
Mixed triorganobismuthines RAr2Bi [Ar = C6F5, 2,4,6-(C6F5)3C6H2] and hypervalent racemic Bi-chiral diorganobismuth(III) bromides RArBiBr (Ar = C6F5, Mes, Ph) with the ligand R = 2-(Me2NCH2)C6H4. Influences of the organic substituent†
Marian
Olaru‡
a,
Mihai G.
Nema‡
a,
Albert
Soran
a,
Hans J.
Breunig
*b and
Cristian
Silvestru
*a
aDepartamentul de Chimie, Centrul de Chimie Supramoleculară Organică şi Organometalică (CCSOOM), Facultatea de Chimie şi Inginerie Chimică, Universitatea Babeş-Bolyai, 400028 Cluj-Napoca, Romania. E-mail: cristian.silvestru@ubbcluj.ro; Fax: (+40) 264-590818; Tel: (+40) 264-593833
bInstitut für Anorganische und Physikalische Chemie, Universität Bremen, D-28334 Bremen, Germany. E-mail: hbreunig@uni-bremen.de; Fax: (+49) 421-21862809; Tel: (+49) 421-21863150
First published on 18th February 2016
Abstract
Triorganobismuthines R(C6F5)2Bi (1) and R[2,4,6-(C6F5)3C6H2]2Bi (2) [R = 2-(Me2NCH2)C6H4] were synthesized by reaction of RBiBr2 with C6F5MgBr and 2,4,6-(C6F5)3C6H2Li, respectively, in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio. The Bi-chiral bromides R(C6F5)BiBr (3), R(Mes)BiBr (4), and R(Ph)BiBr (5) were obtained from RBiBr2 and C6F5MgBr, MesMgBr or PhMgBr, or from PhBiBr2 and RLi, in a 1:1 molar ratio. The molecular structures of 1–5 were determined by single-crystal X-ray diffraction and are discussed taking into account the chirality of the species. Supramolecular aspects in the solid state are presented. The solution behaviour of the title compounds, including dynamic aspects, are discussed on the basis of multinuclear (1H, 13C, 19F) NMR spectroscopy.
:2 molar ratio. The Bi-chiral bromides R(C6F5)BiBr (3), R(Mes)BiBr (4), and R(Ph)BiBr (5) were obtained from RBiBr2 and C6F5MgBr, MesMgBr or PhMgBr, or from PhBiBr2 and RLi, in a 1:1 molar ratio. The molecular structures of 1–5 were determined by single-crystal X-ray diffraction and are discussed taking into account the chirality of the species. Supramolecular aspects in the solid state are presented. The solution behaviour of the title compounds, including dynamic aspects, are discussed on the basis of multinuclear (1H, 13C, 19F) NMR spectroscopy.
Introduction
Bismuth(III) salts and organobismuth(III) compounds continue to receive considerable attention regarding their use in a growing number of applications including reagents involved in various organic transformations, precursors for the development of materials, and catalysis,1–6 but also fundamental aspects with respect to the M–bismuth (M = transition metals such as Cr, Mo, W, Mn, Fe, Pt, Cu, Ag, Au) interactions in complexes containing organobismuth(III) species as ligands.7–13 From this point of view the chemistry of bismuth is still far from being exhausted. Owing it to several advantages (e.g. low toxicity, commercial availability of starting materials) the use of bismuth compounds in catalysis and particularly asymmetric catalysis is highly attractive.2,14Several different types of chirality can be recognized in heavy group 15 element organometallic compounds. The most often encountered are: (i) M-center chirality in organopnicogen compounds with three different substituents (e.g. R1R2R3M, RR′MX; M = pnicogen, X = halide); (ii) chelate-induced metal chirality (e.g. RC,EMR12 or RC,EMX2, RC,E = substituent capable of E→M intramolecular coordination, E = donor atom such as N, P, O, S, etc.); (iii) helical chirality (observed in the solid state for some (RC,E)3M compounds when all three substituents are chelating the metal centre); (iv) planar chirality arising from the non-planarity of the chelate ring formed by intramolecular coordination of RC,E substituents to the metal; (v) substituent- or ligand-chiral compounds (e.g. in compounds containing chiral substituents or ligands).3
Although many reports discuss chiral organophosphorus(III), organoarsenic(III), and organoantimony(III) compounds, solid state structural investigations of chiral organobismuth(III) derivatives are rather scarce.3,15–22 This has been accounted to a lack of appropriate synthetic protocols and to the greater instability of organobismuthines.21 However, since the first reports on chiral organobismuth compounds,23,24 advances have been made e.g. several strategies that allowed the synthesis of organobismuthines bearing three different substituents were developed,21,25–29 and the synthesis of optically pure Bi-chiral diastereomers has been achieved.30,31
Murafuji et al. reported the only optically pure Bi-chiral triorganobismuthine characterized by single-crystal X-ray diffraction, obtained diastereoselectively as Bi-(R) and Bi-(S) diastereomers, namely RFc(4-MeC6H4)(4-ClC6H4)-Bi-(R) and RFc(4-MeC6H4)(4-ClC6H4)-Bi-(S), RFc = (S)-2-[(R)-1-(dimethylamino)ethyl]ferrocenyl.30 Bismuth-chiral diorganobismuth(III) halides are better represented in the literature. Examples of compounds structurally characterized in the solid state include, but may not be limited to, R(4-MeC6H4)BiCl [R = 2-(Me2NCH2)C6H4],27 RFc(4-ClC6H4)BiI (R = (R)-(+)-N,N-dimethyl-1-ferrocenylethylamine),31 2,6-[(4-MeC6H4)(Cl)Bi]2C6H3SO2(t-Bu)32 RC,NBi(Ph)Cl (RC,N = [2-(CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N-2,6-iPr2C6H3)C6H4]),33 RC,O(4-MeC6H4)BiBr (RC,O = 2-MeC(O)C6H4),29 and others.26,34
N-2,6-iPr2C6H3)C6H4]),33 RC,O(4-MeC6H4)BiBr (RC,O = 2-MeC(O)C6H4),29 and others.26,34
A significant number of organobismuth(III) compounds with intramolecular coordination-induced chirality at the Bi centre and planar chirality have been reported to date.3,4
We report here on the structural characterisation of R(C6F5)2Bi (1), R[2,4,6-(C6F5)3C6H2]2Bi (2), and racemic Bi-chiral diorganobismuth(III) bromides R(C6F5)BiBr (3), R(Mes)BiBr (4), and R(Ph)BiBr (5) [R = 2-(Me2NCH2)C6H4]. An improved method of synthesis for the previously reported compound 527 and its complete NMR characterisation is also reported in this work.
Results and discussion
Synthesis
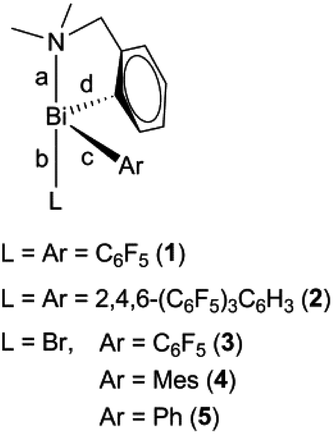
The triorganobismuth(III) derivatives R(C6F5)2Bi (1) and R[2,4,6-(C6F5)3C6H2]2Bi (2) were synthesized in the reaction of [2-(Me2NCH2)C6H4]BiBr2 with C6F5MgBr or 2,4,6-(C6F5)3C6H2Li, respectively, in a 1:2 molar ratio. The reaction of [2-(Me2NCH2)C6H4]BiBr2 with one equivalent of the corresponding organomagnesium bromide afforded the chiral organobismuth(III) bromide, [2-(Me2NCH2)C6H4](Ar)BiBr [Ar = C6F5 (3), Mes (4)] (Scheme 1).
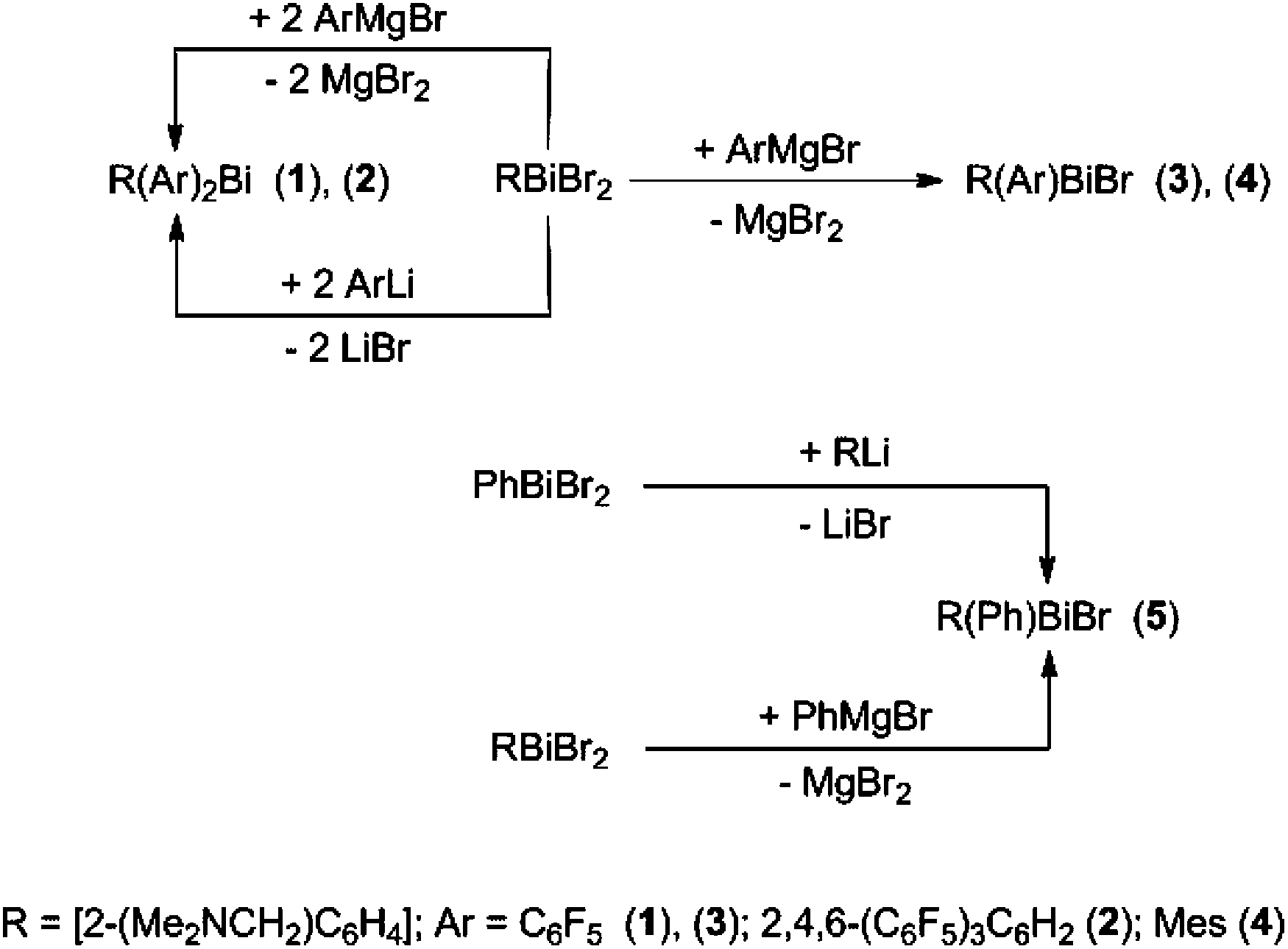 | ||
| Scheme 1 Preparation of compounds 1–5. | ||
Compound 5 was previously synthesized by reaction of Ph2BiCl with [2-(Me2NCH2)C6H4]Li followed by treatment of RPh2Bi first with BF3·Et2O, then with NaCl. The subsequent halide exchange reaction between the resulting R(Ph)BiCl and NaBr gave 5 with an overall yield of 47%.27 We prepared compound 5 by two other routes. The salt elimination reaction between [2-(Me2NCH2)C6H4]Li and phenylbismuth(III) dibromide in THF (Scheme 1) afforded 5 with a rather low yield (34%). The slow addition of a solution of PhMgBr to a suspension of [2-(Me2NCH2)C6H4]BiBr2 in THF improved considerably the yield of 5 (67%).
Compounds 1–5 are colourless crystalline solids, soluble in common organic solvents. Compounds 2, 4 and 5 are air-stable for weeks, while 1 and 3 decompose very quickly when their solutions are exposed to traces of water, with formation of C6F5H and poorly soluble (in CDCl3 or C6D6) by-products. In contrast with 1, compound 2 is air and moisture stable although it contains two electron-withdrawing substituents, i.e. 2,4,6-(C6F5)3C6H2. The steric protection provided by the two m-terphenyl-like substituents increases the kinetic stability of 2.
Solid state structures
Single-crystal X-ray diffraction studies were carried out in order to establish the molecular structures of 1–5 which are depicted in Fig. 1, 2 and 4–6. Selected molecular parameters are given in Table 1. | ||
| Fig. 1 Molecular structure of isomer (ABi,pSN)-1. Thermal ellipsoids are drawn at the 30% probability. | ||
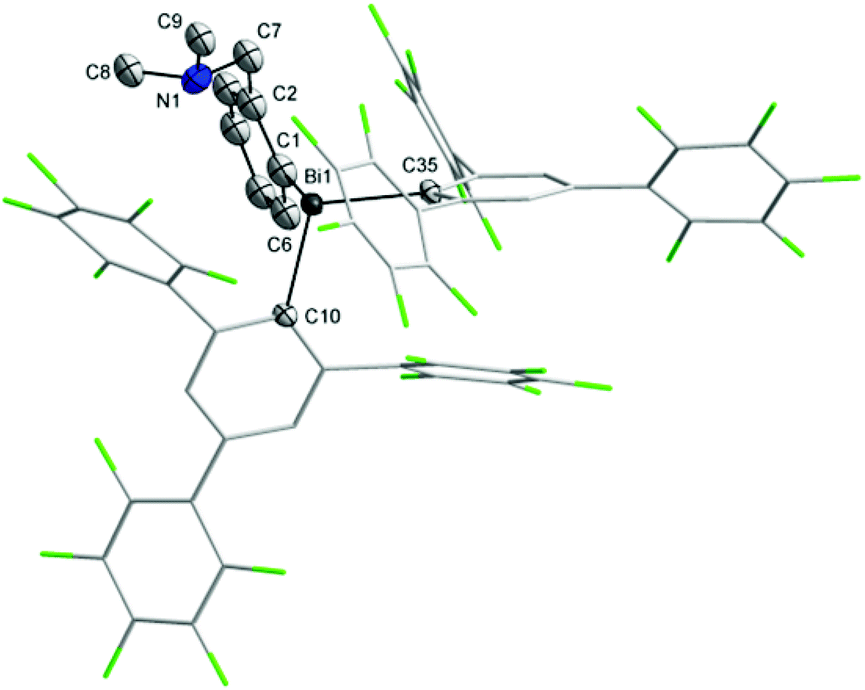 | ||
| Fig. 2 Molecular structure of 2. Thermal ellipsoids are drawn at the 20% probability. Hydrogen atoms are omitted for clarity. | ||
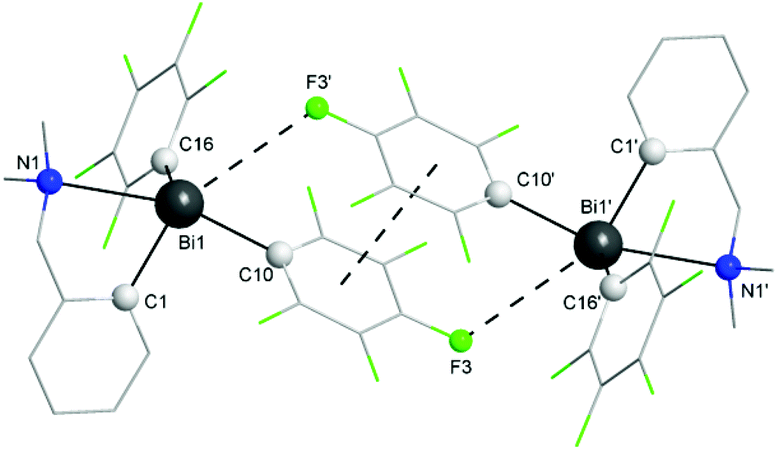 | ||
| Fig. 3 Dimer associations between (ABi,pSN) and (CBi,pRN) isomers in the crystal of 1. Hydrogen atoms are omitted for clarity. | ||
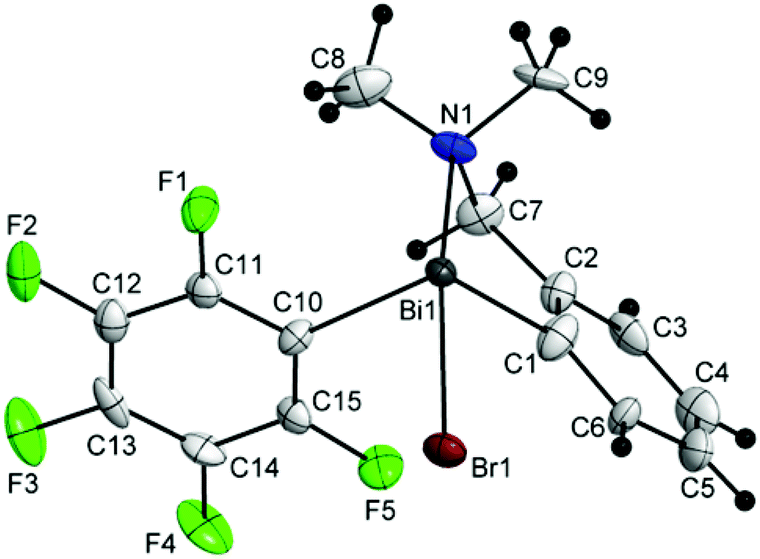 | ||
| Fig. 4 Molecular structure of isomer (ABi1,pSN1)-3a. Thermal ellipsoids are drawn at the 40% probability. | ||
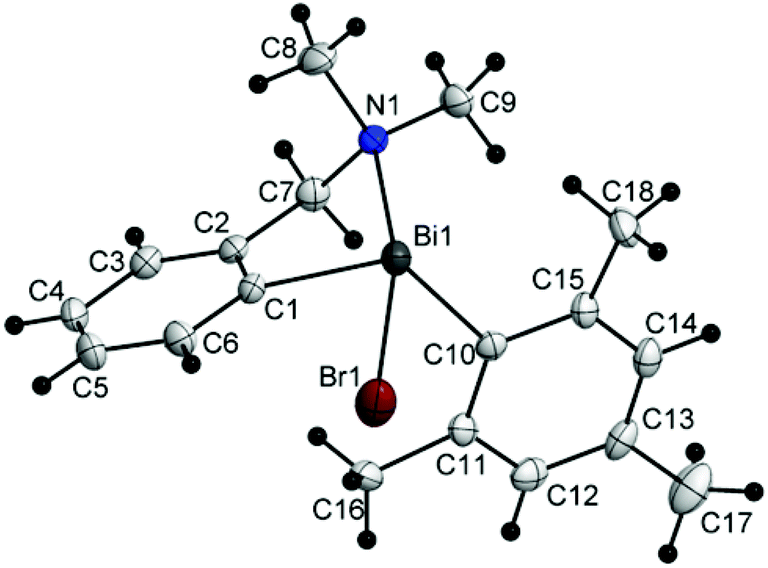 | ||
| Fig. 5 Molecular structure of isomer (CBi,pRN)-4. Thermal ellipsoids are drawn at the 40% probability. | ||
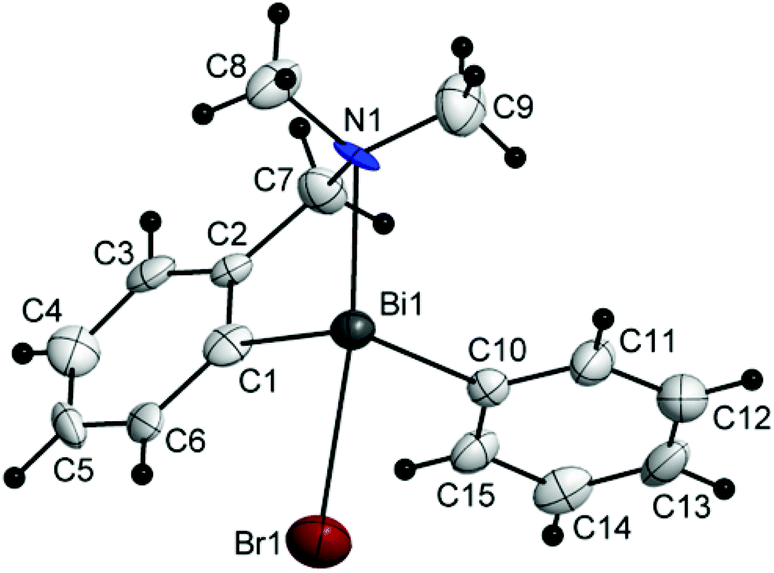 | ||
| Fig. 6 Molecular structure of isomer (ABi,pRN)-5. Thermal ellipsoids are drawn at the 40% probability. | ||
| 1 | 2 | 3 | 4 | 5 | |||
|---|---|---|---|---|---|---|---|
| 3a | 3b | ||||||
| a Non-bonding distance. | |||||||
|
a | 2.725(6) | [3.334(16)]a | 2.563(9) | 2.511(9) | 2.570(5) | 2.536(13) |
| b | 2.363(7) | 2.309(11) | 2.747(2) | 2.807(2) | 2.7702(9) | 2.789(2) | |
| c | 2.287(7) | 2.338(12) | 2.352(11) | 2.277(11) | 2.282(5) | 2.241(16) | |
| d | 2.230(7) | 2.239(18) | 2.250(13) | 2.233(9) | 2.251(5) | 2.224(19) | |
| a–b | 162.1(2) | — | 161.3(2) | 162.2(2) | 165.39(10) | 164.6(3) | |
| a–c | 85.5(2) | — | 84.5(4) | 85.9(3) | 87.21(16) | 86.9(5) | |
| a–d | 70.9(2) | — | 72.5(4) | 74.2(4) | 72.46(17) | 72.8(6) | |
| b–c | 94.4(2) | 108.8(4) | 88.4(3) | 87.0(3) | 94.89(13) | 89.9(4) | |
| b–d | 91.6(3) | 89.3(6) | 91.0(4) | 90.2(3) | 92.94(14) | 92.4(5) | |
| c–d | 100.0(3) | 104.4(6) | 95.3(4) | 95.2(4) | 99.52(19) | 94.0(6) | |
The Bi–C bond distances are similar in the heteroleptic bismuthines 1 [Bi(1)–C(1) 2.230(7) Å, Bi(1)–C(10) 2.363(7) Å, Bi(1)–C(16) 2.287(7) Å] and 2 [Bi(1)–C(1) 2.24(2) Å, Bi(1)–C(10) 2.309(11) Å, Bi(1)–C(35) 2.339(12) Å]. However, there are important structural differences between these two compounds. In the molecule of 1 the nitrogen of the pendant arm is strongly coordinated to the metal [Bi(1)–N(1) 2.725(6) Å] trans to a C6F5 group [N(1)–Bi(1)–C(10) 162.1(2)°] (Fig. 1) [cf. sum of the van der Waals radii for the corresponding atoms, ∑rvdW(Bi,N) 3.94 Å].35 The Bi–N distance in 1 is longer than that observed in the related complex containing the Martin ligand [2-(Me2NCH2)C6H4]Bi[C6H4{C(CF3)2O}-2] [2.629(95) Å].36
Although the pendant arm brings its nitrogen atom in a quite close proximity of the bismuth atom [Bi(1)–N(1) 3.33(2) Å], the NMe2 group is twisted and its lone pair is not oriented towards the metal centre (an angle of 34.9° between the Bi–N vector and the normal to the plane defined by the three carbons attached to nitrogen) to allow a N→Bi interaction in the molecule of 2 (Fig. 2).
The coordination of the NMe2 group effectively results in the formation of a hypervalent 10-Bi-4 [(C,N)BiC2 core] species37,38 in the case of 1, and the overall geometry at the bismuth centre becomes distorted pseudo-trigonal bipyramidal (“see-saw”), with the C(1) and C(16) atoms in equatorial positions. The C–Bi–C angles are in the range 91.6(3)–100.0(3)°, close to the CPh–Bi–CPh angles in triphenylbismuthine (92–96°).39 The intramolecular N→Bi interaction results in chelate-induced Bi-chirality3,40 (in the molecule of 1 the C6F5 groups are different, being placed in axial and equatorial positions, respectively) which can be described in terms of ABi and CBi isomers.41 The C3BiN chelate cycle in 1 is not planar, the nitrogen atom being out of the plane defined by the C3Bi unit. This gives rise to coordination-induced planar chirality, with the aromatic ring as the chiral plane and the N atom as the pilot atom (for planar chirality the enantiomers are given as pSN and pRN).42 The crystal of 1 contains a racemic mixture of isomers. Dimeric associations between (ABi,pSN)-1 and (CBi,pRN)-1 (Fig. 3) are formed through weak intermolecular Bi⋯F contacts [3.562(5) Å; cf. sum of the corresponding van der Waals radii, ∑rvdw(Bi,F) 3.75 Å (ref. 35)] and π⋯π interactions [Arcentroid{C(10)–C(15)}⋯Arcentroid{C(10′)–C(15′)} distance 3.41 Å].43 Such dimers are further connected through additional weak intermolecular F⋯H interactions involving an ortho-F of the axial C6F5 group from a neighbouring dimer [F(5)⋯H(3d) 2.54 Å; cf. sum of the corresponding van der Waals radii, ∑rvdW(F,H) 2.55 Å (ref. 35)], resulting in a layer network (Fig. S7, ESI†). No interactions between parallel layers were observed.
The coordination geometry around the bismuth atom in 2 (Fig. 2) can be described as distorted pyramidal, with two enlarged C–Bi–C angles [C(1)–Bi(1)–C(35) 104.4(5)°, for C(10)–Bi(1)–C(35) 108.8(4)°] as a consequence of the steric pressure imposed by the polyfluorinated substituents.
There are no interactions between heavy atoms in the crystal of 2, but the molecules are connected through intermolecular C–F⋯π (Arcentroid) interactions [C(24)–F(24)⋯Arcentroid{C(47)–C(52)} 3.16 Å] to form chain polymers (Fig. S13 in ESI†), without further inter-chain contacts.
The chiral bromides R(Ar)BiBr [R = 2-(Me2NCH2)C6H4; Ar = C6F5 (3) (Fig. 4), Mes (4) (Fig. 5), Ph (5) (Fig. 6)] exhibit similar molecular structures in the solid state. The crystal of the bromide 3 contains two independent molecules (3a and 3b) which exhibit only small differences in the magnitude of the molecular parameters (Table 1).
Some common trends observed at the molecular level for compounds 3–5 are: (i) the Bi–Br bond lengths are shorter than in the related bromide [2-(Me2NCH2)C6H4]2BiBr [Bi(1)–Br(1) 2.845(7) Å];44 (ii) the primary coordination sphere consists of a trigonal pyramidal C2BiBr core; (iii) a strong intramolecular N→Bi interaction [2.563(9) Å for (3a) and 2.511(9) Å for (3b); 2.570(5) Å for (4); 2.536(13) Å for (5); cf. sum of the corresponding covalent radii, ∑rcov(Bi,N) 2.27 Å (ref. 35)] of similar magnitude as found in [2-(Me2NCH2)C6H4]2BiBr [Bi(1)–N(1) 2.534(5) Å],44 is placed trans to the Bi–Br bond; (iv) taking into account the N→Bi interaction the overall geometry around bismuth is distorted pseudo-trigonal bipyramidal [(C,N)CBiBr core; hypervalent 10-Bi-4 species37,38], with the axial positions occupied by the nitrogen and bromine atoms (see Table 1 for the N–Bi–Br angles); (v) the resulting folded BiC3N chelate ring induces planar chirality; (vi) the compounds crystallize as racemates: (ABi1,pSN1)-3a/(CBi1,pRN1)-3a and (ABi2,pSN2)-3b/(CBi2,pRN2)-3b isomers for 3, (ABi,pSN)-4/(CBi,pRN)-4 isomers for 4, and (ABi,pRN)-5 and (CBi,pSN)-5 isomers for 5.
The N–Bi distances found in compounds 3–5 are rather similar and do not reflect a clear trend depending on the electronic properties of the second organic substituent (with C6F5, Ph and Mes substituents as electron-deficient, electron-neutral, and electron-rich groups, respectively). The strength of the N→Bi interaction depends more on the number of organic ligands attached to the metal and parallels the increased Lewis acidity of the bismuth centre in the series R3Bi < R2BiX < RBiX2 [e.g. N–Bi distances in [2-(Me2NCH2)C6H4](4-MeC6H4)2Bi 2.902(4) Å,45 [2-(Me2NCH2)C6H4](4-MeC6H4)BiCl 2.525(6) Å,27 and [2-(Me2NCH2)C6H4]BiCl2 2.458(6) Å (ref. 44)].
The deviations of the bond angles at the bismuth centre from the ideal values are mainly due to the constraint imposed by the small bite of the C,N-chelating ligand and in the cases of 3 and 4 also due to the bulkier C6F5 and mesityl groups, respectively, as compared to the phenyl substituent in 5.
A closer investigation of the crystal packing of 3 revealed that intermolecular Bi⋯Br interactions generate a ribbon-like chain polymeric arrangement which consists of binuclear units of (ABi2,pSN2)-3b/(CBi2,pRN2)-3b isomers [Bi(2)⋯Br(2′) 3.608(2) Å] connected through bridging (ABi1,pSN1)-3a and (CBi1,pRN1)-3a isomers [Bi(1)⋯Br(2) 3.439(2) Å and Bi(2b)⋯Br(1) 3.893(2) Å; cf. the sum of the corresponding van der Waals radii, ∑rvdW(Bi,Br) 4.35 Å (ref. 35)] (Fig. 7). This chain polymer is supported by additional intra-chain, weak intermolecular F⋯H and Br⋯H interactions (Fig. S20, ESI†). Further weak intermolecular F⋯H, C–H⋯π (Arcentroid) and C–F⋯π (Arcentroid) contacts (Fig. S21, ESI†) connect such chains in a 3D supramolecular architecture.
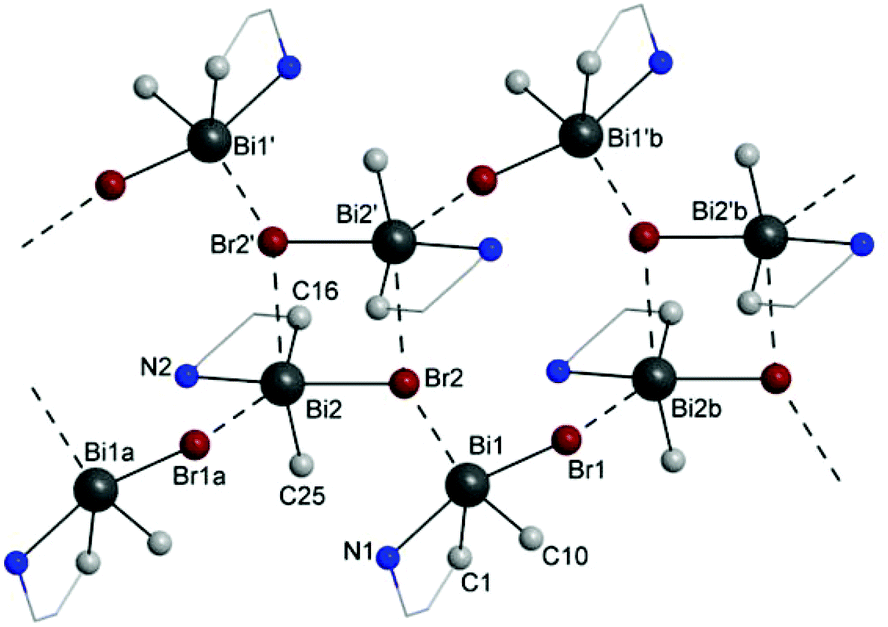 | ||
| Fig. 7 Fragment of the chain polymeric arrangement with a skeleton based on intermolecular Bi⋯Br interactions between (ABi1,pSN1)-3a/(CBi1,pRN1)-3a and (ABi2,pSN2)-3b/(CBi2,pRN2)-3b isomers in the crystal of 3 (only atoms directly connected to the metal centres and carbons of the chelate rings are shown). | ||
In the crystals of the bromides 4 and 5 there are no interactions between heavy atoms. Chains of either (ABi,pSN)-4 or (CBi,pRN)-4 (Fig. S26, ESI†) isomers are formed through Br⋯H and C–H⋯π (Arcentroid) contacts. Alternating such parallel chains which develop along the b axis are connected through Br⋯H interactions [Br(1)⋯H(14b′′) 2.99 Å; cf. the sum of the corresponding van der Waals radii, ∑rvdW(Br,H) 3.15 Å (ref. 35)] to give layers (Fig. S28, ESI†) which stack into a three-dimensional arrangement based on C–Haryl⋯π (Arcentroid) interactions [i.e. H⋯Arcentroid contacts shorter than 3.1 Å and the angle γ between the normal to the aromatic ring and the line through the H atom and Arcentroid smaller than 30°:46 C(3)–H(3)⋯Arcentroid{C(10d)–C(15d)} 2.78 Å, γ = 13.9°] (Fig. S30, ESI†).
No C–H⋯π (Arcentroid) or π–π interactions are observed in the crystal of 5. However, several weak intermolecular Br⋯H interactions result in a 3D architecture based on parallel, alternating chain polymers built from (ABi,pRN)-5 and (CBi,pSN)-5 isomers, respectively (Fig. S34–40, ESI†).
Solution behaviour
All compounds 1–5 were investigated by multinuclear NMR spectroscopy in solution. The assignment of the resonance signals in the 1H and 13C NMR spectra was based on the H–H (COSY) and H–C (HSQC, HMBC) correlation spectra. The numbering scheme used to designate the 1H and 13C NMR resonance signals is presented in Table 2. The numbering of the hydrogen atoms follows that of the carbon atom to which they are attached.| Compound | L | RX | RY | |
|---|---|---|---|---|
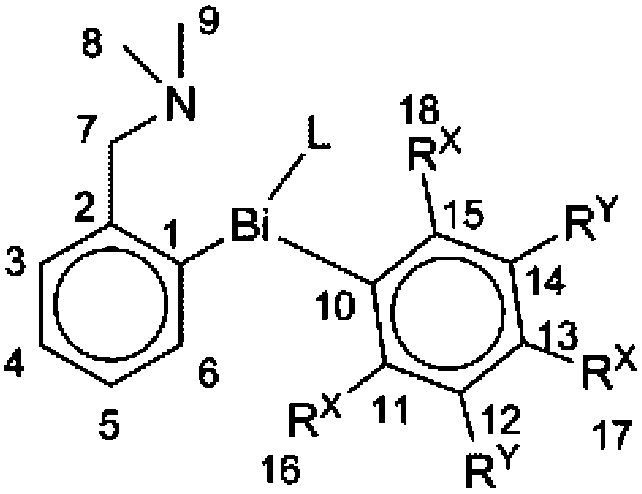
|
1 | C6F5 | F | F |
| 2 | 2,4,6-(C6F5)3C6H2 | C6F5 | H | |
| 3 | Br | F | F | |
| 4 | Br | CH3 | H | |
| 5 | Br | H | H |
The room temperature 1H NMR spectra of 1 and 2 in solution exhibit two sharp singlet resonances for the NMe2 and CH2 protons, which is consistent either with the absence of intramolecular N→Bi coordination or a fast coordination–dissociation process between nitrogen and bismuth, therefore yielding averaged NMR resonances. The most downfield shifted doublet resonances correspond to the aromatic protons H6 (δ 8.34 ppm for 1 and 7.69 ppm for 2). The 1H NMR spectrum of 1 at −60 °C (in CDCl3) exhibited sharp resonances for the methyl and methylene protons similar to those observed in the spectrum measured at room temperature, which is consistent with a similar dynamic behaviour of the compound even at this low temperature.
In the case of the bromides 3–5 the 1H NMR spectra recorded at room temperature in CDCl3 show two singlet signals for the dimethylamino group and an AB spin system for the methylene protons, indicating that in solution the pendant arm is coordinated to the chiral bismuth centre through the nitrogen atom. These spectral features have been reported before for the phenyl-substituted compound 5 and related bismuthines.27 The resonance of the H6 proton is further shifted downfield in the series of compounds 3–5, i.e. δ 9.20 ppm (3) > 9.05 ppm (4) > 9.03 ppm (5). For compound 4 two singlet resonances for the methyl protons from the ortho and para positions of the mesityl moiety and one singlet resonance for the aromatic protons of the mesityl group were observed, consistent with a free rotation of the mesityl group around the Bi–C bond.
The dynamic behaviour of the bromides 3–5 was studied by 1H NMR experiments at higher temperature. The resonance signals corresponding to the protons of the methyl groups of 3 reached coalescence at 30 °C in C6D6 (ΔG‡ = 16.5 kcal mol−1). The AB system characteristic for the diastereotopic methylene protons broadened significantly at 70 °C but did not coalesce. For compound 4, coalescence of the NMe2 proton resonances was observed at 48 °C (ΔG‡ = 16.6 kcal mol−1) in the 1H NMR spectrum (in C6D6), while the AB system corresponding to the methylene protons remained unchanged even at 60 °C. The coalescence temperatures of the NMe2 proton resonances of 5 and of the related chloride [2-(Me2NCH2)C6H4](Ph)BiCl were determined by Suzuki et al. to be 65 °C and 70 °C, respectively, in toluene-d8.27 We observed for 5 the coalescence of the NMe2 proton resonances at 62 °C (ΔG‡ = 16.5 kcal mol−1) in C6D6. The behaviour of the 1H NMR resonances of the NMe2 groups in 3, 4 and 5 indicate that the dissociation of the N→Bi interaction takes place followed by a vertex inversion at the N atom and subsequent restoration of the N→Bi coordinative bond. However, an inversion at the Bi centre (indicated by the coalescence of the methylene proton resonances) requires more energy to take place.
The 13C NMR spectra of 1 and 2 exhibit two sharp singlet resonances, i.e. one for the NMe2 group and one for the methylene carbon. The ipso carbon atoms of the C6F5 groups of 1 give rise to a broad triplet at δ 125.40 ppm. The other 13C resonances of the C6F5 groups are observed as doublets of multiplets with 1JFC coupling constants of ca. 250 Hz. The fine splitting of these signals caused by further couplings with fluorine nuclei at two, three, and four bonds distance considerably diminish their relative intensity. A similar pattern was observed in the 13C NMR spectrum of 3 for the resonance signals corresponding to the C6F5 group attached to the bismuth atom. In the 13C NMR spectrum of 2, two different sets of resonance signals can be distinguished which correspond to the non-equivalent pentafluorophenyl groups belonging to the 2,4,6-(C6F5)3C6H2 substituent (i.e. two C6F5 groups in positions 2 and 6, and one C6F5 group in position 4). The 13C NMR spectrum of compound 4 exhibits in the aliphatic region two singlet resonances (δ 21.37 ppm for p-CH3 and 27.32 ppm for o-CH3) for the carbon atoms of the ortho and para methyl groups from the mesityl moiety. Two sharp singlet resonances corresponding to the NMe2 groups and one sharp signal for the CH2 group were assigned to the pendant arm of the [2-(Me2NCH2)C6H4] moiety. The same pattern was observed for the [2-(Me2NCH2)C6H4] group in the 13C NMR spectra of 3 and 5.
In the 19F NMR spectrum of 1, three resonances for the two equivalent C6F5 groups bound to bismuth were observed at δ −118.14, −153.82, and −160.45 ppm for the ortho, para and meta fluorine atoms, respectively. The three resonances corresponding to the ortho, meta and para fluorine atoms of 3 were observed at chemical shifts close to those of 1.
The 19F NMR spectrum of 2 exhibits three resonance signals for the ortho fluorine atoms and three resonances for the meta fluorine atoms of the C6F5 moieties. This indicates that the rotation of the C6F5 groups in positions 2 and 6 is hindered, and thus one half of a C6F5 group becomes non-equivalent with the other half. The resonance signals corresponding to the para fluorine atoms of all C6F5 groups overlap at δ −153.2 ppm.
Experimental
General measurements and analysis instrumentation
The melting points are not corrected. The 1H, 13C, and 19F NMR spectra were recorded at room temperature on Bruker Avance III 400 or Bruker Avance III 600 instruments. Low-temperature NMR spectra were recorded on the Bruker Avance III 400 apparatus. The 1H chemical shifts are reported in δ units (ppm) relative to the residual peak of the deuterated solvent (CHCl3, 7.26 ppm; C6D6, 7.16 ppm). The 13C chemical shifts are reported in δ units (ppm) relative to the peak of the deuterated solvent (CDCl3, 77.16 ppm; C6D6, 128.06 ppm).47 The 1H and 13C resonances were assigned using H–H and H–C correlation NMR experiments (COSY, HSQC, HMBC). The 19F chemical shifts are quoted in δ units (ppm) relative to CFCl3 as the external standard. The NMR spectra were processed using the MestReNova software.48 Free energy of activation values were calculated using the Gutowsky–Holm equations: Kc [s−1] = πΔν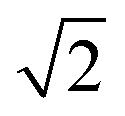 and ΔG‡ [kcal mol−1] = 4.58Tc10−3 (10.32 + logTc/Kc).49 The EI mass spectra were recorded on a Finnigan MAT 8200 instrument, while HRMS APCI(+) spectra were recorded on a Thermo Scientific Orbitrap XL mass spectrometer equipped with a standard ESI/APCI source. Data analysis and calculations of the theoretical isotopic patterns were carried out with the Xcalibur software package.50
and ΔG‡ [kcal mol−1] = 4.58Tc10−3 (10.32 + logTc/Kc).49 The EI mass spectra were recorded on a Finnigan MAT 8200 instrument, while HRMS APCI(+) spectra were recorded on a Thermo Scientific Orbitrap XL mass spectrometer equipped with a standard ESI/APCI source. Data analysis and calculations of the theoretical isotopic patterns were carried out with the Xcalibur software package.50
Crystal structure determination
Single crystals were obtained at −28 °C from concentrated solutions in hexane (for 1) or diethyl ether (for 3). Very slow evaporation of heptane from a concentrated solution of 2 gave single crystals of the compound, while slow evaporation of a CHCl3 solution gave crystals of 4. Suitable crystals of 5 were obtained by hexane diffusion into a solution of the compound in toluene (toluene–hexane: 1/5, v/v). The details of the crystal structure determination and refinement for compounds 1–5 are given in Table S1 (see ESI†). Data were collected on Siemens P4 (1, 3, and 4) and Bruker SMART APEX (2 and 5) diffractometers, using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). For this purpose the crystals were attached with Kel-F oil (1, 3, 4) to a glass fiber or with Paratone oil (2, 5) to a cryo-loop. The crystals of 1, 3, and 4 were cooled under a nitrogen stream, while data for 2 and 5 was collected at room temperature. The structures were refined with anisotropic thermal parameters. All hydrogen atoms were refined with a riding model and a mutual isotropic thermal parameter. For structure solving and refinement the software package SHELX-97 and SHELXL-2014 were used.51,52 The drawings were created with the Diamond program.53 The CCDC reference numbers are 833208 (1), 1430429 (2), 833207 (3), 833209 (4) and 833206 (5).Materials and methods
All reactions were carried out under argon using standard Schlenk techniques. Solvents were dried by standard procedures and were freshly distilled under argon prior to use. Bismuth(III) trichloride, N,N-dimethylbenzylamine, bromopentafluorobenzene, 2-bromo-1,3,5-trimethylbenzene and butyllithium (15% in n-hexane) were purchased from commercial suppliers and used without further purification. The other starting materials were prepared according to literature methods: [2-(Me2NCH2)C6H4]Li,54 [2-(Me2NCH2)C6H4]BiBr2,55 2,4,6-(C6F5)3C6H2Br.56 The concentration of the organomagnesium derivatives used for the synthesis of 1 and 3–5 was determined by iodimetric titration as described by Krasovskiy and Knochel.57:0.03 as calculated from the integral ratio in the 19F NMR spectrum) and another minor unidentified impurity. Recrystallization by diffusion of hexane into a solution of 3 in CH2Cl2 did not improve significantly the purity of 3. 1H NMR (CDCl3, 400.13 MHz, r.t.): δ 2.52 (s, 3H, H-8), 2.58 (s, 3H, H-9), AB spin system with A at 3.79 ppm and B at 4.19 ppm (2H, 2JHH = 14.2 Hz, H-7), 7.50 (ddd, 1H, 3JHH = 7.5 Hz, 4JHH = 1.3 Hz, H-4), 7.60–7.67 (m, 2H, H-3 and H-5), 9.20 (d, 1H, 3JHH = 7.6 Hz, H-6). 1H NMR (C6D6, 400.13 MHz, r.t.): δ 1.53 (s, 3H, H-8 or H-9), 1.54 (s, 3H, H-9 or H-8), AB spin system with A at 2.90 ppm and B at 3.12 ppm (2H, 2JHH = 14.2 Hz, H-7), 7.10–7.21 (m, 2H, resonance signals for H-3 and H-4 overlapped by the residual C6D5H resonance signal), 7.39 (ddd, 1H, 3JHH = 7.4 Hz, 4JHH = 1.4 Hz, H-5), 9.65 (d, 1H, 3JHH = 7.5 Hz, H-6). 1H NMR (C6D6, 400.13 MHz, 30 °C): δ 1.55 (s, br, 6H, H-8 and H-9), AB spin system with A at 2.92 ppm and B at 3.13 ppm (2H, 2JHH = 14.2 Hz, H-7), 7.10–7.21 (m, 2H, resonance signals for H-3 and H-4 overlapped by the residual C6D5H resonance signal), 7.39 (ddd, 1H, 3JHH = 7.4 Hz, 4JHH = 1.4 Hz, H-5), 9.64 (d, 1H, 3JHH = 7.6 Hz, H-6). 13C{1H} NMR (CDCl3, 100.61 MHz, r.t.): δ 45.60 (s, C-9), 46.98 (s, C-8), 69.84 (s, C-7), 129.00 (s, C-3), 129.14 (s, C-4), 131.56 (s, C-5), 134.94 (t, br, 2JFC = 43 Hz, C-10) 138.39 (dm, 1JFC = 257 Hz, C6F5), 142.13 (dm, 1JFC = 254 Hz, C6F5), 142.40 (s, C-6), 147.68 (dm, 1JFC = 238 Hz, C6F5), 148.00 (s, C-2), 174.59 (s, C-1). 19F{1H} NMR (CDCl3, 376.46 MHz, r.t.): δ (−116.61)–(−116.94) (m, 2F, ortho-F, C6F5), −150.87 (t, 1F, 3JFF = 19.8 Hz, para-F, C6F5), (−158.00)–(−158.44) (m, 2F, meta-F, C6F5). MS (EI, 70 eV, 200 °C): m/z, (relative intensity, %) 589 (0.2) [M+], 510 (100) [M+ − Br], 422 (30) [M+ − C6F5].
Method B. A suspension of [2-(Me2NCH2)C6H4]Li (1 g, 7.09 mmol) in THF (50 mL) was added dropwise to a suspension of PhBiBr2 (3.16 g, 7.09 mmol) in THF (25 mL), at −78 °C. The reaction mixture was stirred at this temperature for 2 h, then allowed to warm to room temperature and stirred for another 72 h. The solvent was removed under vacuum and the remaining solid was treated with hot toluene. The suspension was filtered and the solvent removed under vacuum to give 5 as a white solid (1.20 g, 34%), m.p. 158–160 °C (lit.27 163–165 °C). 1H NMR (CDCl3, 400.13 MHz, r.t.): δ 2.30 (s, 3H, H-8), 2.54 (s, 3H, H-9), AB spin system with A at 3.58 ppm and B at 3.88 ppm (2H, 2JHH = 14.0 Hz, H-7), 7.33 (tt, 1H, 3JHH = 7.0 Hz, 4JHH = 3.0 Hz, H-13), 7.45–7.56 (m, 4H, H-3, H-4, H-12 and H-14), 7.59 (ddd, 1H, 3JHH = 7.0 Hz, 4JHH = 3.0 Hz, H-5), 8.10 (dd, 2H, 3JHH = 6.0 Hz, 4JHH = 3.0 Hz, H-11 and H-15), 9.03 (d, 1H, 3JHH = 7.2 Hz, H-6). 1H NMR (C6D6, 400.13 MHz, r.t.): δ 1.54 (s, 3H, H-8), 1.68 (s, 3H, H-9), AB spin system with A at 3.03 ppm and B at 2.96 ppm (2H, 2JHH = 14.0 Hz, H-7), 7.05 (t, 1H, 3JHH = 7.5 Hz, H-13), 7.10 (d, 1H, 3JHH = 7.5 Hz, H-3), 7.18–7.26 (m, 3H, H-4, H-12 and H-14), 7.41 (dd, 1H, 3JHH = 7.4 Hz, H-5), 8.11 (d, 2H, 3JHH = 7.4 Hz, H-11 and H-15), 9.57 (d, 1H, 3JHH = 7.4 Hz, H-6). 1H NMR (C6D6, 400.13 MHz, 62 °C): δ 1.66 (s, br, 6H, H-8 and H-9), 3.07 (br, 2H, H-7), 7.06 (t, 1H, 3JHH = 7.4 Hz, H-13), 7.11 (d, 1H, 3JHH = 7.5 Hz, H-3), 7.18–7.26 (m, 3H, H-4, H-12 and H-14), 7.41 (ddd, 1H, 3JHH = 7.4 Hz, 4JHH = 1.3 Hz, H-5), 8.07 (d, 2H, 3JHH = 6.4 Hz, H-11 and H-15), 9.49 (d, 1H, 3JHH = 7.5 Hz, H-6). 13C{1H} NMR (CDCl3, 100.61 MHz, r.t.): δ 45.85 (s, C-9), 46.63 (s, C-8), 69.18 (s, C-7), 128.56 (s, C-4), 128.81 (s, C-13), 129.23 (s, C-3), 131.29 (s, C-5), 131.49 (s, C-12 and C-14), 137.58 (s, C11 and C15), 140.72 (s, C-6), 147.51 (s, C-2), 174.16 (s, C-10), 177.93 (s, C-1). MS (EI, 70 eV, 200 °C): m/z, (relative intensity, %) 499 (1) [M+], 422 (90) [M+ − C6H5], 420 (100) [M+ − Br], 343 (54) [RBi]+, 286 (5) [PhBi+] [R = 2-(Me2NCH2)C6H4]. HRMS (APCI+): [M+ − Br] calcd for C15H17BiN 420.11749. Found: 420.11593.
Conclusions
In contrast to 1, for which the nitrogen of the pendant arm is coordinated intramolecularly to the metal centre in the solid state, the bulkiness of the polyfluorinated substituents in the molecule of 2 prevents the formation of a similar coordinative interaction in the solid state or in solution. Variable temperature 1H NMR spectra of the heteroleptic triorganobismuthine 1 indicate either a lack of coordination of nitrogen to bismuth or the existence of a fast coordination–dissociation process in solution even at −60 °C. This behaviour may be caused by the fact that the two C6F5 substituents do not increase effectively the Lewis acidity of the metal centre.The chiral bromides 3–5 exhibit in the solid state a strong intramolecular N→Bi coordination established trans to the Bi–Br bond. The N–Bi bond distances in 3–5, although smaller than in 1, are similar, indicating that the second organic substituent (C6F5, Ph and Mes, respectively) does not influence considerably the length of the coordinative bond. The room temperature 1H NMR spectra of 3–5 exhibit an AB system for methylene protons and two resonances for the NMe2 groups, consistent with the coordination of nitrogen to bismuth in solution.
The heteroleptic triorganobismuthine 1 and the bromides 3–5 crystallize as racemates, i.e. 1:1 mixtures of (ABi,pSN) and (CBi,pRN) isomers for 1, 3 and 4, and (ABi,pRN) and (CBi,pSN) isomers for 5. Chain polymeric and layered structures, as well as 3D supramolecular architectures, based on different type of interactions [e.g. Bi⋯F, Bi⋯Br, F⋯H, Br⋯H, C–H⋯π (Arcentroid), C–F⋯π (Arcentroid) or π⋯π], are evidenced in the solid state.
Acknowledgements
Financial support from the National Research Council of Romania (CNCS, Research Project No. PN-II-ID-PCE-2011-3-0933) is greatly appreciated. We also thank Deutsche Forschungsgemeinschaft and DAAD for financial support and Universität Bremen for providing research facilities during the research stays of M.G.N. and C.S. The support provided by Alexandra Pop and Richard A. Varga (National Center for X-Ray Diffraction, Babeş-Bolyai University, Cluj-Napoca, Romania) for the solid state structure determinations is highly acknowledged.References
- Organobismuth Chemistry, ed. H. Suzuki and Y. Matano, Elsevier Science B.V., Amsterdam, 2001 Search PubMed.
- Bismuth-Mediated Organic Reactions, in Topics in Current Chemistry, ed. T. Ollevier, Springer, Berlin, Heidelberg, 2012, vol. 312 Search PubMed.
- C. I. Raţ, C. Silvestru and H. J. Breunig, Coord. Chem. Rev., 2013, 257, 818–879 CrossRef.
- C. Silvestru, H. J. Breunig and H. Althaus, Chem. Rev., 1999, 99, 3277–3328 CrossRef CAS PubMed.
- H. Suzuki, T. Ikegami and Y. Matano, Synthesis, 1997, 249–267 CrossRef CAS.
- M. Mehring, Coord. Chem. Rev., 2007, 251, 974–1006 CrossRef CAS.
- E. J. Fernández, A. Laguna, J. M. López-de-Luzuriaga, M. Monge, M. Nema, M. E. Olmos, J. Pérez and C. Silvestru, Chem. Commun., 2007, 571–573 RSC.
- H. J. Breunig, E. Lork, C. I. Raţ and R. P. Wagner, J. Organomet. Chem., 2007, 692, 3430–3434 CrossRef CAS.
- H. J. Breunig, T. Borrmann, E. Lork, O. Moldovan, C. I. Raţ and R. P. Wagner, J. Organomet. Chem., 2009, 694, 427–432 CrossRef CAS.
- T.-P. Lin, I.-S. Ke and F. P. Gabbaï, Angew. Chem., Int. Ed., 2012, 51, 4985–4988 CrossRef CAS PubMed.
- C. Tschersich, C. Limberg, S. Roggan, C. Herwig, N. Ernsting, S. Kovalenko and S. Mebs, Angew. Chem., Int. Ed., 2012, 51, 4989–4992 CrossRef CAS PubMed.
- I.-S. Ke and F. P. Gabbai, Aust. J. Chem., 2013, 66, 1281–1287 CAS.
- I.-S. Ke and F. P. Gabbai, Inorg. Chem., 2013, 52, 7145–7151 CrossRef CAS PubMed.
- T. Ollevier, Org. Biomol. Chem., 2013, 11, 2740–2755 CAS.
- H. B. Kagan and M. Sasaki, in The Chemistry of Organophosphorus Compounds, ed. F. R. Hartley, John Wiley & Sons, Chichester, 1990, vol. 1, pp. 51–102 Search PubMed.
- S. B. Wild, in The Chemistry of Organic Arsenic, Antimony and Bismuth Compounds, ed. S. Patai, John Wiley & Sons, Chichester, 1994, pp. 89–152 Search PubMed.
- S. Okajima, S. Yasuike, N. Kakusawa, A. Osada, K. Yamaguchi, H. Seki and J. Kurita, J. Organomet. Chem., 2002, 656, 234–242 CrossRef CAS.
- D. Copolovici, F. Isaia, H. J. Breunig, C. I. Raţ and C. Silvestru, RSC Adv., 2014, 4, 26569–26576 RSC.
- S. L. Benjamin and G. Reid, Coord. Chem. Rev., 2015, 297–298, 168–180 CrossRef CAS.
- W. Levason and G. Reid, Coord. Chem. Rev., 2006, 250, 2565–2594 CrossRef CAS.
- Y. Matano, T. Miyamatsu and H. Suzuki, Organometallics, 1996, 15, 1951–1953 CrossRef CAS.
- Y. Matano, S. A. Begum, T. Miyamatsu and H. Suzuki, Organometallics, 1999, 18, 5668–5681 CrossRef CAS.
- P. Bras, H. Herwijer and J. Wolters, J. Organomet. Chem., 1981, 212, C7–C9 CrossRef CAS.
- P. Bras, A. Van Der Gen and J. Wolters, J. Organomet. Chem., 1983, 256, C1–C4 CrossRef CAS.
- H. Suzuki and T. Murafuji, J. Chem. Soc., Chem. Commun., 1992, 1143–1144 RSC.
- H. J. Breunig, I. Ghesner, M. E. Ghesner and E. Lork, Inorg. Chem., 2003, 42, 1751–1757 CrossRef CAS PubMed.
- H. Suzuki, T. Murafuji, Y. Matano and N. Azuma, J. Chem. Soc., Perkin Trans. 1, 1993, 2969–2973 RSC.
- H. Suzuki, T. Murafuji and N. Azuma, J. Chem. Soc., Perkin Trans. 1, 1993, 1169–1175 RSC.
- T. Murafuji, T. Mutoh, K. Satoh, K. Tsunenari, N. Azuma and H. Suzuki, Organometallics, 1995, 14, 3848–3854 CrossRef CAS.
- T. Murafuji, I. Makabe, K. Nishio, Y. Sugihara, Y. Mikata and S. Yano, J. Organomet. Chem., 2000, 611, 100–105 CrossRef CAS.
- T. Murafuji, K. Satoh, Y. Sugihara and N. Azuma, Organometallics, 1998, 17, 1711–1715 CrossRef CAS.
- Y. Matano, H. Kurata, T. Murafuji, N. Azuma and H. Suzuki, Organometallics, 1998, 17, 4049–4059 CrossRef CAS.
- I. Urbanová, R. Jambor, A. Růžička, R. Jirásko and L. Dostál, Dalton Trans., 2014, 43, 505–512 RSC.
- L. Balázs, O. Stânga, H. J. Breunig and C. Silvestru, Dalton Trans., 2003, 2237–2242 RSC.
- J. Emsley, Die Elemente, Walter de Gruyter, Berlin, 1994 Search PubMed.
- Y. Yamamoto, X. Chen and K.-y. Akiba, J. Am. Chem. Soc., 1992, 114, 7906–7907 CrossRef CAS.
- Chemistry of Hypervalent Compounds, ed. K.-y. Akiba, Wiley-VCH, New York, 1999 Search PubMed.
- The N-X-L nomenclature system has been described previously: N valence shell electrons about a central atom X with L ligands; C. W. Perkins, J. C. Martin, A. J. Arduengo III, W. Lau, A. Alegria and K. Kochi, J. Am. Chem. Soc., 1980, 102, 7753–7759 CrossRef CAS.
- D. M. Hawley and G. Ferguson, J. Chem. Soc. A, 1968, 2059–2063 RSC.
- D. Copolovici, V. R. Bojan, C. I. Raţ, A. Silvestru, H. J. Breunig and C. Silvestru, Dalton Trans., 2010, 39, 6410–6418 RSC.
- Nomenclature of Inorganic Chemistry–IUPAC Recommendations 2005, ed. N. G. Connelly, T. Damhus, R. M. Hartshorn and A. T. Hutton, RSC Publishing, Cambridge, 2005 Search PubMed.
- Nomenclature of Organic Chemistry, Sections A, B, C, D, E, F and H, ed. J. Rigaudy and S. P. Klesney, Pergamon Press, Oxford, 1979 Search PubMed.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC.
- H. J. Breunig, L. Königsmann, E. Lork, M. Nema, N. Philipp, C. Silvestru, A. Soran, R. A. Varga and R. Wagner, Dalton Trans., 2008, 1831–1842 RSC.
- M. Kawahata, S. Yasuike, I. Kinebuchi, K. Yamaguchi and J. Kurita, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, m25 CAS.
- M. Nishio, Phys. Chem. Chem. Phys., 2011, 13, 13873–13900 RSC.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- MestReNova, Mestrelab Research S.L. Feliciano Barrera 9B, Bajo, 15706 Santiago de Compostela, Spain., 2015.
- H. Friebolin, Basic One- and Two-Dimensional NMR Spectroscopy, Wiley-VCH Verlag GmbH, 3rd edn, 1998 Search PubMed.
- Qual Browser Thermo Xcalibur, Thermo Fisher Scientific Inc., Waltham, MA, 02454, 2011 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- K. Brandenburg, DIAMOND-Visual Crystal Structure Information System, Crystal Impact, Postfach 1251, D-53002 Bonn, Germany, 2015 Search PubMed.
- A. Meller, H. Hoppe, W. Maringgele, A. Haase and M. Noltemeyer, Organometallics, 1998, 17, 123–124 CrossRef CAS.
- H. Breunig, M. Nema, C. Silvestru, A. Soran and R. Varga, Z. Anorg. Allg. Chem., 2010, 636, 2378–2386 CrossRef CAS.
- M. Olaru, J. Beckmann and C. I. Rat, Organometallics, 2014, 33, 3012–3020 CrossRef CAS.
- A. Krasovskiy and P. Knochel, Synthesis, 2006, 890–891 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra; X-ray crystallographic data in CIF format for 1–5; figures representing the optical isomers as well as the supramolecular architectures in the crystals of these compounds. CCDC 833206–833209 and 1430429. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt05074j |
| ‡ These co-authors have contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2016 |