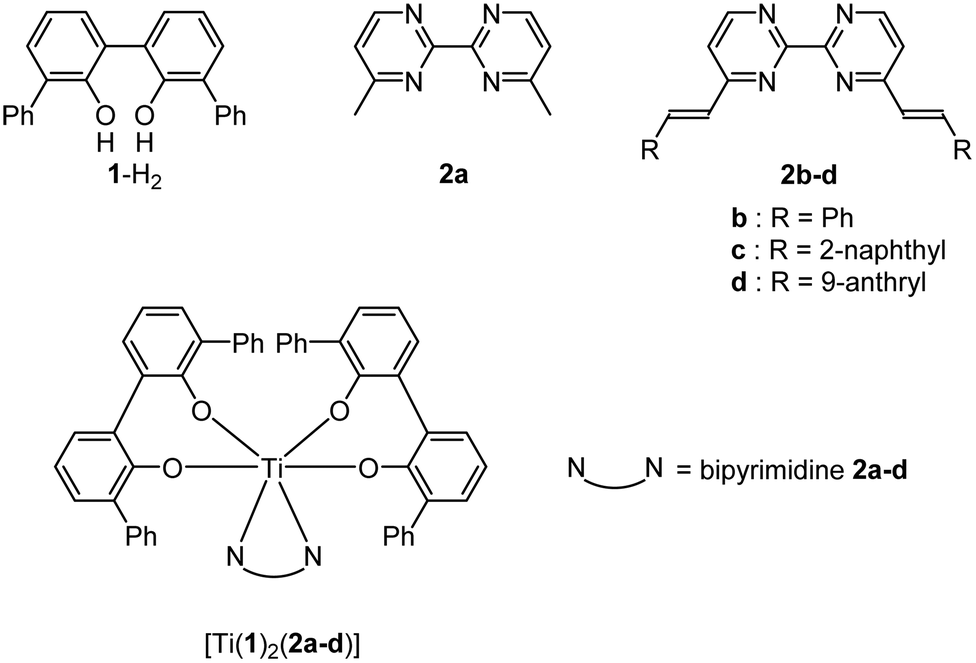
DOI:
10.1039/C6DT03477B
(Paper)
Dalton Trans., 2016,
45, 19072-19085
Monomeric Ti(IV)-based complexes incorporating luminescent nitrogen ligands: synthesis, structural characterization, emission spectroscopy and cytotoxic activities†
Received
6th September 2016
, Accepted 29th October 2016
First published on 18th November 2016
Abstract
This manuscript describes the synthesis of a series of neutral titanium(IV) monomeric complexes constructed around a TiO4N2 core. The two nitrogen atoms that compose the coordination sphere of the metallic center belong to 2,2′-bipyrimidine ligands homo-disubstituted in the 4 and 4′ positions by methyl (2a), phenylvinyl (2b), naphthylvinyl (2c) or anthrylvinyl (2d) groups. The crystal structures of these complexes named [Ti(1)2(2a)], [Ti(1)2(2b)], [Ti(1)2(2c)] and [Ti(1)2(2d)] (where 1 is a 2,2′-biphenolato ligand substituted in the 6 and 6′ positions by phenyl groups) are reported. The hydrolytic stability of the four complexes is evaluated by monitoring the evolution of the free 2a–d signals by 1H NMR spectroscopy. For the conditions tested (6 mM, DMSO-d6/D2O: 8/1), a rather good stability with t1/2 ranging from 180 to 300 min is determined for the complexes. In the presence of an acid (DCl), the hydrolysis of [Ti(1)2(2a)] is faster than without an acid. The cytotoxic activity against gastric cancer cells of the titanium-based compounds and the free disubstituted 2,2′-bipyrimidine ligands is tested, showing IC50 ranging from 6.2 ± 1.2 μM to 274 ± 56 μM. The fluorescence studies of the ligands 2a–d, and the complexes [Ti(1)2(2a–d)] reveal an important fluorescence loss of the ligands 2c and 2d upon coordination with the Ti(1)2 fragment. Frontier orbitals obtained by DFT calculations permit us to explain this fluorescence quenching.
Introduction
Luminescence is a recurrent property displayed by transition metal complexes.1 Luminescent transition metal complexes are particularly attractive for applications as chemosensors,2 probes for biological molecules3 or organic light emitting diodes.4 Luminescent transition metal complexes such as zinc,5 copper,6 gold,7 platinum8 or d6 transition metal complexes (especially ruthenium)9 have also been extensively applied for fluorescence cell imaging.
Titanium(IV) complexes are very diverse and represent a major class of compounds widely used in many fields of modern molecular science. Organometallic and coordination titanium(IV) compounds are powerful reagents for catalytic enantioselective processes,10 organic transformations11 and as initiators for polymerization reactions.12 Alkoxides or aryloxides titanium species are precursors to generate titanium-based materials through sol–gel processes.13 In metallo-supramolecular chemistry, titanium(IV) centers are intensively employed to create self-assembled architectures from oxygen ligands14 or from multiple components.15 Applications of titanium(IV) complexes in bioinorganic chemistry are also deeply investigated, as long as titanium(IV) is recognized for its low toxicity for living matter. Therefore, titanium is found in many biomaterials and numerous applications of Ti(IV) in a medical or biological context have been described.16,17 Particularly, titanium(IV) complexes are promising candidates for developing new anti-cancer drugs with minor side effects.18,19
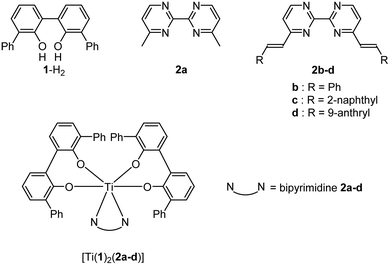
As we were intrigued by the rarity of reports detailing the coordination chemistry of titanium(IV) complexes bearing luminescent ligands,20 this work aimed to study the ligating properties to a titanium(IV) center of fluorescent ligands and their resulting emissions. Thus, we report the synthesis and the structural characterization of a series of TiO4N2-based monomeric octahedral complexes bearing various substituted 2,2′-bipyrimidines (Chart 1). These complexes are built from two biphenolato derivatives and one substituted 2,2′-bipyrimidine unit as depicted in Fig. 1. The choice of these nitrogen ligands is motivated by the fact that 2,2′-bipyrimidine has shown to be a well-adapted chelate to successfully generate TiO4N2-based complexes.15,21 Also, 4,4′-dimethyl-2,2′-bipyrimidine can be easily functionalized by various groups through Knoevenagel reactions.22 Therefore, the synthesis of a TiO4N2-based monomeric octahedral complex bearing 2,2′-bipyrimidines substituted by “classical” fused polycyclic aromatic emitters, as the naphthyl or anthryl group,23 was envisaged. Additionally, we also detail in this manuscript some biological applications of these Ti(IV)-based species.
 |
| | Chart 1 | |
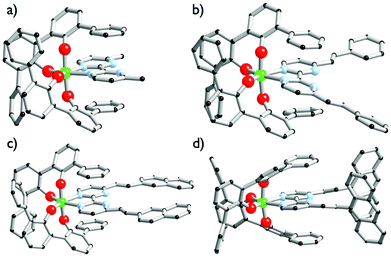 |
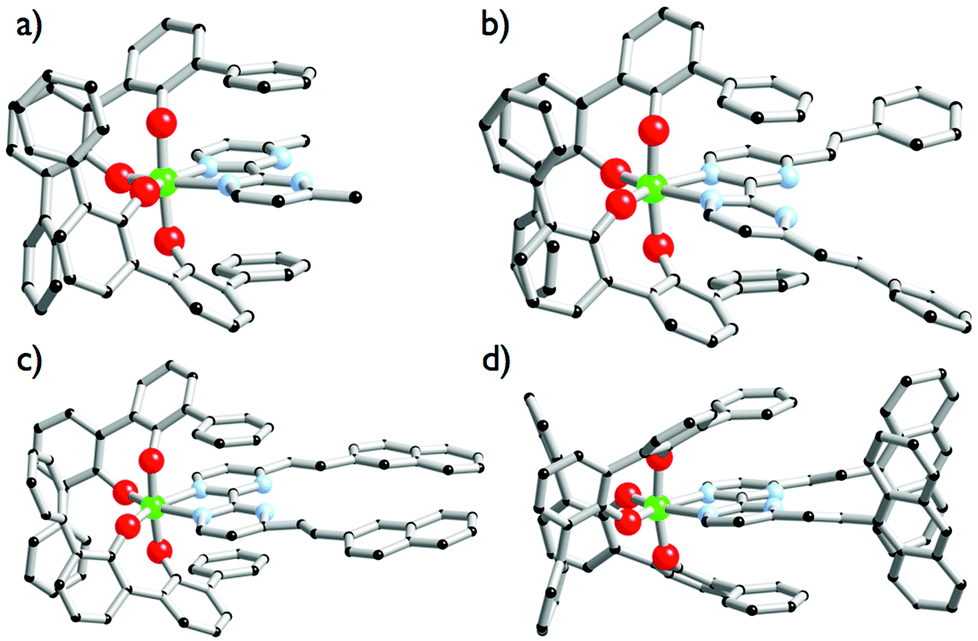
| | Fig. 1 Solid-state molecular structures of [Ti(1)2(2a)] (a), [Ti(1)2(2b)] (b), [Ti(1)2(2c)] (c) and [Ti(1)2(2d)] (d). Ti atoms are in green, C atoms in black, O atoms in red, N atoms in blue. Hydrogen atoms are omitted for clarity. | |
Results and discussion
Synthesis of ligands and complexes
The ligands 2b–d were synthesized according to Scheme 1 in a one-step double Knoevenagel condensation starting from the 4,4′-dimethyl-2,2′-bipyrimidine precursor 2a.22 Compound 2a was condensed with benzaldehyde, 2-naphthaldehyde or 9-anthraldehyde using tBuOK to afford the targeted bipyrimidine derivatives 2b–d (yields ∼ 55%). The 1H NMR spectra for 2b–d highlighted doublets with trans vinylic coupling (3J ∼ 16 Hz) confirming the formation of the desired bipyrimidine compounds. Having in hand this set of bipyrimidine derivatives, the synthesis of the targeted octahedral monomeric complexes [Ti(1)2(2a–d)] constructed around a TiO4N2 core was undertaken. The complexes were obtained by following a well-established procedure21 based on the exchange of the labile monodentate ligands coordinating the metal center in [Ti(1)2(HOiPr)2]24 by a nitrogen bidentate ligand as proposed in eqn (1).| | | [Ti(1)2(HOiPr)2] + 2a–d → [Ti(1)2(2a–d)] + 2HOiPr | (1) |
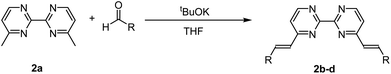 |
| | Scheme 1 General synthetic scheme followed to generate compounds 2b–d. | |
Complexes [Ti(1)2(2a–d)] were isolated as single crystals suitable for X-ray analysis by dissolving in a chlorinated solvent the [Ti(1)2(HOiPr)2] precursor with one equivalent of the 2a–d ligand after slow vapour diffusion of n-pentane. The molecular structures of complexes [Ti(1)2(2a–d)] are shown in Fig. 1. As expected, the structures highlight monomeric C2-symmetric complexes incorporating an octahedral TiO4N2 unit. Interestingly, these complexes crystallize in achiral space groups with the exception of [Ti(1)2(2a)]. [Ti(1)2(2a)] crystallizes in the noncentrosymmetric P212121 space group when the formation of the complex is performed in 1,2-dichloroethane and n-pentane is used as a precipitant. The Λ-[Ti(1)2(2a)] enantiomer is observed in the analyzed crystal. The Flack parameter of 0.006(15) determined for the Λ-[Ti(1)2(2a)] structure resolution confirms the absolute configuration attribution. Concerning the molecular structures description, it is worth noticing that the aromatic units substituting the bipyrimidine ligands in [Ti(1)2(2b)] and [Ti(1)2(2c)] are coplanar to the neighbouring pyrimidine ring. The situation is different for [Ti(1)2(2d)] since the two anthryl groups adopt an almost orthogonal arrangement with respect to the pyrimidine rings. In all structures, the double bonds adopt a trans isomerism as indicated by the 1H NMR analysis of the free ligands. The metrical description of the structures is unexceptional. The Ti–O (d(Ti–O) = 1.86 ± 0.06 Å) and Ti–N (d(Ti–N) = 2.238 ± 0.004 Å) bond distances measured in these complexes are rather similar and identical, respectively. The coordination polyhedra found in these structures are distorted as attested by the mean angles measured around the titanium atoms (N–Ti–N = 71.4° ± 1.2, O–Ti–O(O trans to N) = 111.3° ± 1.6). We also notice in [Ti(1)2(2d)] that the two anthryl groups are rather distant from one another, as the closest C–C distance measured between the two anthryl units is found to be d(Canthryl–Canthryl) = 6.66 Å.
Hydrolytic stability
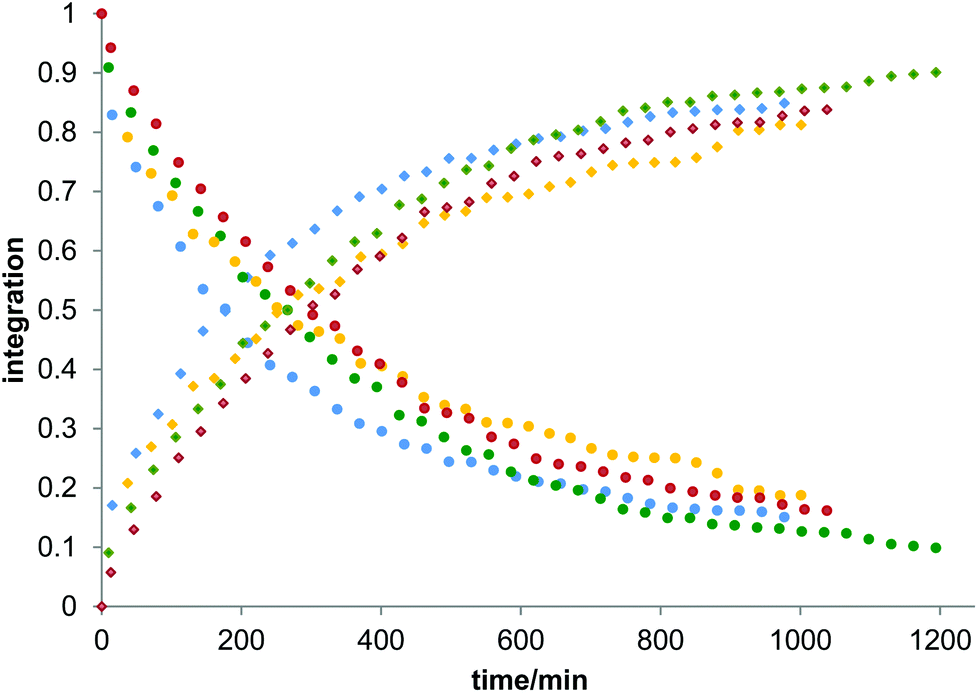
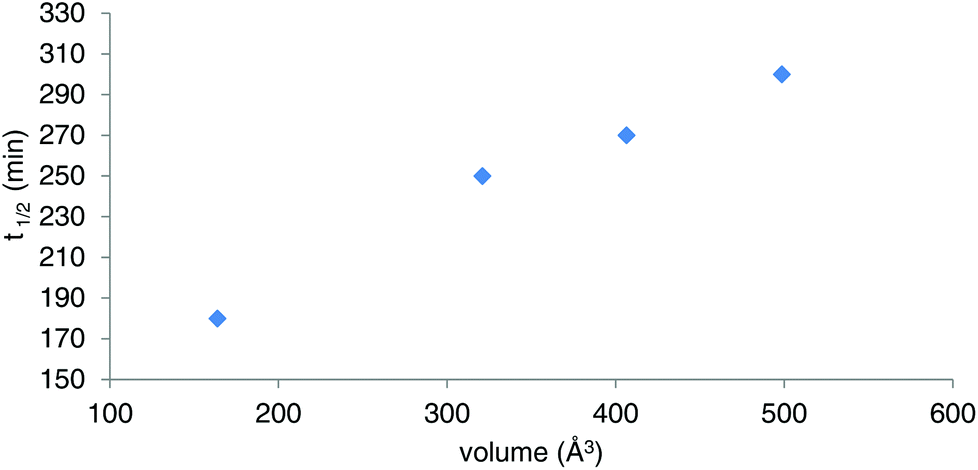
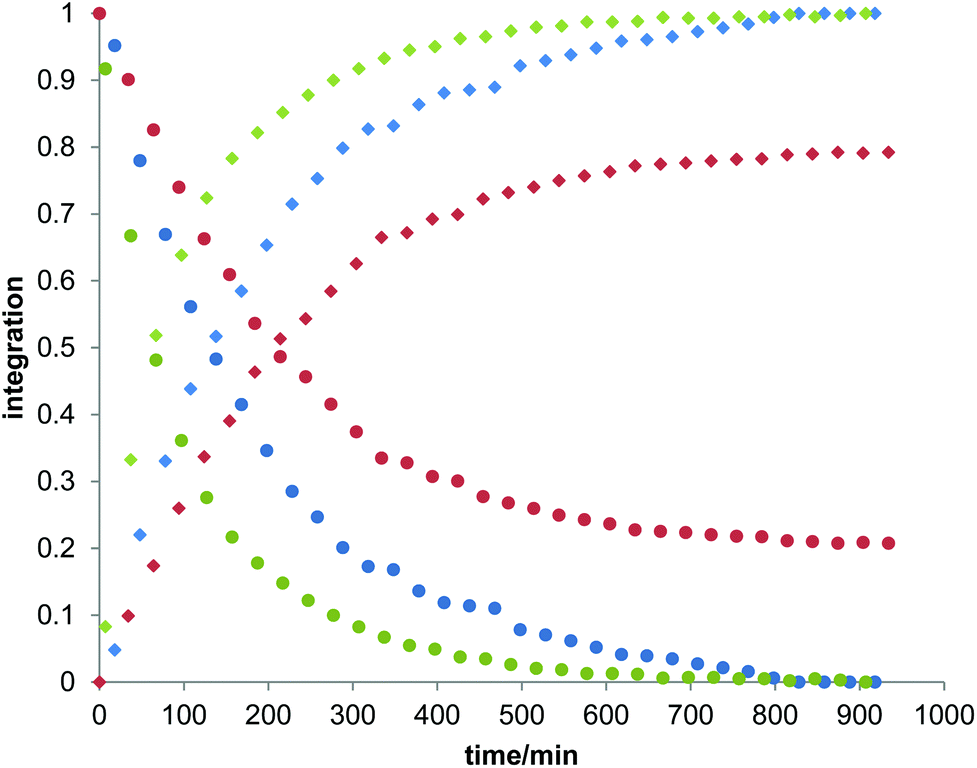
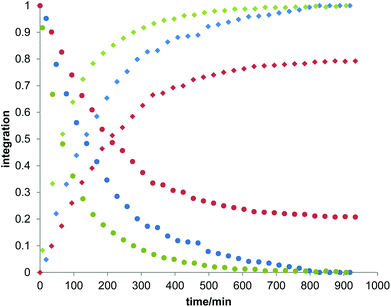
Owing to the poor or moderate hydrolytic stability usually observed for molecular Ti(IV) species such as classical anticancer titanium-based compounds like budotitane and titanocene dichloride,25a–c the behaviour of compounds [Ti(1)2(2a–d)] towards hydrolysis has been evaluated by following a methodology similar to the one already reported for titanium complexes.25d–f These studies were conducted by 1H NMR analysis of solutions in DMSO-d6 of the [Ti(1)2(2a–d)] complexes over time, upon addition of D2O at room temperature. For these analyses, a large amount of D2O (1000 equivalents versus the complex) was added to the complex solution so that a final complex solution concentration of 6 mM was obtained. It should be noted first that when the concentration of the complex solutions was doubled, an orange precipitate formed instantaneously, reflecting the moderate solubility of these species in polar media. Secondly, when the same experiment was performed without D2O, a slight degradation of the initial complex was noticed in DMSO. Next, the resulting 6 mM samples were analyzed by 1H NMR. The evolution of the mixtures composition was monitored for [Ti(1)2(2a)], [Ti(1)2(2b)] and [Ti(1)2(2c)] by integrating the bipyrimidine 6,6′ proton resonances for the complex and for the free diimine ligand. For compound [Ti(1)2(2d)], the situation was different, as the vinylic proton signals were monitored. The integration plots of these signals overtime for complexes [Ti(1)2(2a–d)] are shown in Fig. 2. The complete transformation of the complexes over a period of time of more than 16 hours is not observed. By analyzing the mixtures after one week of their preparation, the presence of a not negligible amount of the starting complexes is still noticed. Also, the absence of free 2,2′-biphenol derivative in solution is confirmed. Therefore, the formation of titanium(IV) oxo-aggregates incorporating 1 through an olation or oxolation reaction mechanism could be envisaged since oxo-clusters are common products when low nuclearity titanium(IV) complexes react with water.26 The presence of novel species formed upon hydrolysis in solution is confirmed by NMR analysis. The aromatic region of the 1H NMR spectrum indicates broad signals different from the 1H NMR signature of 1-H2. ES-MS analysis of an aliquot also proves the formation of unknown hydrolytic products displaying molecular weights not compatible with the initial complex and the free ligands (see the ESI†). Plotting ln[complex] or 1/[complex] with respect to time indicated highly complex kinetics for these hydrolytic processes. The t1/2 values found for these experiments demonstrated significant differences between each reaction (Table 1). Complex [Ti(1)2(2a)] exhibits a faster hydrolysis rate compared to [Ti(1)2(2b)], [Ti(1)2(2c)] or [Ti(1)2(2d)]. A linear relationship was found between these t1/2 values and the volumes of the bipyrimidine ligands were computed from the crystal structures of [Ti(1)2(2a–d)] with the PACHA software27,28 as shown in Fig. 3. This relationship reveals the shield effect played by the aromatic groups substituting the bipyrimidine ligands against water. Next, the role played by an acid on the complex [Ti(1)2(2a)] stability was examined by repeating the hydrolysis experiment in the presence of DCl (38 wt% in D2O). It appears that with DCl, the transformation of the starting [Ti(1)2(2a)] complex is nearly quantitative after 10 hours for the two DCl concentrations tested as shown in Fig. 4. With an excess of DCl (9 equivalents versus [Ti(1)2(2a)]), the t1/2 value of the complex is divided by two in comparison to an experiment conducted with 1.8 equivalent of DCl vs. [Ti(1)2(2a)]. Altogether, with an acid, the disappearance of the complex is faster than the same reaction performed without DCl.
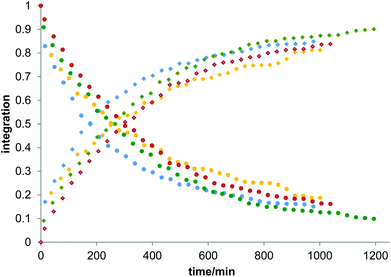 |
| | Fig. 2 Plots of the integration of the bipyrimidine 6,6′ proton signals related to the bound diimine ligands and the free ligands for complexes [Ti(1)2(2a)], [Ti(1)2(2b)] and [Ti(1)2(2c)]. For compound [Ti(1)2(2d)], the vinylic proton signals were monitored. In these experiments, D2O (50 μl) was added to the solution of the complex in DMSO-d6 to give a final complex concentration of 6 mM. The blue, yellow, green and red dots correspond to the measurements made for [Ti(1)2(2a)], [Ti(1)2(2b)], [Ti(1)2(2c)] and [Ti(1)2(2d)], respectively. Dots are the integrations corresponding to the coordinated diimine ligands, squares are the integrations of the free bipyrimidine ligands. The t1/2 values of hydrolysis calculated from these plots are compiled in Table 1. | |
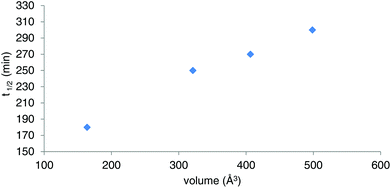 |
| | Fig. 3 Plot of the t1/2 values determined for [Ti(1)2(2a–d)] in the course of the hydrolytic stability studies vs. the molecular volumes of 2a–d calculated from the [Ti(1)2(2a–d)] crystal structures. | |
 |
| | Fig. 4 Plots of the integration of the bipyrimidine 6,6′ proton signals related to the bound diimine ligands (dots) and the free ligands (squares) in the spectra of [Ti(1)2(2a)]. The red curves correspond to the measurement made for a solution of [Ti(1)2(2a)] in DMSO-d6 (450 μl) where D2O (50 μl) was added. The green and blue curves correspond to the measurements made for a solution of [Ti(1)2(2a)] in DMSO-d6 (450 μl) where D2O and DCl were added (50 μl in total for the volume of D2O and DCl). The green curves correspond to the experiment conducted with 2.5 μl of DCl solution (38 wt% in D2O) whereas the blue curves correspond to the experiment conducted with 0.5 μl of DCl solution (38 wt% in D2O). t1/2 = 67 min ± 5% for the experiment conducted with 2.5 μl of DCl; t1/2 = 138 min ± 5% for the experiment conducted with 0.5 μl of DCl. | |
Table 1
t
1/2 Values determined for the hydrolytic processes. Ligand volumes computed from the crystal structures [Ti(1)2(2a–d)]
| |
t
1/2 (min) (±5%) |
Volume of ligands 2a–d (Å3) (±10%) |
| [Ti(1)2(2a)] |
180 |
163 |
| [Ti(1)2(2b)] |
250 |
320 |
| [Ti(1)2(2c)] |
270 |
406 |
| [Ti(1)2(2d)] |
300 |
498 |
Photophysical analyses
To the best of our knowledge, there are very few reports of titanium(IV) coordination complexes that would be photoluminescent in solution at room temperature, if we exclude titanium phthalocyanines, which are very specific species.29 Yet some other group 4 metal complexes (Zr, Hf) exhibit such properties.30 However, the luminescence of several Ti complexes at 77 K has been investigated.31 Besides, substituted pyrimidines are a class of organic molecules, which often display light-emitting properties in solution at ambient temperature, and which have even been considered as candidates for OLEDs.32 Unsubstituted 2,2′-bipyrimidine has been used as a bridging ligand in photoluminescent binuclear lanthanide complexes.33 Moreover, various substituted 2,2′-bipyrimidines and their coordination complexes have been reported in the literature, which display interesting photophysical properties (fluorescence and non-linear optics).34
In the present section, we describe the optical properties, viz. absorption and emission spectroscopy, of solutions in dichloromethane of the ligands 1-H2 and 2a–d, and of the derived titanium complexes [Ti(1)2(2a–d)]. The data obtained in solution at room temperature are presented in Fig. 5 and 6 and summarized in Table 2. Incidentally, the UV-vis spectrum of 1-H2 has been reported in the literature, but in a different solvent.35
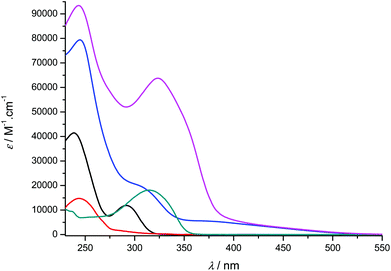 |
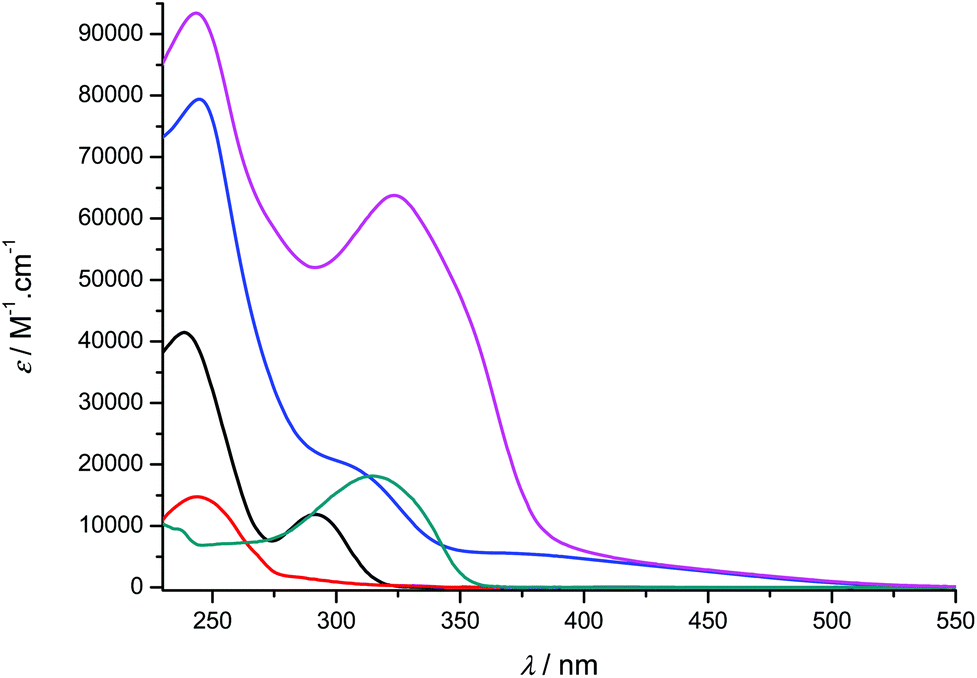
| | Fig. 5 UV-visible absorption spectra of compounds 1-H2 (black), 2a (red), [Ti(1)2(2a)] (blue), 2b (green) and [Ti(1)2(2b)] (pink) recorded at room temperature in CH2Cl2 (concentration range: 9–55 μM). | |
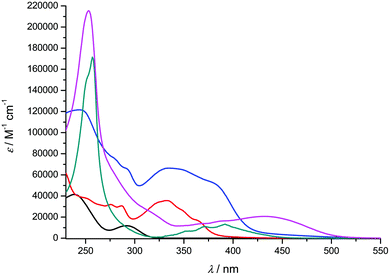 |
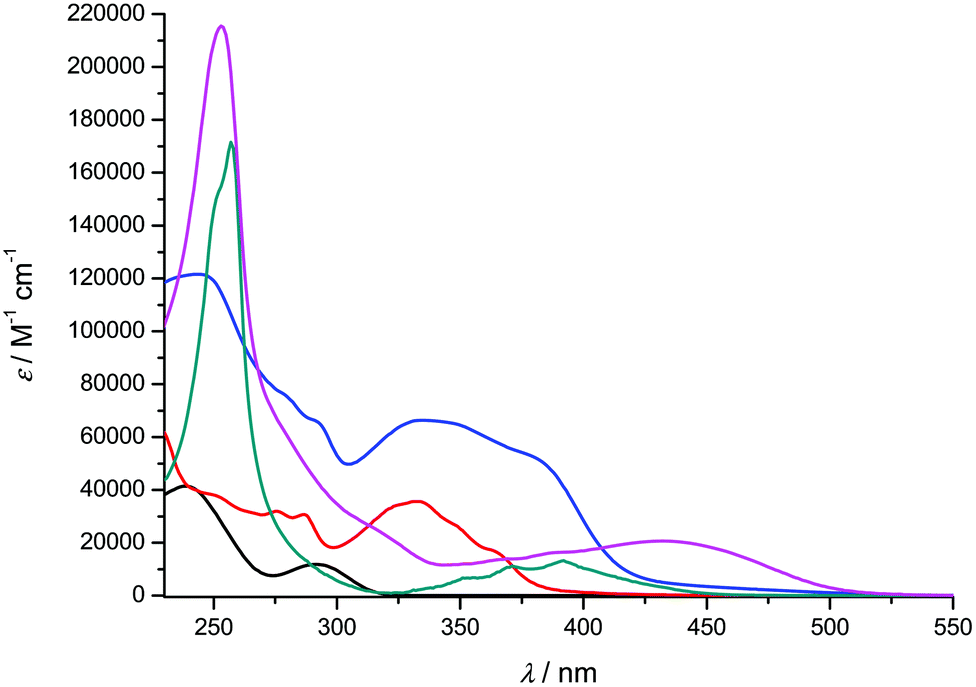
| | Fig. 6 UV-visible absorption spectra of compounds 1-H2 (black), 2c (red), [Ti(1)2(2c)] (blue), 2d (green) and [Ti(1)2(2d)] (pink) recorded at room temperature in CH2Cl2 (concentration range: 2–18 μM). | |
Table 2 Photophysical data of the ligands and titanium complexesa
| Compound |
Absorption |
Photoluminescence |
λ
max/nm (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε)b ε)b |
λ
excitation/nm |
Emission: λmax/nm (Φ) |
|
Conditions: solutions in dichloromethane at room temperature (concentration range: absorption = 9–55 μM; emission = 1–3 μM); Φ = quantum yield; sh = shoulder.
ε in M−1 cm−1.
Not photoluminescent.
Not determined (very weak luminescence).
|
|
1-H2 |
239 (4.62), 291 (4.07) |
290 |
353 (9 × 10−2) |
|
2a
|
244 (4.17) |
— |
—c |
|
2b
|
315 (4.26) |
315 |
405 (n.d.d) |
|
2c
|
276 (4.50), 287 (4.49), 332 (4.55), 350sh (4.42), 365sh (4.23) |
332 |
411 (6 × 10−3) |
|
2d
|
256 (5.35), 354sh (3.94), 372 (4.14), 392 (4.20) |
392 |
525 (7 × 10−3) |
| [Ti(1)2(2a)] |
245 (4.90), 312sh (4.26), 380sh (3.73) |
— |
—c |
| [Ti(1)2(2b)] |
244 (4.97), 323 (4.80) |
— |
—c |
| [Ti(1)2(2c)] |
244 (5.08), 280sh (4.88), 293sh (4.81), 334 (4.82), 380sh (4.72) |
380 |
477 (<10−3) |
| [Ti(1)2(2d)] |
253 (5.33), 365sh (4.13), 385sh (4.20), 433 (4.32) |
365 |
490 (<10−3) |
Overall, the compounds absorb UV-vis light over the 240–500 nm range. The λmax value of compound 2a in CH2Cl2 (244 nm) is close to the value reported in EtOH (248 nm).36 A significant bathochromic shift is observed for the phenylvinyl-substituted bipyrimidine 2b, in agreement with an extended electronic delocalization. The spectrum of 2c exhibits several bands of similar intensities, ranging from 276 to 365 nm; strikingly, the UV-vis spectrum of a reported bis(naphthylvinyl)-substituted pyrimidine also exhibits signals at 276 and 332 nm with comparable absorption coefficients.22b Following the interpretation of Le Bozec et al. with regard to other related molecules, viz. tetrakis(arylvinyl)-substituted bipyrimidines,22a these absorptions can be assigned to intramolecular charge transfer (ICT) from the phenyl or naphthyl group towards the electron-deficient bipyrimidine nucleus. This statement is supported by the coplanarity of each aryl group with a vinyl bridge and with the pyrimidine cycle, which is observed on the X-ray structures shown in Fig. 1 (assuming that coordination to titanium does not affect the conformation of the ligand); this coplanarity allows a delocalization of the π electrons. However, as far as [Ti(1)2(2b)] and [Ti(1)2(2d)] bear only two substituents instead of four, and as the aryl groups bear no donor atom, the transitions that we observe lie at a higher energy than the reported transition.22a
Finally, the absorption spectrum of compound 2d contrasts with that of 2b and 2c, with an intense blue-shifted band at 256 nm (probably assigned to a vinylpyrimidine-centered transition) and less intense bands at 354, 372 and 392 nm. The pattern of the latter is reminiscent of the absorption spectrum of anthracene itself.37a Therefore, the spectrum of 2d suggests the absence of electronic connection with the anthracene moieties and the rest of the molecule. Again, this is in agreement with the conformation of this crystal structure of the titanium derivative [Ti(1)2(2d)] (Fig. 1) where the anthryl mean planes make an angle of 67.25° with the vinyl planes.
The UV-vis spectra of the complexes [Ti(1)2(2a–d)] display various intense bands (4.13 < logε < 5.33) between 244 and 433 nm that can be reasonably assigned to ligand-centered (π → π*) transitions. For complexes [Ti(1)2(2c)] and [Ti(1)2(2d)], the pattern of the free ligands 2c and 2d is roughly reproduced (Fig. 6). In addition, they exhibit a very broad and less intense band; this band is often obscured by the other transitions, but it is revealed by its tail in the visible region (roughly up to 520 nm) and is responsible for the red or orange color of the solutions. In the case of complex [Ti(1)2(2a)], it is fairly detectable and is centered at ca. 380 nm. It can be assigned to an aryloxide-to-Ti LMCT transition, in agreement with the literature data.30d,38
The photoluminescence analyses were conducted at low concentrations in order to avoid self-absorption effects. Compound 1-H2 exhibits a strong luminescence at 353 nm with a quantum yield slightly lower than 10%. In comparison, the light-induced emission properties of phenol-derived organic molecules related to compound 1-H2 have been reported.39 Ligand 2a is not light-emissive in solution at RT, which is not surprising because 2,2′-bipyrimidine is not either;33a it is noteworthy that even the π-delocalized 4,4′-diphenyl-6,6′-dimethyl-2,2′-bipyrimidine ligand is not photoluminescent at room temperature.34a2b is only slightly emissive, and the corresponding titanium complexes [Ti(1)2(2a)] and [Ti(1)2(2b)] are not. Conversely, and as we expected, the 2-naphthyl and 9-anthryl-substituted ligands 2c and 2d are characterized by distinct emission signals respectively centered at 411 and 525 nm (Table 2 and Fig. 7). It is again interesting to compare the luminescence properties of these 4,4′-di(arylvinyl)-2,2′-bipyrimidines with those of related compounds: 4,6-di(arylvinyl)pyrimidines, with aryl = 2-naphthyl or 9-anthryl, respectively, emitting at λmax = 424 and 525 nm;22b or 4,4′,6,6′-tetra(arylvinyl)-2,2′-bipyrimidines (aryl = N,N-disubstituted aminophenyl), mentioned above, whose emissions are red-shifted with respect to our data.22a With regard to quantum yields, 2c and 2d are less emissive (0.6 and 0.7%) than these species, especially the latter. We also note that under aerobic conditions, the quantum yield of anthracene itself reaches 21%, whereas it is much lower with 4,6-di(anthrylvinyl)pyrimidine (2%) or with 2d; obviously the pyrimidine ring(s) partly quench the fluorescence of anthracene.
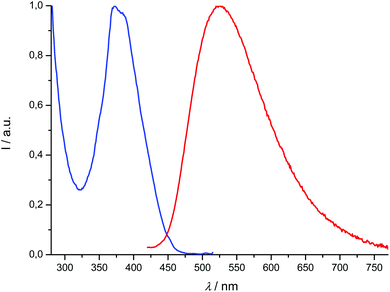 |
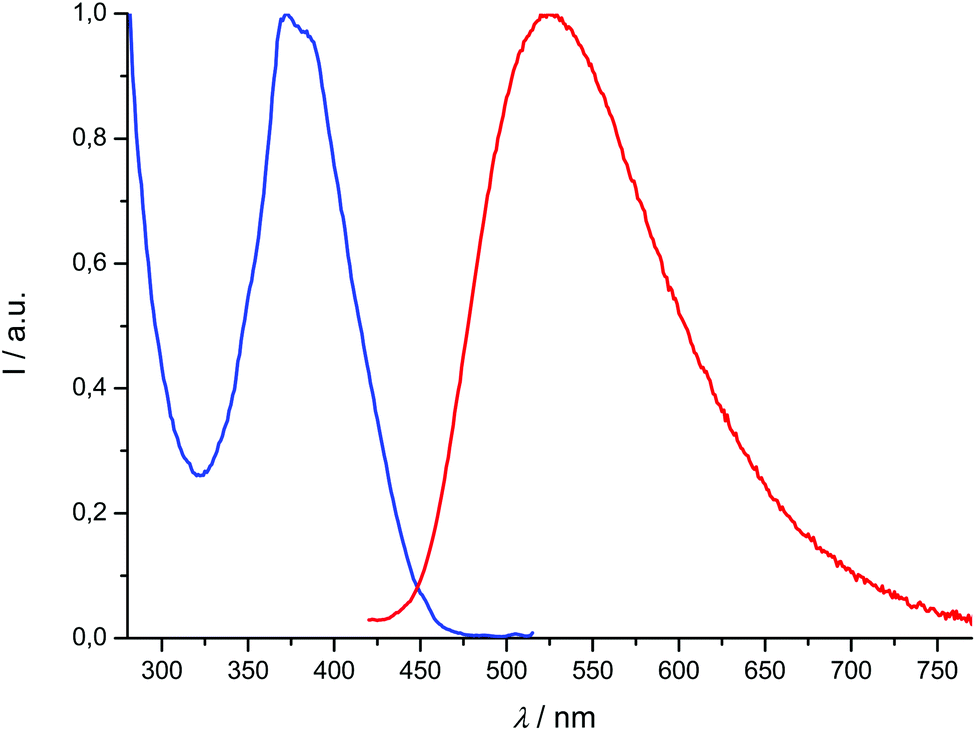
| | Fig. 7 Normalized excitation spectrum (λem = 525 nm, blue) and emission spectrum (λexc = 392 nm, red) of compound 2d recorded at room temperature in CH2Cl2 (concentration: 1.91 μM). | |
Of the four titanium complexes [Ti(1)2(2a–d)], only [Ti(1)2(2c)] and [Ti(1)2(2d)] exhibit an emission, which is very weak, so that the quantum yields cannot be accurately determined; several excitation wavelengths were tested before we could detect a significant signal. Therefore, it appears that coordination to titanium(IV) quenches the fluorescence of the ligands almost completely. The emission band of compound [Ti(1)2(2c)] undergoes a bathochromic shift with regard to 2c (Δλmax = 66 nm), while [Ti(1)2(2d)] is blue-shifted by Δλmax = 35 nm with regard to 2d. Overall, the lifetimes of the excited states are very low, within a 0.1–10 ns range (see the ESI†). In addition, we checked that degassing a solution of [Ti(1)2(2d)] has no influence on the emission (which remains very weak), excluding thus any interaction with dioxygen. These data are in agreement with a fluorescence process (singlet excited states). The excitation spectra corresponding to the emission maxima reported in Table 2 were recorded and were usually in agreement with absorption spectra, with regard to the wavelength maxima observed in the spectra (compounds 1-H2, 2c, 2d, [Ti(1)2(2c)]). The fit was not as good for compound [Ti(1)2(2d)], but many overlapping transitions were observed in the absorption spectrum; it is possible that some would be emissive and others would not.
DFT calculations, frontier orbitals and discussion
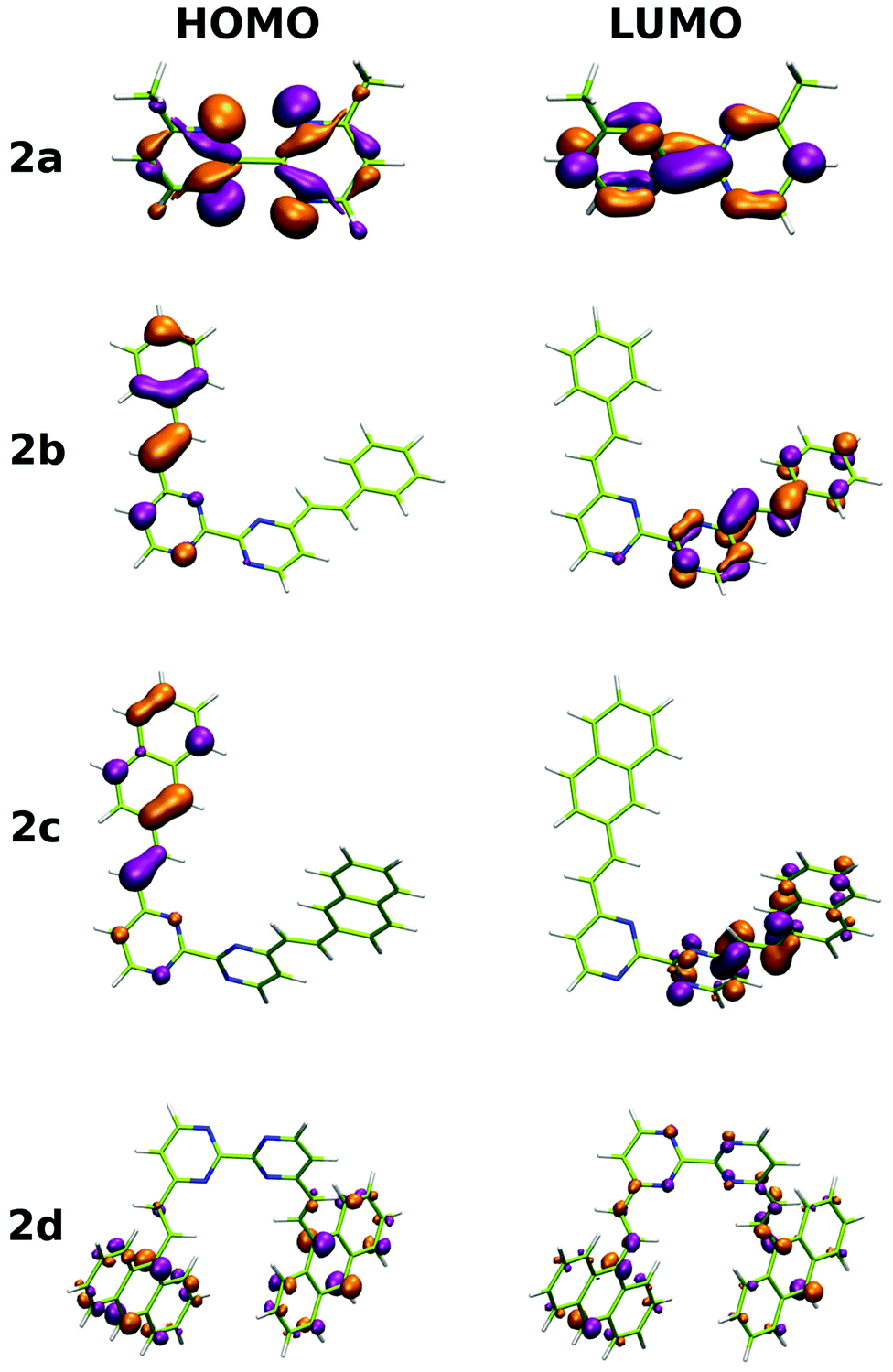
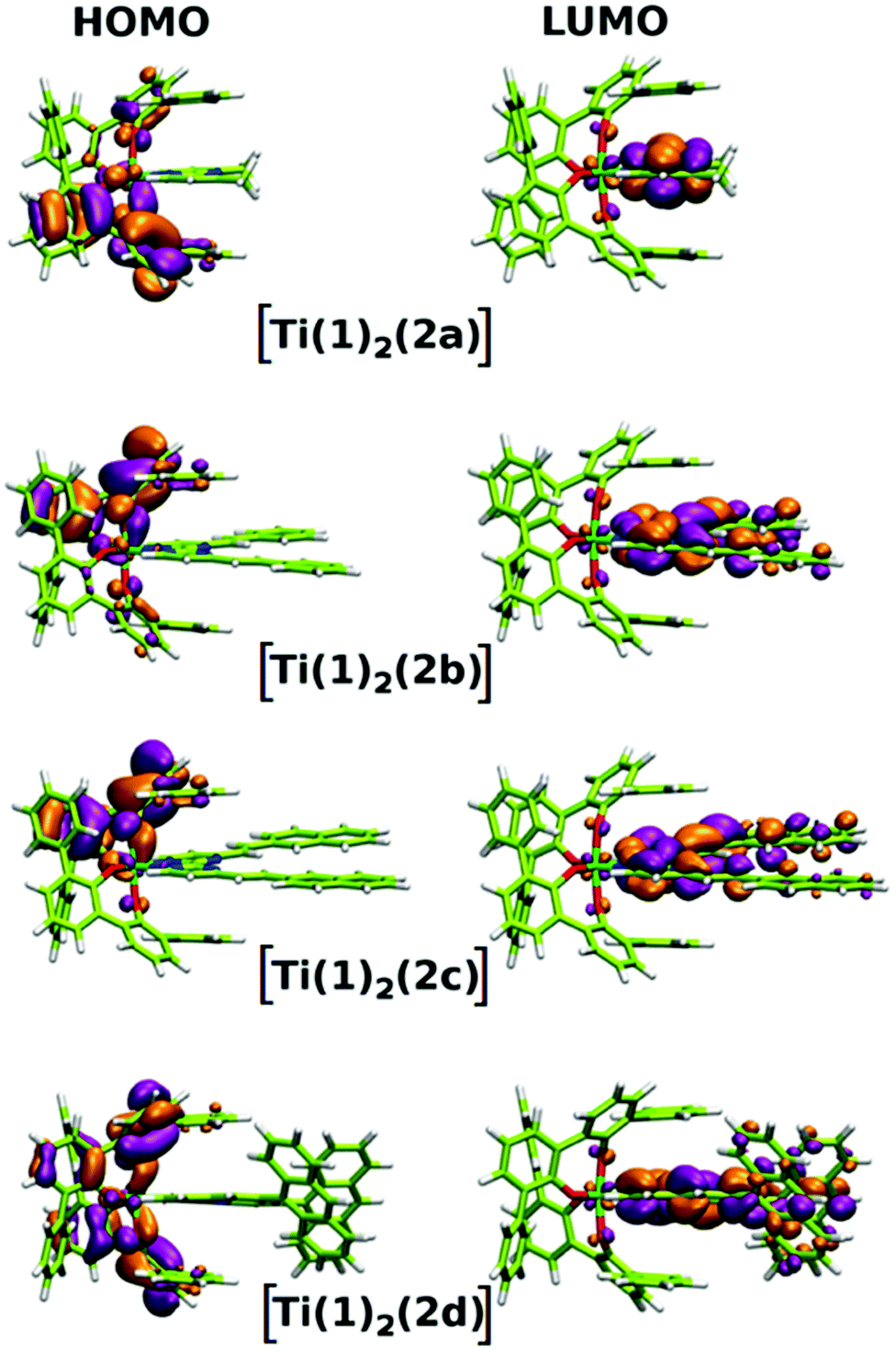
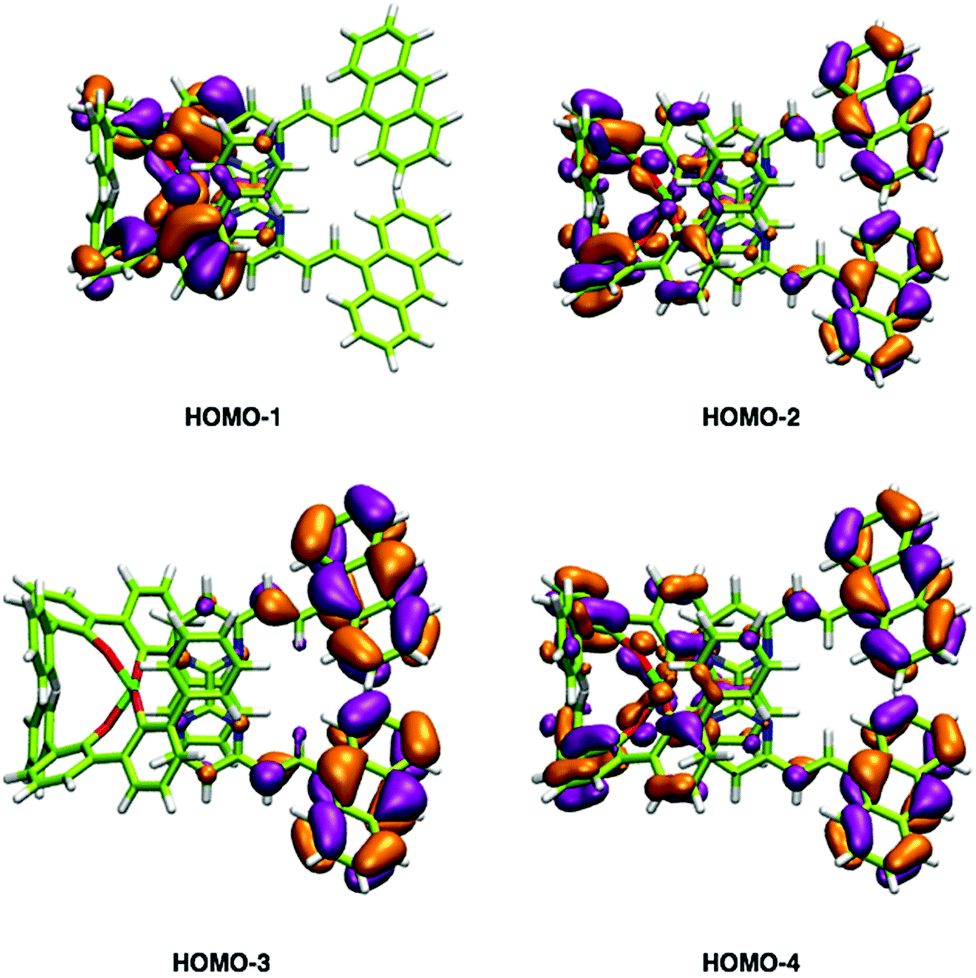
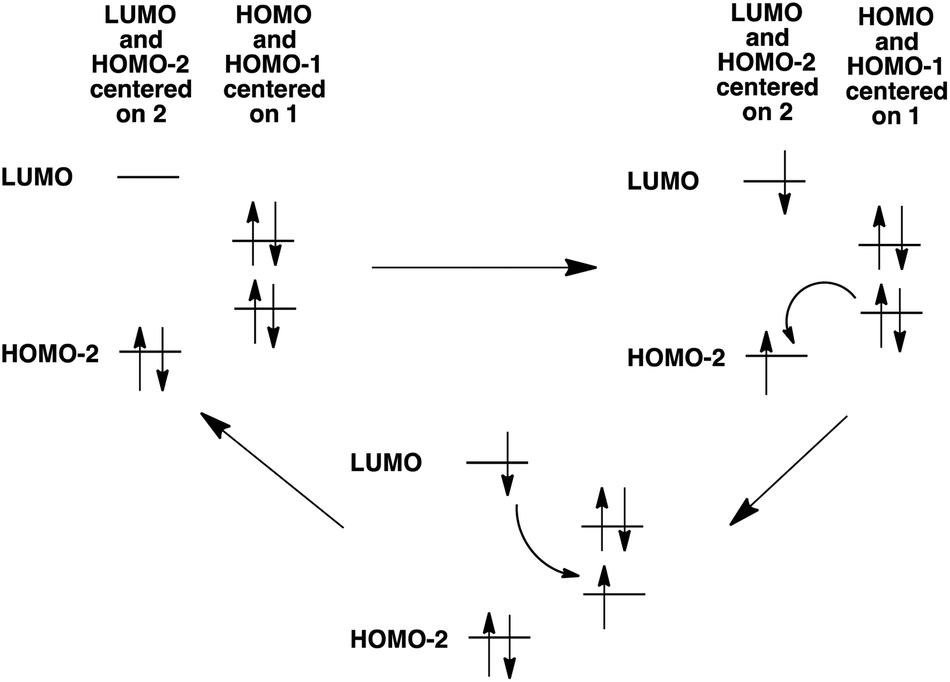
In addition to the experimental optical and emission properties evaluation, the frontier orbitals of the ligands and the complexes were computed using DFT calculations with the B3LYP hybrid functional and cc-pVTZ basis functions. Calculations were performed with the GAUSSIAN09 Revision D01 software.40 Energies associated with these frontier orbitals are listed in Table 3. First of all, for the ligands an excellent match is found between the HOMO–LUMO gaps and the lower energy bands characterized by UV-visible absorption spectroscopy. The HOMO and LUMO representations obtained for the ligands 2a–d are shown in Fig. 8. For ligands 2b and 2c, the HOMOs and the LUMOs are localized on one half of the molecules, indicating an electronic communication between the pyrimidine unit and the cyclic aromatic hydrocarbon part of the molecules. The situation is different for ligand 2d, as the HOMO and to a lesser extent the LUMO are almost centred on the anthryl fragment. This is in good agreement with the 2d absorption spectrum in the region between 350 nm and 450 nm, which resembles strongly the spectrum of anthracene (vide supra). This originated from a tilted arrangement of the anthryl units related to the pyrimidine units in 2d, as shown in the [Ti(1)2(2d)] crystal structure (Fig. 1). Concerning frontier orbitals of the complexes shown in Fig. 9, similar tendencies are noticed between each complex. All HOMOs and HOMO−1 are localized on the substituted biphenolato ligand (HOMO−1 are given in the ESI†). The LUMOs are all centred both on the metal and the nitrogen ligands. Indeed, the simple inspection of HOMOs, HOMO−1 and LUMO orbitals do not permit us to give clues about the origin of the emission properties modulations of 2c and 2d upon coordination to the titanium(IV) center. However, the examination of deeper orbitals for [Ti(1)2(2c)] and [Ti(1)2(2d)] is much more enlightening (the representations of the [Ti(1)2(2c)] HOMO−6 and HOMO−7 are given in the ESI†). For [Ti(1)2(2d)] as shown in Fig. 10, the anthryl fragments start to be involved for the HOMO−2 since the HOMO and the HOMO−1 are only centered on the biphenolato ligands. Furthermore, the energies associated with these orbitals are given in Table 4. Herein, an excellent match is found between the theoretical wavelength corresponding to the ΔE(LUMO–HOMO−2) and the λmax determined experimentally in the visible region (λ(calcd) = 438 nm, λ(exp) = 433 nm). Therefore, it can be reasonably proposed that upon excitation, an excited state is gained through the electronic promotion of an electron of a 2d-centered occupied orbital to the 2d-centered LUMO orbital. As the 1-centered HOMO orbitals lie between the HOMOs and the LUMO involving 2d, the fluorescence loss of 2d upon coordination is explained by an electron transfer of one electron of the 1-centered HOMO to the hole in the 2d-centered HOMOs. Transfer of the initially excited electron to the hole in the 1-centered HOMO follows this step. This intramolecular photoinduced electron transfer (PET) mechanism41 is shown in Fig. 11. An identical PET mechanism is proposed to explain the fluorescence quenching in [Ti(1)2(2c)]. For [Ti(1)2(2c)], an excellent match is also found between the experimental λmax (380 nm) and the wavelength λ(calcd) = 369 nm associated with the gap determined between the frontier orbitals centered on the naphthyl fragment (LUMO–HOMO−6).
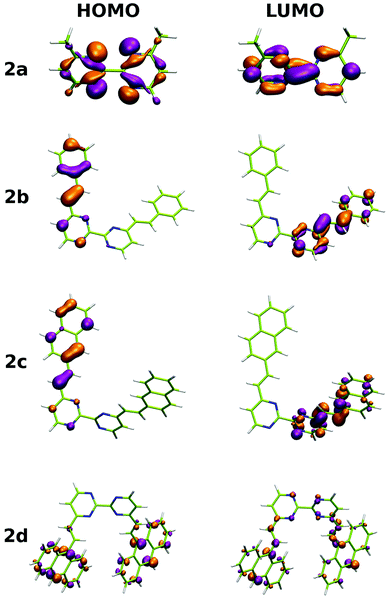 |
| | Fig. 8 HOMO and LUMO representations for ligands 2a–d. | |
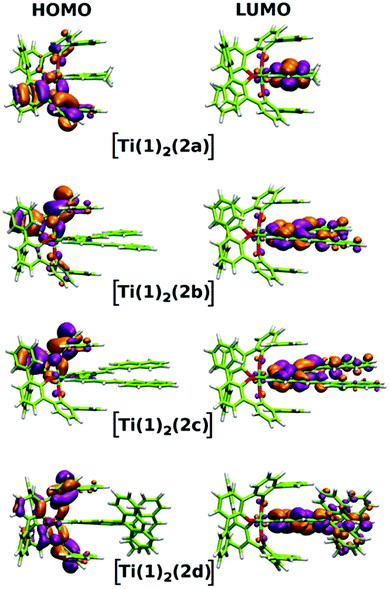 |
| | Fig. 9 HOMO and LUMO representations for complexes [Ti(1)2(2a–d)]. | |
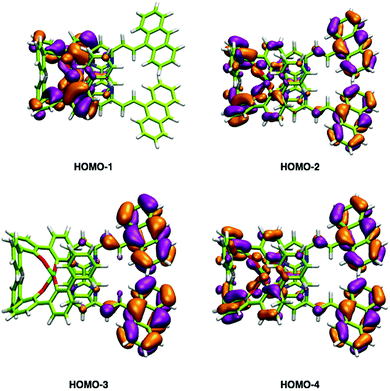 |
| | Fig. 10 HOMO−1, HOMO−2, HOMO−3 and HOMO−4 representations of the [Ti(1)2(2d)] complex. | |
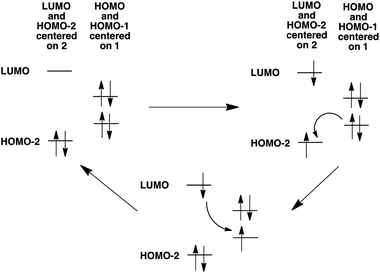 |
| | Fig. 11 Mechanism for PET explaining the luminescence quenching of 2d in [Ti(1)2(2d)]. A similar mechanism is proposed concerning the luminescence quenching of 2c in [Ti(1)2(2c)]. | |
Table 3 LUMO+1, LUMO, HOMO, HOMO−1 energies for ligands 2a–d and complexes [Ti(1)2(2a–d)]
| |
2a (eV) |
2b (eV) |
2c (eV) |
2d (eV) |
| LUMO+1 |
−1.152 |
−2.103 |
−2.163 |
−2.305 |
| LUMO |
−1.599 |
−2.202 |
−2.319 |
−2.327 |
| HOMO |
−6.591 |
−6.227 |
−5.981 |
−5.465 |
| HOMO−1 |
−7.013 |
−6.333 |
−6.024 |
−5.503 |
| Δ(LUMO–HOMO)a |
4.992 |
4.025 |
3.662 |
3.138 |
| |
[Ti(1)2(2a)] (eV) |
[Ti(1)2(2b)] (eV) |
[Ti(1)2(2c)] (eV) |
[Ti(1)2(2d)] (eV) |
|
Corresponding values of λ: 2a, 248 nm; 2b, 308 nm; 2c, 338 nm; 2d, 395 nm. The experimental values of λ are: 2a, 244 nm; 2b, 315 nm; 2c, 332 nm; 2d, 392 nm.
|
| LUMO+1 |
−2.169 |
−2.633 |
−2.684 |
−2.722 |
| LUMO |
−2.695 |
−2.922 |
−2.978 |
−2.903 |
| HOMO |
−5.552 |
−5.439 |
−5.400 |
−5.487 |
| HOMO−1 |
−5.580 |
−5.464 |
−5.453 |
−5.489 |
| Δ(LUMO–HOMO) |
2.857 |
2.517 |
2.422 |
2.584 |
Table 4 Selected LUMO and HOMO energies for complexes [Ti(1)2(2c)] and [Ti(1)2(2d)]. Values in bold correspond to the HOMOs centered on the anthryl fragments and the naphthyl fragments in [Ti(1)2(2d)] and [Ti(1)2(2c)] respectively
| |
[Ti(1)2(2c)] (eV) |
[Ti(1)2(2d)] (eV) |
| LUMO+1 |
−2.684 |
−2.722 |
| LUMO |
−2.978 |
−2.903 |
| HOMO |
−5.400 |
−5.487 |
| HOMO−1 |
−5.453 |
−5.489 |
| HOMO−2 |
−5.465 |
−5.728
|
| HOMO−3 |
−5.466 |
−5.737
|
| HOMO−4 |
−5.887 |
−5.758
|
| HOMO−5 |
−5.922 |
|
| HOMO−6 |
−6.341
|
|
| HOMO−7 |
−6.370
|
|
Biological properties
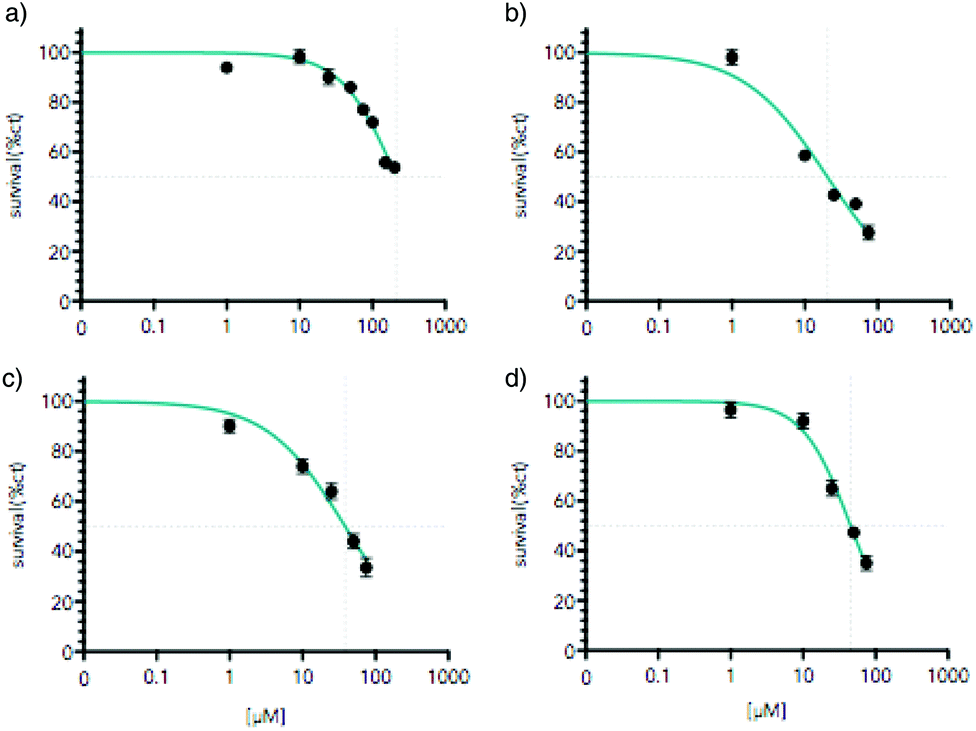
As mentioned above, a promising issue for Ti(IV) complexes concerns some applications in biology and more particularly as antitumor agents. Therefore, the biological activity of the titanium-based compounds was tested on cell viability in the AGS gastric cancer cells. The cells were treated for 48 hours with increased concentrations of the drugs ranging from 1 to 200 μM (Fig. 12). The compounds were first solubilized in DMSO at a concentration of 10 mM for [Ti(1)2(2a)] and 2.5 mM for [Ti(1)2(2b)], [Ti(1)2(2c)] and [Ti(1)2(2d)] before a final dilution in the culture medium. The cell viability was measured using MTT assay. [Ti(1)2(2a)] showed a moderate reduction of AGS viability reaching the IC50 at about 200 μM. In contrast, [Ti(1)2(2b)], [Ti(1)2(2c)] and [Ti(1)2(2d)] caused a marked diminution of cell viability already at 75 μM. Higher doses of [Ti(1)2(2b)], [Ti(1)2(2c)] and [Ti(1)2(2d)] were not possible to test due to the poor solubility of these compounds in DMSO and the toxic effect of high doses of DMSO. The concentrations able to reduce by 50% the cell viability (IC50) were determined using as maximal effect wells with no cells alive due to high cisplatin doses (Fig. 13) in order to compare the relative cytotoxicity of the various drugs. Systematically, the impact of DMSO alone was tested in parallel, and its lack of toxicity checked at the concentrations of the tests. Similar IC50 results were obtained when the drugs were incubated in a medium for 4 hours and before a fresh medium without drugs was replaced for the remaining 44 hours before MTT assay. These results indicate that the structure of the titanium complex has a direct impact on the biological activity of the compound.
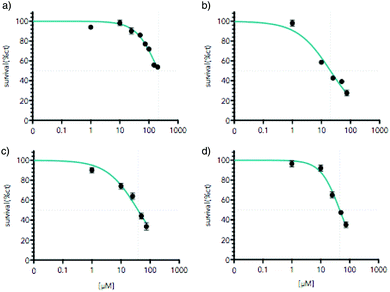 |
| | Fig. 12 Titanium complexes reduce AGS gastric cancer cells viability. AGS cells were treated with increasing concentration of the titanium complex ranging from 1 μM to 200 μM for 48 hours; (a) [Ti(1)2(2a)], (b) [Ti(1)2(2b)], (c) [Ti(1)2(2c)], (d) [Ti(1)2(2d)]. Cell viability was assessed by MTT assay. Graphs represent mean (n = 8) with standard deviation of one representative experiment (n = 3). *p < 0.01 as calculated by a one-way ANOVA test followed by a Tukey post-test. Lines indicate IC50 calculated taking as maximal effect wells with cells completely dead due to the high dose of cisplatin. For each dose, the equivalent dose of DMSO without a titanium compound was tested to verify the lack of toxicity caused by DMSO. | |
 |
| | Fig. 13 IC50 for [Ti(1)2(2a–d)] and ligands 2b–d calculated as described in Fig. 12. | |
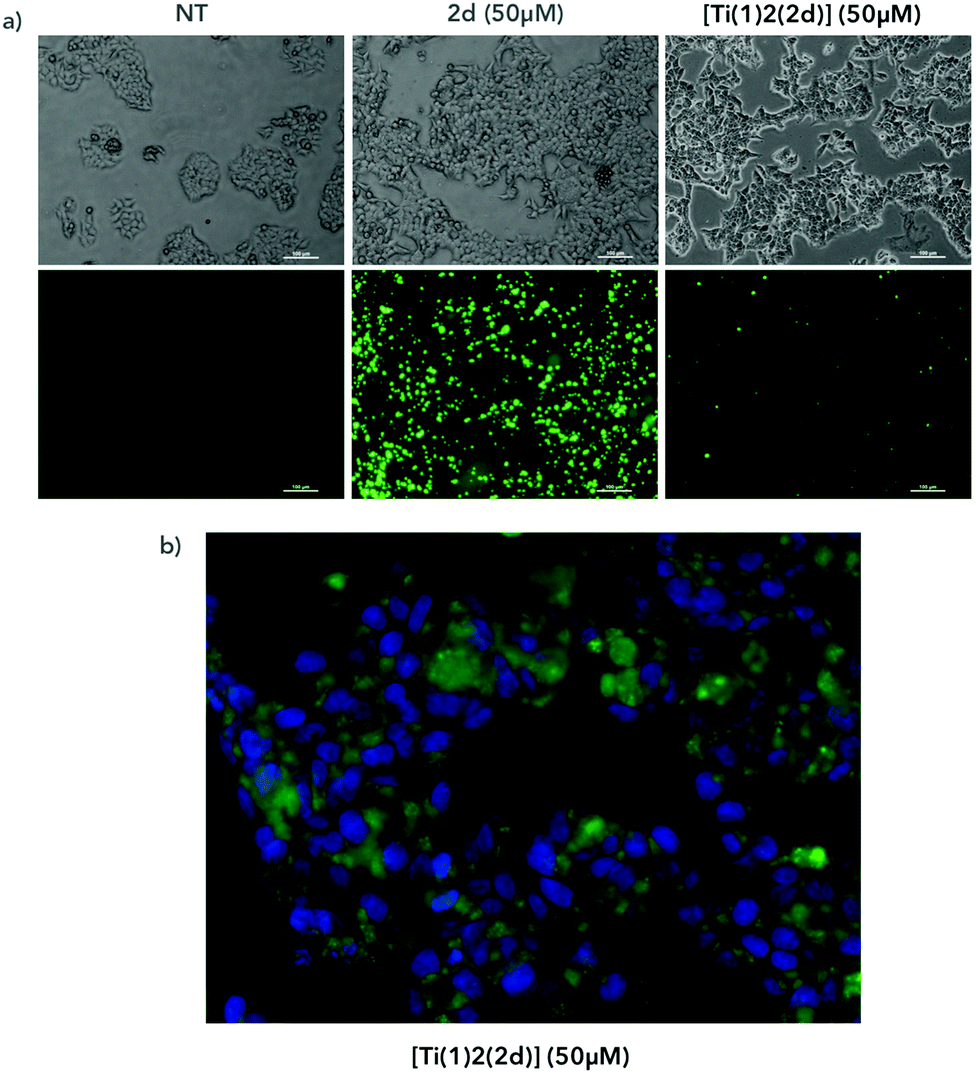
To follow the localization of the compounds in cells, we incubated for 4 hours the compounds with AGS cells and we observed the fluorescence of the compounds under a fluorescent microscope (Fig. 14a). We compared the cells treated with the [Ti(1)2(2d)] versus the cells treated with the fluorescent ligand alone (2d). In both conditions, we observed fluorescent aggregates, reflecting the poor solubility of the compound in the medium. At a similar concentration (50 μM), the overall fluorescence was reduced with the titanium complex compared to the ligand alone, which is in full accordance with the conclusions drawn from the fluorimetric assay. At a higher magnification, we observed the presence of fluorescence inside the cells and around the nucleus labeled with the fluorescent nucleus dye DAPI (Fig. 14b). However, at this stage, we are not able to conclude if the complex has crossed the cellular membrane and liberated the diimine ligand into the cells or if the free ligand has been generated in the extracellular medium before entering into the cells. Further investigations are planned to elucidate that point.
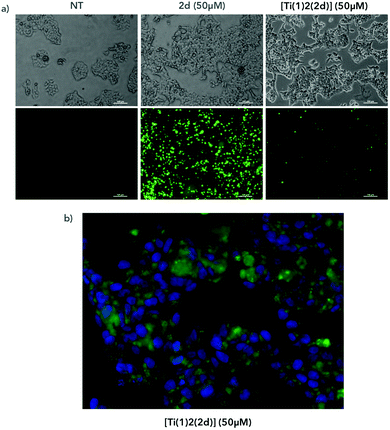 |
| | Fig. 14 AGS cells were treated with [Ti(1)2(2d)] containing a fluorescent ligand or with the ligand alone (2d) at 50 μM for 4 hours (a) or with DMSO (NT). Cells were then observed under a microscope in a bright field (a, top panels) or at excitation 433 nm and emission 532 nm and 461 nm (a, bottom panels and b). (b) Bottom panels show magnifications of cells treated with [Ti(1)2(2d)] (in green) and labelled with DAPI to visualize nuclei (in blue). | |
Conclusions and perspectives
We have reported herein the synthesis, using a well-established procedure, of a series of octahedral TiO4N2-based complexes bearing substituted 2,2′-bipyrimidine ligands starting from [Ti(1)2(HOiPr)2]. Two of these complexes, i.e. [Ti(1)2(2c)] and [Ti(1)2(2d)], incorporate fluorescent nitrogen ligands which contain naphthyl and anthryl fragments, respectively. We have demonstrated that the luminescent properties of 2c and 2d are strongly affected by their coordination to the Ti(1)2 unit, as a strong fluorescence loss is observed for the [Ti(1)2(2c)] and [Ti(1)2(2d)] complexes relative to the free nitrogen ligands. The explanation of the emission quenching has been provided by the examination of the HOMO and LUMO levels. We have also noticed a rather good hydrolytic stability for these Ti(IV)-based species that is correlated to the size of the polyaromatic substituents decorating the nitrogen ligand backbone. Interestingly, this stability is affected by the addition of an acid. Additionally, preliminary biological studies were carried out. The toxicity of these compounds has been evaluated, highlighting that the complexes and the nitrogen ligands are moderately toxic. Also, we observed fluorescence inside the cells when they were treated with the complex [Ti(1)2(2d)]. The long-term applications of the behaviour of this family of complexes in a medical or biological context are envisaged since pH-responsive systems are highly attractive for developing novel drug delivery systems or analytical tools.42 However, before envisaging such applications, the solubility of these Ti(IV)-based compounds in aqueous media must be significantly improved.
Experimental part
General procedures
Bruker Avance-300 and Avance-500 spectrometers were used for solution NMR spectroscopy analysis. Mass spectroscopy and elemental analyses were performed at the Service Commun d'Analyses, University of Strasbourg (France). The electrospray analyses were performed on Micro-TOF (Bruker) apparatus equipped with an electrospray (ES) source. The elemental analyses were performed on a Flash 2000 (Thermo Fisher Scientific) for C, H, and N elements. The analyzed mass peaks refer to the most intense peaks (unless mentioned otherwise in the discussion). 2-Chloro-4-phenyl-6 methyl-pyrimidine, triphenylphosphine, NiCl2·6H2O, zinc powder, and tBuOK were used as received from Alfa-Aesar or Sigma Aldrich. All solvents were dried before use with activated 3 Å molecular sieves. Tetrahydrofuran (THF) was dried by distillation using sodium/benzophenone.
4,4′-Dimethyl-2,2′-bipyrimidine (2a).
Under N2 protection, in a mixture of 2-chloro-4-phenyl-6 methyl-pyrimidine (2 g, 15.56 mmol), NiCl2·6H2O (924 mg, 3.89 mmol), triphenylphosphine (4.08 g, 15.56 mmol), and zinc powder (508 mg, 7.78 mmol) was added 50 mL of degassed DMF. The mixture was heated at 70 °C overnight. The resulting dark red solution was poured into a solution of 2 M aqueous ammonia (100 mL). The solution was extracted in the first place with diethyl ether to eliminate triphenylphosphine residues and then with CH2Cl2. The organic layer (CH2Cl2) was combined and evaporated to dryness. The resulting yellow solid was purified by flash chromatography on a silica gel using CH2Cl2/MeOH (97/3) as an eluent, which provided BpymMe as a yellow solid (704 mg, 68% yield). 1H NMR (CDCl3, 500 MHz): δ (ppm) 8.82 (d, J = 4.5 Hz, 2H), 7.24 (d, J = 2.7 Hz, 2H), 2.69 (s, 6H); 13C NMR (CDCl3, 125 MHz): δ (ppm) 168.6, 162.5, 157.6, 121.1, 24.7. Mp = 117 °C; Anal. Calcd for C10H10N4: C% 64.50 N% 30.09 H% 5.41; found C% 64.40, N% 30.01, H% 5.33. MS (ESI) m/z calcd for [2a + Na]+ = 209.0803 found 209.0790.
Preparation of bipyrimidine compounds – general procedure.
Under N2 protection, 4,4′-dimethyl-2,2′-bipyrimidine (50 mg, 0.218 mmol) was dissolved in distilled THF (20 mL) in a bicol flask at −78 °C. tBuOK (90 mg, 0.8 mmol) was slowly added, the solution turned brown after a few minutes. Then the aldehyde derivative (2.3 equiv.) was added in small portions. After 10 min, the mixture was stored at room temperature for 1 h. Then, 10 mL of water was added to the solution and the THF was evaporated. The precipitate was removed by filtration. The resulting solid was purified by flash chromatography using CH2Cl2/MeOH (97/3) as an eluent.
4,4′-Di((E)-styryl)-2,2′-bipyrimidine (2b).
Yield = 58%. 1H NMR (CD2Cl2, 500 MHz): δ (ppm) 8.93 (d, 3J = 3.9 Hz, 2H), 8.02 (d, 3J = 16.1 Hz, 2H), 7.70 (m, 4H), 7.51 (d, 3J = 5 Hz, 2H), 7.45–7.37 (complex, 6H), 7.29 (d, 3J = 16.0 Hz, 2H). 13C NMR (CD2Cl2, 125 MHz): δ (ppm) 163.5, 162.8, 157.8, 137.9, 135.7, 129.6, 129.0, 127.8, 126.0, 118.0. Mp = 214 °C. Anal. Calcd for C24H18N4·1/3CH2Cl2: C% 74.80, N% 14.34, H% 4.82; found C% 75.47, N% 13.42, H% 4.97. MS (ESI) m/z calcd for [2d + H]+ = 363.1531 found 363.1587.
4,4′-Bis((E)-2-(naphthalen-2-yl)vinyl)-2,2′-bipyrimidine (2c).
Yield = 55%. 1H NMR (CDCl3, 500 MHz): δ (ppm) 8.99 (d, 3J = 4.7 Hz, 2H), 8.12 (d, 3J = 16.5 Hz, 2H), 8.05 (s, 2H), 7.88 (m, 8H), 7.59 (d, 3J = 5.2 Hz, 2H), 7.51 (m, 4H), 7.47 (d, 3J = 16.5 Hz, 2H). 13C NMR (CDCl3, 125 MHz): δ (ppm) 164.1, 163.0, 158.3, 138.2, 134.0, 133.5, 133.2, 129.3, 128.8, 128.5, 127.8, 127.0, 126.7, 126.6, 123.6, 117.6. Mp = 227 °C. Anal. Calcd for C32H22N4·CH2Cl2: C% 72.40 N% 12.95 H% 4.42; found C% 72.49, N% 12.36, H% 4.90. MS (ESI) m/z calcd for [BpymNapht + Na]+ = 485.1742 found 485.1699.
4,4′-Bis((E)-2-(anthracen-9-yl)vinyl)-2,2′-bipyrimidine (2d).
Yield = 54%. 1H NMR (CDCl3, 500 MHz): δ (ppm) 9.07 (d, 3J = 5.1 Hz, 2H), 8.94 (d, 3J = 16.0 Hz, 2H), 8.44 (s, 2H), 8.36 (dd, 3J = 7.0 Hz, 4J = 1.7 Hz, 4H), 8.01 (dd, 3J = 6.0 Hz, 4J = 1.7 Hz, 4H), 7.64 (d, 3J = 5.1 Hz, 2H), 7.51 (m, 8H), 7.23 (d, 3J = 16.0 Hz, 2H); 13C NMR (CDCl3, 125 MHz): δ (ppm) 163.5, 163.3, 158.6, 135.7, 134.8, 131.4, 130.7, 129.6, 128.9, 127.9, 126.1, 125.7, 125.4, 118.2. Mp = 239 °C. Anal. Calcd for C40H26N4·9/5CH2Cl2: C% 70.16, N% 7.83, H% 4.17; found C% 69.75, N% 7.90, H% 4.27. MS (ESI) m/z calcd for [2d + Na]+ = 585.2055 found 585.2100.
Preparation of titanium(IV) complexes – general procedure
Under N2 protection (glove bag), a counter solvent was allowed to slowly diffuse into a solution containing the bipyrimidine ligand (1 equiv.) and the cis-[Ti(1)2(HOiPr)2] complex24 (10 mg) in a minimum amount of dry CH2Cl2/CHCl3 in a small vial. Crystals were isolated by filtration.
[Ti(1)2(2a)].
Crystals were obtained from 1,2 dichloroethane/n-pentane. C58H42N4O4Ti; T = 173(2) K; orthorhombic; space group P2(1)2(1)2(1); a = 11.8033(3) Å, b = 16.0200(4) Å, c = 23.9080(6) Å; V = 4520.7(2) Å3; Z = 4; Dcalcd = 1.332 g cm−3; reflections collected: 90802; Rint = 0.0572; R1(F) (I > 2σ(I)) = 0.0402, wR2(F2) (all data) = 0.0917; GOF(F2) = 1.027. CCDC number 1468167. Yield = 61%. 1H NMR (CD2Cl2, 500 MHz): δ (ppm) 8.33 (d, 3J = 5.4 Hz, 2H), 7.44 (dd, 3J = 7.3 Hz, 4J = 3.3 Hz, 4H), 7.29 (td, 3J = 7.5 Hz, 4J = 1.9 Hz, 4H), 7.13 (dd, 3J = 7.4 Hz, 4J = 2.0 Hz, 2H), 7.09 (t, 3J = 7.6 Hz, 2H), 7.04 (m, 6H), 6.81 (dd, 3J = 8.4 Hz, 4J = 1.5 Hz, 6H), 6.73 (m, 10H), 6.66 (t, 3J = 7.4 Hz, 2H), 3.76 (s, 6H). 13C NMR (CD2Cl2, 125 MHz): δ (ppm) 170.9, 160.7, 160.0, 157.2, 155.2, 138.8, 138.4, 131.8, 131.4, 129.8, 129.7, 129.6, 129.3, 129.1, 129.0, 128.7, 128.0, 127.5, 127.0, 126.0, 125.3, 122.1, 120.9, 119.7, 24.4. MS (ESI) m/z calcd for [[Ti(1)2(2a)] + H]+ = 907.27 found 907.28.
[Ti(1)2(2b)].
Crystals were obtained from CH2Cl2/n-pentane. C72H50N4O4Ti, T = 173(2) K; monoclinic; space group P21/n; a = 13.5432(3) Å, b = 14.0231(4) Å, c = 29.0361(8) Å; β = 93.2494(14)°; V = 5505.6(3) Å3; Z = 4; Dcalcd = 1.307 g cm−3; reflections collected: 65399; Rint = 0.0763; R1(F) (I > 2σ(I)) = 0.0606, wR2(F2) (all data) = 0.1947; GOF(F2) = 1.036. CCDC number 1468801. Yield = 51%. 1H NMR (CDCl3, 500 MHz): δ (ppm) 8.36 (d, 3J = 5.8 Hz, 2H), 7.81 (d, 3J = 16.1 Hz, 2H), 7.74 (d, 3J = 7.4 Hz, 4H), 7.49 (m, 10H), 7.25 (m, 4H), 7.14 (dd, 3J = 7.4 Hz, 4J = 1.4 Hz, 2H), 7.02 (m, 10H), 6.86 (m, 6H) 6.07 (m, 5H), 6.59 (m, 3H). 13C NMR (CDCl3, 125 MHz): δ (ppm) 165.3, 160.8, 160.2, 157.7, 155.5, 140.6, 138.7, 138.6, 135.2, 131.5, 131.3, 130.7, 130.4, 129.9, 129.6, 129.4, 129.3, 129.2, 129.0, 128.8, 128.2, 127.7, 127.4, 126.9, 125.9, 125.5, 125.4, 121.9, 119.4, 116.8. MS (ESI) m/z calcd for [[Ti(1)2(2b)] + H]+ = 1083.3312 found 1083.3406.
[Ti(1)2(2c)].
Crystals were obtained from CHCl3/n-pentane. 2(C80H54N4O4Ti), C5H12; T = 173(2) K; monoclinic; space group P21/c; a = 20.622(2) Å, b = 13.9118(16) Å, c = 23.906(3) Å; β = 96.372(4)°; V = 6816.0(14) Å3; Z = 2; Dcalcd = 1.188 g cm−3; reflections collected: 13151; Rint = 0.1213; R1(F) (I > 2σ(I)) = 0.1288, wR2(F2) (all data) = 0.3330; GOF(F2) = 1.137. CCDC number 1468168. Yield = 47%. 1H NMR (CDCl3, 500 MHz): δ (ppm) 8.39 (d, 3J = 5.2 Hz, 2H), 8.16 (s, 2H), 7.95 (m, 10H), 7.58 (dd, 3J = 5.2, 4J = 2.9 Hz, 4H), 7.48 (d, 3J = 7.5 Hz, 4H), 7.35 (d, 3J = 15.6 Hz, 2H), 7.28 (m, 4H), 7.16 (dd, 3J = 8.1, 4J = 1.7 Hz, 2H), 7.03 (m, 9H), 6.90 (m, 6H), 6.73 (m, 6H), 6.62 (m, 3H). 13C NMR (CDCl3, 125 MHz): δ (ppm) 170.9, 165.7, 165.4, 160.8, 160.7, 160.2, 160.1, 157.8, 157.7, 157.5, 155.5, 155.4, 155.3, 140.8, 138.8, 138.7, 138.6, 138.5, 134.3, 133.5 (2 signals), 132.7, 132.6, 131.6, 131.5, 131.3, 131.1, 130.7, 130.2, 129.9, 129.7, 129.6, 129.5, 129.4 (2 signals), 129.3, 129.2, 129.0 (2 signals), 128.9, 128.8, 128.7, 128.4, 128.3, 128.0, 127.9, 127.5, 127.4, 127.0, 126.9, 126.0, 125.9, 125.6, 125.5, 125.0, 123.6 (2 signals), 122.0, 121.9, 121.4, 120.8, 119.4, 116.7, 116.4. MS (ESI) m/z calcd for [[Ti(1)2(2c)] + Na]+ = 1205.3625 found 1205.3411.
[Ti(1)2(2d)].
Crystals were obtained from CHCl3/n-pentane. 4(C88H58N4O4Ti), CHCl3, 4(H2O); T = 173(2) K; monoclinic; space group P2/c; a = 21.6458(9) Å, b = 18.2186(10) Å, c = 19.9733(11) Å; β = 112.479(2)°; V = 7278.1(6) Å3; Z = 1; Dcalcd = 1.223 g cm−3; reflections collected: 19714; Rint = 0.0869; R1(F) (I > 2σ(I)) = 0.0912, wR2(F2) (all data) = 0.3117; GOF(F2) = 1.051. CCDC number 1468169. Yield = 41%. 1H NMR (CDCl3, 500 MHz): δ (ppm) 9.14 (d, 3J = 15.8 Hz, 2H), 8.48 (m, 4H), 8.42 (d, 3J = 7.9 Hz, 4H), 8.03 (dd, 3J = 7.9 Hz, 4J = 1.8 Hz, 4H), 7.48 (m, 12H), 7.31 (m, 4H), 7.20 (dd, 3J = 7.3 Hz, 4J = 1.2 Hz, 2H), 7.05 (m, 14H), 6.86 (m, 10H), 6.72 (t, 3J = 7.3 Hz, 2H). Dept 13C NMR (CDCl3, 125 MHz): CH δ (ppm) 156.1, 138.1, 132.7, 131.8, 131.4, 129.9, 129.7, 129.6, 129.2, 129.0, 128.6, 127.5, 127.0, 126.6, 126.0, 125.5 (2 signals), 125.0, 122.0, 119.5, 118.0. MS (ESI) m/z calcd for [[Ti(1)2(2d)] + H]+ = 1283.3938 found 1283.4100.
Preparation of the NMR samples for hydrolytic stability assay without and with DCl
The preparation of the samples was conducted under an argon atmosphere. Complexes [Ti(1)2(2a)], [Ti(1)2(2b)], [Ti(1)2(2c)] and [Ti(1)2(2d)] were dissolved in dry DMSO-d6. Then, the resulting solutions were transferred into an NMR tube. To these solutions were added D2O (50 μl). The final concentrations were 6 mM. The NMR tubes were closed with a Teflon cap. The final solutions were analyzed using a 600 MHz spectrometer.
For the experiments conducted with DCl, the final concentrations were also 6 mM. Complex [Ti(1)2(2a)] was dissolved in dry DMSO-d6. To this solution was added 50 μl of an aqueous (D2O) solution of DCl. The more acidic solution contains 2.5 μl of DCl solution (38 wt% in D2O) (3 × 10−5 mol, 9 equivalents versus [Ti(1)2(2a)]) whereas the second solution contains 0.5 μl of DCl solution (38 wt% in D2O) (6 × 10−6 mol, 1.8 equivalent versus [Ti(1)2(2a)]).
Cell cultures
Human AGS gastric cancer cells were obtained from ATCC. AGS cells were manipulated and cultured in DMEM with 10% FCS (Dominique Dutcher™) and 1% penicillin + streptomycin (Sigma) at 37 °C under a 5% CO2 atmosphere as previously described.43
Cell survival
5000 cells were seeded per well in 96-well microplates (Falcon Multiwell), 24 hours prior to any treatment. The complexes were applied for 48 h in fresh medium. MTT assay was performed as previously described by replacing the medium with a fresh medium supplemented with 5 mg L−1 MTT (Sigma) for 1 h.44 The cells were lysed in isopropanol with 0.04 N HCl. Measurements were performed at 550 nm. IC50 were calculated using as maximal value wells with complete loss of cell survival due to the high dose of cisplatin (50 μM).
Microscopy
The cells were grown on cover glasses in a growth medium and incubated for 4 hours with the complexes at various dilutions. The cells were then incubated with DAPI (Thermo Fisher) for 15 min. After incubation, the cells were observed directly under an epifluorescence microscope (Zeiss Apotome V2 axiozoom) at excitation 433 nm and emission 532 nm and 461 nm (DAPI).
X-ray crystallography
The X-ray diffraction data were collected on a Bruker Smart CCD diffractometer with Mo-Kα radiation (λ = 0.71073 Å). The structures were solved and refined using the Bruker SHELXTL Software Package using SHELXS-97 (Sheldrick, 2014) and refined by full matrix least-squares on F2 using SHELXL-97 (Sheldrick, 2014) with anisotropic thermal parameters for all non-hydrogen atoms.45 The hydrogen atoms were introduced at calculated positions and not refined (riding model). For the structures of compounds [Ti(1)2(2c)] and [Ti(1)2(2d)], we used the squeeze command because the solvents could not be identified.
Photophysical measurements
The UV-visible absorption spectra were recorded using a Perkin Elmer Lambda 650S spectrometer at room temperature with a 1 cm path cell. The luminescence spectra were recorded at room temperature using a Fluorolog FL3-22 spectrofluorometer (Horiba Jobin Yvon) equipped with a TBX-04 or an R928 detector. All of the luminescence measurements were performed on optically dilute solutions with spectrophotometry-grade dichloromethane, with a maximum absorbance of 0.08. For the luminescence steady-state measurements, the spectrometer was equipped with a 450 W xenon lamp excitation source and excitation and emission double-grating monochromators with bandpass set at 2 nm for the experiments. The spectra were corrected for the lamp, the monochromators, and the detector responses; blanks (solvent alone) were recorded and subtracted from the raw spectra, unless otherwise stated. The luminescence quantum yields were determined in spectrophotometry-grade dichloromethane solutions using either terphenyl in cyclohexane (Φ = 0.93, for compound 1-H2) or quinine sulfate in aerated 0.05 M H2SO4 (Φ = 0.55, for the other compounds) as luminescence standards.37b
Conflict of interest
The authors declare no competing financial interest.
Acknowledgements
We thank Martine Heinrich for technical assistance and helpful discussions, and Benoît Colasson for a critical reading of the manuscript. We are also thankful for the technical support of E. Martin. This project was supported by the Centre National pour la Recherche Scientifique (CNRS, France), ARC, Ligue contre le Cancer, European action COST CM1105 (C. G.), the Laboratory of Excellence (LABEX) “Chemistry of Complex Systems” (scholarship to G. K.) and IDEX grant (Strasbourg University).
References
-
(a) R. Visbal and M. C. Gimeno, Chem. Soc. Rev., 2014, 43, 3551–3574 RSC;
(b) V. W.-W. Yam and K. M.-C. Wong, Chem. Commun., 2011, 47, 11579–11592 RSC;
(c) Y. Chi and P.-T. Chou, Chem. Soc. Rev., 2007, 36, 1421–1431 RSC.
-
(a) D.-L. Ma, V. P.-Y. Ma, D. S.-H. Chan, K.-H. Leung, H.-Z. He and C.-H. Leung, Coord. Chem. Rev., 2012, 256, 3087–3113 CrossRef CAS;
(b) X. He and V. W.-W. Yam, Coord. Chem. Rev., 2011, 255, 2111–2123 CrossRef CAS;
(c) B. A. DeGraff and J. N. Demas, Coord. Chem. Rev., 2001, 211, 317–351 CrossRef.
- E. Baggaley, J. A. Weinstein and J. A. G. Williams, Coord. Chem. Rev., 2012, 256, 1762–1785 CrossRef CAS.
-
(a) Y. Chi and P.-T. Chou, Chem. Soc. Rev., 2010, 39, 638–655 RSC;
(b) J. A. G. Williams, S. Develay, D. L. Rochester and L. Murphy, Coord. Chem. Rev., 2008, 252, 2596–2611 CrossRef.
- S. I. Pascu, P. A. Waghorn, T. D. Conry, B. Lin, H. M. Betts, J. R. Dilworth, R. B. Sim, G. C. Churchill, F. I. Aigbirhio and J. E. Warren, Dalton Trans., 2008, 2107–2110 RSC.
- S. I. Pascu, P. A. Waghorn, T. D. Conry, H. M. Betts, J. R. Dilworth, G. C. Churchill, T. Pokrovska, M. Christlieb, F. I. Aigbirhio and J. E. Warren, Dalton Trans., 2007, 4988–4997 RSC.
- E. E. Langdon-Jones, D. Lloyd, A. J. Hayes, S. D. Wainwright, H. J. Mottram, S. J. Coles, P. N. Horton and S. J. A. Pope, Inorg. Chem., 2015, 54, 6606–6615 CrossRef CAS PubMed.
- S. W. Botchway, M. Charnley, J. W. Haycock, A. W. Parker, D. L. Rochester, J. A. Weinstein and J. A. Williams, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16071–16076 CrossRef CAS PubMed.
-
(a) V. Fernández-Moreira, F. L. Thorp-Greenwood and M. P. Coogan, Chem. Commun., 2010, 46, 186–202 RSC;
(b) M. Klajner, C. Licona, L. Fetzer, P. Hebraud, G. Mellitzer, M. Pfeffer, S. Harlepp and C. Gaiddon, Inorg. Chem., 2014, 53, 5150–5158 CrossRef CAS PubMed.
-
(a) D. J. Ramón and M. Yus, Chem. Rev., 2006, 106, 2126–2208 CrossRef PubMed;
(b) R. O. Duthaler and A. Hafner, Chem. Rev., 1992, 92, 807–832 CrossRef CAS.
- S. A. Ryken and L. L. Schafer, Acc. Chem. Res., 2015, 48, 2576–2586 CrossRef CAS PubMed.
-
(a) E. Le Roux, Coord. Chem. Rev., 2016, 306, 65–85 CrossRef CAS;
(b) V. C. Gibson and S. K. Spitzmesser, Chem. Rev., 2003, 103, 283–315 CrossRef CAS PubMed;
(c) L. Resconi, L. Cavallo, A. Fait and F. Piemontesi, Chem. Rev., 2000, 100, 1253–1345 CrossRef CAS PubMed;
(d) L. Azor, C. Bailly, L. Brelot, M. Henry, P. Mobian and S. Dagorne, Inorg. Chem., 2012, 51, 10876–10883 CrossRef CAS PubMed.
- U. Schubert, J. Mater. Chem., 2005, 15, 3701–3715 RSC.
-
(a) M. Albrecht, I. Janser, S. Kamptmann, P. Weis, B. Wibbeling and R. Fröhlich, Dalton Trans., 2004, 37–43 RSC;
(b) M. Albrecht, I. Janser, A. Lützen, M. Hapke, R. Fröhlich and P. Weis, Chem. – Eur. J., 2005, 11, 5742–5748 CrossRef CAS PubMed;
(c) M. Scherer, D. L. Caulder, D. W. Johnson and K. N. Raymond, Angew. Chem., 1999, 111, 1689–1694 (
Angew. Chem., Int. Ed.
, 1999
, 38
, 1587–1592
) CrossRef;
(d) M. Albrecht, H. Röttele and P. Burger, Chem. – Eur. J., 1996, 2, 1264–1268 CrossRef CAS;
(e) M. Albrecht, S. Kamtmann and R. Fröhlich, Polyhedron, 2003, 22, 643–647 CrossRef CAS;
(f) M. Albrecht, M. Schneider and R. Fröhlich, New J. Chem., 1998, 22, 753–754 RSC;
(g) M. Albrecht and S. Kotila, Angew. Chem., 1996, 108, 1299–1300 (
Angew. Chem., Int. Ed. Engl.
, 1996
, 35
, 1208–1210
) CrossRef;
(h) Y. Sakata, S. Hiraoka and M. Shionoya, Chem. – Eur. J., 2010, 16, 3318–3325 CrossRef CAS PubMed;
(i) C. Diebold, P. Mobian, C. Huguenard, L. Allouche and M. Henry, Inorg. Chem., 2010, 49, 6369–6371 CrossRef CAS PubMed;
(j) D. M. Weekes, C. Diebold, P. Mobian, C. Huguenard, L. Allouche and M. Henry, Chem. – Eur. J., 2014, 20, 5092–5101 CrossRef CAS PubMed.
- P. Mobian, N. Baradel, N. Kyritsakas, G. Khalil and M. Henry, Chem. – Eur. J., 2015, 21, 2435–2441 CrossRef CAS PubMed.
- C. W. Schwietert and J. P. McCue, Coord. Chem. Rev., 1999, 184, 67–89 CrossRef CAS.
-
(a) K. M. Buettner and A. M. Valentine, Chem. Rev., 2011, 112, 1863–1881 CrossRef PubMed;
(b) T. B. Parks, Y. M. Cruz and A. D. Tinoco, Inorg. Chem., 2014, 53, 1743–1749 CrossRef CAS PubMed.
-
(a) For a review discussing the search of alternatives for administering Ti(IV)-based metallo-drugs, see: W. A. Wani, S. Prashar, S. Shreaz and S. Gómez-Ruis, Coord. Chem. Rev., 2016, 312, 67–98 CrossRef CAS. For general reviews on Ti(IV)-based metallo-drugs, see:
(b) K. Strohfeldta and M. Tackeb, Chem. Soc. Rev., 2008, 37, 1174–1187 RSC;
(c) F. Caruso and M. Rossi, Mini-Rev. Med. Chem., 2004, 4, 49–60 CrossRef CAS PubMed;
(d) E. Meléndez, Crit. Rev. Oncol. Hematol., 2002, 42, 309–315 CrossRef.
-
(a) H. Glasner and E. Y. Tshuva, Inorg. Chem. Commun., 2015, 53, 31–33 CrossRef CAS;
(b) M. Grützke, T. Zhao, T. A. Immel and T. Huhn, Inorg. Chem., 2015, 54, 6697–6706 CrossRef PubMed;
(c) H. Glasner, S. Meker and E. Y. Tshuva, J. Organomet. Chem., 2015, 788, 33–35 CrossRef CAS;
(d) T. Zhao, M. Grützke, K. H. Götz, T. Druzhenko and T. Huhn, Dalton Trans., 2015, 44, 16475–16485 RSC;
(e) M. Lin, Y. Cao, H. Pei, Y. Chen, J. Wu, Y. Li and W. Liu, RSC Adv., 2014, 4, 9255–9260 RSC;
(f) T. B. Parks, Y. M. Cruz and A. D. Tinoco, Inorg. Chem., 2014, 53, 1743–1749 CrossRef CAS PubMed;
(g) S. Barroso, A. M. Coelho, S. Gómez-Ruiz, M. J. Calhorda, Ž. Žižak, G. N. Kaluđerović and A. M. Martins, Dalton Trans., 2014, 43, 17422–17433 RSC;
(h) R. Hernández, J. Méndez, J. Lamboy, M. Torres, F. R. Román and E. Meléndez, Toxicol. in Vitro, 2010, 24, 178–183 CrossRef PubMed;
(i) A. D. Tinoco, H. R. Thomas, C. D. Incarvito, A. Saghatelian and A. M. Valentine, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5016–5021 CrossRef CAS PubMed;
(j) E. Y. Tshuva and J. A. Ashenhurst, Eur. J. Inorg. Chem., 2009, 2203–2218 CrossRef CAS.
- For rare reports of luminescent titanium(IV)-based complexes, see: B. P. Baranwal, A. K. Singh and A. Varna, Spectrochim. Acta, Part A, 2011, 125–129 CrossRef CAS PubMed.
- D. M. Weekes, N. Baradel, N. Kyritsakas, P. Mobian and M. Henry, Eur. J. Inorg. Chem., 2012, 34, 5701–5713 CrossRef.
-
(a) H. Akdas-Kilig, T. Roisnel, I. Ledoux and H. Le Bozec, New J. Chem., 2009, 33, 1470–1473 RSC;
(b) S. Achelle, I. Nouira, B. Pfaffinger, Y. Ramondenc, N. Plé and J. Rodríguez-López, J. Org. Chem., 2009, 74, 3711–3717 CrossRef CAS PubMed.
-
(a)
J. B. Birks, Photophysics of Aromatic Molecules, Wiley-Interscience, London, 1970 Search PubMed;
(b) H. Park, J. Lee, I. Kang, H. Y. Chu, J. I. Lee, S. K. Kwon and Y. H. Kim, J. Mater. Chem., 2012, 22, 2695–2700 RSC;
(c) P. Rajamalli and E. Prasad, Org. Lett., 2011, 13, 3714–3717 CrossRef CAS PubMed;
(d) H. Huang, Q. Fu, S. Zhuang, Y. Liu, L. Wang, J. Chen, D. Ma and C. Yang, J. Phys. Chem. C, 2011, 115, 4872–4878 CrossRef CAS;
(e) Y. Y. Lyu, J. Kwak, O. Kwon, S. H. Lee, D. Kim, C. Lee and K. Char, Adv. Mater., 2008, 20, 2720–2729 CrossRef CAS PubMed;
(f) H. Ihmels, A. Meiswinkel, C. J. Mohrschladt, D. Otto, M. Waidelich, M. Towler, R. White, M. Albrecht and A. Schnurpfeil, J. Org. Chem., 2005, 70, 3929–3938 CrossRef CAS PubMed.
- C. Diebold, P. Mobian, C. Huguenard, L. Allouche and M. Henry, Dalton Trans., 2009, 10178–10180 RSC.
-
(a) F. Caruso and M. Rossi, Mini-Rev. Med. Chem., 2004, 4, 49–60 CrossRef CAS PubMed;
(b) E. Melendez, Crit. Rev. Oncol. Hemat., 2002, 42, 309–315 CrossRef;
(c) J. H. Toney and T. J. Marks, J. Am. Chem. Soc., 1985, 107, 947–953 CrossRef CAS;
(d) A. Tzubery and E. Tshuva, Inorg. Chem., 2012, 51, 1796–1804 CrossRef CAS PubMed;
(e) D. Peri, S. Meker, C. M. Manna and E. Y. Tshuva, Inorg. Chem., 2011, 50, 1030–1038 CrossRef CAS PubMed;
(f) E. Y. Tshuva and D. Peri, Coord. Chem. Rev., 2008, 253, 2098–2115 CrossRef.
- D. Peri, S. Meker, M. Shavit and E. Y. Thuva, Chem. – Eur. J., 2009, 15, 2403–2415 CrossRef CAS PubMed.
- These volumes are estimated according to A. Gavezotti, J. Am. Chem. Soc., 1983, 105, 5220–5225 CrossRef CAS . van der Waals radii used: H = 1.2 Å, C = 1.7 Å, N = 1.55 Å (see A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef ).
-
(a)
M. Henry, in Encyclopedia of Nanoscience and Nanotechnology, ed. H. S. Nalwa, American Scientific Publishers, 2011, vol. 14, pp. 1–43 Search PubMed;
(b)
M. Henry, in Advances in Quantum Chemical Bonding Structures, ed. M. V. Putz, Transworld Research Network, Kerala, India, 2009, pp. 153–211 Search PubMed;
(c) M. Henry, ChemPhysChem, 2002, 3, 561–569 CrossRef CAS PubMed;
(d) M. Henry, ChemPhysChem, 2002, 3, 607–616 CrossRef CAS PubMed.
- A. Erdoğmuş, M. Durmuş, A. L. Uur, O. Avciata, U. Avciata and T. Nyokong, Synth. Met., 2010, 160, 1868–1876 CrossRef.
-
(a) V. W.-W. Yam, G.-Z. Qi and K.-K. Cheung, J. Chem. Soc., Dalton Trans., 1998, 1819–1823 Search PubMed;
(b) G. V. Loukova, W. Huhn, V. P. Vasiliev and V. A. Smirnov, J. Phys. Chem. A, 2007, 111, 4117–4121 CrossRef CAS PubMed;
(c) V. E. Pritchard, F. L. Thorp-Greenwood, R. G. Balasingham, C. F. Williams, B. M. Kariuki, J. A. Platts, A. J. Hallett and M. P. Coogan, Organometallics, 2013, 32, 3566–3569 CrossRef CAS;
(d) C. Romain, S. Choua, J.-P. Collin, M. Heinrich, C. Bailly, L. Karmazin-Brelot, S. Bellemin-Laponnaz and S. Dagorne, Inorg. Chem., 2014, 53, 7371–7376 CrossRef CAS PubMed.
-
(a) J. W. Kenney, D. R. Boone, D. R. Striplin, Y.-H. Chen and K. B. Hamar, Organometallics, 1993, 12, 3671–3676 CrossRef CAS;
(b) G. V. Loukova and V. A. Smirnov, Chem. Phys. Lett., 2000, 329, 437–442 CrossRef CAS;
(c) A. Vogler and H. Kunkely, Top. Curr. Chem., 2001, 213, 143–182 CrossRef CAS;
(d) E. L. Patrick, C. J. Ray, G. D. Meyer, T. P. Ortiz, J. A. Marshall, J. A. Brozik, M. A. Summers and J. W. Kenney, J. Am. Chem. Soc., 2003, 125, 5461–5470 CrossRef CAS PubMed.
-
(a) K.-T. Wong, T. S. Hung, Y. Lin, C.-C. Wu, G.-H. Lee, S.-M. Peng, C. H. Chou and Y. O. Su, Org. Lett., 2002, 4, 513–516 CrossRef CAS PubMed;
(b) K. Itami, D. Yamazaki and J. Yoshida, J. Am. Chem. Soc., 2004, 126, 15396–15397 CrossRef CAS PubMed;
(c) S. Achelle, Y. Ramondenc, G. Dupas and N. Plé, Tetrahedron, 2008, 64, 2783–2791 CrossRef CAS;
(d) S. Achelle, I. Nouira, B. Pfaffinger, Y. Ramondenc, G. Dupas, N. Plé and J. Rodríguez-López, J. Org. Chem., 2009, 74, 3711–3717 CrossRef CAS PubMed;
(e) E. V. Verbitskiy, E. M. Cheprakova, J. O. Subbotina, A. V. Schepochkin, P. A. Slepukhin, G. L. Rusinov, V. N. Charushin, O. N. Chupakhin, N. I. Makarova, A. V. Metelitsa and V. I. Minkin, Dyes Pigm., 2014, 100, 201–214 CrossRef CAS;
(f) S. Achelle, S. Kahlal, A. Barsella, J.-Y. Saillard, X. Che, J. Vallet, F. Bureš, B. Caro and F. Robin-le Guen, Dyes Pigm., 2015, 113, 562–570 CrossRef CAS.
-
(a) H. Jang, C.-H. Shin, B.-J. Jung, D. Kim, H.-K. Shim and Y. Do, Eur. J. Inorg. Chem., 2006, 718–725 CrossRef CAS;
(b) S. Swavey, J. A. Krause, D. Collins, D. D'Cunha and A. Fratini, Polyhedron, 2008, 27, 1061–1069 CrossRef CAS;
(c) T. Lazarides, H. Adams, D. Sykes, S. Faulkner, G. Calogero and M. D. Ward, Dalton Trans., 2008, 691–698 RSC.
-
(a) Q.-D. Liu, R. Wang and S. Wang, Dalton Trans., 2004, 2073–2079 RSC;
(b) H. Akdas-Kilig, T. Roisnel, I. Ledoux and H. Le Bozec, New J. Chem., 2009, 33, 1470–1473 RSC;
(c) P. Savel, H. Akdas-Kilig, J.-P. Malval, A. Spangenberg, T. Roisnel and J.-L. Fillaut, J. Mater. Chem. C, 2014, 2, 295–305 RSC;
(d) H. Akdas-Kilig, M. Godfroy, J.-L. Fillaut, B. Donnio, B. Heinrich, P. Kędziora, J.-P. Malval, A. Spangenberg, S. van Cleuvenbergen, K. Clays and F. Camerel, J. Phys. Chem. C, 2015, 2, 3697–3710 CrossRef.
- M. Sarakha, M. Bolte and H. D. Burrows, J. Photochem. Photobiol., A, 1997, 107, 101–106 CrossRef CAS.
- V. M. Mukkala, C. Sund, M. Kwiatkowski, P. Pasanen, M. Högberg, J. Kankare and H. Takalo, Helv. Chim. Acta, 1992, 75, 1621–1632 CrossRef CAS.
-
(a)
M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of Photochemistry, Taylor & Francis, London, 2006, p. 570 Search PubMed;
(b)
M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of Photochemistry, Taylor & Francis, London, 2006, p. 574 Search PubMed.
- D. Lionetti, A. J. Medvecz, V. Ugrinova, M. Quiroz-Guzman, B. C. Boll and S. N. Brown, Inorg. Chem., 2010, 49, 4687–4697 CrossRef CAS PubMed.
-
(a) S. K. Nayaki and M. Swaminathan, J. Indian Chem. Soc., 2000, 77, 376–379 CAS;
(b) Y. Kitayama, T. Amako, N. Suzuki, M. Fujiki and Y. Imai, Org. Biomol. Chem., 2014, 12, 4342–4346 RSC;
(c) I. Yamaguchi, K. Sato and M. Okuno, J. Phys. Org. Chem., 2014, 27, 622–627 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven Jr., J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
-
(a) J. Yoon and A. W. Czarnik, Bioorg. Med. Chem., 1993, 1, 267–271 CrossRef CAS PubMed;
(b) J. S. Kim and D. T. Quang, Chem. Rev., 2007, 107, 3780–3799 CrossRef CAS PubMed.
-
(a) S. Park, E. Kim, W. Y. Kim, C. Kang and J. S. Kim, Chem. Commun., 2012, 51, 9343–9345 RSC;
(b) A. V. Saura, M. J. Marin, M. I. Burguete, D. A. Russell, F. Galindo and S. V. Luis, Org. Biomol. Chem., 2015, 13, 7736–7749 RSC;
(c) J. Hu, W. Zhao, M. Sun, X. Liu and W. Gao, Chem. Commun., 2015, 1–4 Search PubMed.
- F. Sohm, C. Gaiddon, M. Antoine, A. L. Boutillier and J. P. Loeffler, Oncogene, 1999, 18, 2762–2769 CrossRef CAS PubMed.
- C. Gaiddon, M. de Tapia and J. P. Loeffler, Mol. Endocrinol., 1999, 13, 742–751 CrossRef CAS PubMed.
-
(a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed;
(b) G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures and data, including photoluminescence data (spectra and lifetimes), and DFT calculations concerning the [Ti(1)2(2c)] complex (PDF), X-ray crystal structure details (CIF). CCDC 1468167–1468169 and 1468801. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt03477b |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a