
A high-nuclearity metal-cyanide cluster [Mo6Cu14] with photomagnetic properties†
N.
Bridonneau
a,
L.-M.
Chamoreau
a,
G.
Gontard
a,
J.-L.
Cantin
b,
J.
von Bardeleben
b and
V.
Marvaud
*a
aIPCM – CNRS UMR-8232, UPMC – Univ. Paris 6, 4 place Jussieu, 75252 Paris cedex 05, France. E-mail: valerie.Marvaud@upmc.fr; Fax: +33 (0)1 44 27 38 41
bINSP – CNRS UMR-7588, UPMC – Univ. Paris 6, 4 place Jussieu, 75252 Paris cedex 05, France
First published on 27th April 2016
Abstract
A high-nuclearity metal-cyanide cluster [Mo6Cu14] has been prepared and its photomagnetic properties investigated. The photoswitchable magnetic phenomenon observed is thermally reversible (T ≈ 230 K). In the field of photomagnetism, [Mo6Cu14] represents a unique example of a nanocage and the highest nuclearity observed so far.
Introduction
There is an increased awareness of high-nuclearity clusters1 and smart multi-metallic architectures, as they may substantially exhibit magnetic2 or photo-magnetic properties.3 Thus, an extraordinary variety of molecular clusters containing paramagnetic transition metal ions have been obtained and have attracted a great deal of interest for their specific properties, such as single-molecule magnets,4 high spin molecules,5 or magnetic coolers.6 On the other hand, photomagnetic materials draw special attention due to their potential application.Besides the synthetic challenge of obtaining such molecular architectures, one could benefit from the important number of metal centers to improve magnetic properties by optimizing spin and anisotropy. Furthermore, designing multi-metallic compounds might be a fruitful strategy for reaching multifunctional architectures. The first examples of polynuclear complexes with attractive magnetic properties are [Mn12]5 and [Fe19]4 published by Gatteschi's and Powell's groups respectively. Following this approach, high spin molecules and single-molecule magnets have been described in the past few years by several teams.7–11 Recently, Whitehead and Winpenny showed that heterometallic rings could be assembled in different supramolecular architectures, like the [(Ni12)(Cr7Ni)6]12 complex, where the central [Ni12] ring acts as a Lewis acid bearing terminal THF ligands that can be removed by the [Cr7Ni] Lewis base building blocks to form the “ring of ring” structure. Many other examples of high-nuclearity clusters might be found in the literature.13–17 Among them, several groups have taken advantage of the cyanide ligand that is known to mediate strong magnetic coupling between spin carriers, allowing us to get various heterometallic structures such as [M6Mn9]18 (M = Mo, W), [M6Co9]19 (M = Mo, W), [W7Cu13],20 [Fe8Co6],21 [Co6Fe4]21 or the [Fe42] nanocage.22 Very few switchable architectures have been also reported in the literature, especially by Holmes,23 Oshio24 and Sieklucka.25
In our group, we have demonstrated the feasibility of synthesizing discrete polynuclear complexes based on polycyanometallate cores (M = Co(CN)6, Cr(CN)6, Mo(CN)8, W(CN)8) surrounded by external metallic building blocks (M = Cu, Ni, Mn, Co) bearing a capping ligand.26 In particular, the use of octacyanometallate centers has proven to be an effective way of obtaining multiple kinds of architectures with promising photomagnetic properties27 due to its versatile accessible coordination polyhedra (square base antiprism, dodecahedron or mixed geometries).
The design of these assemblies enabled a precise control of nuclearity and magnetic properties. In particular, the role of the blocking ligand has proven to be of crucial influence on the metal-ion environment and consequently on the overall topology of the final product. By using blocking ligands that leave only one accessible position on the metal ions, we succeeded in obtaining high nuclearity complexes such as [CrMn6]26 or [MoCu6].27 This latter example exhibits impressive photomagnetic response up to room temperature. However, the steric hindrance coupled to charge repulsion between the peripheral metal atoms prevent increasing the nuclearity of the [MoCu6] complex, despite the eight cyanide ligands available for complexation. In order to obtain higher nuclearity complexes, we selected blocking ligands with potentially two free reactive positions instead of one: tacn (1,4,7-triazacyclononane) and Me3tacn (1,4,7-trimethyl-1,4,7-triazacyclononane). Following such an approach, we succeeded in synthesizing two new high nuclearity complexes based on the octacyanometallate core and peripheral copper atoms: [Mo6Cu14-tacn] (1) and [Mo3Cu4-Me3tacn] (2). In addition, both complexes exhibit photomagnetic behavior, the [Mo6Cu14-tacn] (1) being the most effective one with a relaxation temperature above 200 K.
Results and discussion
Synthesis and X-ray structures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :v = 1:2). Slow evaporation of the solution gave purple diamond shaped crystals. X-ray diffraction analysis shows that 1 crystallizes in the triclinic space group P
:v = 1:2). Slow evaporation of the solution gave purple diamond shaped crystals. X-ray diffraction analysis shows that 1 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . Cell parameters are a = 19.1825(10) Å, b = 19.3041(10) Å, c = 19.3498(10) Å, α = 76.569(3)°, β = 75.823(3)° and γ = 75.935(3)°, with a cell volume of V = 6624.1(6) Å3.
. Cell parameters are a = 19.1825(10) Å, b = 19.3041(10) Å, c = 19.3498(10) Å, α = 76.569(3)°, β = 75.823(3)° and γ = 75.935(3)°, with a cell volume of V = 6624.1(6) Å3.
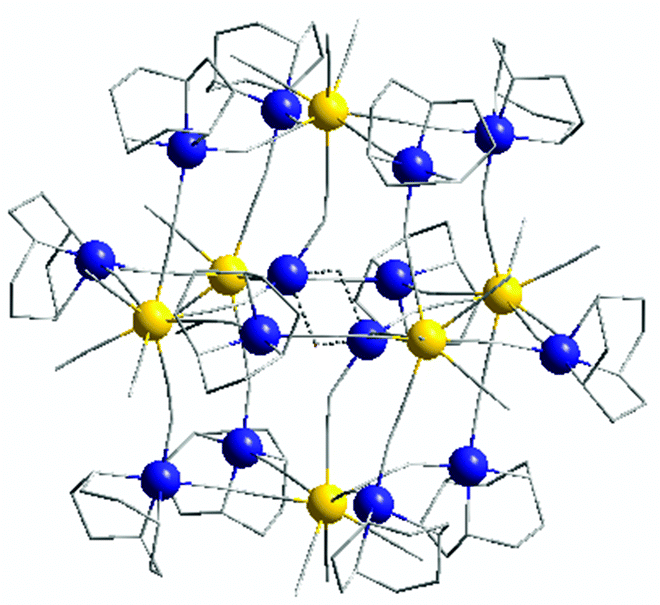 | ||
| Fig. 1 Crystal structure of the [Mo6Cu14] cluster (1) (yellow: Mo; dark blue: Cu; grey: C, N). | ||
Under similar conditions, an apparent capping ligand (Me3tacn), leads to a different compound, [Mo3Cu4–Me3tacn] noted as 2 in the present paper (Fig. 2). Taking into account that the quality of the X-ray structure can lead to a discussion (vide supra), we shall focus mainly on compound 1. Nevertheless, it is important to note that compound 2 has been identified without ambiguity and the proposed structure is wholly consistent with the analytical, spectroscopic and magnetic data.
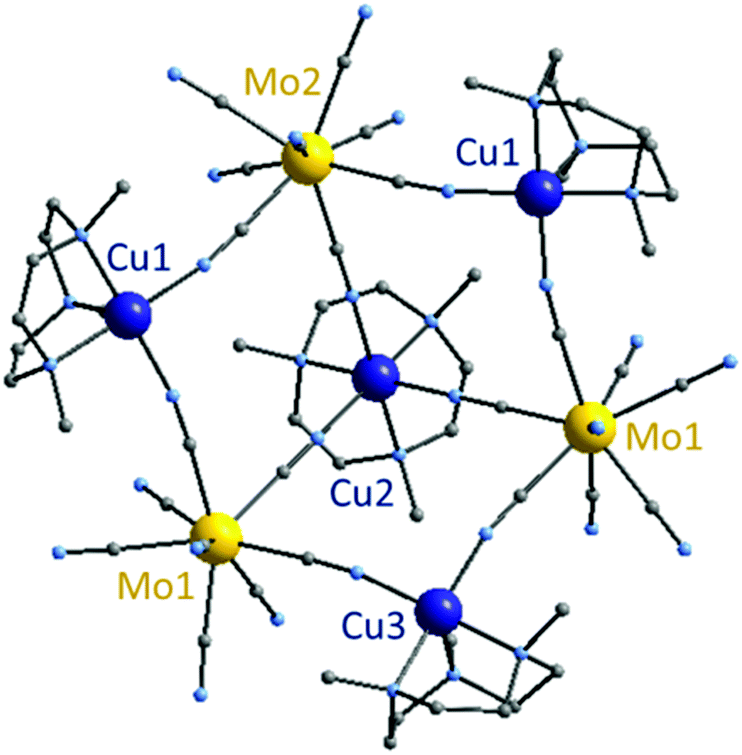 | ||
| Fig. 2 Crystal structure of 2 (yellow: Mo; dark blue: Cu; grey: C, N). | ||
The characterization of the polynuclear complexes 1 and 2 performed by infrared spectroscopy indicates similarities between the samples. In the 2000–2200 cm−1 range, characteristic of the anti-symmetric stretching vibration of the cyanido groups, two bands are observed for 1: at 2152 cm−1 relative to the bridging cyanide and another at 2124 cm−1 assigned to the free cyanides (2153 and 2123 cm−1 for 2 respectively, with a slightly different ratio).
[Mo 6 Cu 14 -tacn] (1) is composed of six octacyanomolybdate centers Mo1, Mo2 and Mo3 forming an octahedron, each molybdenum apex being linked together through a copper atom via cyanide bonds. Finally, two supplementary centrosymmetric copper atoms (Cu7) occupy the center of the structure (Fig. 1), giving an overall positively charged architecture with the following chemical formula: [(Mo(CN)7)6{(μ-CN)(CuC6H15N3)12(CuOH)2}](ClO4)2·xH2O and (x ∼ 23).
Molybdenum atoms thus connect four different copper ions plus the central ones and have three free cyanides directed outwards from the structure. The outer copper atoms (Cu1 to Cu6) have a coordination number of five and adopt a square based pyramidal environment. The central copper atom Cu7 is disordered (2 crystallographic positions in an approx. 70/30 proportion, Cu7/Cu7b), with a Cu7⋯Cu7 distance of 3.074(2) Å. Cu7 adopts a particular 4-coordinated environment, connecting three molybdenum centers and an oxygen atom located close to the Cu7b crystallographic position, with a distorted geometry already observed in the literature.28 Hence a different way of describing the [Mo6Cu14] structure is to consider four [Mo3Cu4] moieties connected together by two copper atoms in the center of the octahedron (see ESI, Fig. S4†). This particular arrangement can be explained by the great similarity of the two capping ligands Me3tacn and tacn, which impose the same geometry onto the copper ions.
Molybdenum atoms in the [Mo6Cu14] complex (1) adopt a mixed geometry between the square based antiprism and dodecahedron (as shown by Shape30 analysis, see ESI, Table S1†). The Mo⋯Cu distances are comprised between 5.108(2) (Mo3–Cu2) and 5.269(1) Å (Mo3–Cu1). Mo–C(![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) bond lengths are similar to the previous Mo3Cu4 structure and range from 2.116(9) Å to 2.189(12) Å and the C–N ones from 1.112(13) Å to 1.171(15) Å. Cyano bridges are linear on the molybdenum side with Mo–CN angles ranging from 172.5(8)° to 178.1(10)°. CN–Cu angles are bent and range from 157.5(9)° to 177.1(10)°. [Mo6Cu14] complexes are well separated from each other by perchlorate anions and solvent molecules; the shortest distance separating two metal atoms from different molecules is 7.448(1) Å, reported between molybdenum and copper ions.
N) bond lengths are similar to the previous Mo3Cu4 structure and range from 2.116(9) Å to 2.189(12) Å and the C–N ones from 1.112(13) Å to 1.171(15) Å. Cyano bridges are linear on the molybdenum side with Mo–CN angles ranging from 172.5(8)° to 178.1(10)°. CN–Cu angles are bent and range from 157.5(9)° to 177.1(10)°. [Mo6Cu14] complexes are well separated from each other by perchlorate anions and solvent molecules; the shortest distance separating two metal atoms from different molecules is 7.448(1) Å, reported between molybdenum and copper ions.
488(1) Å3. 2 is composed of three molybdenum and three copper atoms alternatively linked by cyanide bonds forming a ring that exhibits a chair conformation, in the center of which a fourth copper atom connects the three octacyanomolybdate centers (Fig. 2). This last copper atom Cu2 is offset to the side of the hexagonal ring, giving a bowl shape to the whole molecule. All molybdenum centers in 2 adopt a similar environment with a mixed geometry between a square based antiprism and dodecahedron (as shown by Shape analysis, see ESI, Table S3†). Each molybdenum atom connects two different copper ions and the central one Cu2. The Mo⋯Cu distances are comprised between 5.208(2) Å and 5.408(1) Å. Due to the Me3tacn ligand, copper atoms forming the ring, Cu1 and Cu3, adopt a square base pyramid environment. The last copper atom Cu2 situated in the center of the ring is linked to the three molybdenum atoms as well as the ligand, thus adopting a slightly distorted octahedron geometry (N–Cu2–N angles are 81.368° on the ligand side and 92.011° on the ring side). The hexacoordinated geometry imposes sensibly longer Cu2–N(C) bond lengths (2.0037(9) to 2.118(6) Å) than the ones observed for Cu1–N(C) and Cu3–N(C). Cyano bridge angles are linear on the molybdenum side with Mo–CN angles ranging from 176.3(6)° to 177.8(7)°; but slightly distorted on the copper side with CN–Cu angles comprised between 167.7(8)° and 177.6(8)°. The Mo–C(N) bond lengths range from 2.138(8) Å to 2.177(9) Å and the CN ones from 1.130(12) Å to 1.157(13) Å. See ESI, Table S2† for additional interatomic distances and angles.
Within the crystal, the complexes are ordered in a head to tail manner, forming large channels along the b-axis, with a diameter of approximately 19 Å, that contain highly disordered molecules (see ESI, Fig. S6†). The presence of this disorder thus reduces resolution of the overall MOF-like structure as already observed in the literature.29 As the unit cell contains four negatively charged [Mo3Cu4] complexes alongside two perchlorate anions, we can therefore expect the presence of potassium and residual copper ions in the cavity. Unfortunately, despite all our efforts, it was not possible to accurately locate them and we preferred to leave the channels empty. However, with an approximate volume of 3800 Å3 per unit cell, the channel seems large enough to accommodate potassium and copper complexes without any organisation. The resulting chemical formula for the crystal would be [(Mo(CN)7)3{(μ-CN)CuC9H21N3}4][CuC9H21N3]2(ClO4)2K2·20H2O, in accordance with the elementary analysis as well as the magnetic and EPR experiments.
Photomagnetic properties
Photomagnetic properties were investigated for the two complexes 1 and 2 and showed that both compounds exhibit photomagnetic behavior. Properties recorded for 1 are depicted hereafter (see ESI Fig. S10–S12 for 2†). The temperature dependence of magnetic susceptibility was measured on crystalline samples under a 1000 Oe field in the 300–2 K range. At room temperature the χmT values 5.22 cm3 mol−1 K for 1 are in good agreement with the expected theoretical values of 5.52 cm3 mol−1 K corresponding to fourteen independent copper ions (CuII, d9, S = 1/2, g = 2.05), MoIV centers being diamagnetic. At low temperature, a decrease is observed that might be explained by the two central copper ions (Cu7), bridged by the hydroxo groups and antiferromagnetically coupled. On the whole, before irradiation, χmT plots show the expected paramagnetic behavior for isolated CuII centers. M versus H plot recorded for 1 (ESI, Fig. S7†) showed a saturation value of 13.5μB at 50 kOe and is well simulated with Brillouin's law for fourteen independent CuII atoms (gCu = 2.06).The samples were then irradiated for 4 hours at 18 K using a 405 nm laser beam (20 mW) equipped with a fiber. A clear increase of magnetization was immediately observed upon light irradiation and saturation occurring typically after 3 to 4 hours (ESI, Fig. S8 for 1 and S11 for 2†). For both complexes, stopping the irradiation causes no decrease of the recorded magnetization, proving that a long-lived metastable state is obtained. After switching off the laser the temperature was cooled down to 2 K, then the magnetization data were recorded on the heating mode (2–300 K). Magnetic susceptibility data recorded after irradiation showed a clear increase, as depicted in Fig. 3 for complex 1 (see ESI, Fig. S12 for 2†). The maximum of the observed χmT product for 1 evolved from 5.22 to 8.97 cm3 mol−1 K at 5 K. This positive deviation from Curie's law indicates ferromagnetic interactions between the spin carriers. With increasing temperature the χmT product gradually decreases and both curves merge to the same value at a temperature approaching 230 K for 1 (∼90 K for 2), showing unambiguously that the system is thermally reversible. A small evolution of the magnetization curves recorded after irradiation was also reported (ESI Fig. S9 and S12†), the increase being completely reversible after heating the compound to 300 K and cooling it, demonstrating again the thermal reversibility of the process.
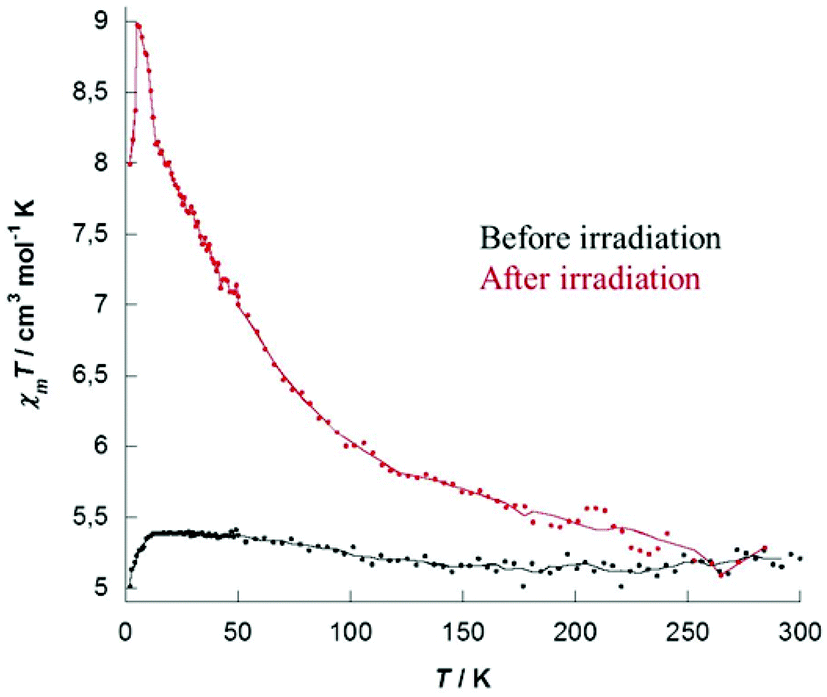 | ||
| Fig. 3 χ m T versus T plots of complex 1 before (black) and after (red) irradiation with a 405 nm laser for 3.5 h (lines are guides for the eye). | ||
Similar results have been obtained for compound (2) and on desolvated samples. See details in the ESI.†
EPR spectroscopy
EPR spectroscopy measurements were performed on crystalline samples of 1 and 2 (X-band studies, frequency of 9.35 GHz). Crystals were placed in a tube and directly irradiated at 4 K with the same 405 nm laser light. Before irradiation, the spectra showed the expected CuII signal centered in Bres = 3149 Oe for 2 (giso = 2.13) and Bres = 3148 Oe for 1 (giso = 2.13), as shown in Fig. 4 for complex 1 (see ESI, Fig. S13 for 2†). Upon irradiation the signal immediately decreased, reflecting the coupling of the CuII ions. Complex 1 exhibited an almost complete quenching of the CuII signal (Fig. 4, red curve) that persists even when irradiation is switched off. The samples were heated to room temperature, before being cooled again to show complete relaxation of the signal (Fig. 4, green curve), hence supporting the total thermal reversibility of the phenomenon.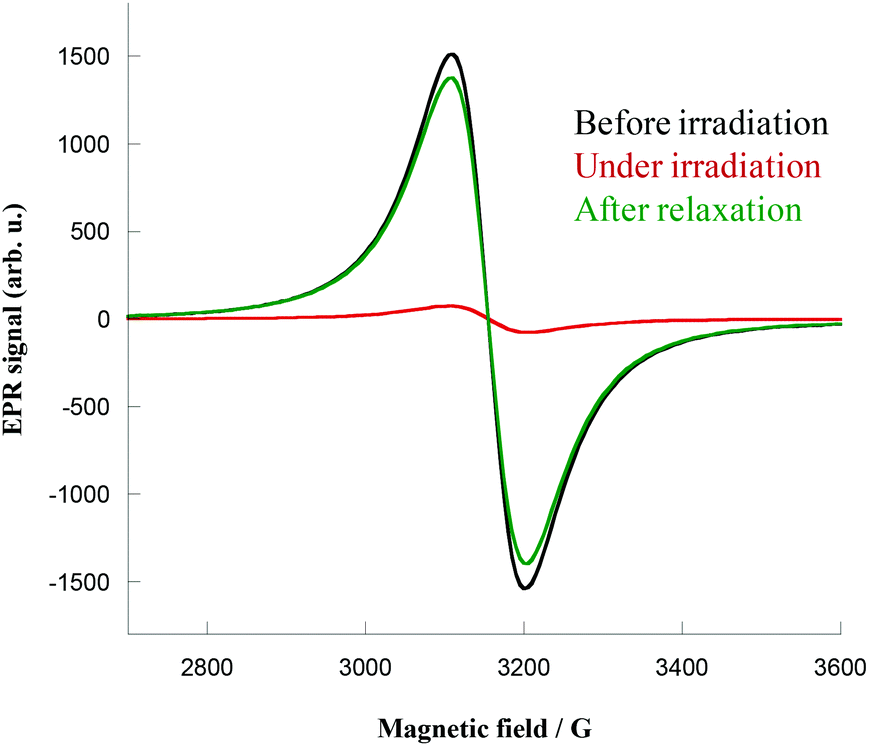 | ||
| Fig. 4 EPR spectra of complex 1 before irradiation (black), under irradiation (red) and after relaxation (green) at 4 K. | ||
Complexes 1 and 2 presented herein exhibit an unambiguously photomagnetic response. Magnetic and EPR experiments support the existence of a long-lived photo-induced metastable state for both compounds. The photomagnetic properties of these compounds can be rationalized considering the two usually assumed hypotheses for “molybdenum–copper” complexes, as suggested by us and others:3,31 spin transition on the molybdenum center (MoIV-LS (d2, S = 0) → MoIV-HS (d2, S = 1)) and a photo-induced electron transfer between molybdenum and copper atoms (MoIV-LS–CuII → MoV–CuI). In the present study, and especially for the [Mo6Cu14] (1) complex, the remarkably high relaxation temperature observed from the magnetic data (Trelax ∼ 230 K) is consistent with the electron transfer hypothesis31 and is also supported by previous calculations realized on [MoCux] complexes32 and the preliminary results on the [Mo6Zn14] analogue with no photomagnetic effect. Considering the hypothesis of an electron transfer involving the CuII centers located at the center of the structure (i.e. a diamagnetic copper dimer leading to a paramagnetic center), the expected χmT value would then be 10.25 cm3 mol−1 K at a low temperature (considering two MoVCuICu4II and four isolated CuII centers), which is relatively close to the observed value of 8.97 cm3 mol−1 K. In contrast, considering only a high spin state would lead to a considerably higher χmT value.
Conclusion
In the present paper, two new high nuclearity photomagnetic complexes [Mo6Cu14] (1) and [Mo3Cu4] (2) have been synthesized and characterized. In particular, complex 1 shows an impressive photomagnetic response with an almost doubled χmT value recorded after irradiation and a relaxation temperature as high as 230 K, which is not common for photo-switchable [MoCux] complexes. Additional experiments were conducted on analoguous compounds based on octacyanotungstate precursors: [W6Cu14] and [W3Cu4], showing only partial photo-transformation of the compounds, probably caused by the too short lifetime of the excited states as well as a low relaxation temperature, as has been seen on other tungsten compounds. Due to the complexity of the [Mo6Cu14] structure, all the performed analyses were not sufficient enough to unambiguously demonstrate the photomagnetic mechanism occurring. However, no photo-magnetic response was observed when conducting experiments on the analogous zinc compound Mo6Zn14, which tends to favor the photo-induced electron transfer process, as suggested above.From a synthetic point of view, these new compounds open the route to high nuclearity functional complexes. Furthermore, bearing either a positive or negative charge, the complexes 1 and 2, might be viewed as very promising clusters for the design of even more complex architectures.
Experiment section
Synthetic procedures
Tacn (1,4,7-triazacyclononane) ligand was synthesized as described in the literature,33,34 following slightly modified procedures, as presented in ESI-Fig. S1.† Me3tacn (1,4,7-trimethyl-1,4,7-triazacyclononane) ligand was commercially available and used without purification. Caution! Cyanides are very toxic and must be handled with care.IR spectra were obtained between 4000 and 250 cm−1 on a Bio-Rad FTS 165 FT-IR spectrometer on KBr pellets.
Crystallography
Suitable crystals for X-ray crystallography were directly obtained from the reaction medium. A single crystal of the compounds was selected rapidly, mounted onto a cryoloop, and transferred in a cold nitrogen gas stream. Intensity data were collected with a Bruker Kappa-APEXII with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). Data collections were performed with APEX2 suite (Bruker). Unit-cell parameter refinement, integration and data reduction were carried out with the SAINT program (Bruker). SADABS (Bruker) was used for scaling and multi-scan absorption corrections. In the WinGX suite of programs,35 the structures were solved with SIR9236 and SUPERFLIPS37 programs respectively and refined by full-matrix least-squares methods using SHELXL-97 and SHELXL-14.38DC magnetic susceptibility measurements
DC magnetic susceptibility measurements were carried out on a Quantum Design MPMS SQUID susceptometer equipped with a 7 T magnet and operating in a range of temperature from 1.8 to 400 K. The powdered samples (10 ± 50 mg) were placed in a diamagnetic sample holder and the measurements realised in a 1000 Oe applied field using the extraction technique. Before analysis, the experimental susceptibility was corrected from diamagnetism using Pascal constants.Photomagnetic experiments
Photomagnetic experiments were performed on ground crystals deposited on the film. Irradiation was realized with a 405 nm laser placed at approx. 2 cm from the samples. Samples were typically irradiated at 10 K under a 1 kOe field for a few hours until saturation of the signal was observed. The temperature was then decreased to 2 K and magnetic data recorded in the heating mode (under a 500 Oe or 1000 Oe field). To check the reversibility of the process, the same workup was applied, then the compound was kept for a few hours at room temperature (typically 3 h) and the magnetic data were recorded again in the 300 K–2 K range.EPR measurements
EPR measurements were performed on a single crystal with a standard X-band spectrometer and a 4–300 K variable temperature cryostat under irradiation. EPR measurements were performed with a Bruker ESP 300 spectrometer, at X Band frequency (9.5 GHz). The magnetic field modulation frequency was set at 100 kHz and the modulation amplitude and the microwave power were both adjusted to avoid saturation. The spectra were measured at temperatures between 4 K and 300 K. The excitation of the samples at low temperature was performed with laser sources at 405 and 488 nm.Elemental analyses
Elemental analyses were performed at the Service Central d'Analyse in the CNRS center (Vernaison. France).Syntheses of complexes
:v = 1:2). Slow evaporation of the blue solution afforded purple crystals shaped as diamonds after a few days.
Yield: 15%. IR (KBr): 2152, 2124, 1626, 1451, 1100, 1090, 916, 618. Anal. Calc. for (Mo(CN)8)6(CuC6H15N3)12(Cu2(OH)2)(ClO4)2(H2O)23 (KClO4)2: Mo 11.09; Cu 17.15; C 27.77; H 4.43; N 22.68; Cl 2.73. Found: Mo 10.76; Cu 17.3; C 28.05; N 22.84; H 4.37; Cl 2.93.
Yield: 40%. IR (KBr): 2153, 2123, 1639, 1461, 1080, 1009, 629. Anal. Calc. for Mo3Cu6C78H166N42Cl2K2O28: Mo 9.39; Cu 12.44; C 30.94; N 19.74; H 5.09; Cl 3.47. Found: Mo 8.54; Cu 13.52; C 32.92; N 19.50; H 5.44; Cl 4.25.
Structural description
A first set of data was collected for 1 considering the rhombohedral R symmetry of the system. However, the crystal structure solution was not convincing and the ligands capping copper atoms were highly disordered. We then worked in the P space group, which led to a lower R1 factor and no disorder of the ligands. A second dataset was thus collected on a second sample and the results, considering a triclinic symmetry, are reported herein. However, even if results are better than the ones from the first dataset some residual density remains which can be due to the very large solvent region. All non-hydrogen atoms of the cation were refined anisotropically while some solvent molecules were refined isotropically. Hydrogen atoms were placed at calculated positions for tacn ligands and acetonitrile molecules. No hydrogen atoms were introduced for water molecules. The inside part of the complex is highly disordered and we were not able to fully characterize it. The sum of sof for Cu7/Cu7b was restrained to be equal to 1. Much information on the environment of this copper atom is still missing, as we were not able to model the residual density located between two central copper atoms. We suppose they are bridged with a O(H) atom but were not able, with SC-XRD, to really confirm that hypothesis. Two solvent molecules, located near the cation, were refined as a disorder of water and acetonitrile. The perchlorate anion is disordered over two orientations and undergoes restraints. Most of the final residual density is located near metallic atoms. We also notice a non-negligible amount of density in the large solvent region.
Acknowledgements
The research has been supported by the CNRS, UPMC (University Pierre et Marie Curie), the French Ministry of Research, ANR Switch (2010-Blan-712) and ANR E-storic (14-CE05-0002).Notes and references
- (a) J. Ferrando-Soria, A. Fernandez, E. Moreno Pineda, S. A. Varey, R. W. Adams, I. J. Vitorica-Yrezabal, F. Tuna, G. A. Timco, C. A. Muryn and R. E. P. Winpenny, J. Am. Chem. Soc., 2015, 137(24), 7644–7647 CrossRef CAS PubMed and references therein; (b) For an in-depth discussion of the term cluster in inorganic chemistry, see: M. H. Chisholm, Polyhedron, 1998, 17, 2773–3044 CrossRef CAS.
- (a) J. M. Clemente-Juan and E. Coronado, Coord. Chem. Rev., 1999, 193(5), 361–394 CrossRef; (b) L. M. C. Beltran and J. R. Long, Acc. Chem. Res., 2005, 38(4), 325–334 CrossRef CAS PubMed.
- (a) O. Sato, J. Tao and Y. Z. Zhang, Angew. Chem., Int. Ed., 2007, 46(13), 2152–2187 CrossRef CAS PubMed; (b) A. Bleuzen, V. Marvaud, C. Mathonière, B. Sieclucka and M. Verdaguer, Inorg. Chem., 2009, 48(8), 3453–3466 CrossRef CAS PubMed.
- R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141 CrossRef CAS.
- A. K. Powell, S. L. Heath, D. Gatteschi, L. Pardi, R. Sessoli, G. Spina, F. Del Giallo and F. Pieralli, J. Am. Chem. Soc., 1995, 117, 2491–2502 CrossRef CAS.
- Y.-Z. Zheng, G.-Z. Zhou, Z. Zheng and R. Winpenny, Chem. Soc. Rev., 2014, 43, 1462–1475 RSC.
- M. Charalambous, E. E. Moushi, C. Papatriantafyllopoulou, W. Wernsdorfer, V. Nastopoulos, G. Christou and A. J. Tasiopoulos, Chem. Commun., 2012, 48(44), 5410–5412 RSC.
- E. E. Moushi, C. Lampropoulos, W. Wernsdorfer, V. Nastopoulos, G. Christou and A. J. Tasiopoulos, J. Am. Chem. Soc., 2010, 132(45), 16146–16155 CrossRef CAS PubMed.
- R. T. W. Scott, S. Parsons, M. Murugesu, W. Wernsdorfer, G. Christou and E. K. Brechin, Angew. Chem., Int. Ed., 2005, 44(40), 6540–6543 CrossRef CAS PubMed.
- (a) G. Karotsis, S. Kennedy, S. J. Teat, C. M. Beavers, D. A. Fowler, J. J. Morales, M. Evangelisti, S. J. Dalgarno and E. K. Brechin, J. Am. Chem. Soc., 2010, 132(37), 12983–12990 CrossRef CAS PubMed; (b) S. M. Taylor, R. D. McIntosh, J. Reze, S. J. Dalgarno and E. K. Brechin, Chem. Commun., 2012, 48(74), 9263–9265 RSC.
- V. A. Milway, F. Tuna, A. R. Farrell, L. E. Sharp, S. Parsons and M. Murrie, Angew. Chem., Int. Ed., 2013, 52(7), 1949–1952 CrossRef CAS PubMed.
- (a) G. F. S. Whitehead, F. Moro, G. A. Timco, W. Wernsdorfer, S. J. Teat and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2013, 52(38), 9932–9935 CrossRef CAS PubMed; (b) J. Ferrando-Soria, J. A. Fernandez, E. M. Pineda, S. A. Varey, R. W. Adams, I. J. Vitorica-Yrezabal, F. Tuna, G. A. Timco, C. A. Muryn and R. E. P. Winpenny, J. Am. Chem. Soc., 2015, 137(24), 7644–7647 CrossRef CAS PubMed.
- A. M. Ako, I. J. Hewitt, V. Mereacre, R. Clerac, W. Wernsdorfer, C. E. Anson and A. K. Powell, Angew. Chem., Int. Ed., 2006, 45(30), 4926–4929 CrossRef CAS PubMed.
- (a) J. J. Sokol, M. P. Shores and J. R. Long, Inorg. Chem., 2002, 41(12), 3052–3054 CrossRef CAS PubMed; (b) P. A. Berseth, J. J. Sokol, M. P. Shores, J. L. Heinrich and J. R. Long, J. Am. Chem. Soc., 2000, 122(40), 9655–9662 CrossRef CAS; (c) L. M. C. Beltran, J. J. Sokol and J. R. Long, J. Cluster Sci., 2007, 18(3), 575–596 CrossRef CAS; (d) L. Jiang, H. J. Choi, X.-L. Feng, T.-B. Lu and J. R. Long, Inorg. Chem., 2007, 46(6), 2181–2186 CrossRef CAS PubMed.
- C. P. Berlinguette and K. R. Dunbar, Chem. Commun., 2005, 19, 2451 RSC.
- A. Müller, F. Peters, M. T. Pope and D. Gatteschi, Chem. Rev., 1998, 98(1), 239–272 CrossRef.
- J.-B. Peng, Q.-C. Zhang, X.-J. Kong, Y.-Z. Zheng, Y.-P. Ren, L.-S. Long, R.-B. Huang, L.-S. Zheng and Z. Zheng, J. Am. Chem. Soc., 2012, 134, 3314–3317 CrossRef CAS PubMed.
- (a) J. Larionova, M. Gross, M. Pilkington, H. Andres, H. Stoeckli-Evans, H. U. Gudel and S. Decurtins, Angew. Chem., Int. Ed., 2000, 39(9), 1605 CrossRef CAS; (b) Z. J. Zhong, H. Seino, Y. Mizobe, M. Hidai, A. Fujishima, S. Ohkoshi and K. Hashimoto, J. Am. Chem. Soc., 2000, 122(12), 2952–2953 CrossRef CAS.
- Y. Song, P. Zhang, X. M. Ren, X. F. Shen, Y. Z. Li and X. Z. You, J. Am. Chem. Soc., 2005, 127(11), 3708–3709 CrossRef CAS PubMed.
- J. Wang, Z.-C. Zhang, H.-S. Wang, L.-C. Kang, H.-B. Zhou, Y. Song and X.-Z. You, Inorg. Chem., 2010, 49(7), 3101–3103 CrossRef CAS PubMed.
- (a) G. N. Newton, K. Mitsumoto, R.-J. Wei, F. Iijima, T. Shiga, H. Nishikawa and H. Oshio, Angew. Chem., Int. Ed., 2014, 53(11), 2941–2944 CrossRef CAS PubMed; (b) T. Shiga, T. Tetsuka, K. Sakai, Y. Sekine, M. Nihei, G. N. Newton and H. Oshio, Inorg. Chem., 2014, 53(12), 5899–5901 CrossRef CAS PubMed.
- S. Kang, H. Zheng, T. Liu, K. Hamachi, S. Kanegawa, K. Sugimoto, Y. Shiota, S. Hayami, M. Mito, T. Nakamura, M. Nakano, M. L. Baker, H. Nojiri, K. Yoshizawa, C. Duan and O. Sato, Nat. Commun., 2015, 6, 5955 CrossRef PubMed.
- D. F. Li, R. Clerac, O. Roubeau, E. Harte, C. Mathoniere, R. Le Bris and S. M. Holmes, J. Am. Chem. Soc., 2008, 130(1), 252–258 CrossRef CAS PubMed.
- K. Mitsumoto, E. Oshiro, H. Nishikawa, T. Shiga, Y. Yamamura, K. Saito and H. Oshio, Chem. – Eur. J., 2011, 17(35), 9612–9618 CrossRef CAS PubMed.
- R. Podgajny, S. Chorazy, W. Nitek, M. Rams, A. M. Majcher, B. Marszalek, J. Zukrowski, C. Kapusta and B. Sieklucka, Angew. Chem., Int. Ed., 2012, 52(3), 896–900 CrossRef PubMed.
- (a) V. Marvaud, C. Decroix, A. Scuiller, C. Guyard-Duhayon, J. Vaissermann, F. Gonnet and M. Verdaguer, Chem. – Eur. J., 2003, 9(8), 1677–1691 CrossRef CAS PubMed; (b) F. Tuyeras, A. Scuiller, C. Duhayon, M. Hernandez-Molina, F. Fabrizi de Biani, M. Verdaguer, T. Mallah, W. Wernsdorfer and V. Marvaud, Inorg. Chim. Acta, 2008, 361, 3508–3518 CrossRef.
- J. M. Herrera, V. Marvaud, M. Verdaguer, J. Marrot, M. Kalisz and C. Mathonière, Angew. Chem., Int. Ed., 2004, 43(41), 5468–5471 CrossRef CAS PubMed.
- (a) M. J. Katz, C. J. Shorrock, R. J. Batchelor and D. B. Leznoff, Inorg. Chem., 2006, 45, 1757–1765 CrossRef CAS PubMed; (b) L. P. Wu, M. Yamamoto, T. Kuroda-Sowa, M. Maekawa, Y. Suenaga and M. Munakata, J. Chem. Soc., Dalton Trans., 1996, 2031–2037 RSC.
- M. Kurmoo, Chem. Soc. Rev., 2009, 38, 1353–1379 RSC.
- (a) S. Alvarez, P. Alemany, D. Casanova, J. Cirera, M. Llunell and D. Avnir, Coord. Chem. Rev., 2005, 249(17–18), 1693–1708 CrossRef CAS; (b) D. Casanova, J. Cirera, M. Llunell, P. Alemany, D. Avnir and S. Alvarez, J. Am. Chem. Soc., 2004, 126(6), 1755–1763 CrossRef CAS PubMed; (c) J. Cirera, E. Ruiz and S. Alvarez, Organometallics, 2005, 24(7), 1556–1562 CrossRef CAS.
- (a) M. A. Arrio, J. Long, C. C. D. Moulin, A. Bachschmidt, V. Marvaud, A. Rogalev, C. Mathoniere, F. Wilhelm and P. Sainctavit, J. Phys. Chem. C, 2010, 114(1), 593–600 CrossRef CAS; (b) N. Bridonneau, J. Long, J.-L. Cantin, J. von Bardeleben, S. Pillet, E.-E. Bendeif, D. Aravena, E. Ruiz and V. Marvaud, Chem. Commun., 2015, 51, 8229–8232 RSC; (c) S. Brossard, F. Volatron, L. Lisnard, M. A. Arrio, L. Catala, C. Mathoniere, T. Mallah, C. C. D. Moulin, A. Rogalev, A. Smekhova and P. Sainctavit, J. Am. Chem. Soc., 2012, 134(1), 222–228 CrossRef CAS PubMed; (d) S. Ohkoshi, Y. Hamada, T. Matsuda, Y. Tsunobuchi and H. Tokoro, Chem. Mater., 2008, 20(9), 3048–3054 CrossRef CAS; (e) S.-I. Ohkoshi, K. Imoto, Y. Tsunobuchi, S. Takano and H. Tokoro, Nat. Chem., 2011, 3(7), 564–569 CrossRef CAS PubMed.
- R. Raghunathan, S. Ramasesha, C. Mathonière and V. Marvaud, Phys. Rev. B: Condens. Matter, 2006, 73(4), 045131 CrossRef.
- C. A. Barta, S. R. Bayly, P. W. Read, B. O. Patrick, R. C. Thompson and C. Orvig, Inorg. Chem., 2008, 47(7), 2280–2293 CrossRef CAS PubMed.
- J. Kang and J. H. Jo, Bull. Korean Chem. Soc., 2003, 24(9), 1403–1406 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343–350 CrossRef.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1052206 and 1426776 for 1 and 2. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt00743k |
| This journal is © The Royal Society of Chemistry 2016 |