
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioisosteric ferrocenyl-containing quinolines with antiplasmodial and antitrichomonal properties†
Muneebah
Adams
a,
Tameryn
Stringer
a,
Carmen
de Kock
b,
Peter J.
Smith
b,
Kirkwood M.
Land
c,
Nicole
Liu
c,
Christina
Tam
d,
Luisa W.
Cheng
d,
Mathew
Njoroge
e,
Kelly
Chibale
aef and
Gregory S.
Smith
*a
aDepartment of Chemistry, University of Cape Town, Rondebosch 7701, Cape Town, South Africa. E-mail: Gregory.Smith@uct.ac.za
bDivision of Clinical Pharmacology, Department of Medicine, University of Cape Town, Observatory 7925, South Africa
cDepartment of Biological Sciences, University of the Pacific, Stockton, CA 95211, USA
dFoodborne Toxin Detection and Prevention Research Unit. Agricultural Research Service, United States Department of Agriculture, Albany, CA 94710, USA
eInstitute of Infectious Disease and Molecular Medicine, University of Cape Town, Rondebosch 7701, South Africa
fSouth African Medical Research Council Drug Discovery & Development Research Unit, University of Cape Town, Rondebosch 7701, South Africa
First published on 15th November 2016
Abstract
Bioisosteric ferrocenyl-containing quinolines and ferrocenylamines containing organosilanes and their carbon analogues, were prepared and fully characterised. The molecular structures of two ferrocenyl-containing quinolines, determined using single-crystal X-ray diffraction, revealed that the compounds crystallise in a folded conformation. The compounds were screened for their antiplasmodial activity against the chloroquine-sensitive (NF54) and CQ-resistant (Dd2) strains of P. falciparum, as well as for their cytotoxicity against Chinese Hamster Ovarian (CHO) cells. The ferrocenyl-containing quinolines displayed activities in the low nanomolar range (6–36 nM), and showed selectivity towards parasites. β-Haematin inhibition assays suggest that the compounds may in part act via the inhibition of haemozoin formation, while microsomal metabolic stability studies reveal that the ferrocenyl-containing quinolines are rapidly metabolised in liver microsomes. Further, antitrichomonal screening against the metronidazole-sensitive (G3) strain of the mucosal pathogen T. vaginalis revealed that the quinoline-based compounds displayed superior parasite growth inhibition when compared to the ferrocenylamines. The library was also tested E. coli and on Lactobacilli spp. found as part of the normal flora of the human microbiome and no effect on growth in vitro was observed, supporting the observation that these compounds are specific for eukaryotic pathogens.
Introduction
The development of resistance to known drug treatments has become problematic across a wide range of diseases. The infectious parasitic diseases malaria and trichomoniasis have certainly not escaped this trend.Malaria is an infectious disease which has been responsible for a significant number of childhood deaths in Africa.1 Chloroquine (CQ; I; Fig. 1) has been one of the most widely-used antimalarial drugs.2 The identification of mutations in various Plasmodium falciparum genes and proteins has been attributed to rising drug resistance. For example, the development of the P. falciparum CQ resistance transporter (PfCRT) led to the occurrence of CQ-resistant strains.3,4 Therefore, due to the presence of these transporters, there is a pronounced decrease in accumulated CQ and a concomitant decrease in activity.
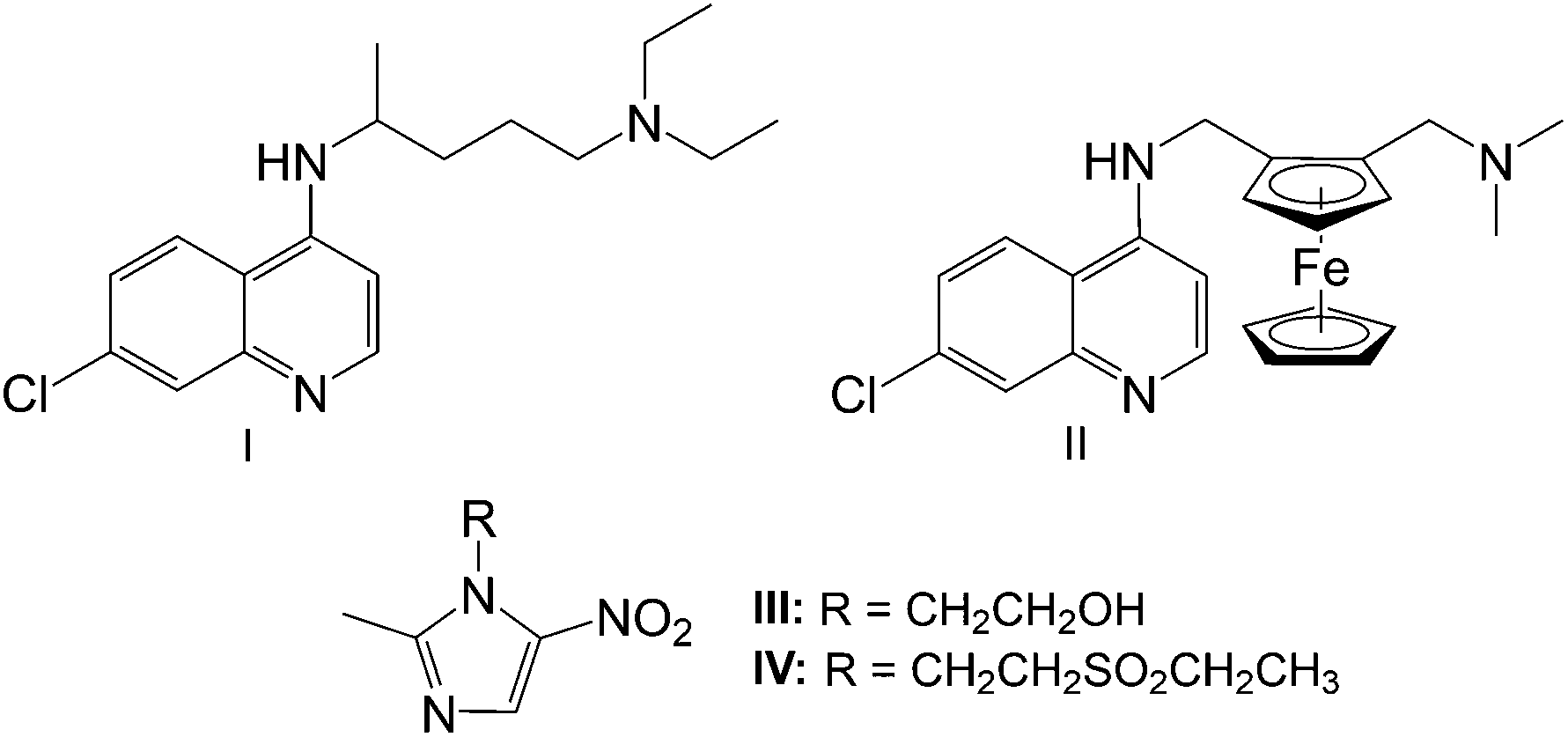 | ||
| Fig. 1 Antiparasitic agents chloroquine (I), ferroquine (II), metronidazole (III) and tinidazole (IV).5,16,17 | ||
CQ resistance led to the development of ferroquine (II, Fig. 1), a metal-containing quinoline-based compound, containing a ferrocenyl entity in the lateral side-chain. Ferroquine received a lot of attention due to its noteworthy potency against CQ-resistant strains of P. falciparum.5–8 The difference in basicity and lipophilicity between ferroquine and chloroquine at cytosolic and digestive vacuole pH, renders a more efficient accumulation of ferroquine than chloroquine, enhancing its effectiveness.9 Ferroquine is also thought to promote oxidative stress due to its redox activity. It is able to generate reactive oxygen species (ROS) via a Fenton-like reaction, which results in irreversible damage towards the parasite.
Human trichomoniasis, caused by the mucosal pathogen Trichomonas vaginalis, is the most common non-viral sexually transmitted disease and is normally treated using 5-nitroimidazole-based compounds such as metronidazole (III) and tinidazole (IV), shown in Fig. 1.10–12 These compounds passively diffuse into the parasite as prodrugs, wherein the nitro group is reduced, resulting in the formation of cytotoxic nitro radical-ion intermediates which are able to interact with DNA.13,14 This interaction is believed to result in DNA cleavage, disruption of mitosis and eventual cell death.13 However, T. vaginalis drug resistant parasites have started to emerge and despite efforts to determine the mechanism of resistance, no conclusive mechanism has been identified.15
Consequently, the identification of compounds which are able to circumvent parasite drug resistance is of great importance.
Within the context of our research aimed at identifying new chemical drug leads without cross resistance to existing agents, previous work in our laboratories have explored organosilane quinolines and a ferroquine derivative.18,19 The quinoline derivatives exhibited activity in the nanomolar range, with the compound containing a propyl spacer displaying ca. 2-fold higher activity over the methylene spacer. However, a ca. 2-fold decrease in activity was observed against the CQ-resistant (Dd2) strain in comparison with the CQ-sensitive (NF54) strain.18 The ferroquine derivative, containing a methylene spacer, exhibited activity in the low nanomolar range with activities comparable to or significantly greater than both chloroquine and ferroquine.19
Herein, we now report on the synthesis and biological activities of a series of ferrocenyl-containing quinolines based on the incorporation of amine-terminated organosilanes, and their carbon bioisosteres.
Results and discussion
Synthesis and characterisation of ferrocenyl-containing quinolines (1–5) and ferrocenylamines (6 & 7)
Ferroquine, along with its quaternised form, were prepared following published methods.5,20 The ferrocenyl-containing quinolines (1–5) were prepared via nucleophilic substitution of the trimethylammonium [–N(CH3)3+] group using the appropriate amines (Scheme 1). As seen in Scheme 1, compounds 1 and 3 are the silicon analogues of compounds 2 and 4, respectively. Furthermore, compounds 3 and 4 were prepared in order to determine how a propyl spacer would affect the pharmacological activity in comparison to those containing a methylene spacer (1, 2). In order to determine the effect of replacing a methyl group in compounds 1–4 with a phenyl ring, compound 5 was prepared. The previously published compound 119 was prepared following the slightly modified method described herein.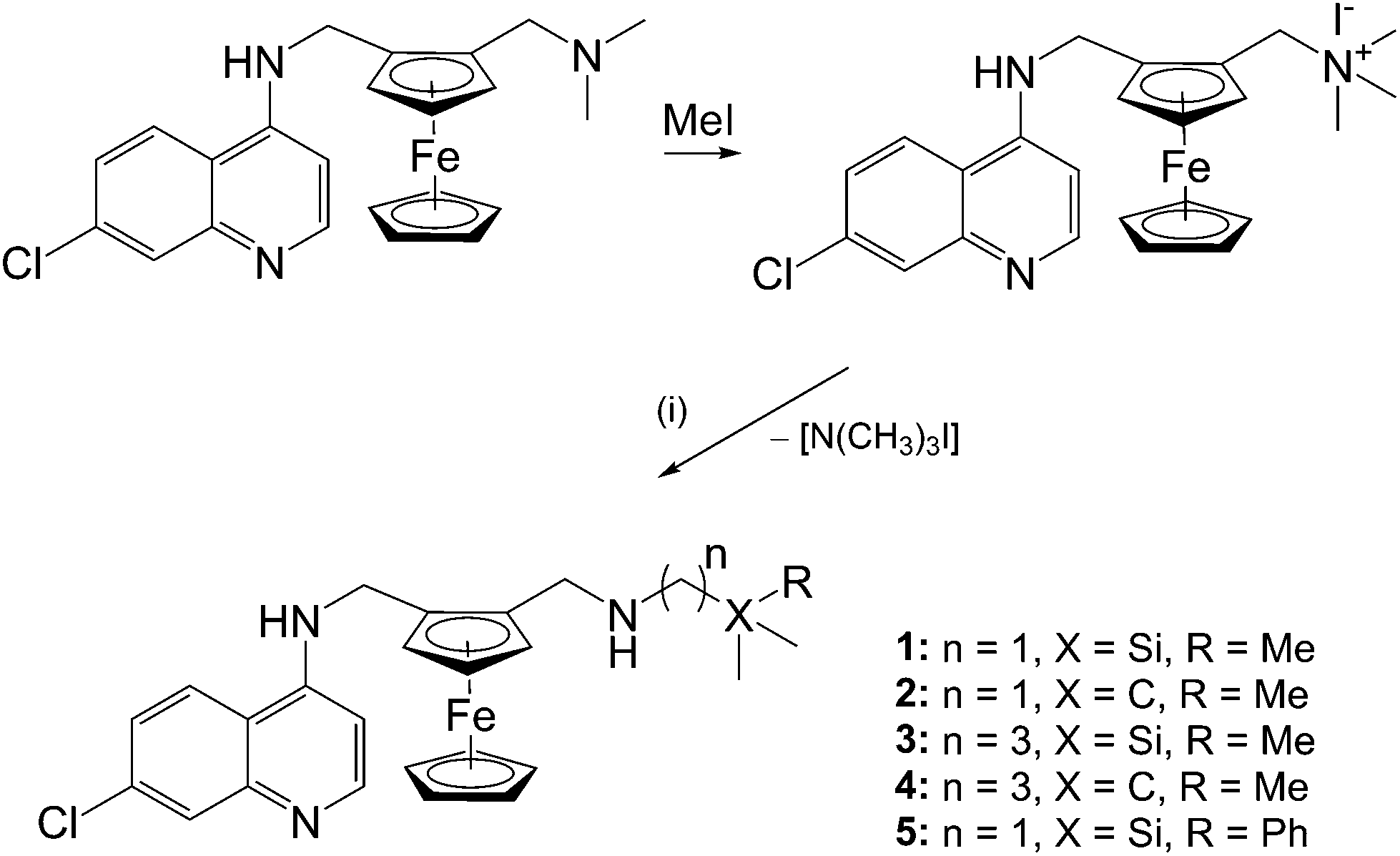 | ||
| Scheme 1 Reagents and conditions: (i) (aminomethyl)trimethylsilane (1), 2,2′-dimethyl-propan-1-amine (2), (3-aminopropyl)trimethylsilane (3), 4,4′-dimethylpentan-1-amine (4) or (aminomethyl)dimethylphenylsilane (5); CH3CN, K2CO3, N2, reflux, 72 h. | ||
In addition, ferrocenylamines (6, 7) lacking the 4-aminoquinoline entity were prepared in order to determine whether the ferrocenyl moiety alone could impart the desired pharmacological activity. Therefore, preparation of the ferrocenylamines 6 and 7 was carried out via a one-pot reductive amination of ferrocene carboxaldehyde with the appropriate amine (Scheme 2).
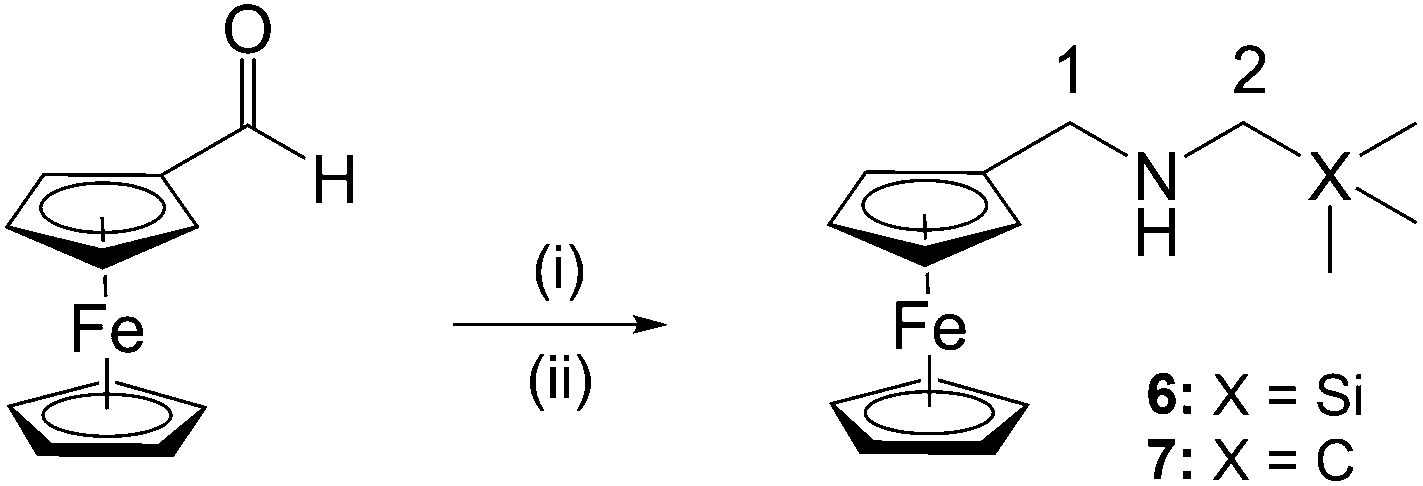 | ||
Scheme 2 Reagents and conditions: (i) (aminomethyl)trimethylsilane (6) or 2,2′-dimethylpropan-1-amine (7), DCM, 35 °C, 5 h, N2, in dark; (ii) NaBH4, DCM–MeOH (v/v, 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2), rt, overnight, N2, in dark. :2), rt, overnight, N2, in dark. | ||
In the 1H NMR spectra of compounds 1–5, the absence of a signal for the trimethylammonium group, and the presence of signals for the amine side-chain confirmed successful synthesis of the proposed compounds. This is particularly evident for compounds 1, 2 and 5, where the methylene protons of the newly incorporated amine side-chain resonate as two sets of doublets (diastereotopic protons), which confirms the inclusion in the planar chiral compound.
For the ferrocenylamines 6 and 7, the expected ferrocenyl signals were observed as two triplets (J ∼ 1.6 Hz) at 4.11 and 4.21 ppm for the substituted Cp ring and a singlet (4.13 ppm) for the unsubstituted cyclopentadienyl ring. In addition to the ferrocenyl signals, two singlets corresponding to the methylene protons were observed, as well as a signal for the protons of the methyl groups.
The 13C{1H} NMR spectra for compounds 1–5 show the quinoline carbon atoms resonating between 98.6 and 152.1 ppm. The peaks for the ferrocenyl carbon atoms were observed in the range from 65 to 86.1 ppm. As expected, due to the electropositive nature of silicon, the methyl carbons of the compounds (1, 3, 5) which contain the [Si(CH3)3] carbon atoms were heavily shielded in comparison to those compounds (2, 4) which contain the [C(CH3)3] group. For the ferrocenylamines 6 and 7, the ferrocenyl, methylene and methyl carbon atoms were all accounted for in the 13C{1H} NMR spectra. As seen for compounds 1–5, the methylene and methyl carbons of the silicon-containing compound are more shielded than the carbon analogue.
Infrared analysis of compounds 1–7 was carried out using attenuated total reflectance (ATR) infrared spectrsocopy. Preparation of compounds 1–5 was confirmed by the two absorptions bands for the N–H stretches observed at around 2950 and 3200 cm−1. For compounds 6 and 7, the absence of absorption bands for the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (aldehyde) or CN (imine) moieties confirmed preparation of the amines. The presence of an absorption band for the N–H was observed at 3093 and 3088 cm−1 for 6 and 7, respectively.
O (aldehyde) or CN (imine) moieties confirmed preparation of the amines. The presence of an absorption band for the N–H was observed at 3093 and 3088 cm−1 for 6 and 7, respectively.
The identity of compounds 2–7 were further confirmed using electron impact mass spectrometry. The molecular ion peaks were observed at m/z 475.09, 519.14, 503.12 and 553.14 for the ferrocenyl-containing quinolines 2–5, respectively. For the ferrocenylamines 6 and 7, the molecular ion peaks were observed at m/z 301.09 and 285.02, respectively.
Stability of compound 3
It is important to monitor metal-based compounds in solution in order to either ensure stability of the compound or identify the active species. As a model system, the stability of compound 3 was investigated and monitored by 1H NMR spectroscopy over a 72 h period at 37 °C. The 1H NMR spectra (Fig. 2) of compound 3 was recorded in DMSO-d6:D2O (9:1, v/v) and remains unchanged over the study period.
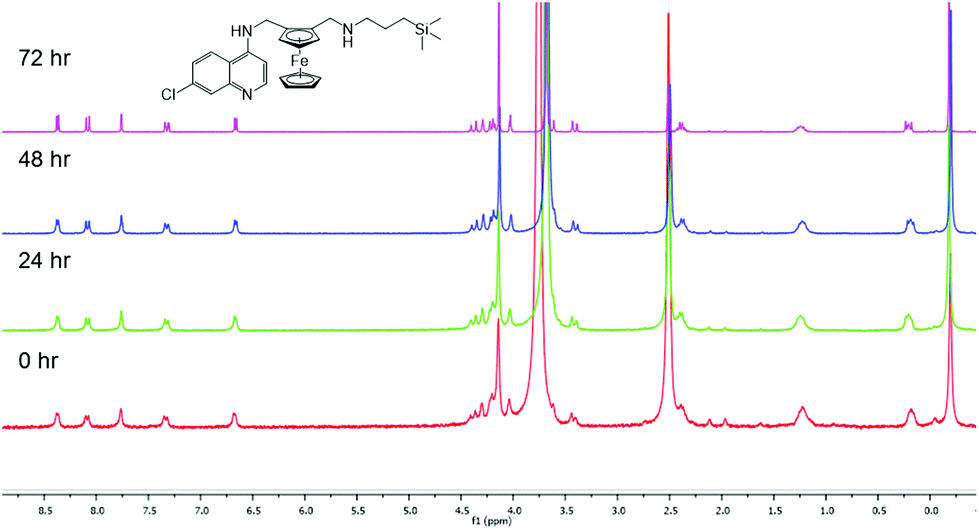 | ||
| Fig. 2
1H NMR spectrum of compound 3 in DMSO-d6:D2O heated at 37 °C over 72 h. | ||
Molecular structure of compounds 2 and 3
Crystals suitable for single-crystal X-ray diffraction were grown by the slow evaporation of solutions of 2 and 3 in dichloromethane and acetone, respectively. Both compounds crystallised as red blocks in a triclinic crystal system with the centrosymmetric space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (Table 2). Selected bond lengths and angles are listed in Table 1.
(Table 2). Selected bond lengths and angles are listed in Table 1.
| 2 | 3 | |
|---|---|---|
| Bond lengths (Å) | ||
| C(22)–C(23) | 1.531(3) | — |
| C(24)–Si(1) | — | 1.8722(19) |
| N(3)–C(22) | 1.465 | 1.467(2) |
| C(23)–C(24) | 1.538(3) | 1.522 |
| Si(1)–C(25) | — | 1.872(3) |
| Hydrogen bond lengths (Å) | ||
| N(2)–H(2)⋯N(3) | 3.160(2) | 2.993(2) |
| N(3)–H(3)⋯N(1) | 3.404(3) | — |
| Bond angles (°) | ||
| C(9)–N(2)–C(10) | 119.54(17) | 120.85(14) |
| C(21)–N(3)–C(22) | 111.53(17) | 113.05(14) |
| N(3)–C(22)–C(23) | 115.1(2) | 111.44(16) |
| C(23)–C(24)–Si(1) | — | 114.89(13) |
| Hydrogen bond angles (°) | ||
| N(3)–C(22)–C(23) | 145.9 | 155.7(19) |
| N(3)–H(3)⋯N(1) | 159 | — |
| Dihedral bond angles (°) | ||
| C(10)–C(11)–C(12)–C(21) | −7.7(3) | 1.5(2) |
| N(2)–C(10)–C(11)–C(12) | 70.5(2) | 67.39(19) |
| C(11)–C(12)–C(21)–N(3) | −77.4(2) | −74.4(2) |
| 2 | 3 | |
|---|---|---|
| Chemical formula | C26H30ClFeN3 | C27H34ClFeN3Si |
| Formula weight | 475.83 | 519.96 |
| Crystal system | Triclinic | Triclinic |
| Space group |
P |
P |
| Crystal size (mm) | 0.05 × 0.09 × 0.15 | 0.21 × 0.32 × 0.34 |
| a, b, c (Å) | 8.6284(12), 10.9159(15), 26.353(4) | 6.6872(6), 7.7098(7), 25.388(2) |
| α, β, γ (°) | 83.680(3), 84.639(3), 73.336(3) | 92.4700(10), 94.337(2), 95.214(2) |
| V/Å3 | 2358.4(6) | 1298.13(19) |
| Z | 4 | 2 |
| T/K | 173 | 173 |
| D c/g cm−3 | 1.340 | 1.330 |
| μ/mm−1 | 0.771 | 0.750 |
| Scan range/° | 1.96 < θ < 28.4 | 1.66 < θ < 27.2 |
| Unique reflections | 11751 |
5756 |
| Reflections used [I > 2σ(I)] | 8046 | 4992 |
| R int | 0.057 | 0.035 |
| R indices (all data) | 0.0413, wR2 0.0903, S 1.03 | 0.0339, wR2 0.0809, S 1.03 |
| Goodness-of-fit | 1.026 | 1.027 |
| Max, Min Δρ/e Å | −0.34; 0.40 | −0.31; 0.33 |
It is evident from the crystal structures of both compounds 2 and 3 that the compounds have a folded conformation due to intramolecular hydrogen bonding between the secondary amine nitrogen atoms N(2) and N(3) (Fig. 3). This folded conformation has previously been observed for ferroquine and similar ferrocenyl-containing aminoquinoline compounds.6,19,21–23 As expected, the terminal C–C bonds [C(22)–C(23) and C(23)–C(24)] of compound 2 are shorter than the corresponding C–Si bonds [C(24)–Si(1) and Si(1)–C(25)] of compound 3, with lengths of ∼1.53 and ∼1.87 Å, respectively (Table 1).
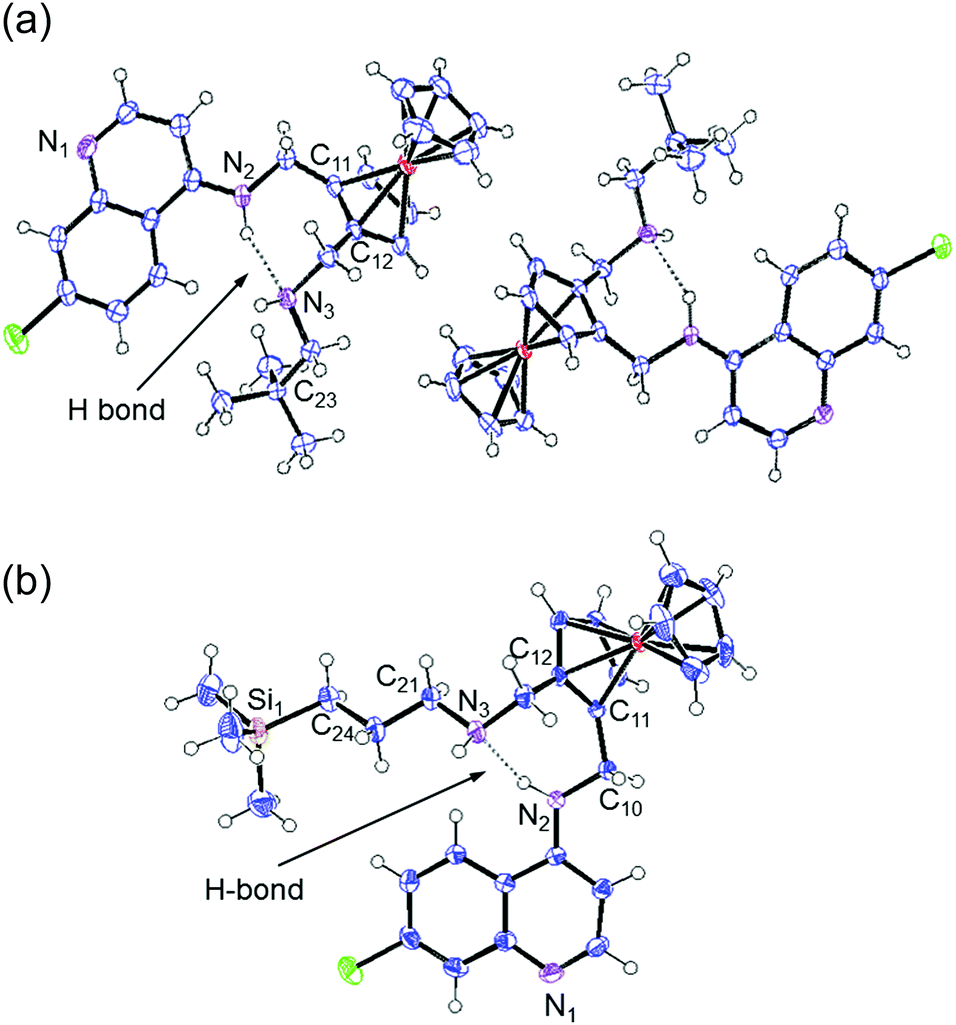 | ||
| Fig. 3 The molecular structure of compounds 2 (a) and 3 (b) with ellipsoids drawn at the 50% probability level. | ||
In vitro antiplasmodial evaluation
The bioisosteric ferrocenyl-containing quinolines 1–4 and the non-quinoline based ferrocenylamines 6 and 7, along with ferroquine, were screened for their antiplasmodial activity against the chloroquine-sensitive (NF54) and chloroquine-resistant (Dd2) strains of P. falciparum. Chloroquine and artesunate were used as control drugs. Compound 5 was not tested due to its poor solubility. Furthermore, compounds 1–4, as well as ferroquine were tested as racemates.As seen in Table 3, the ferrocenyl-containing quinolines 1–4 (IC50 < 36 nM) displayed improved activities over the ferrocenylamines 6–7, confirming the inclusion of the quinoline entity to maintain good antiplasmodial activity and to act as an inhibitor of haemozin formation (vide infra). The carbon analogues (2, 4) were slightly more active than the silicon analogues (1, 3) against the chloroquine-sensitive (NF54) strain. However, as opposed to the carbon analogues, the silicon analogues were equipotent against both the sensitive and resistant strains. For the carbon analogues a slight loss of activity was observed against the chloroquine-resistant (Dd2) strain, as illustrated in Fig. 4 (RI > 1). Incorporating organosilanes therefore appears to improve activity, particularly in the chloroquine-resistant strain.
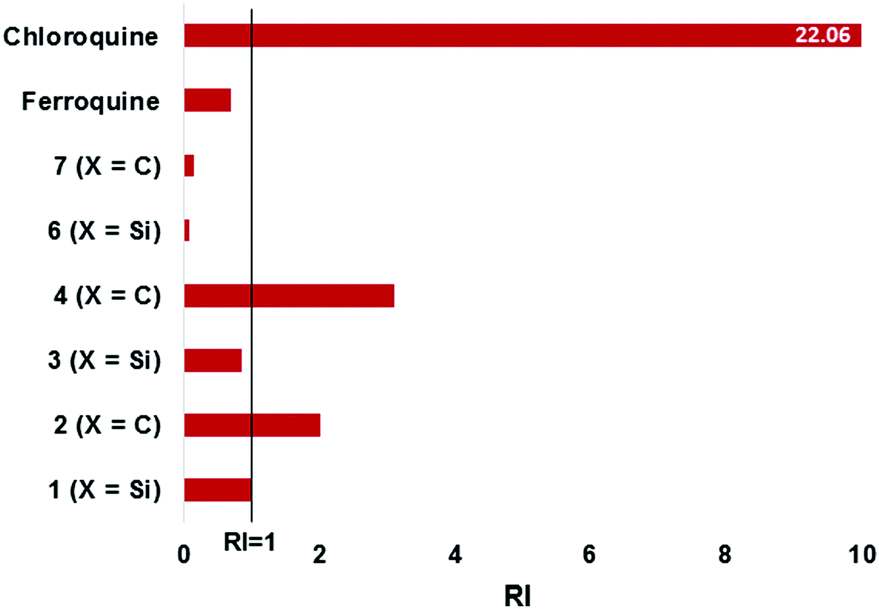 | ||
| Fig. 4 Illustrating the resistance indices (RI) for the tested compounds. RI = [IC50(Dd2)]/[IC50(NF54)]. | ||
| Compound | IC50 (nM) NF54 | IC50 (nM) Dd2 | IC50 (μM) CHO |
|---|---|---|---|
| ND = not determined. | |||
| Ferroquine | 24.90 ± 2.84 | 17.34 ± 5.90 | 24.44 ± 0.65 |
| 1 (X = Si) | 13.32 ± 2.46 | 13.40 ± 0.69 | 3.87 ± 1.43 |
| 2 (X = C) | 6.90 ± 0.86 | 13.87 ± 2.82 | 15.36 ± 0.90 |
| 3 (X = Si) | 31.37 ± 10.45 | 27.13 ± 3.58 | 9.64 ± 1.25 |
| 4 (X = C) | 11.45 ± 0.79 | 35.57 ± 6.16 | 3.62 ± 0.32 |
| 6 (X = Si) | 3168.59 ± 76.39 | 59.45 ± 10.30 | 276.01 ± 53.14 |
| 7 (X = C) | 23580.37 ± 526.11 |
3584.58 ± 343.73 | 212.90 ± 30.51 |
| Chloroquine | 13.66 ± 3.25 | 301.37 ± 9.07 | ND |
| Artesunate | <5.2 | 16.91 ± 2.86 | ND |
| Emetine | ND | ND | 0.23 ± 0.01 |
Although not very active in the NF54 strain, the ferrocenylamines were more potent against the resistant Dd2 strain, with RI values below 0.2 (Fig. 4). However, despite this, the activities of the ferrocenylamines are not as significant as that observed for the quinoline compounds. The quinoline-based compounds displayed activities comparable to chloroquine and artesunate against the NF54 strains, whereas chloroquine (IC50 = 301.37 nM), as expected, displayed poor activities against the Dd2 strain. The organosilane ferrocenyl quinolines clearly show a similar activity profile when compared to chloroquine and ferroquine. The resistance indices for the organosilanes display RI values close to 1 (or lower), indicative that these entities are more beneficial for maintaining or enhancing activity in the resistant strain.
The compounds (1–4, 6, 7), along with ferroquine, were also screened for their cytotoxicity against the Chinese Hamster Ovarian (CHO) cell-line (Table 3). Emetine was used as the control drug. This data was used to calculate selectivity indices which are illustrated in Fig. 5. The tested compounds displayed selectivity towards the parasite strains, with selectivity indices of the quinoline compounds ranging between 101 and 2227. The silicon-containing ferrocenylamine 6, in particular, displayed the greatest selectivity towards the parasites over CHO cells, with a selectivity index of 4642.66 relative to the CQ-resistant parasites.
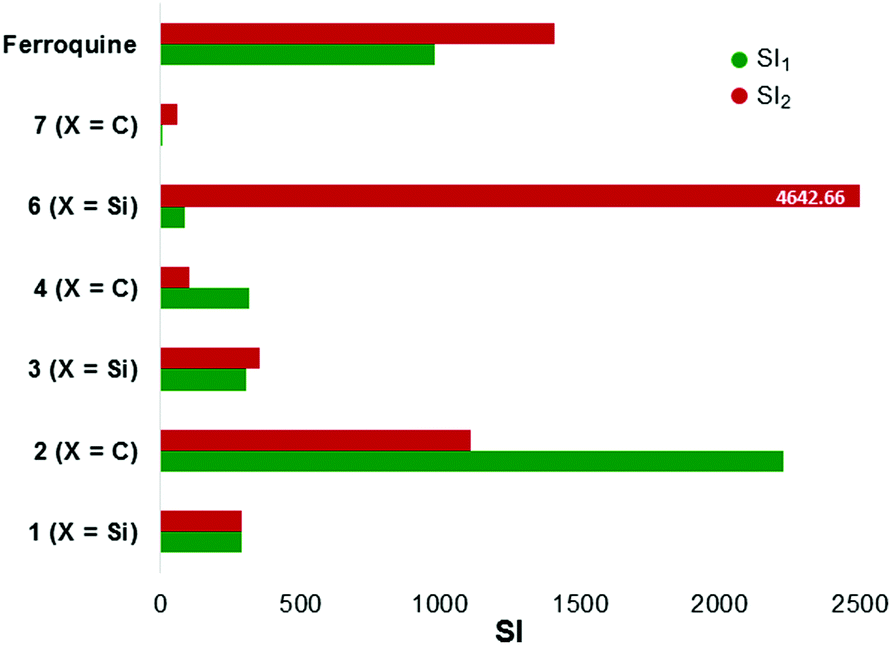 | ||
| Fig. 5 Illustrating the selectivity indices (RI) for the tested compounds. SI = [IC50(CHO)]/[IC50(NF54 or Dd2)]. SI1 (relative to NF54); SI2 (relative to Dd2). | ||
β-Haematin inhibition studies
It has been widely established that antimalarials, particularly quinoline-based compounds such as chloroquine, target and inhibit the formation of haemozoin (and β-haematin) crystal growth.24,25 Upon infection with the parasite, haemoglobin degradation in the digestive vacuole releases a toxic by-product known as haematin (ferriprotoporphyrin IX) to which the antimalarials bind, thus preventing the conversion to the less toxic haemozoin.26,27 An accumulation of large amounts of haematin will result in parasite damage. In this NP-40 detergent mediated assay,28,29 compounds 3 and 4 were selected for investigation to determine if extension from the methylene to propyl spacer improves inhibitory effects, and if the presence of silicon is significant for the inhibition of β-haematin formation. Chloroquine was used as the control drug.Both ferrocenyl-containing quinolines 3 and 4 inhibited the formation of β-haematin with IC50 values of 9.03 and 10.02 μM, respectively. These IC50 values are significantly lower than that observed for chloroquine (73.76 μM, Fig. 6). Chellan et al. previously reported that in the NP-40 assay, ferroquine inhibited the formation of β-haematin with an IC50 value of 14.51 μM [confidence interval: 13.72–15.34].30 Compound 1 (methylene spacer) has previously been evaluated for its β-haematin inhibition activity, displaying inhibitory effects with an IC50 value of 16.2 μM.19 Therefore, the ferrocenyl-containing quinoline compounds (1, 3, 4) and ferroquine display inhibition of β-haematin formation at similar IC50 values, and are significantly more effective inhibitors when compared to chloroquine.
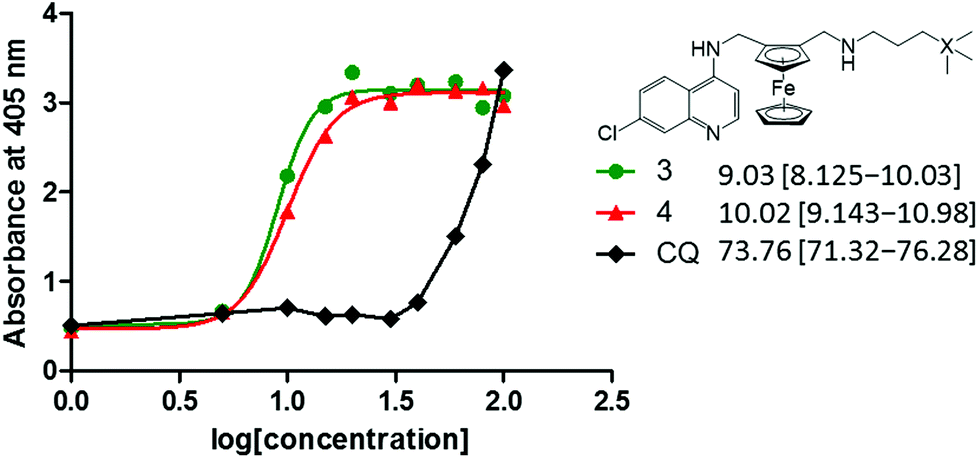 | ||
| Fig. 6 Concentration dose–response curves for NP-40 detergent mediated β-haematin assays of 3, 4 and chloroquine. | ||
Microsomal metabolic stability
Since a good in vitro activity was observed, the microsomal stability of compounds 1–3 and ferroquine were assessed in human (HLM) and mouse (MLM) liver microsomes using the single point assay.31 As seen in Table 4, compounds 1–3 were rapidly metabolised when incubated with both human and mouse liver microsomes. When incubated in HLM, less than 10% of the organosilane-containing compounds (1, 3) remained. The same result was observed for compound 1 in mouse liver microsomes, with compound 3 metabolising more slowly. Metabolite identification studies were performed to investigate the microsomal metabolism of these compounds. The main metabolites of ferroquine were tentatively identified as N-desmethylferroquine and ferroquine-N-oxide, based on their fragmentation patterns and on previous literature.34 Compound 1 was also metabolised by N-dealkylation but no oxidation metabolites were observed. The only metabolite detected for compounds 2 and 3 was a hydroxylation metabolite that was tentatively proposed to occur on the respective side chains, though the exact site of metabolism is unclear from the fragmentation data.| Compound | Percentage remaining | |
|---|---|---|
| HLM | MLM | |
| 1 | <10 | <10 |
| 2 | 26 | 20 |
| 3 | <10 | 26 |
| Ferroquine | 58 | 22 |
In general, the carbon analogue metabolised more slowly than the silicon analogues. In comparison to ferroquine, which has made it to phase IIb clinical trials, compounds 2 and 3 displayed comparable stability in MLM, but ferroquine was significantly more stable than compound 2 when incubated with HLM. Compounds 1–3 would therefore not be expected to perform well in vivo as parent compounds based on the poor microsomal metabolic stability. However, as with ferroquine, the contribution of pharmacologically active metabolites to any in vivo activity cannot be ruled out. For example, the N-dealkylation metabolite of compound 1 is also formed by ferroquine. While it is less active than the parent compound, it still has an IC50 of around 100 nM against P. falciparum 3D7 and W2.34 Thus this work can benefit from further metabolite identification studies followed by the synthesis and evaluation of major metabolites.
In vitro antitrichomonal evaluation
The ferrocenyl-containing quinolines (1–4), along with ferroquine and the ferrocenylamines (6, 7) were screened for general growth inhibition against the metronidazole-sensitive G3 strain of T. vaginalis. Compounds 1–3 and ferroquine were tested at 50 μM concentrations, while the remaining compounds were tested at a 100 μM concentration with DMSO and metronidazole as the controls. The antitrichomonal activity data is listed in Table 5.As seen in Table 5, the quinoline-based compounds display varied inhibitory effects on the parasites. However, the ferrocenyl-containing quinolines were generally better inhibitors than the ferrocenylamines (<30%). The IC50 values were determined for the carbon analogues 2 and 4 which displayed percentage inhibitions above 90%. These compounds displayed moderate activity in the micromolar range, with the carbon analogue 4 being the most active with an IC50 value of 8.30 μM. Despite the superior activity observed for compound 4 over compound 2, no definitive statement could be made when comparing compounds with methylene and propyl spacers. However, incorporation of the side-chain generally appears to improve the inhibition of parasite growth when compared to ferroquine. These compounds were not as effective as the FDA approved drug metronidazole (IC50 = 0.72 μM (ref. 32)). Since T. vaginalis is a mucosal pathogen found within a complex urogenital microbiome, we also tested the library in vitro against E. coli and Lactobacilli spp., both common mucosal normal flora bacteria. None of the compounds, when tested at the highest concentrations, had any inhibitory effect on normal flora bacteria. This additional experiment provides support that these compounds could become scaffolds for further development of new inhibitors against T. vaginalis, and possibly other mucosal pathogens, with minimal effect on the host microbiome.
Conclusions
Bioisosteric ferrocenyl-containing quinolines, along with non-quinoline based ferrocenylamines, were prepared and fully characterised. When evaluated against the NF54 and Dd2 strains of P. falciparum, the quinoline-based compounds displayed significant antiplasmodial activity. The silicon analogues displayed similar activities against both the NF54 and Dd2 strains, whereas an almost 2-fold loss in activity is observed for the carbon analogues against the CQ-resistant strain Dd2. Furthermore, these derivatives displayed improved activities over that observed for chloroquine against the CQ-resistant strain. This suggests that these derivatives may interact with the active site/target in manner similar to that seen for ferroquine, with the silicon analogues, in particularly, interacting differently with the resistant strain.Studies on the mechanism of action of ferroquine revealed that a combination of higher basicity and lipophilicity at pH 7.4, as well as the conformation of ferroquine due to the intramolecular hydrogen bond, improved ferroquine's ability to interact with lipid structures. This contributed to improved ferroquine interaction through π–π stacking with haematin via the quinoline ring,21,33 thus displaying improved inhibition of β-haematin formation. Therefore, due to the similarities between ferroquine and the ferrocenyl-containing quinolines reported herein, it is possible that the same properties lead to the potent β-haematin inhibition observed.
Microsomal metabolic stability studies demonstrated that selected ferrocenyl-containing quinolines were metabolised quickly in both human and mouse liver microsomes. Metabolite identification studies revealed that metabolism occurs on the side chain, either by N-dealkylation (compound 1) or by hydroxylation (compounds 2 and 3).
In terms of the antitrichomonal screening, the quinoline-based compounds generally displayed superior parasite growth inhibition when compared to the ferrocenylamines. The carbon analogues (2, 4) displayed inhibitions above 90% with moderate IC50 values. Quinoline-based treatments for trichomoniasis may prove promising.
Experimental
General remarks
All reagents and solvents were purchased from commercial suppliers. All reagents were purchased from either Sigma Aldrich or the FCH group and used without further purification. 2-[(N,N-Dimethylamino)methyl]-ferrocene carboxaldehyde, 2-[(N,N-dimethyl-amino)methyl]ferrocene oxime, 2-[(N,N-dimethylamino)methyl]ferrocenemethylamine, ferroquine and quaternised ferroquine were prepared via published methods.20,35 The previously reported compound 119 was prepared following the slightly modified method described herein. Nuclear magnetic resonance (NMR) spectra were recorded on either a Varian Mercury 300 spectrometer (1H at 300.08 MHz), a Bruker 400 FT spectrometer (1H at 400.200 MHz, 13C{1H} at 100.600 MHz or a Bruker 600 FT spectrometer (1H at 600.100 MHz, 13C{1H} at 150.60 MHz) at 30.0 °C. 1H and 13C{1H} NMR chemical shifts are referenced to the deuterated solvent. Infrared (IR) spectra were determined using a Perkin-Elmer Spectrum 100 FT-IR spectrometer, and were recorded using ATR (Attenuated Total Reflection). Elemental analyses (C, H and N) were recorded on a Thermo Flash 1112 Series CHNS-O Analyser. Mass spectrometry was either carried out on a JEOL GCmateII and data were recorded using Electron Impact (EI) or on a Waters API Quattro Micro triple quadrupole mass spectrometer (samples injected into a stream of 50% acetonitrile and 0.1% formic acid) and data recorded using Electrospray Ionisation (ESI) mass spectrometry in the positive mode. Melting points were determined on the Büchi Melting Point apparatus B-540. Single-crystal X-ray diffraction data were collected on a Bruker KAPPA APEX II DUO diffractometer using graphite-monochromated Mo-Kα radiation (χ = 0.71073 Å).Ferrocenyl-containing quinolines
:H2O (1:1; 60.0 mL) and extracted using chloroform (2 × 40.0 mL). The organic layer was dried over anhydrous Na2SO4 and the solvent removed. The compound was purified using silica gel chromatography, eluting with ethyl acetate:petroleum ether:triethylamine (7:2:1). The previously published compound 119 was prepared following the method described above.
7-Chloro-4-[2-(N′-substituted aminomethyl)-N-ferrocenylmethyl-(amino-2,2′-dimethylpropyl)]quinoline (2). Quaternised ferroquine (0.201 g, 0.350 mmol); 2,2′-dimethylpropan-1-amine (0.340 mL, 2.90 mmol). Compound 2 was obtained as an orange solid (0.111 g, 67%). Rf value = 0.52. m.p.: 143.1–144.9 °C. 1H NMR (300.08 MHz, CDCl3): δ (ppm) = 8.56 (d, 3JHH = 5.70 Hz, 1H, H-2); 7.94 (d, 4JHH = 2.01 Hz, 1H, H-8); 7.72 (d, 3JHH = 9.00 Hz, 1H, H-5); 7.26 (dd, 4JHH = 2.10 Hz, 3JHH = 8.70 Hz, 1H, H-6); 6.46 (d, 3JHH = 5.10 Hz, 1H, H-3); 6.20 (br s, 1H, NH); 4.35–4.40 (m, 1H, H-11); 4.27–4.28 (m, 1H, C5H3); 4.23–4.24 (m, 1H, C5H3); 4.14–4.21 (m, 6H, C5H5 & H-11); 4.12 (t, 3JHH = 2.40 Hz, 1H, C5H3); 3.72 (d, 3JHH = 12.30 Hz, 1H, H-12); 3.51 (d, 3JHH = 12.60 Hz, 1H, H-12); 2.46 (d, 3JHH = 11.70 Hz, 1H, H-13); 2.39 (d, 3JHH = 11.40 Hz, 1H, H-13); 0.91 (s, 9H, C(CH3)3). 13C{1H} NMR (100.64 MHz, CDCl3): δ (ppm) = 152.1, 149.7, 149.3, 134.7, 128.6, 124.9, 121.9, 117.4, 99.2 (CAr); 86.1, 83.2 (Fcquat.); 70.3, 69.8, 69.1, 66.4 (Fc); 62.5, 49.2, 42.0 (CH2); 31.5 (
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) (CH3)3); 27.9 (C(H3)3). FT-IR (ATR, cm−1): ν = 3270 (br, N–H); 2948 (w, N–H); 1611 (w, CN); 1577 (s, CC). MS (EI+, m/z): 475.09 ([M]+, 81%). Elemental analysis for C26H30ClFeN3 (475.14 g mol−1): Found C 65.30, H 6.59, N 8.32%; Calculated C 65.66, H 6.36, N 8.74%.
(CH3)3); 27.9 (C(H3)3). FT-IR (ATR, cm−1): ν = 3270 (br, N–H); 2948 (w, N–H); 1611 (w, CN); 1577 (s, CC). MS (EI+, m/z): 475.09 ([M]+, 81%). Elemental analysis for C26H30ClFeN3 (475.14 g mol−1): Found C 65.30, H 6.59, N 8.32%; Calculated C 65.66, H 6.36, N 8.74%.
7-Chloro-4-[2-(N′-substituted aminomethyl)-N-ferrocenylmethyl-(aminopropyl)trimethylsilane]quinoline (3). Quaternised ferroquine (0.199 g, 0.347 mmol); (3-aminopropyl)trimethylsilane (0.450 mL, 2.74 mmol). Compound 3 was isolated as a yellow solid (0.129 g, 72%). Rf value = 0.36. m.p.: 149.4–150.9 °C. 1H NMR (300.08 MHz, CDCl3): δ (ppm) = 8.54 (d, 3JHH = 5.40 Hz, 1H, H-2); 7.92 (d, 4JHH = 2.10 Hz, 1H, H-8); 7.81 (d, 3JHH = 9.00 Hz, 1H, H-5); 7.24 (dd, 4JHH = 2.40 Hz, 3JHH = 9.00 Hz, 1H, H-6); 6.48 (d, 3JHH = 5.40 Hz, 1H, H-3); 4.38 (d, 3JHH = 13.20 Hz, 1H, H-11); 4.29–4.30 (m, 1H, C5H3); 4.22–4.24 (m, 1H, C5H3); 4.17–4.20 (m, 6H, C5H5 & H-11); 4.10 (t, 3JHH = 2.40 Hz, 1H, C5H3); 3.71 (d, 3JHH = 12.30 Hz, 1H, H-12); 3.53 (d, 3JHH = 12.00 Hz, 1H, H-12); 2.60–2.70 (m, 2H, H-13); 1.46–1.59 (m, 2H, H-14); 0.42 (t, 3JHH = 8.40 Hz, 2H, H-15); −0.036 (s, 9H, Si(CH3)3). 13C{1H} NMR (100.64 MHz, CDCl3): δ (ppm) = 152.1, 150.1, 149.4, 134.7, 128.4, 124.7, 122.6, 117.8, 98.9 (CAr); 85.6, 83.5 (Fcquat.); 70.7, 70.2, 69.2, 66.1 (Fc); 53.4, 47.8, 42.4, 24.2, 14.2 (CH2); −1.74 (Si(CH3)3). FT-IR (ATR, cm−1): ν = 3198 (br, N–H); 2954 (w, N–H); 1611 (w, C
N); 1570 (s, CC). MS (EI+, m/z): 519.14 ([M]+, 49%). Elemental analysis for C27H34ClFeN3Si (519.15 g mol−1): Found C 62.13, H 6.82, N 7.99%; Calculated C 62.41, H 6.66, N 8.09%.
7-Chloro-4-[2-(N′-substituted aminomethyl)-N-ferrocenylmethyl-(amino-4,4′-dimethylpentyl)]quinoline (4). Quaternised ferroquine (0.194 g, 0.337 mmol); 4,4′-dimethylpentan-1-amine (0.380 mL, 2.64 mmol). Compound 4 was isolated as a yellow solid (0.144 g, 85%). Rf value = 0.28. m.p.: 143.1–144.5 °C. 1H NMR (300.08 MHz, CDCl3): δ (ppm) = 8.55 (d, 3JHH = 5.40 Hz, 1H, H-2); 7.92 (d, 4JHH = 2.40 Hz, 1H, H-8); 7.80 (d, 3JHH = 9.00 Hz, 1H, H-5); 7.25 (dd, 4JHH = 2.40 Hz, 3JHH = 9.00 Hz, 1H, H-6); 7.20 (br s, 1H, NH); 6.48 (d, 3JHH = 5.40 Hz, 1H, H-3); 4.37 (d, 3JHH = 12.90 Hz, 1H, H-11); 4.27–4.28 (m, 1H, C5H3); 4.20–4.21 (m, 1H, C5H3); 4.13–4.18 (m, 6H, C5H5 & H-11); 4.10 (t, 3JHH = 2.40 Hz, 1H, C5H3); 3.70 (d, 3JHH = 12.00 Hz, 1H, H-12); 3.53 (d, 3JHH = 12.00 Hz, 1H, H-12); 2.55–2.66 (m, 2H, H-13); 1.40–1.53 (m, 2H, H-14); 1.11 (t, 3JHH = 8.70 Hz, 2H, H-15); 0.85 (s, 9H, C(CH3)3). 13C{1H} NMR (100.64 MHz, CDCl3): δ (ppm) = 152.1, 150.1, 149.4, 134.6, 128.5, 124.7, 122.6, 117.8, 98.9 (CAr); 85.6, 83.5 (Fcquat.); 70.4, 70.2, 69.2, 66.1 (Fc); 50.9, 47.9, 42.4, 41.6 (CH2); 30.1 (
(CH3)3); 29.3 (C(H3)3); 25.0 (CH2). FT-IR (ATR, cm−1): ν = 3258 (br, N–H); 2956 (w, N–H); 1611 (w, CN); 1578 (s, CC). MS (EI+, m/z): 503.12 ([M]+, 78%). Elemental analysis for C28H34ClFeN3 (503.17 g mol−1): Found C 66.73, H 6.77, N 8.08%; Calculated C 66.78, H 6.81, N 8.35%.
7-Chloro-4-[2-(N′-substituted aminomethyl)-N-ferrocenylmethyl-(aminomethyl)dimethylphenylsilane]quinoline (5). Quaternised ferroquine (0.101 g, 0.176 mmol); (aminomethyl)dimethylphenylsilane (0.25 mL, 1.39 mmol). Compound 5 was isolated as a yellow solid (0.0470 g, 48%). Rf value = 0.66. m.p.: 157.0–159.4 °C. 1H NMR (400.22 MHz, CDCl3): δ (ppm) = 8.52 (d, 3JHH = 5.40 Hz, 1H, H-2); 7.92 (d, 4JHH = 2.10 Hz, 1H, H-8); 7.56 (d, 3JHH = 9.00 Hz, 1H, H-5); 7.49–7.52 (m, 2H, Ph); 7.32–7.34 (m, 3H, Ph); 7.16 (dd, 4JHH = 2.10 Hz, 3JHH = 9.00 Hz, 1H, H-6); 6.50 (br s, 1H, NH); 6.46 (d, 3JHH = 5.40 Hz, 1H, H-3); 4.27–4.33 (m, 2H, C5H3 & H-11); 4.15–4.20 (m, 2H, C5H3 & H-11); 4.09–4.11 (m, 6H, C5H3 & C5H5); 3.75 (d, 3JHH = 12.60 Hz, 1H, H-12); 3.44 (d, 3JHH = 12.60 Hz, 1H, H-12); 2.46 (d, 3JHH = 14.00 Hz, 1H, H-13); 2.37 (d, 3JHH = 14.00 Hz, 1H, H-13); 0.34 (s, 6H, Si(CH3)2). 13C{1H} NMR (100.64 MHz, CDCl3): δ (ppm) = 151.5, 149.4, 148.7, 136.9, 134.3, 133.2, 129.0, 128.0, 127.6, 124.6, 121.5, 117.1, 98.6 (CAr); 84.8, 82.9 (Fcquat.); 70.0, 69.4, 68.7, 66.0 (Fc); 51.6, 41.6, 38.8 (CH2); −3.99 (Si(CH3)). FT-IR (ATR, cm−1): ν = 3298 (br, N–H); 2948 (w, N–H); 1609 (w, C
N); 1578 (s, CC). EI+-MS: m/z 553.14 ([M]+, 12%); 389.07 ([M − NHCH2Si(CH3)2Ph]+, 39.5%); 388.03 ([M − H − NHCH2Si(CH3)2Ph]+, 100%). Elemental analysis for C30H32ClFeN3Si·½H2O (562.14 g mol−1): Found C 63.79, H 5.87, N 7.38%; Calculated C 64.04, H 5.74, N 7.47%.
Ferrocenylamines
N-(Ferrocenylmethyl)-N-(trimethylsilyl)-methanamine (6). Ferrocenecarboxaldehyde (0.145 g, 0.678 mmol); (aminomethyl)trimethylsilane (0.100 mL, 0.747 mmol); NaBH4 (0.0509 g, 1.34 mmol). Compound 6 was isolated as a sticky orange solid (0.196 g, 96%). 1H NMR (300.08 MHz, CDCl3): δ (ppm) = 4.21 (t, 3JHH = 1.50 Hz, 2H, C5H4); 4.12 (s, 5H, C5H5); 4.11 (t, 3JHH = 1.50 Hz, 2H, C5H4); 3.52 (s, 2H, H-1); 2.11 (s, 2H, H-2); 0.057 (s, 9H, Si(CH3)3). 13C{1H} NMR (100.64 MHz, CDCl3): δ (ppm) = 87.1 (Fcquat.); 68.5, 68.3, 67.6 (Fc); 53.4, 40.0 (CH2); −2.51 (Si(
H3)3). FT-IR (ATR, cm−1): ν = 3093 (br, N–H). EI+-MS: m/z 301.09 ([M]+, 73%). Elemental analysis for C15H23FeNSi·⅓H2O (307.08 g mol−1): Found C 58.75, H 7.55, N 4.19%; Calculated C 58.61, H 7.55, N 4.55%.
N-(Ferrocenylmethyl)-N-2,2-dimethylpropan-1-amine (7). Ferrocenecarboxaldehyde (0.149 g, 0.696 mmol); 2,2′-dimethylpropan-1-amine (0.0900 mL, 0.768 mmol); NaBH4 (0.0578 g, 1.53 mmol). Compound 7 was isolated as an orange solid (0.188 g, 95%). m.p.: 55.2–57.2 °C. 1H NMR (300.08 MHz, CDCl3): δ (ppm) = 4.21 (t, 3JHH = 1.80 Hz, 2H, C5H4); 4.13 (s, 5H, C5H5); 4.11 (t, 3JHH = 1.80 Hz, 2H, C5H4); 3.52 (s, 2H, H-1); 2.40 (s, 2H, H-2); 0.93 (s, 9H, C(CH3)3). 13C{1H} NMR (100.64 MHz, CDCl3): δ (ppm) = 87.8 (Fcquat.); 68.4, 68.3, 67.5 (Fc); 62.2, 49.9 (CH2); 31.5 (
(CH3)3); 27.8 (C(H3)3). FT-IR (ATR, cm−1): ν = 3088 (br, N–H). EI+-MS: m/z 285.02 ([M]+, 79%). Elemental analysis for C16H23FeN·⅕H2O (288.71 g mol−1): Found C 66.83, H 8.46, N 4.63%; Calculated C 66.50, H 8.03, N 4.85%.
:D2O (9:1, v/v). The 1H NMR spectra were recorded at 0 h. The solution was warmed at 37 °C, and the stability monitored by 1H NMR spectroscopy at 24, 48 and 72 h time intervals to confirm stability of compound during the in vitro assay time period.
Antiplasmodial assay. Continuous in vitro cultures of asexual erythrocyte stages of P. falciparum were maintained using a modified method of Trager and Jensen.36 Quantitative assessment of antiplasmodial activity in vitro was determined via the parasite lactate dehydrogenase assay using a modified method described by Makler.37 The antiplasmodial assay was conducted according to previously published methods.37 A full dose–response was performed for all compounds to determine the concentration inhibiting 50% of parasite growth (IC50 value). Test samples were tested at a starting concentration of 100 μg ml−1, which was then serially diluted 2-fold in complete medium to give 10 concentrations; with the lowest concentration being 0.2 μg ml−1. Reference drugs were tested at a starting concentration of 1000 ng ml−1. Active compounds were retested at starting concentrations of 10 μg ml−1 or 1000 ng ml−1. The highest concentration of solvent to which the parasites were exposed had no measurable effect on the parasite viability (data not shown). The IC50 values were obtained using a non-linear dose–response curve fitting analysis via Graph Pad Prism v.4.0 software.
Cytotoxicity assay. The MTT-assay is used as a colorimetric assay for cellular growth and survival, and compares well with other available assays.38,39 The test samples were tested in triplicate on one occasion. The same stock solutions prepared for the antiplasmodial activity testing were used for the cytotoxicity tests. Dilutions were prepared on the day of the experiment in complete medium. Emetine was used as the reference drug. The initial concentration of emetine was 100 μg ml−1, which was serially diluted in complete medium with 10-fold dilutions to give 6 concentrations, the lowest being 0.001 μg ml−1. The same dilution technique was applied to the all test samples. The highest concentration of solvent to which the cells were exposed to had no measurable effect on the cell viability (data not shown). The IC50 values were obtained from full dose response curves, using a non-linear dose–response curve fitting analysis via GraphPad Prism v.4 software.
β-Haematin inhibition assay. The β-haematin inhibition assay was adapted from the method described by Wright and co-workers.28 Compounds were prepared as a 10 mM stock solution in DMSO. The samples were tested at various concentrations between 5 and 500 μM. The stock solution was serially diluted to give 12 concentrations in a 96 well flat-bottom assay plate. NP-40 detergent was added to mediate the formation of β-haematin (305.5 μM). A 25 mM stock solution of haematin was prepared by dissolving haemin (16.3 mg) in dimethyl sulfoxide (1 mL). A 177.76 μL aliquot of haematin stock was suspended in 20 ml of a 2 M acetate buffer at pH 4.7. The suspension was then added to the plate to give a final haematin concentration of 100 μM. The plate was then incubated for 16 hours at 37 °C. The assay was analysed using the pyridine-ferrochrome method developed by Ncokazi and Egan.29 32 μL of a solution of 50% pyridine, 20% acetone, 20% water and 10% 2 M HEPES buffer (pH 7.4) was added to each well. To this, 60 μL acetone was added to each well and mixed. The absorbance of the resulting complex was measured at 405 nm on a SpectraMax 340PC plate reader. The IC50 values were obtained using a non-linear dose–response curve fitting analysis via GraphPad Prism v.5.00 software.
Microsomal metabolic stability study. A single-point microsomal stability assay was conducted in 96-well format to determine the microsomal clearance of the compounds.31 Test compounds and controls were prepared from 10 mM DMSO stock solutions. 0.40 mg protein per mL microsomes (pooled Human mixed gender, male Mouse BALB/c; Xenotech) were incubated with 1 μM test compound at 37 °C. Metabolic reactions were initiated by the addition of NADPH and the plates were incubated for 30 min. The reactions were quenched with acetonitrile containing carbamazepine as internal standard. The centrifuged and filtered samples were analysed by HPLC-MS/MS and the analyte
:internal standard ratio at T30 compared to that at T0 to determine % compound remaining. Control standards (midazolam and propranolol) were included in the assay to provide quality control and an indication of the metabolic capacity of the microsomes used. All analyses were performed on a ABSciex 4000 QTrap® coupled to an Agilent 1200 HPLC system.
Metabolite identification. Samples obtained from the microsomal stability assay were analysed on an ABSciex 5500 QTrap® coupled to an Agilent 1200 HPLC system. The analyses were performed in positive electrospray ionisation mode using enhanced mass spectrum and precursor ion scans coupled through information dependent acquisition to enhanced product ion scans. Metabolites were tentatively identified based on their pseudomolecular ions ([M + H]+) and on their fragmentation patterns relative to the parent compound.
Antitrichomonal assay. Cultures of the G3 strain of T. vaginalis were grown in 5 ml complete TYM Diamond's media in a 37 °C incubator for 24 h. 50 mM stocks of the compounds were made by dissolving in DMSO and were screened against G3 strain of T. vaginalis. Untreated cells and those inoculated with 5 μl DMSO (0.1%) were used as controls. 5 μL of 50 mM stocks of the compound library were inoculated for a final concentration of 50 μM. Results were calculated based on cell counts utilising a haemocytometer after 24 h. IC50 values were determined using serial dilution of the compounds and the calculated IC50 values confirmed using the same assay described above.
Human normal flora assays. Cultures of non-pathogenic strains such as Lactobacillus reuteri (ATCC 23272), Lactobacillus acidophilus (ATCC 43560), and Lactobacillus rhamnosus (ATCC 53103) were grown in lactobacilli MRS at 37 °C under anaerobic conditions whereas Escherichia coli K-12 MG 1655 was grown at 37° aerobically. Stock solutions of compounds at 100 mM as well as vehicle control DMSO were diluted to 100 μM in media and incubated with empty BDL-sensi-discs (6 mm) for 20 min at room temperature. Discs containing vehicle control, compounds, or various antibiotic discs [levofloxacin (5 μg), gentamicin (10 μg), and gentamicin (120 μg)] were placed onto the bacterial streaked agar plates and incubated overnight at 37 °C. Vehicle, compound, or antibiotic sensitivity was determined via measurement of zones of inhibition around each disc in mm.
Acknowledgements
Financial support was received from the University of Cape Town, the National Research Foundation (NRF) of South Africa (Grant No. 90500), and the South African Medical Research Council (SAMRC). The South African Research Chairs initiative of the Department of Science and Technology administered by the NRF is gratefully acknowledged for support (KC). Even though the work was supported by the SAMRC, the views and opinions expressed are not those of the SAMRC but of the authors of the material produced or published. Professor Timothy J. Egan (University of Cape Town) is gratefully acknowledged for the use of facilities in his lab for the β-haematin inhibition assays. This work was also funded by the U. S. Department of Agriculture, Agricultural Research Service (National Program 108, Project #5325-42000-049-00D) K. M. L. was supported by the Department of Biological Sciences, the University of the Pacific.Notes and references
- World Health Organisation http://who.int/malaria/media/world_malaria_report_2014/en/ (Accessed June 2016).
- S. Mushtaque, Eur. J. Med. Chem., 2015, 90, 280–295 CrossRef PubMed.
- D. A. Fidock, T. Nomura, A. K. Talley, R. A. Cooper, S. M. Dzekunov, M. T. Ferdig, L. M. Ursos, A. B. Sidhu, B. Naudé, K. W. Deitsch, X. Z. Su, J. C. Wootton, P. D. Roepe and T. E. Wellems, Mol. Cell, 2000, 6, 861–871 CrossRef CAS PubMed.
- A. Ecker, A. M. Lehane, J. Clain and D. A. Fidock, Trends Parasitol., 2012, 28, 504–514 CrossRef CAS PubMed.
- C. Biot, G. Glorian, L. A. Maciejewski and J. S. Brocard, J. Med. Chem., 1997, 40, 3715–3718 CrossRef CAS PubMed.
- F. Dubar, T. J. Egan, B. Pradines, D. Kuter, K. K. Ncokazi, D. Forge, J. F. Paul, C. Pierrot, H. Kalamou, J. Khalife, E. Buisine, C. Rogier, H. Vezin, I. Forfar, C. Slomianny, X. Trivelli, S. Kapishnikov, L. Leiserowitz, D. Dive and C. Biot, ACS Chem. Biol., 2011, 6, 275–287 CrossRef CAS PubMed.
- A. Kreidenweiss, P. G. Kremsner, K. Dietz and B. Mordmüller, Am. J. Trop. Med. Hyg., 2006, 75, 1178–1181 Search PubMed.
- M. Henry, S. Briolant, A. Fontaine, J. Mosnier, E. Baret, R. Amalvict, T. Fusai, L. Fraisse, C. Rogier and B. Pradines, Antimicrob. Agents Chemother., 2008, 52, 2755–2759 CrossRef CAS PubMed.
- C. Biot, D. Taramelli, I. Forfar-Bares, L. A. Maciejewski, M. Boyce, G. Nowogrocki, J. S. Brocard, N. Basilico, P. Olliaro and T. J. Egan, Mol. Pharm., 2005, 2, 185–193 CrossRef CAS PubMed.
- J. R. Schwebke and D. Burgess, Clin. Microbiol. Rev., 2004, 17, 794–803 CrossRef PubMed.
- P. Kissinger, BMC Infect. Dis., 2015, 15, 307–314 CrossRef PubMed.
- H. B. Fung and T. Doan, Clin. Ther., 2005, 27, 1859–1884 CrossRef CAS PubMed.
- M. Müller, Biochem. Pharmacol., 1986, 35, 37–41 CrossRef.
- V. Ali and T. Nozaki, Clin. Microbiol. Rev., 2007, 20, 164–187 CrossRef CAS PubMed.
- S. Gehrig and T. Efferth, Open Bioact. Compd. J., 2009, 2, 21–28 CrossRef CAS.
- J. N. Burrows, K. Chibale and T. N. C. Wells, Curr. Top. Med. Chem., 2011, 11, 1226–1254 CrossRef CAS PubMed.
- D. Petrin, K. Delgaty, R. Bhatt and G. Garber, Clin. Microbiol. Rev., 1998, 11, 300–317 CAS.
- Y. Li, C. de Kock, P. J. Smith, H. Guzgay, D. T. Hendricks, K. Naran, V. Mizrahi, D. F. Warner, K. Chibale and G. S. Smith, Organometallics, 2013, 32, 141–150 CrossRef CAS.
- Y. Li, C. de Kock, P. J. Smith, K. Chibale and G. S. Smith, Organometallics, 2014, 33, 4345–4348 CrossRef CAS.
- C. Biot, L. Delhaes, C. M. N'Diaye, L. A. Maciejewski, D. Camus, D. Dive and J. S. Brocard, Bioorg. Med. Chem., 1999, 7, 2843–2847 CrossRef CAS PubMed.
- F. Dubar, J. Khalife, J. Brocard, D. Dive and C. Biot, Molecules, 2008, 13, 2900–2907 CrossRef CAS PubMed.
- C. Biot, W. Castro, C. Botté and M. Navarro, Dalton Trans., 2012, 41, 6335–6349 RSC.
- M. Navarro, W. Castro and C. Biot, Organometallics, 2012, 31, 5715–5727 CrossRef CAS.
- K. Kaur, M. Jain, R. P. Reddy and R. Jain, Eur. J. Med. Chem., 2010, 45, 3245–3264 CrossRef CAS PubMed.
- T. J. Egan and K. K. Ncokazi, J. Inorg. Biochem., 2005, 99, 1532–1539 CrossRef CAS PubMed.
- S. E. Francis, D. J. Sullivan and D. E. Goldberg, Annu. Rev. Microbiol., 1997, 51, 97–123 CrossRef CAS PubMed.
- T. J. Egan, Mol. Biochem. Parasitol., 2008, 157, 127–136 CrossRef CAS PubMed.
- R. D. Sandlin, M. D. Carter, P. J. Lee, J. M. Auschwitz, S. E. Leed, J. D. Johnson and D. W. Wright, Antimicrob. Agents Chemother., 2011, 55, 3363–3369 CrossRef CAS PubMed.
- K. K. Ncokazi and T. J. Egan, Anal. Biochem., 2005, 338, 306–319 CrossRef CAS PubMed.
- P. Chellan, K. M. Land, A. Shokar, A. Au, S. H. An, D. Taylor, P. J. Smith, K. Chibale and G. S. Smith, Organometallics, 2013, 32, 4793–4804 CrossRef CAS.
- L. Di, E. H. Kerns, N. Gao, S. Q. Li, Y. Huang, J. L. Bourassa and D. M. Huryn, J. Pharm. Sci., 2004, 93, 1537–1544 CrossRef CAS PubMed.
- M. Adams, Y. Li, H. Khot, C. de Kock, P. J. Smith, K. Land, K. Chibale and G. S. Smith, Dalton Trans., 2013, 42, 4677–4685 RSC.
- W. A. Wani, E. Jameel, U. Baig, S. Mumtazuddin and L. T. Hun, Eur. J. Med. Chem., 2015, 101, 534–551 CrossRef CAS PubMed.
- W. Daher, L. Pelinski, S. Klieber, F. Sadoun, V. Meunier, M. Bourrié, C. Biot, F. Guillou, G. Fabre, J. Brocard, L. Fraisse, J.-P. Maffrand, J. Khalife and D. Dive, Drug Metab. Dispos., 2006, 34, 667–682 CrossRef CAS PubMed.
- C. Biot, B. Pradines, M.-H. Sergeant, J. Gut, P. J. Rosenthal and K. Chibale, Bioorg. Med. Chem. Lett., 2007, 17, 6434–6438 CrossRef CAS PubMed.
- W. Trager and J. B. Jensen, Science, 1976, 193, 673–675 CAS.
- M. T. Makler, J. M. Ries, J. A. Williams, J. E. Bancroft, R. C. Piper, B. L. Gibbins and D. J. Hinrichs, Am. Soc. Trop. Med. Hyg., 1993, 48, 739–741 CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- L. V. Rubinstein, R. H. Shoemaker, K. D. Paull, R. M. Simon, S. Tosini, P. Skehan, D. A. Scudiero, A. Monks and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1113–1118 CrossRef CAS PubMed.
Footnote |
| † CCDC 1486958 and 1486956 for 2 and 3. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt03175g |
| This journal is © The Royal Society of Chemistry 2016 |